DOI:
10.1039/D0RA04344C
(Paper)
RSC Adv., 2020,
10, 32069-32077
Heterodinuclear nickel(II)–iron(II) azadithiolates as structural and functional models for the active site of [NiFe]-hydrogenases†
Received
15th May 2020
, Accepted 20th August 2020
First published on 28th August 2020
Abstract
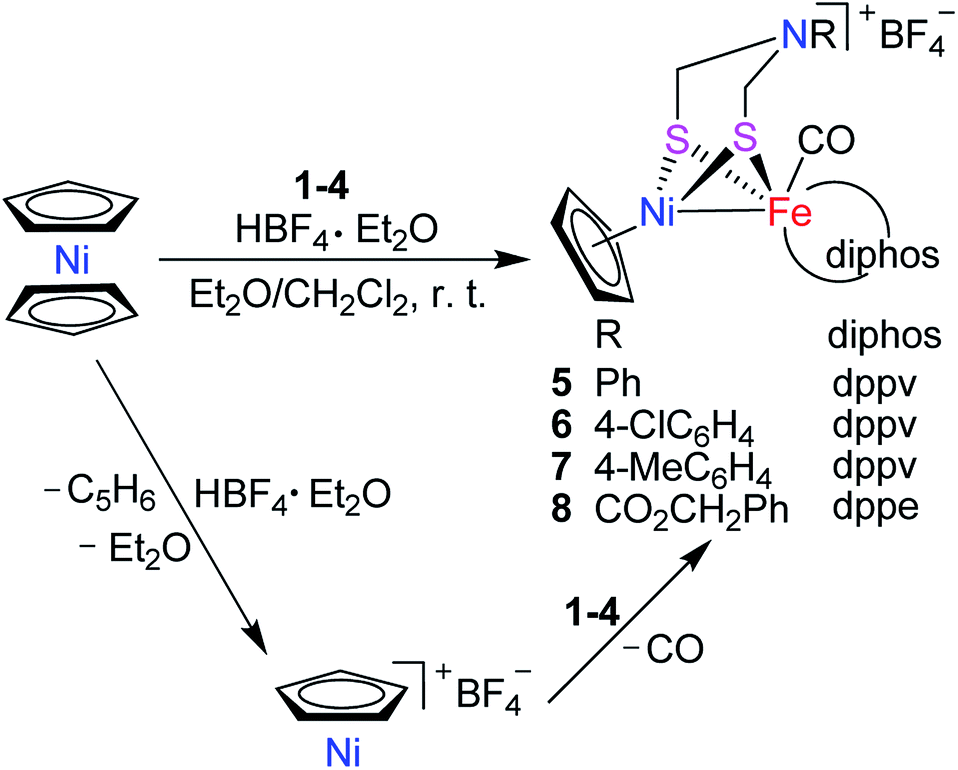
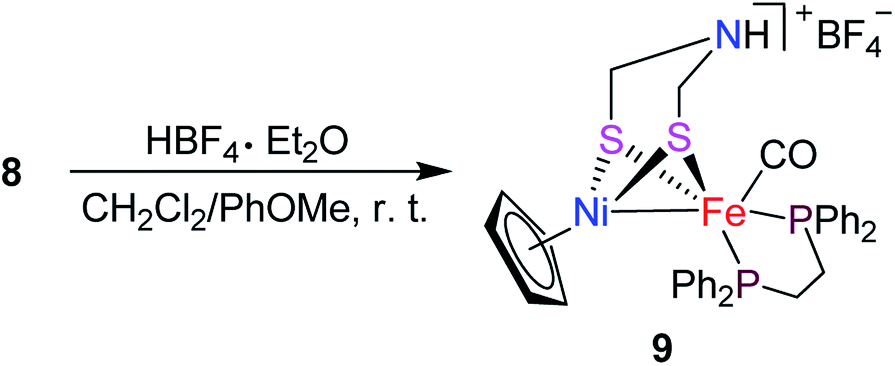
To develop the biomimetic chemistry of [NiFe]-H2ases, the first azadithiolato-bridged NiFe model complexes [CpNi{(μ-SCH2)2NR}Fe(CO)(diphos)]BF4 (5, R = Ph, diphos = dppv; 6, 4-ClC6H4, dppv; 7, 4-MeC6H4, dppv; 8, CO2CH2Ph, dppe; 9, H, dppe) have been synthesized via well-designed synthetic routes. Thus, treatment of RN[CH2S(O)CMe]2 with t-BuONa followed by reaction of the resulting intermediates RN(CH2SNa)2 with (dppv)Fe(CO)2Cl2 or (dppe)Fe(CO)2Cl2 gave the N-substituted azadithiolato-chelated Fe complexes [RN(CH2S)2]Fe(CO)2(diphos) (1, R = Ph, diphos = dppv; 2, 4-ClC6H4, dppv; 3, 4-MeC6H4, dppv; 4, CO2CH2Ph, dppe). Further treatment of 1–4 with nickelocene in the presence of HBF4·Et2O afforded the corresponding N-substituted azadithiolato-bridged NiFe model complexes 5–8, while treatment of 8 with HBF4·Et2O resulted in formation of the parent azadithiolato-bridged model complex 9. While all the new complexes 1–9 were characterized by elemental analysis and spectroscopy, the molecular structures of model complexes 6–8 were confirmed by X-ray crystallographic study. In addition, model complexes 7 and 9 were found to be catalysts for H2 production with moderate icat/ip and overpotential values from TFA under CV conditions.
