DOI:
10.1039/D0RA04306K
(Paper)
RSC Adv., 2020,
10, 27183-27193
Synthesis of bifunctional nanocatalyst from waste palm kernel shell and its application for biodiesel production
Received
14th May 2020
, Accepted 2nd July 2020
First published on 21st July 2020
Abstract
The potential of bifunctional nanocatalysts obtained from waste palm kernel shell (PKS) was investigated for one-step transesterification–esterification under mild conditions. State-of-the-art characterization illustrated that the synthesized catalyst has high stability through the thermal test, high BET surface area of 438.08 m2 g−1, pore volume of 0.367 cm3 g−1 and pore width of 3.8 nm. The high amount of basicity (8.866 mmol g−1) and acidity (27.016 mmol g−1) promoted the successfulness of simultaneous transesterification–esterification. The investigation revealed that the combination of potassium and copper on activated carbon surface showed good catalytic activity by giving 95.0% FAME yield and 97.3% FFA conversion at a relatively mild condition of 5 wt% catalyst loading, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol to oil molar ratio at 80 °C for 4 hours with FAME yield > 80% after 5 reaction cycles. Characterization of the spent catalyst showed that the amount of basicity was reduced to 3.106 mmol g−1, which validated the reduction of the catalytic performance. The usage of waste material was successfully discovered in producing an effective bifunctional catalyst for biodiesel production from waste cooking oil (WCO) and has high potential for commercialization in the future.
:1 methanol to oil molar ratio at 80 °C for 4 hours with FAME yield > 80% after 5 reaction cycles. Characterization of the spent catalyst showed that the amount of basicity was reduced to 3.106 mmol g−1, which validated the reduction of the catalytic performance. The usage of waste material was successfully discovered in producing an effective bifunctional catalyst for biodiesel production from waste cooking oil (WCO) and has high potential for commercialization in the future.
Introduction
Finding alternative fuels to substitute fossil fuels has encouraged Malaysia to conduct research on biodiesel as an alternative energy source to fossil fuels through the National Biomass Strategy (NBS) 2020 launched by the Malaysian government. Nowadays, biodiesel is well-known as a renewable energy source, producing greener and clean-burning fuel. The biodiesel was formally produced from sunflower oil,1 palm oil,2 olive oil,3 Jatropha curcas oil4 and rapeseed oil.5 The utilization of edible oils as feedstock creates an unhealthy competition between food and biodiesel crops, which in extent will increase the price. The discovery of new potential feedstock resulting in the use of waste cooking oil (WCO) is the best candidate for biodiesel production. The non-value by-product from daily cooking and frying activities reported that 15 million tons have been disposed annually worldwide.6 Most of the time, the WCO is drained off into the drainage system which will cause water pollution. This situation had caused difficulties for authorities in waste management, especially when dealing with WCO. Thus, utilizing WCO for biodiesel production in extent will help to reduce the cost of waste management.
The waste cooking oil (WCO) generally consists of high triglycerides content (85%), free fatty acids (FFA, 15%), moisture and solid residue. Previously, the biodiesel production from WCO was catalyzed with a homogeneous catalyst by two-step esterification–transesterification, where esterification is to reduce the FFA prior to transesterification reaction. Thus, the two-step reaction will double the production cost and time and rise up the cost as it needs the addition of a neutralization step to the treatment system before it is safe for the environment.7
To overcome this shortcoming, varies of bifunctional heterogeneous catalysts were discovered. Previously, different types of bifunctional catalysts were synthesized to produce biodiesel from WCO, such as Mo–Mn/γ-Al2O3-15 wt% MgO,8 zirconia supported Fe2O3/MnO–SO4 (ref. 9) and a combination of molybdenum–zirconia oxide supported CaO10 which have successfully synthesized and produced more than 90% biodiesel yield. Besides, the commercially available catalyst supports, such as alumina and silica are highly expensive, thus limiting a wider usage.
Alternatively, the discovery of using waste from palm oil industries can produce a high value activated carbon (AC) as the catalyst support for biodiesel production. Activated carbon from waste palm oil, especially the palm kernel shell (PKS) was widely studied by researchers as catalyst support as it has high caloric value, low sulfur content, low ash content, no species variation, improved shelf-life and excellent year-round availability.11–13 The Malaysian Palm Oil Board (MPOB) reported that each ton of fresh fruit bunch (FFB) in palm oil milling will generate about 5–7% PKS, 21–22% empty fruit bunch (EFB) and 12–16% mesocarp fiber (MF). In 2015, estimated 4.56 million tons of PKS, 21.63 million tons of EFB and 15.73 million tons of MF were generated as by-products as Malaysian palm oil mills processed about 98.34 million tons of FFB.14 Interestingly, activated carbon, especially from PKS could produce a form of amorphous carbon with highly porous carbon-rich material.15
This work aimed to synthesize an efficient heterogeneous bifunctional catalyst from PKS for one-pot transesterification–esterification from WCO as feedstock. Four basic agents were chosen for screening and copper nitrate was the main source of acidic agent for this study. The optimized catalyst was further fully characterized for its physical characteristics through Brunauer–Emmett–Teller (BET) surface area, crystallinity by X-ray diffraction (XRD), amount of basicity and acidity by thermal program desorption (TPD-CO2 and NH3), functional group determination through Fourier transform infrared (FTIR), surface morphology observation by field emission scanning electron microscope (FESEM), elemental distribution using energy-dispersive X-ray (EDX) and studied in composition via thermal gravimetric analysis (TGA). The catalytic performance was evaluated through its efficiency in producing the highest biodiesel product by conventional reflux method. The physicochemical properties of the chosen catalyst were analyzed and the effect of reaction parameters such as catalyst loading, methanol to oil molar ratio, temperature and reaction duration on biodiesel yield and acid value were studied. The reusability of the catalyst was also evaluated.
Materials and methods
Chemicals and materials
Waste cooking oil (WCO) was collected from food stalls located in Section 4, Bandar Baru Bangi, Selangor, Malaysia. The chemicals such as potassium carbonate (K2CO3; 99.5%), sodium hydroxide (NaOH; 98.0%) and copper nitrate (Cu(NO3)2; 99.5%) were provided by R&M Chemicals (Subang, SGR, MY), while phosphoric acid (H3PO4; 85.0%), potassium phosphate tribasic (K3PO4; 98.0%) and potassium hydroxide (KOH; 86.0%) were purchased from J.T. Barker (St. Phillipsburg, NJ, USA, 85.0%). Methanol (CH3OH) was supplied by Systerm (Shah Alam, SGR, MY, 99.8%). Pure fatty acid methyl ester (FAME) standards such as methyl heptadecanoate, methyl oleate, methyl linoleate, methyl palmitate, methyl myristate and methyl stearate, all with 99.9% purity and n-hexane (99.0%) were purchased from Merck (Darmstadt, DE). All of the chemicals obtained in this work were of analytical grade and used directly as received.
Preparation and pretreatment of AC from PKS
The PKS samples were collected from FELDA Mempaga, Bentong, Pahang, Malaysia. The PKS was cleaned and crushed into powder using a grinder. After that, the PKS powder was soaked in 20% H3PO4, followed by activation in a tubular furnace at 700 °C for 3 h with continuous N2 flow. The produced AC was washed until pH 7 and dried in an oven at 105 °C for 12 h and given name as PKS activated carbon or PKSAC.
Catalyst preparation
The catalysts were synthesized by the wet impregnation method through mixing the desired percentages of KOH, K2CO3, K3PO4, NaOH (as the basic agent) and Cu(NO3)2 (as the acidic agent) to the weight of AC. For screening purposes, the basic and acidic agents were fixed at 30 wt% and 5 wt%, respectively. All the chemicals were dissolved in excess distilled water and stirred vigorously for 6 h at room temperature. The mixture was then dried at 105 °C for 24 h prior to the activation step at 700 °C for 3 h with continuous N2 flow in a tubular horizontal furnace. The prepared catalysts were given names of PKSAC-KOH(30%)CuO(5%), PKSAC-K2CO3(30%)CuO(5%), PKSAC-K3PO4(30%)CuO(5%) and PKSAC-NaOH(30%)CuO(5%). After that, the optimization was done for the selected catalyst by preparing a series of different percentages of the chosen basic and acidic agents by using the same procedure. The synthesized catalyst was named PKSAC-K2CO3(x%)CuO(y%).
Catalyst characterization
The physicochemical properties of the PKSAC and PKSAC derived catalysts were characterized. The physical properties were analyzed by Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) (Micromeritics ASAP-2020, Micromeritics Instrument, Norcross, GA, USA). The evaluations were obtained by performing the N2 adsorption–desorption isotherm at low temperature (liquid N2 temperature is −196 °C). Briefly, 0.5 g sample was degassed at 150 °C for 12 h. The surface area of the sample was calculated based on BET equations, while the pore volume diameter was determined by the amount of adsorbed N2 uptake at 1 atm. Powder X-ray diffraction (XRD) was used to determine and obtain information on the existed phase structure, composition and the degree of crystalline of the sample powder. JCPDS-International Center for Diffraction Data was used to provide the information and determine the specific minerals or elements that exist in the sample. In this work, the crystalline structure of the catalysts was analyzed by an XRD diffractometer (CuKα radiation; λ = 0.154 nm; XRD 6000, Shimadzu, Kyoto, JPN) in the range of 5° to 80°. The amounts of basicity and acidity were measured by using the temperature-programed desorption for carbon dioxide and ammonia (TPD-CO2 & TPD-NH3; TPDRO 1100, Thermo Finnigan, Kyoto, JPN), respectively. The sample underwent a one-hour treatment with respective gas (30 ml min−1) and later set to 900 °C. An additional step was needed for acidity determination where the sample was pretreated with nitrogen gas to remove moisture and later underwent prior treatment with ammonia gas. The amounts of desorbed carbon dioxide and ammonia were recorded by a thermal conductivity detector (TCD). The functional group attached on the surface of the prepared catalyst was recorded by using Fourier Transform Infrared spectrometer (FTIR; Thermo Nicolet 7000, Thermo Fisher Scientific, Waltham, MA, USA) equipped with Attenuated Total Reflection (ATR) method. The FTIR spectrum was scanned in the range of 400 to 4000 cm−1. Field emission scanning electron microscope (FESEM; Novananosem-2300) equipped with the energy dispersive X-ray (EDX) analysis were used to capture the morphological characteristics and to determine the elemental composition of the prepared catalysts, respectively. Lastly, the thermogravimetric analysis (TGA) was performed using the Mettler Toledo TG-50 Mettler instrument (Mettler Toledo, Shah Alam, SGR, MY) to observe the thermal decomposition of PKSAC and optimum catalyst. The sample was heated from room temperature to 1000 °C at a heating rate of 10 °C min−1 with nitrogen flow of 200 ml min−1.
The catalytic activity of the catalyst
Experiments were designed to ascertain the effect of each prepared catalyst type and loading percentages, methanol-to-oil molar ratio, reaction temperature and duration on the percentage of produced biodiesel or namely FAME from waste cooking oil (WCO) by conventional reflux method. The optimization was done as follows: methanol to WCO (MeOH:oil) molar ratio (6:1 to 21:1), catalyst loading (1–8 wt%), reaction temperature (50–100 °C) and reaction duration (1–6 h). Briefly, 10 g of WCO was preheated to the desired temperature before the catalyst and methanol were mixed into the reaction flask. The reaction mixture was allowed to cool and the solid catalyst was separated from the FAME by centrifugation at 5000 rpm for 15 min. The unreacted methanol was removed by using rotary evaporator for 10 min. After that, the FAME layer was purified by washing thrice with hot water using a pear-shaped separating funnel. Afterward, the yield of FAME was analyzed to monitor the efficiency of the reaction.
Fatty acid methyl ester (FAME) analysis
Analysis of FAME was conducted by mixing the product containing FAME with 1 ml 0.05% (w/v) methyl heptadecanoate/hexane solution. The 1 μl of the mixture was then injected to a gas chromatography equipped with a flame ionization detector (GC-FID; Agilent 7890A, BPX 70 column, length: 60 m, ID: 0.25 mm and capillary thickness 0.25 μm) (Trajan Scientific, Ringwood, VIC, AUS). A series of reference standards were prepared at 500 ppm for each standard of methyl palmitate, methyl oleate, methyl myristate, methyl stearate and methyl linoleate. The oven was programed at an initial temperature of 100 °C and then heated up to 250 °C with a heating rate of 10 °C min−1. The temperature of the injector port and detector were maintained at 230 °C and 270 °C, respectively, through the analysis. The calculation of yield percentage was done by using the following eqn (1):| | |
FAME yield (%) = (∑A − Ameh)/Ameh × (Cmeh × Vmeh)/Wt × 100
| (1) |
where, ∑A is the total area for FFA peaks, Ameh is the peak area of the internal standard, Cmeh is the concentration of the methyl heptadecanoate, Vmeh is the volume of the methyl heptadecanoate and Wt is the mass of the produced FAME.
The % free fatty acid (FFA) conversion to FAME was calculated based on the difference of the acid value of the feedstock before and after the reaction according to AOCS 5a-40 standard method by using eqn (2), where AVf stand for the acid value of the feedstock before the reaction, while AVp stands for the acid value of the product containing FAME.
| | |
FFA conversion, % = (AVf − AVp)/AVf × 100%
| (2) |
Catalyst reusability study
Reusability study was performed by employing the optimum reaction condition of 5 wt% of catalyst loading and 12:1 MeOH:oil molar ratio at 80 °C for 4 h. For this test, the spent catalyst used in the first cycle was centrifuged, recovered with methanol and hexane and dried in the oven at 105 °C for 4 h. The dried catalyst was re-calcined at 700 °C for 3 hours before it was used for the next reaction cycle. The spent catalyst was characterized using BET, XRD and TPD-CO2 and NH3.
Results and discussion
Properties of waste cooking oil (WCO)
Properties of the WCO were identified before it could be used for FAME production. The obtained saponification value was 183.96 mg g−1 with an average molecular weight of 915.04 g mol−1. The acid value was 2.058 mg g−1 and the content of FFA was calculated to be 1.029% w/w.
Catalyst characterization for screening
The results for physical characteristics are presented in Table 1. For the PKSAC, the BET surface area is 1411.16 m2 g−1 and the pore volume is 0.961 mm3 g−1. After impregnated with K2CO3 and Cu(NO3)2, the BET surface area and pore volume decreased. This phenomenon occurred as a result of the acidic and basic components filled and incorporated into the available pores provided by the PKSAC.16 Among all of the synthesized catalysts, the PKSAC-NaOH(30%)/CuO(5%) provided the highest BET surface area and pore volume at 589.44 m2 g−1 and 0.3330 mm3 g−1, respectively. All the prepared catalysts had relatively large pore diameter, giving advantages in providing extra accessibility for triglycerides molecules to the inner pore structure network since the typical triglycerides have a diameter of 2.5 nm. Therefore, this may contribute to the catalytic performance during the reaction.17
Table 1 Physicochemical properties of the PKSAC and PKSAC derived catalysts
| Catalyst |
BET surface area (m2 g−1) |
BJH pore volume (mm3 g−1) |
BJH pore diameter (nm) |
Amount of CO2 desorbed (mmol g−1) |
Amount of NH3 desorbed (mmol g−1) |
| PKSAC |
1411.16 |
0.9613 |
3.1 |
3.644 |
15.726 |
| PKSAC-KOH(30%)CuO(5%) |
250.92 |
0.4730 |
3.6 |
4.398 |
23.976 |
| PKSAC-K2CO3(30%)CuO(5%) |
438.08 |
0.3674 |
3.8 |
8.866 |
27.016 |
| PKSAC-K3PO4(30%)CuO(5%) |
104.13 |
0.0700 |
5.25 |
6.081 |
25.323 |
| PKSAC-NaOH(30%)CuO(5%) |
589.44 |
0.3330 |
4.92 |
3.990 |
20.463 |
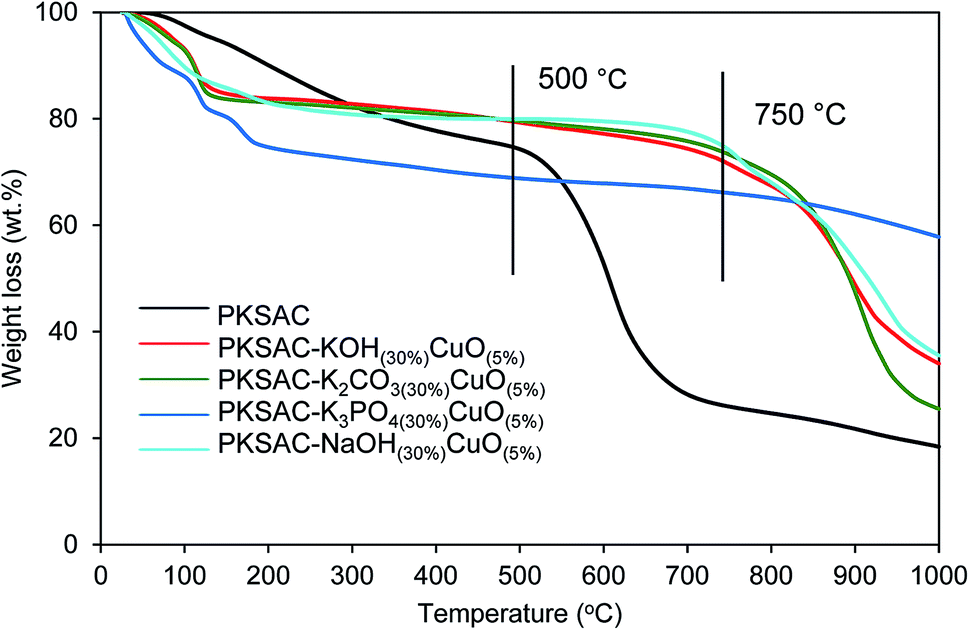
Thermogravimetric analysis (TGA) showed the thermal degradation of the prepared catalyst. The first weight loss ascribed to the loss of moisture adsorbed from the atmosphere during the sample preparation, while the second was because of the decomposition of the catalyst. From Fig. 1, it is shown that all the prepared catalysts were relatively stable up to 750 °C compared to PKSAC which degraded at 500 °C.