Introduction
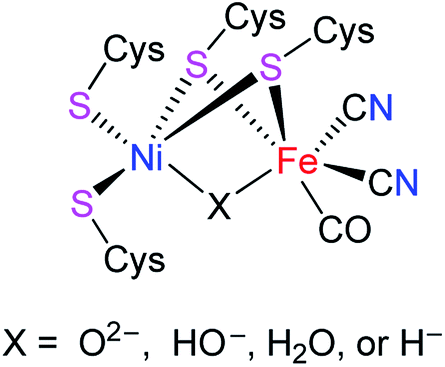
[NiFe]-hydrogenases ([NiFe]-H2ases) are a class of biological enzymes that catalyze the reversible redox reaction between molecular H2 and protons in a variety of microorganisms such as bacteria, archaea, and cyanobacteria.1–3 An X-ray crystallographic study revealed that the active site of [NiFe]-H2ases features a Ni(tetracysteinate) moiety, in which the two cysteinate atoms are connected to the Fe atom of an Fe(CN)2(CO) moiety. In addition, the oxidized state of [NiFe]-H2ases includes an additional bridging oxygenous ligand, while the reduced state of [NiFe]-H2ases contains an additional bridging hydride ligand or a Ni–Fe metal–metal bond (Fig. 1).4–6
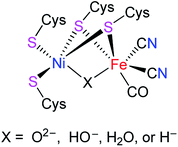 |
| | Fig. 1 Active site of [NiFe]-H2ases. | |
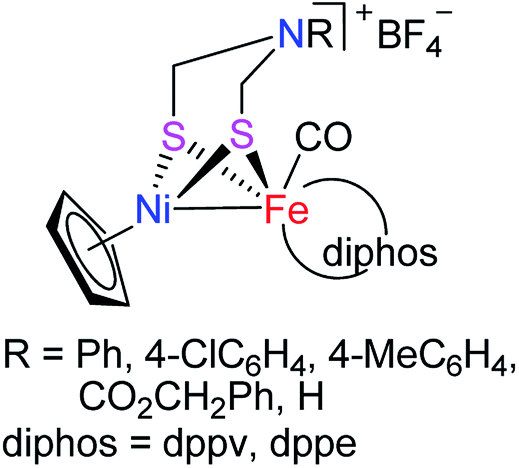
In recent years, molecular H2 has been considered as an ideal energy source to replace fossil fuels to solve the current energy shortage and environmental pollution problems. This is because dihydrogen is a high energy-dense, clean-burning, and sustainable energy.7–9 In addition, although H2 production can be carried out in an industry scale from fossil fuel by petroleum reforming, the utilized catalyst is the expensive and relatively scarce noble metal Pt. To develop the biomimetic chemistry of [NiFe]-H2ases and to search for the water splitting H2-producing catalysts that contain the earth-abundant and cheap metals such as iron and nickel present in the active site of [NiFe]-H2ases, synthetic chemists have prepared a wide variety of biomimetic models for [NiFe]-H2ases.9–29 However, among the prepared models no one bears a bridging azadithiolato ligand between their NiFe centers. In view of the important catalytic role played by the central N atom of the bridging azadithiolato ligand in the active site of [FeFe]-H2ases,30–32 we recently launched a study on the preparation of the first azadithiolato ligand-containing [NiFe]-H2ase models. Fortunately, we have successfully prepared and characterized such a type of azadithiolato-bridged models in which each contains an azadithiolato RN(CH2S)2 ligand bridged between their CpNi and (diphos)(CO)Fe units (Fig. 2). In addition, the two representative models have been found to be catalysts for proton reduction to hydrogen under electrochemical conditions. Herein, we report the interesting results obtained by this study.
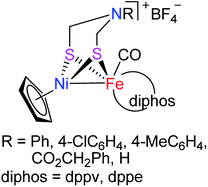 |
| | Fig. 2 Models of [NiFe]-H2ases reported in this article. | |
Results and discussion
Synthesis and characterization of precursor complexes [RN(CH2S)2]Fe(CO)2(diphos) (1–4)
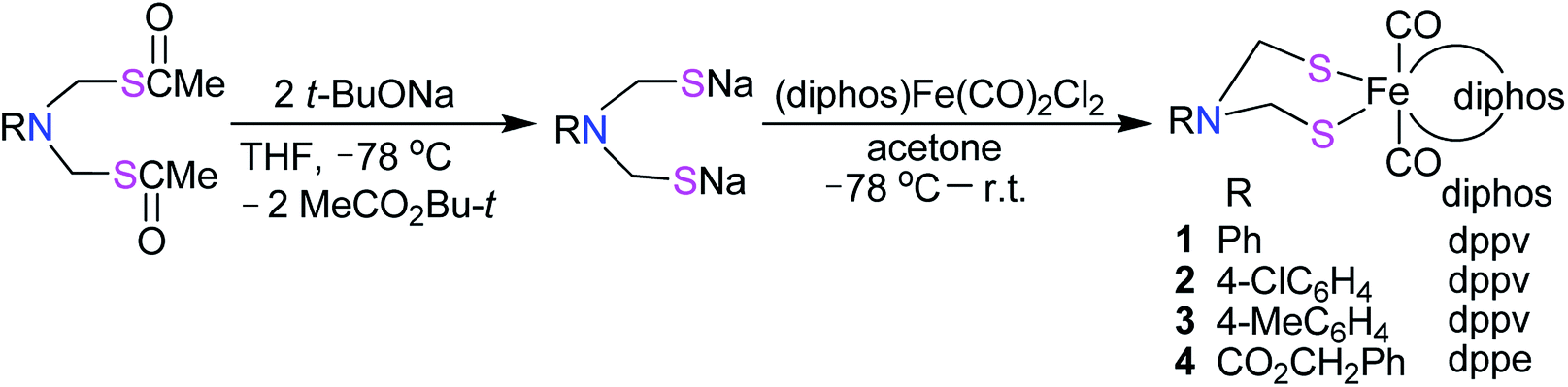
The N-substituted azadithiolato ligand-chelated mononuclear Fe complexes 1–4 utilized for the preparation of the targeted [NiFe]-H2ase models could be prepared by one-pot reactions of the bis(thioester) compounds RN[CH2S(O)CMe]2 (R = Ph, 4-ClC6H4, 4-MeC6H4, CO2CH2Ph) with tert-butoxysodium in THF at −78 °C followed by treatment of the resulting disodium mercaptides RN(CH2SNa)2 with (dppv)Fe(CO)2Cl2 or (dppe)Fe(CO)2Cl2 generated in situ by reaction of an acetone solution of FeCl2 with the corresponding diphosphine and bubbling CO gas33,34 in 18–35% yields (Scheme 1).
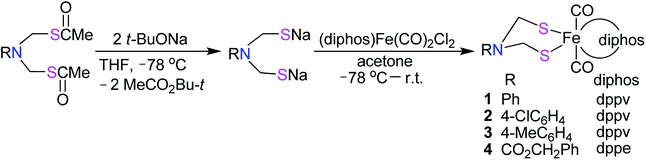 |
| | Scheme 1 Synthesis of precursor complexes 1–4. | |
Precursor complexes 1–4 are air-stable, brown-red solids and have been characterized by elemental analysis and various spectroscopic methods. For example, the 1H NMR spectra of 1–3 each displayed one singlet in the region 4.56–4.63 ppm for the four H atoms in their azadithiolato methylene groups, whereas the 1H NMR spectrum of 4 displayed a multiplet in the range 4.89–5.30 ppm for the four H atoms in its azadithiolato methylene groups. The 13C{1H} NMR spectra of 1–4 exhibited one or two singlets in the range 200–214 ppm for their two terminal carbonyl C atoms, whereas the 31P{1H} NMR spectra of 1–3 displayed one singlet at about 79 ppm for the two P atoms in their dppv ligands and the 31P{1H} NMR spectrum of 4 exhibited one singlet at 44.3 ppm for the two P atoms in its dppe ligand. In addition, the IR spectra of 1–4 showed two absorption bands in the range 2035–1968 cm−1 for their two terminal carbonyls.
Synthesis and characterization of targeted [NiFe]-H2ase models [CpNi{(μ-SCH2)2NR}Fe(CO)(diphos)]BF4 (5–9)
Interestingly, the targeted N-substituted dithiolato-bridged NiFe-based models 5–8 were found to be prepared by treatment of the mononuclear Fe complexes 1–4 with nickelocene/HBF4·Et2O in a mixed solvent CH2Cl2/Et2O at room temperature in 21%–62% yields (Scheme 2). Further treatment of the N-substituted dithiolato-bridged model complex 8 with HBF4·Et2O in a mixed solvent CH2Cl2/PhOMe at room temperature gave rise to the parent azadithiolato-bridged model 9 in 42% yield (Scheme 3). As shown in Schemes 2 and 3, while model 9 can be formally viewed as produced through the selective acidolysis of the N–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond rather than O–C(O) bond of model 8, model complexes 5–8 might be regarded as produced via two elementary reaction steps. The first step involves protonation of one anionic cyclopentadienyl ligand in Cp2Ni to give cyclopentadiene and cyclopentadienylnickel tetrafluoroborate.35,36 The second step involves coordination of the two S atoms in complexes 1–4 to Ni atom of the in situ formed intermediate CpNi(BF4) followed by loss of one CO ligand from 1–4 to give the final products.