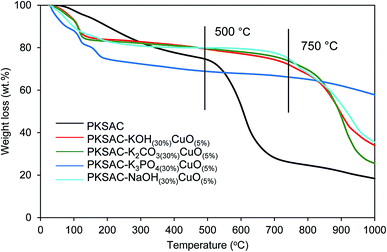 |
| | Fig. 1 TGA profile for PKSAC-KOH(30%)CuO(5%), PKSAC-K2CO3(30%)CuO(5%), PKSAC-K3PO4(30%)CuO(5%) and PKSAC-NaOH(30%)CuO(5%). | |
Unfortunately, even though PKSAC-KOH(30%)CuO(5%) provided relatively good characteristics for physical and stability properties, the amounts of basicity and acidity were also important characteristics to produce high FAME yield. The basicity and acidity from TPD-CO2 and TPD-NH3 results are summarized in Table 1. The results revealed that the amount of CO2 desorbed by the parent catalyst (PKSAC) catalyst indicated very low density for both basicity and acidity, which were 3.644 mmol g−1 and 15.726 mmol g−1, respectively. However, the basicity and acidity were further increased with the introduction of basic and acidic components onto the parent catalyst. The TPD-CO2 and TPD-NH3 results further determined that the amount of CO2 and NH3 uptake was greater for PKSAC-K2CO3(30%)CuO(5%), showing that the amounts of basicity and acidity were the highest on the surface of the catalyst.
All the prepared catalysts were tested on their effectiveness in producing FAME from WCO. For this purpose, the reaction condition was set up as methanol-to-oil molar ratio of 15:1 and 5 wt% catalyst loading at 70 °C for 4 hours. The produced FAME yield percentages were calculated through GC-FID. The results showed that no FAME was produced without the existence of a catalyst. The results also showed that the catalytic performance increased drastically after the addition of active sites onto the parent catalyst. However, the PKSAC-K2CO3(30%)CuO(5%) exhibited better catalytic activity with the highest FAME percentage yield of 84.97% compared to PKSAC-KOH(30%)CuO(5%), PKSAC-K3PO4(30%)CuO(5%) and PKSAC-NaOH(30%)CuO(5%) which provided FAME percentage yields of 76.27%, 77.92% and 77.67%, respectively under identical reaction conditions.
These results indicated that the amounts of basicity and acidity on the catalyst surface could be the most important role in producing FAME at a relatively mild reaction condition. The highest FAME percentage yield can be obtained with the optimum number of active components available on the catalyst surface for one-pot esterification–transesterification reaction suggested by Farooq et al.18 From the above characterization and catalytic testing, PKSAC-K2CO3(30%)CuO(5%) was chosen for further investigation by varying the wt% of K2CO3 with 5 wt% of CuO.
Characterization of the PKSAC-K2CO3(x%)CuO(y%) catalyst
Physical characteristics. The N2 adsorption/desorption isotherm and pore width volume of the synthesized catalysts are illustrated in Fig. 2 and 3, respectively. All the prepared catalysts had hysteresis loop-type shape of N2 physisorption isotherms which can be classified as type IV, a notable characteristic of a type 4 isotherm, with hysteresis loop type H3 in which the pores are aggregates of plate-like particles forming slit-like pores.
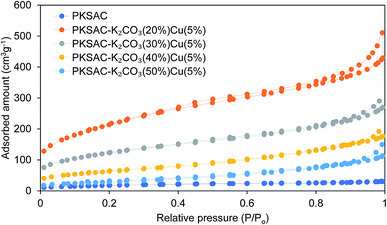 |
| | Fig. 2 Nitrogen adsorption/desorption isotherms of the raw PKS, PKSAC and PKSAC derived catalysts. | |
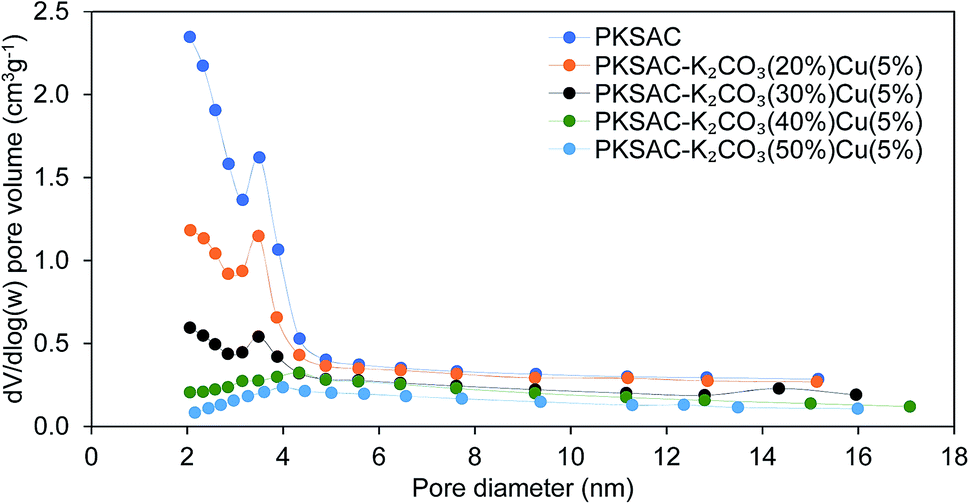
The physical surface characteristics of the PKSAC and PKSAC derived catalysts were measured. The activation of carbon was done to improve the BET surface area by treatment with H3PO4. After activation was completed, the elimination of H3PO4 from the surface of activated carbon allowed immobilization of selected active components during the calcination process. The curve of pore diameter distribution of the raw PKS, PKSAC and PKSAC derived catalysts is presented in Fig. 3. All the samples had a maximum pore diameter at <18 nm, indicating the presence of mesopores. Mesopore volume was estimated from the BJH model at the relative pressure of 0.1–1.0 p/p0 and it was demonstrated that all the catalysts had well-developed mesopore volume. The summary of BET surface area, pore volume and diameter of raw PKS, PKSAC and PKSAC derived catalysts is presented in Table 2. The results presented that the BET surface area and mesopore volume decreased when the PKSAC was modified with varied wt% of K2CO3. Other than that, it was also found that the results of BET surface area mesopores volume decreased from 770.31 m2 g−1 to 104.35 m2 g−1 with the increase of K2CO3 wt% from 20 wt% to 50 wt%, respectively. These results can validate the impregnation of K2CO3 and Cu(NO3)2 onto the parent catalyst, and the decrement was due to the fact that the original empty pores on the PKSAC were partially clogged by the loading of active components.19
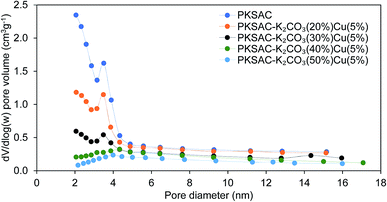 |
| | Fig. 3 Pore diameter distribution of the raw PKS, PKSAC and PKSAC derived catalysts. | |
Table 2 Physicochemical properties of raw PKS, PKSAC and PKSAC derived catalysts
| Catalyst |
BET surface area (m2 g−1) |
BJH pore volume (mm3 g−1) |
BJH pore diameter (nm) |
Amount of CO2 desorbed (mmol g−1) |
Amount of NH3 desorbed (mmol g−1) |
| Raw PKS |
3.57 |
0.0360 |
5.01 |
N.A. |
N.A. |
| PKSAC |
1411.16 |
0.961 |
3.1 |
3.644 |
15.726 |
| PKSAC-K2CO3(20%)CuO(5%) |
770.31 |
0.605 |
3.5 |
1.390 |
20.017 |
| PKSAC-K2CO3(30%)CuO(5%) |
438.08 |
0.367 |
3.8 |
8.866 |
27.016 |
| PKSAC-K2CO3(40%)CuO(5%) |
222.99 |
0.235 |
4.5 |
0.878 |
44.614 |
| PKSAC-K2CO3(50%)CuO(5%) |
104.35 |
0.144 |
4.9 |
0.936 |
19.761 |
X-ray diffraction (XRD)
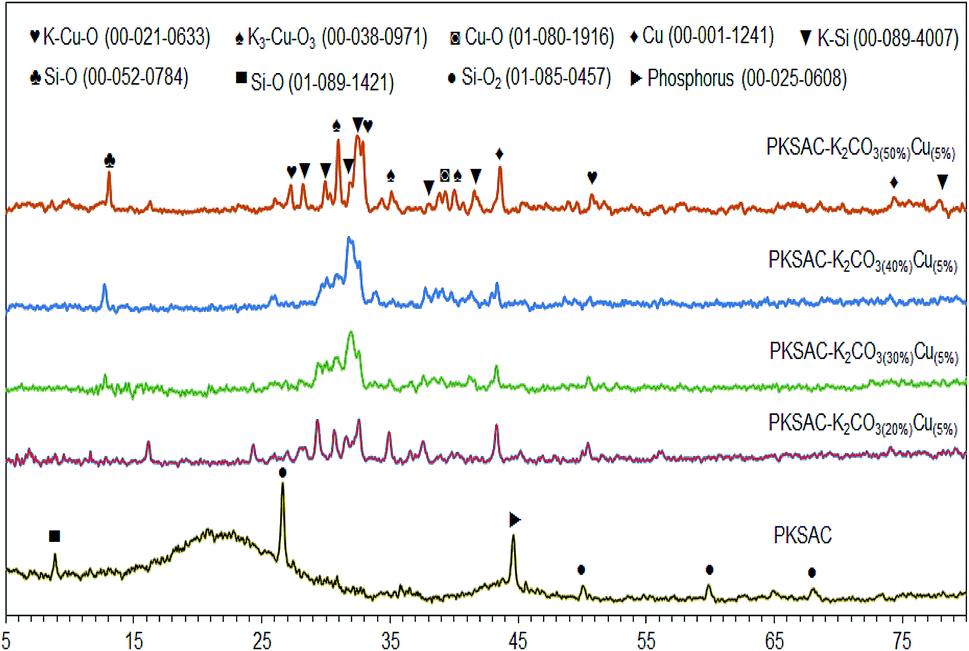
The XRD analysis gives information on the degree of crystallinity of the PKSAC and PKSAC derived catalysts, as presented in Fig. 4. The XRD diffractogram of the PKSAC showed an amorphous structure peak centered at 2θ = 21.0° and additional peaks at 2θ = 26.6°, 50.1°, 59.9° and 68.1° (JCPDS file no. 01-085-1457), which indicated that the carbonaceous materials were amorphous in nature.20 Other than that, there were a zeolite peak at 2θ = 8.8° (JCPDS file no. 01-089-1421) and phosphorus peaks at 2θ = 44.6° (JCPDS file no. 00-025-0608) due to chemical treatment during the preparation of activated carbon. However, the non-crystallite phase and all the peaks at the parent catalyst disappeared after impregnation with both K2CO3 and Cu(NO3)2. The peaks for Cu as cubic phases appeared at 2θ = 43.6° and 74.7° (JCPDS file no. 00-001-1241), while monoclinic Cu–O phase was found at 2θ = 39.1° (JCPDS file no. 01-080-1916). K–Si species was found at 2θ = 28.2°, 29.9°, 32.4°, 39.1°, 41.5° and 77.9° (JCPDS file no. 01-089-4007) and Si–O was found at 2θ = 13.1° (JCPDS file no. 00-052-0784). The successful incorporation of K and Cu species in the catalyst was confirmed with the presence of tetragonal K3–Cu–O3 phases at 2θ = 30.9°, 35.1°, 39.9° (JCPDS file no. 00-038-0971) and K–Cu–O phases at 2θ = 27.2°, 32.9° and 51.1° (JCPDS file no. 00-012-0633).
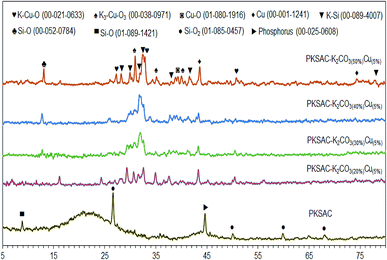 |
| | Fig. 4 XRD diffractogram for PKSAC and PKSAC derived catalysts. | |
Thermal program desorption of CO2 and NH3 (TPD-CO2 and TPD-NH3)
The amounts of basicity and acidity were studied by the desorption amounts of CO2 and NH3 from the catalyst surface. Generally, a higher temperature is needed to desorb CO2 by the strong basic components available on the catalyst surface. All of the prepared catalysts exhibited a peak at Tmax higher than 800 °C, attributed to the strong basic characteristics. Moreover, transesterification needs a relatively high basicity to convert triglycerides to FAME and relatively weak to medium acidic strength for esterification of low FFA feedstock. Table 2 presents the total amount of CO2 desorbed by the synthesized catalyst. The results showed two peaks in which the first small peak occurred at a lower temperature (Tmax = <200 °C), while the second peak occurred at a higher temperature (Tmax = >800 °C). From the results, the total amounts of basicity and acidity were shown to play a significant factor that reflected the catalytic activity. The production of FAME was most pronounced at 30 wt% by weight of K2CO3 at which the total amount CO2 desorbed was the highest (8.866 mmol g−1). These results were supported by the increasing FAME yield which is later discussed in the catalytic activity testing. Unfortunately, the percentage loading of K2CO3 was higher than 30 wt% onto the parent catalyst, showing the decrement of the amount of CO2 desorbed by the catalyst. This phenomenon could be explained by the overloading of active components which would hinder the adsorption of CO2 onto the catalyst surface due to particle agglomeration.21
Fourier transform infrared (FTIR)
The functional groups attached to the surface of the PKSAC and PKSAC derived catalysts were examined by Fourier transform infrared (FTIR) transmission spectra, as depicted in Fig. 5. The PKSAC was found to have a broad band at 3200 to 3600 cm−1, which was attributed to the hydroxyl group (–OH) adsorbed onto the surface of the catalysts. The spectra also showed two peaks at 2921 cm−1 and 2855 cm−1 which have been assigned as the aliphatic structure of C–H, while C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from the carboxyl group that can be observed at 1735 cm−1. The appearance of CC aromatic can be found at 1600 cm−1 and fingerprint peaks of C–H groups were obtained in the range of 700–885 cm−1. All of these peaks were the basic unit of PKS.22 The effects of acid treatment can be observed at 900–1300 cm−1, which belonged to phosphorus group such as O–C in P–O–C, PO in POOH.23 The appearance of new peaks at 1440 cm−1 and 873 cm−1 for PKSAC-K2CO3(40%)CuO(5%) and PKSAC-K2CO3(50%)CuO(5%) suggested the abundance of carbonate group (CO32−) on the surface which promoted a reaction with CO2 in the air during preparation.2 The successful activation step was verified by the disappearance of the absorption band at 2921 cm−1 (C–H) and 2855 cm−1 from PKSAC derived catalysts, which indicated that a significant amount of hydrogen was removed after the carbonization.24 For PKSAC derived catalysts, the peak at 1346 cm−1 was attributed to the existence of bulk K2CO3, which was most intense for PKSAC-K2CO3(30%)CuO(5%) and played an important role in the reaction. The absorption band at 1735 cm−1 (CO) also disappeared for PKSAC derived catalysts, suggesting that the carbonization process at 700 °C has broken the C–O bonds for both aliphatic and aromatic groups.25
O from the carboxyl group that can be observed at 1735 cm−1. The appearance of CC aromatic can be found at 1600 cm−1 and fingerprint peaks of C–H groups were obtained in the range of 700–885 cm−1. All of these peaks were the basic unit of PKS.22 The effects of acid treatment can be observed at 900–1300 cm−1, which belonged to phosphorus group such as O–C in P–O–C, PO in POOH.23 The appearance of new peaks at 1440 cm−1 and 873 cm−1 for PKSAC-K2CO3(40%)CuO(5%) and PKSAC-K2CO3(50%)CuO(5%) suggested the abundance of carbonate group (CO32−) on the surface which promoted a reaction with CO2 in the air during preparation.2 The successful activation step was verified by the disappearance of the absorption band at 2921 cm−1 (C–H) and 2855 cm−1 from PKSAC derived catalysts, which indicated that a significant amount of hydrogen was removed after the carbonization.24 For PKSAC derived catalysts, the peak at 1346 cm−1 was attributed to the existence of bulk K2CO3, which was most intense for PKSAC-K2CO3(30%)CuO(5%) and played an important role in the reaction. The absorption band at 1735 cm−1 (CO) also disappeared for PKSAC derived catalysts, suggesting that the carbonization process at 700 °C has broken the C–O bonds for both aliphatic and aromatic groups.25
 |
| | Fig. 5 FTIR for PKSAC and PKSAC derived catalysts. | |
Field emission scanning electron microscope (FESEM)
A series of FESEM images are presented in Fig. 6(a)–(f), which show the morphological surface of raw PKS, PKSAC and PKSAC derived catalysts. Fig. 6(a) shows that the raw PKS illustrated a dense, neat and rough surface with no visible cracks.26,27 The shape and particle size changed dramatically after treatment with 20% H3PO4 and carbonized at 700 °C for 3 hours. The treatment with H3PO4 has developed a well-formed variety of pore sizes during carbonization process due to the breakdown of the lignocellulosic composition. The H3PO4 later evaporated, thus leaving created pores which can be observed in Fig. 6(b).22 Further changes in the structure and particle sizes were also observed after the introduction of K2CO3 and CuO onto the surface of PKSAC. It was found that the pores were filled with K2CO3 and CuO particles, as shown in Fig. 6(d)–(f) and slowly disappeared with increasing percentage loading of K2CO3 and CuO, finally resulting in the absence of pores which indicated that the K2CO3 and CuO particles were highly dispersed and occupied into the pore matrix.28 These results were in line with the BET surface area results that indicated the decreasing BET surface area with increasing K2CO3 percentage loading.
 |
| | Fig. 6 FESEM images for (a) raw PKS, (b) PKSAC, (c) PKSAC-K2CO3(20%)CuO(5%), (d) PKSAC-K2CO3(30%)CuO(5%), (e) PKSAC-K2CO3(40%)CuO(5%) and (f) PKSAC-K2CO3(50%)CuO(5%) analysed at 20000× magnification. | |
Elemental composition analysis
The elemental composition of the raw PKS, PKSAC and PKSAC derived catalysts were determined by an EDX analyzer equipped with a microscope to verify the presence of potassium, copper, oxygen and carbon on the catalyst's surface. Table 3 tabulates the elemental compositions of raw PKS, PKSAC and PKSAC derived catalysts. The results showed that the percentages of the calculated weight for potassium and copper which were initially used in the preparation of the catalysts were found to be in agreement with the theoretical weight percentage. In addition, phosphorus was also detected for PKSAC and PKSAC derived catalysts which originated from the pretreatment step at the earlier stage.
Table 3 Elemental composition of raw PKS, PKSAC and PKSAC derived catalysts
| Catalysta |
Element compositionb (%) |
| C |
O |
P |
K |
Cu |
| Theoretical K and Cu weight%. Experimental K and Cu weight% in the raw PKSAC derived catalyst analyzed by EDX. |
| Raw PKS |
53.55 |
46.45 |
— |
— |
— |
| PKSAC |
83.43 |
16.57 |
7.53 |
— |
— |
| PKSAC-K2CO3(20%)CuO(5%) |
59.40 |
8.43 |
6.44 |
21.42 |
4.31 |
| PKSAC-K2CO3(30%)CuO(5%) |
51.88 |
9.32 |
6.70 |
29.51 |
2.59 |
| PKSAC-K2CO3(40%)CuO(5%) |
46.31 |
5.12 |
6.32 |
38.55 |
3.70 |
| PKSAC-K2CO3(50%)CuO(5%) |
31.88 |
12.87 |
6.59 |
45.63 |
3.03 |
Thermal gravimetric analyzer (TGA)
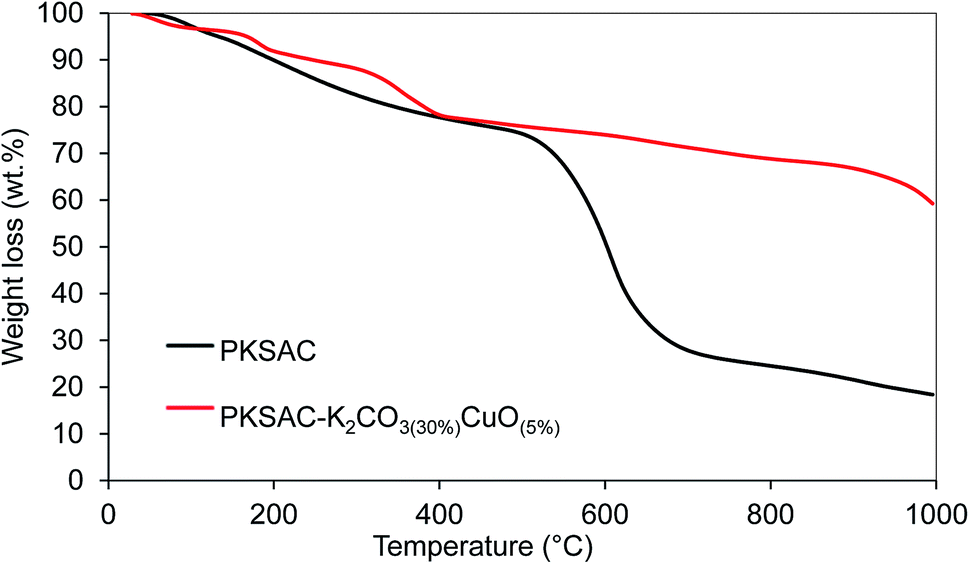
The thermal degradation of the PKSAC-K2CO3(30%)CuO(5%) catalyst as representative was analyzed by the thermogravimetric analyzer (TG-50 Mettler instrument). The weight loss of the sample in the entire process was recorded and plotted versus the temperature of the oven, as displayed in Fig. 7. Both PKSAC and PKSAC-K2CO3(30%)CuO(5%) had an initial weight loss at temperature up to 150 °C due to the elimination of hydroxyl group with a weight loss of 3.4–4.8%. The second weight loss was identified in the range of 150 °C to 300 °C and can be ascribed to the loss of carboxyl groups.29
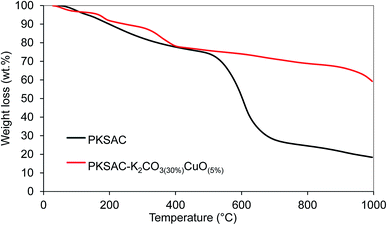 |
| | Fig. 7 TGA profile of PKSAC and PKSAC-K2CO3(30%)CuO(5%) catalyst. | |
In the case of PKSAC, the pyrolysis of hemicellulose and cellulose was focused at a temperature range of 129 °C to 463 °C with maximum weight loss at a very slow rate of 0.0586 wt% °C−1 obtained at Tmax = 170.4 °C. The decomposition of lignin can be seen clearly in the figure as it shows a significant weight loss of 51.0% with a maximum weight loss rate of 0.1521 wt% °C−1 obtained at Tmax = 605 °C. This maximum weight loss could be attributed to slow carbonization of lignin that is responsible in char production. When the temperature is more than 798 °C, the slight weight loss of 6.2% that occurred until 995.6 °C could be due to transformation of residual lignin to carbon monoxide and also release of CO2 and H2O, as mentioned in previous work.30,31 The final residue left after the thermal treatment was 18.4%. For the prepared catalyst, after the removal of adsorbed water at the temperature range of up to 118.6 °C, the decomposition of Cu(NO3)2 showed two peaks which were at Tmax = 182 °C and 369 °C. The nitrate degraded at Tmax = 369 °C until 435 °C and the carbonate compounds started to decompose at 800 °C. At the end of the process, there was about 59.8% residue left.
Effect of K2CO3 wt% loading on the catalytic activity
The catalytic activity of the prepared catalysts was tested in the esterification–transesterification reaction with WCO. The optimization to determine the optimum metal ratio loads onto the PKSAC was done to get the best metal percentage combination for maximum FAME yield percentage and lowest final acid value. Fig. 8(a) and (b) illustrate the variation of percentage for K2CO3 and Cu(NO3)2, respectively. For the determination of optimum wt% percentage of K2CO3, the wt% of Cu(NO3)2 was fixed to 5 wt%. After getting the optimum wt% for K2CO3 loading, the percentage was then fixed for the determination of Cu(NO3)2 wt% load onto the PKSAC. The reaction condition for optimization was set up as 5 wt% catalyst loading, 15:1 methanol to oil molar ratio at 70 °C for 4 hours. From Fig. 8(a), it shows that the reaction catalyzed by PKSAC-K2CO3(30%)CuO(5%) rendered the highest FAME yield percentage of 87.7% and the % FFA conversion was increased from 25.5% to 93.1%. The FAME yield percentage was lower when the K2CO3 wt% was increased from 40 wt% to 50 wt%, as the formation of soap started due to the excess of total basicity. However, the addition of Cu(NO3)2 at more than 5 wt% did not give a significant effect on the FAME yield percentage. Thus, the PKSAC-K2CO3(30%)CuO(5%) was chosen for further investigation.
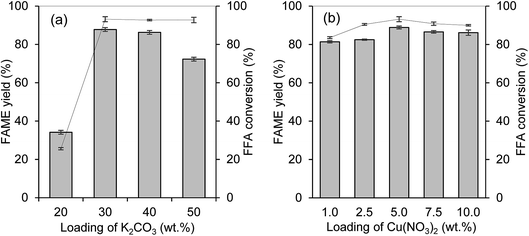 |
| | Fig. 8 (a) Effect of K2CO3 wt% with Cu(NO3)2 fixed at 5 wt% for transesterification–esterification of WCO and (b) effect of Cu(NO3)2 wt% with K2CO3 fixed at 30 wt% for transesterification–esterification of WCO. Reaction condition: 5 wt% catalyst loading, 15:1 methanol-to-oil ratio at 70 °C for 4 hours. | |
Transesterification–esterification of WCO over PKSAC-K2CO3(30%)CuO(5%) catalyst
Investigations on the effects of the wt% catalyst loading, temperature, methanol-to-oil molar ratio and duration on FAME yield percentage and % FFA conversion are shown in Fig. 9(a)–(d).
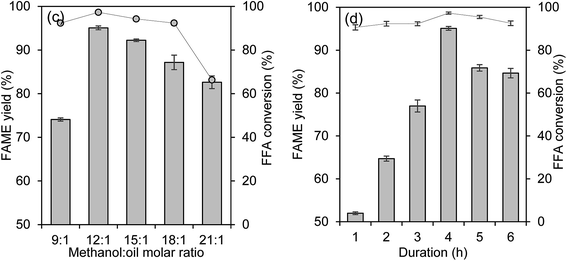 |
| | Fig. 9 (a) The effect of catalyst loading on the FAME yield percentage (methanol:oil molar ratio of 15:1 at 70 °C for 4 hours). (b) The effect of reaction temperature on the FAME yield percentage (catalyst loading of 5 wt%, methanol:oil molar ratio of 15:1 for 4 hours). (c) The effect of methanol-to-oil molar ratio on the FAME yield percentage (catalyst loading of 5 wt% at 80 °C for 4 hours). (d) The effect of duration on the FAME yield percentage (catalyst loading of 5 wt%, methanol:oil molar ratio of 12:1 at 80 °C). | |
Effect of catalyst loading
The effect of wt% catalyst loading on the FAME yield percentage and the % FFA conversion is shown in Fig. 9(a). The reaction was tested in the catalyst loading range of 1 wt% to 8 wt% at 15:1 methanol-to-oil ratio at 70 °C for 4 hours. The FAME yield percentage was improved from 52.5% to 87.7% and the % FFA conversion was increased from 24.4% to 93.1% as the catalyst loading was increased from 1 wt% to 5 wt%. By further increasing the catalyst loading to 8 wt%, the FAME yield percentage decreased as thick emulsion occurred which complicated the separation of FAME and glycerol layer. This result indicated that the sufficient catalyst loading for the reaction was 5 wt%.
Effect of reaction temperature
The temperature of the reaction was also important for the reaction as it proportionally affected the methanol, as methanol has a boiling point of 65 °C. From Fig. 9(b), the increment of temperature over 80 °C gave negative results for FAME yield percentage due to optimum collision energy gained by the methanol to overcome the mass transfer barrier between the three-phase reactant to get the maximum FAME yield percentage.32 Unfortunately, the FAME yield percentage slightly decreased when the temperature was over 80 °C. The reason for this decrement might be due to the adverse effect of methanol vaporization.33 Most of the transesterification was done before requiring an optimum temperature of 65 °C;34,35 unfortunately, the addition of simultaneous esterification reaction requires a higher temperature.36 Thus, the optimum reaction temperature of 80 °C was chosen for the next investigation.
Effect of methanol-to-oil molar ratio
The effect of the methanol-to-oil molar ratio is a crucial parameter for the reaction. This is because the reaction required an adequate amount of methanol to produce a high percentage of FAME yield. The reaction was done by the addition of 5 wt% catalyst loading at 80 °C for 4 hours. The effect of methanol-to-oil molar ratio is illustrated in Fig. 9(c). The figure shows that the highest percentage of FAME yield was obtained at 12:1 and the result for % FFA conversion was improved from 92.3% to 98.1% with the combination of 12:1 methanol–oil molar ratio at 80 °C. By increasing the methanol-to-oil molar ratio over 12:1, the FAME yield percentage deceased. The high amount of methanol loaded into the reaction media caused flooding, thus diluting the catalyst wt%.37 In addition, a higher ratio of methanol-to-oil delayed the separation of glycerol due to the appearance of the thick emulsion during the washing step.38 From the results, the methanol-to-oil ratio of 12:1 was found to be the optimum ratio, with the FAME yield percentage of 95.0%.
Effect of reaction duration
The effect of reaction duration on the reaction was investigated. Fig. 9(d) presents the reaction duration of 1 to 6 hours with the other optimum reaction of 5 wt% catalyst loading with 12:1 methanol-to-oil molar ratio at 80 °C. A low FAME yield percentage was discovered at a short time of 1 hour and slowly increased up to from 51.9% to 76.9% FAME yield after 3 hours of reaction. The maximum FAME yield percentage and the acid value recorded after 4 hours were 95.0% and 97.3% FFA conversion respectively. The reaction was prolonged to 5 and 6 hours and resulted in the decrement of FAME yield. This might be due to the formation of emulsion and could eventually cause difficulties to separate the FAME and glycerol layer, thus some of the FAME layers might be discharged together with glycerol or water during the separation and washing process.39
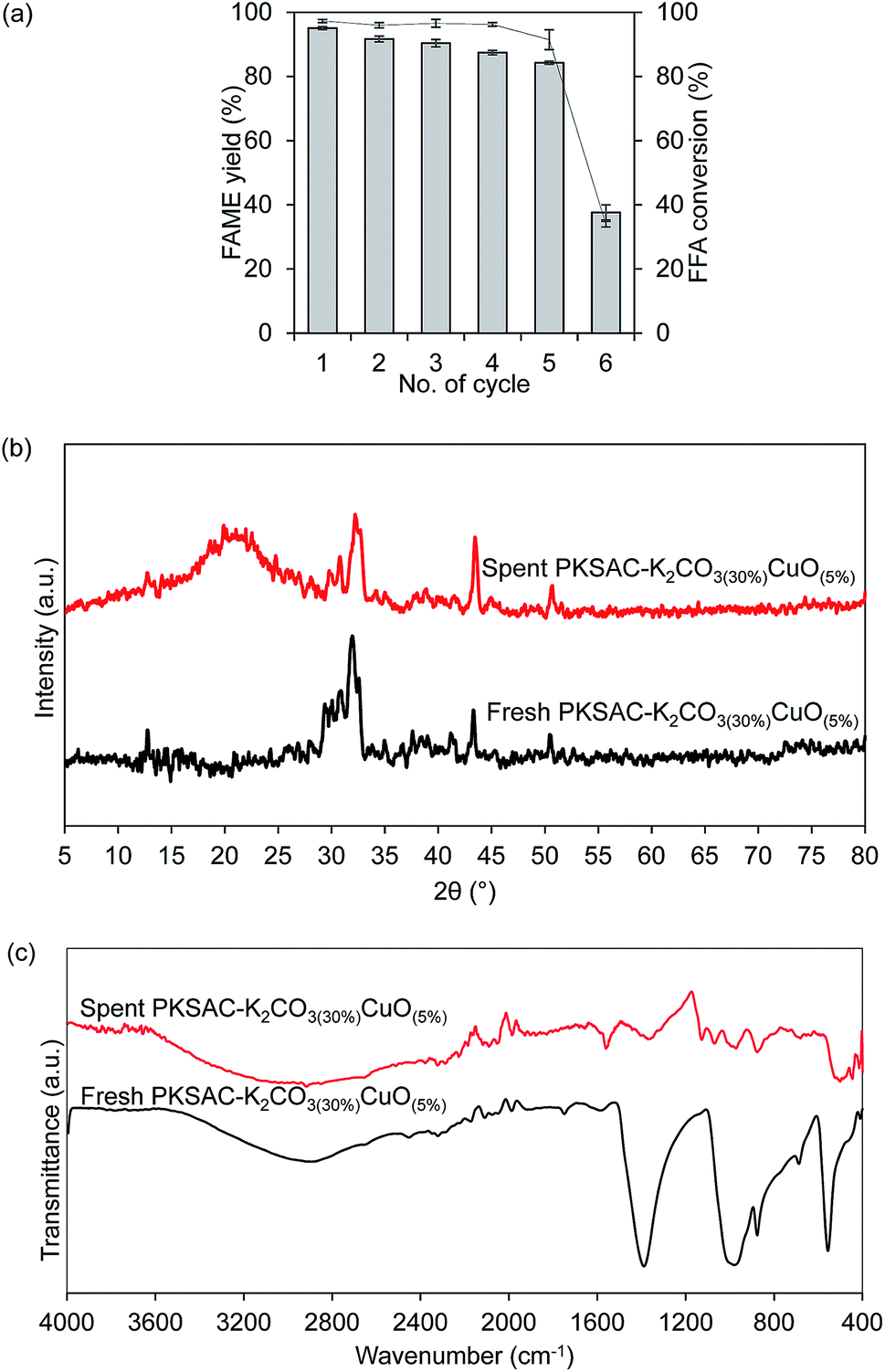
Reusability study and characterization of the spent catalyst
Reusability study is a significant characteristic of the introduction of heterogeneous. Successfulness in the regeneration of the catalyst significantly offers economic impact, as it affects the whole cost of the operation and overall price of the final product. Therefore, repeated reaction cycles were done by using the optimum catalyst and were carried out at the optimum condition of 5 wt% catalyst loading and 12:1 methanol to oil molar ratio at 80 °C for 4 hours. After completing each reaction cycle, the spent catalyst from the first reaction cycle was recovered through centrifugation and washed with methanol and hexane to remove polar and non-polar impurities on the catalyst surface. The re-calcination of the spent catalyst helps to reinstate its catalytic activity.40,41 From Fig. 10(a), the results showed that the FAME yield reduced from 95.2% to 91.1% after the second reaction cycle. In the third and fourth reaction cycles, the FAME yield further reduced to 90.3% and 87.4%, respectively, and 84.2% after the fifth cycle. The reaction further proceeded to another reaction cycle but unfortunately, the FAME yield dropped down to 55.3% only after the sixth reaction cycle. Thus, this study confirmed that the synthesized catalyst can be reused for at least five reaction cycles with relatively good catalytic activity. In addition, a series of characterization for spent catalyst after the sixth reaction cycle was done to validate the statement.
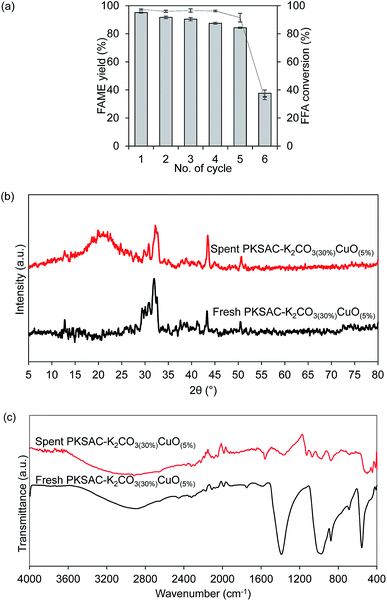 |
| | Fig. 10 (a) The effect of reusability study on the FAME yield percentage. Reaction condition; catalyst loading of 5 wt%, MeOH:oil molar ratio of 12:1, 80 °C for 4 hours. (b) XRD for fresh and spent PKSAC-K2CO3(30%)CuO(5%) catalyst. (c) FTIR for fresh and spent PKSAC-K2CO3(30%)CuO(5%) catalyst. | |
By comparing the XRD patterns after the sixth cycle of the catalyst PKSAC-K2CO3(30%)CuO(5%), as presented in Fig. 10(b), it was detected that the intensity of the diffraction patterns had significantly decreased. The decreasing catalyst activity could be attributed to the carbon structure which collapsed due to repeated carbonization step at high temperature.7 As a result, the number of active components available for the next reaction cycle reduced. Other than that, surface properties, including surface defects of the fresh and spent catalysts would cause catalyst poisoning since the glycerol and unreacted oil blocked the active components, thus making the catalyst inactive.42,43 This statement could also be verified by the results on TPD, as depicted in Table 4. The results showed that the amount of basicity and acidity were decreased. The decreasing of basic active sites led to exposure of acid active components on the catalyst surface, thus affecting the amount of NH3 desorption to slightly increase at the high temperature, as presented.18
Table 4 TPD results for the fresh PKSAC-K2CO3(30%)CuO(5%) and spent PKSAC-K2CO3(30%)CuO(5%) catalysts after the sixth reaction cycle
| Catalyst |
Amount of CO2 desorbed (mmol g−1) |
Amount of NH3 desorbed (mmol g−1) |
| Fresh PKSAC-K2CO3(30%)CuO(5%) |
8.866 |
29.472 |
| Spent PKSAC-K2CO3(30%)CuO(5%) |
3.106 |
27.016 |
The comparison was also conducted for FTIR spectrum between the fresh catalyst and spent PKSAC-K2CO3(30%)CuO(5%) catalyst after the sixth reaction cycle. From Fig. 10(c), it shows that the peak intensities at 1400 cm−1, 667 cm−1 (C–O–K) and 873 cm−1 (C–O–Cu) were reduced due to the reduction of FAME yield after the sixth reaction cycle. This result was in line with the XRD diffractogram result which showed that the intensities of all peaks for the spent catalyst were reduced and formation of amorphous structure clearly appeared after the sixth reaction cycle. Thus, this investigation revealed that the synthesized catalyst can be used up to five cycles with minimal loss of its catalytic activity.
The catalytic activity of PKSAC-K2CO3(30%)CuO(5%) under optimum reaction condition was compared with other similar studies done by other researchers and the results are tabulated in Table 5. It can be seen that the synthesized catalyst had a comparable catalytic activity to the listed catalysts under the relatively mild condition for the production of FAME from WCO. It was also noted that the synthesized catalyst was efficient for one-pot transesterification–esterification reaction in producing FAME and had a great potential in industrial application.
Table 5 Comparison of the catalytic activities of the previously synthesized catalysts
| No. |
Catalyst |
Feedstock |
Reaction conditions |
FAME yield (%) |
Ref. |
| 1 |
Zn doped CaO nanocatalyst |
WCO |
5 wt%, 20:1, 65 °C, 4 h |
96.74 |
44 |
| 2 |
Fe2O3/MnO–SO4/ZrO2 |
WCO |
3 wt%, 20:1, 180 °C, 4 h |
96.50 |
9 |
| 3 |
K+ trapped kaolinite |
WCO |
15 wt%, 14:1, 70 °C, 2 h |
94.76 |
45 |
| 4 |
4Mn–6Zr/CaO |
WCO |
3 wt%, 15:1, 80 °C, 3 h |
92.10 |
10 |
| 5 |
PKSAC-K2CO3(30%)CuO(5%) |
WCO |
5 wt%, 12:1, 80 °C, 4 h |
95.00 |
This study |
Conclusion
In summary, waste biomass was successfully turned into valuable activated carbon via acid treatment and carbonization at 700 °C. The functionalization with K2CO3 and Cu(NO3)2 through wet impregnation produced a bifunctional catalyst for one-step reaction. The high BET surface area and high amount of basicity and acidity played the most significant roles to produce high FAME yield from WCO. The efficiency of the catalytic activity of the catalyst was proven, as it successfully produced 95.0% FAME yield with 97.3% FFA conversion at optimum condition of 5 wt% catalyst loading and reaction temperature of 80 °C with methanol-to-oil molar ratio of 12:1 for 4 hours. The catalyst is also proven to give a good economic impact in the biodiesel industry as it can be reused for five reaction cycles with minimal loss of its catalytic activity. It was proposed that the bifunctional catalyst derived from PKS has a high potential in producing biodiesel from WCO.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to extend their sincere appreciation to Universiti Putra Malaysia for funding this research through GP-IPS/2016/9580300 research grant. The authors would like to extend their sincere appreciation to King Saud University (Riyadh, Saudi Arabia) for the support of this research through Researchers Supporting Project number (RSP-2020/147).
References
- M. Thirumarimurugan, V. M. Sivakumar, A. M. Xavier, D. Prabhakaran and T. Kannadasan, Int. J. Biosci., Biochem. Bioinf., 2013, 2, 441–444 Search PubMed.
- C. Zhao, P. Lv, L. Yang, S. Xing, W. Luo and Z. Wang, Energy Convers. Manage., 2018, 160, 477–485 CrossRef CAS.
- E. Rashtizadeh, F. Farzaneh and Z. Talebpour, Bioresour. Technol., 2014, 154, 32–37 CrossRef CAS PubMed.
- N. Mansir, S. H. Teo, U. Rashid, M. I. Saiman, Y. P. Tan, G. A. Alsultan and Y. H. Taufiq-Yap, Renewable Sustainable Energy Rev., 2018, 82, 3645–3655 CrossRef CAS.
- T. Furusawa, J. Jpn. Pet. Inst., 2017, 60, 170–185 CrossRef CAS.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- S. Baroutian, M. K. Aroua, A. A. A. Raman and N. M. N. Sulaiman, Fuel Process. Technol., 2010, 91, 1378–1385 CrossRef CAS.
- M. Farooq, A. Ramli and D. Subbarao, J. Cleaner Prod., 2013, 59, 131–140 CrossRef CAS.
- F. H. Alhassan, U. Rashid and Y. H. Taufiq-Yap, Fuel, 2015, 142, 38–45 CrossRef CAS.
- N. Mansir, S. H. Teo, I. Rabiu and Y. H. Taufiq-Yap, Chem. Eng. J., 2018, 347, 137–144 CrossRef CAS.
- D. Nuradila, W. A. W. A. K. Ghani and A. B. Alias, Malaysian J. Anal. Sci., 2017, 21, 197–203 CrossRef.
- J. Andas, M. L. A. Rahman and M. S. M. Yahya, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 226, 012156 Search PubMed.
- M. S. A. Farabi, M. L. Ibrahim, U. Rashid and Y. H. Taufiq-Yap, Energy Convers. Manage., 2019, 181, 562–570 CrossRef CAS.
- N. S. A. Wafti, H. L. N. Lau, S. K. Loh, A. A. Aziz, Z. A. Rahman and C. Y. May, J. Oil Palm Res., 2017, 29, 278–290 CrossRef.
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546–564 CrossRef CAS.
- J. Yang and K. Qiu, Chem. Eng. J., 2010, 165, 209–217 CrossRef CAS.
- N. Boz, N. Degirmenbasi and D. M. Kalyon, Appl. Catal., B, 2013, 138–139, 236–242 CrossRef CAS.
- M. Farooq, A. Ramli, A. Naeem and M. Saleem khan, RSC Adv., 2016, 6, 872–881 RSC.
- G. A. Alsultan, N. Asikin-Mijan, H. V. Lee, A. S. Albazzaz and Y. H. Taufiq-Yap, Energy Convers. Manage., 2017, 151, 311–323 CrossRef CAS.
- S. I. Akinfalabi, U. Rashid, R. Yunus and Y. H. Taufiq-Yap, Renewable Energy, 2017, 111, 611–619 CrossRef CAS.
- N. Mansir, S. H. Teo, U. Rashid and Y. H. Taufiq-Yap, Fuel, 2018, 211, 67–75 CrossRef CAS.
- A. F. Nicholas, M. Z. Hussein, Z. Zainal and T. Khadiran, Nanomaterials, 2018, 8, 689 CrossRef PubMed.
- A. M. Puziy, O. I. Poddubnaya, A. Martínez-Alonso, F. Suárez-García and J. M. D. Tascón, Carbon, 2002, 40, 1493–1505 CrossRef CAS.
- A. Heidari, H. Younesi, A. Rashidi and A. A. Ghoreyshi, Chem. Eng. J., 2014, 254, 503–513 CrossRef CAS.
- F. Cheng and X. Li, Catalysts, 2018, 8, 346 CrossRef.
- C. L. Lee, P. S. H’ng, M. T. Paridah, K. L. Chin, U. Rashid, M. Maminski, W. Z. Go, R. A. R. Nazrin, S. N. A. Rosli and P. S. Khoo, R. Soc. Open Sci., 2018, 5, 180775 CrossRef CAS PubMed.
- N. A. Rashidi and S. Yusup, J. Cleaner Prod., 2017, 168, 474–486 CrossRef CAS.
- J. Kazmierczak-Razna, P. Nowicki, M. Wiśniewska, A. Nosal-Wiercińska and R. Pietrzak, J. Taiwan Inst. Chem. Eng., 2017, 80, 1006–1013 CrossRef CAS.
- S. Soltani, U. Rashid, I. A. Nehdi, S. I. Al-Resayes and A. H. Al-Muhtaseb, J. Taiwan Inst. Chem. Eng., 2017, 70, 219–228 CrossRef CAS.
- T. Qu, W. Guo, L. Shen, J. Xiao and K. Zhao, Ind. Eng. Chem. Res., 2011, 50, 10424–10433 CrossRef CAS.
- M. M. Yashim, N. Razali, N. Saadon and N. A. Rahman, J. Eng. Appl. Sci., 2016, 11, 6389–6392 CAS.
- L. Du, S. Ding, Z. Li, E. Lv, J. Lu and J. Ding, Energy Convers. Manage., 2018, 173, 728–734 CrossRef CAS.
- H. M. Kefas, R. Yunus, U. Rashid and Y. H. Taufiq-Yap, Fuel, 2018, 229, 68–78 CrossRef CAS.
- M. Yadav, V. Singh and Y. C. Sharma, Energy Convers. Manage., 2017, 148, 1438–1452 CrossRef CAS.
- K. Malins, Fuel Process. Technol., 2018, 179, 302–312 CrossRef CAS.
- A. Alsultan, A. Mijan and T. Yap, Mater. Sci. Forum, 2016, 840, 348–352 Search PubMed.
- S. Soltani, U. Rashid, R. Yunus and Y. H. Taufiq-Yap, Fuel, 2016, 178, 253–262 CrossRef CAS.
- K. Krisnangkura and R. Simamaharnnop, J. Am. Oil Chem. Soc., 1992, 69, 166–169 CrossRef CAS.
- A. K. Ayoob and A. B. Fadhil, Energy Convers. Manage., 2019, 201, 112149 CrossRef CAS.
- D. Madhu and Y. C. Sharma, Resour.-Effic. Technol., 2017, 3, 144–157 Search PubMed.
- I. Ambat, V. Srivastava, E. Haapaniemi and M. Sillanpää, Renewable Energy, 2019, 139, 1428–1436 CrossRef CAS.
- A. Kumar, S. Osembo, S. Namango and K. Kiriamiti, Mech. Eng. Conf. Sustain. Res. Innov., 2012, 4, 59–68 Search PubMed.
- M. L. Ibrahim, N. N. A. Nik Abdul Khalil, A. Islam, U. Rashid, S. F. Ibrahim, S. I. Sinar Mashuri and Y. H. Taufiq-Yap, Energy Convers. Manage., 2020, 205, 112445 CrossRef CAS.
- M. J. Borah, A. Das, V. Das, N. Bhuyan and D. Deka, Fuel, 2019, 242, 345–354 CrossRef CAS.
- M. R. Abukhadra and M. A. Sayed, Energy Convers. Manage., 2018, 177, 468–476 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yun Hin Taufiq-Yapbc,
Mohd Lokman Ibrahimde,
Chawalit Ngamcharussrivichaifg and
Muhammad Azamh
*a,
Yun Hin Taufiq-Yapbc,
Mohd Lokman Ibrahimde,
Chawalit Ngamcharussrivichaifg and
Muhammad Azamh