O) bond rather than O–C(O) bond of model 8, model complexes 5–8 might be regarded as produced via two elementary reaction steps. The first step involves protonation of one anionic cyclopentadienyl ligand in Cp2Ni to give cyclopentadiene and cyclopentadienylnickel tetrafluoroborate.35,36 The second step involves coordination of the two S atoms in complexes 1–4 to Ni atom of the in situ formed intermediate CpNi(BF4) followed by loss of one CO ligand from 1–4 to give the final products.
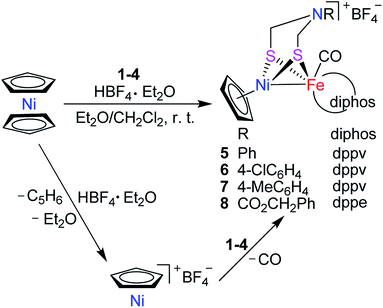 |
| | Scheme 2 Synthesis of targeted models 5–8. | |
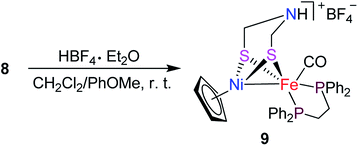 |
| | Scheme 3 Synthesis of targeted model 9. | |
Model complexes 5–9 are also air-stable, brown-red solids. They were characterized by the IR, 1H NMR, 13C{1H} NMR, and 31P{1H} NMR spectroscopic techniques and elemental analysis. For instance, the IR spectra of 5–9 showed one strong absorption band in the region 1956–1939 cm−1 for their terminal carbonyls, which are close to that (1944 cm−1) displayed by the [NiFe]-H2ase from D. Vulgaris Miyazaki F.37 The 1H NMR spectra of 5–9 displayed one singlet in the range 4.42–4.61 ppm for the five H atoms in their Cp rings. In addition, the 13C{1H} NMR spectra of 5–9 exhibited one singlet in the region 206–220 ppm for their terminal carbonyl C atoms, whereas the 31P{1H} NMR spectra of 5–7 displayed one singlet in the region 82.7–83.9 ppm for the two P atoms in their dppv ligands and those of 8 and 9 exhibited the corresponding signals in the range 78.1–79.1 ppm for the two P atoms in their dppe ligands.
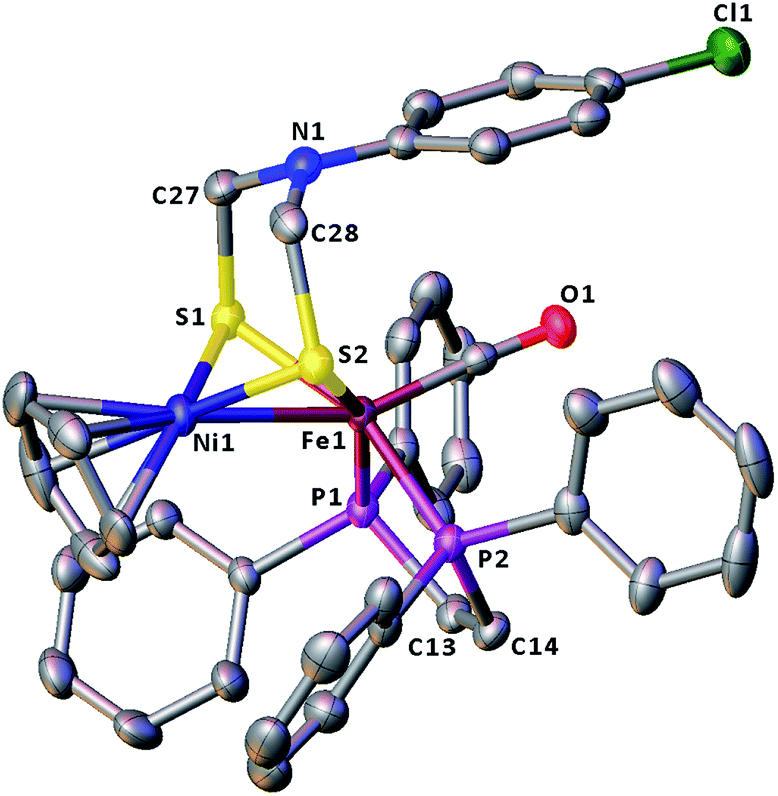
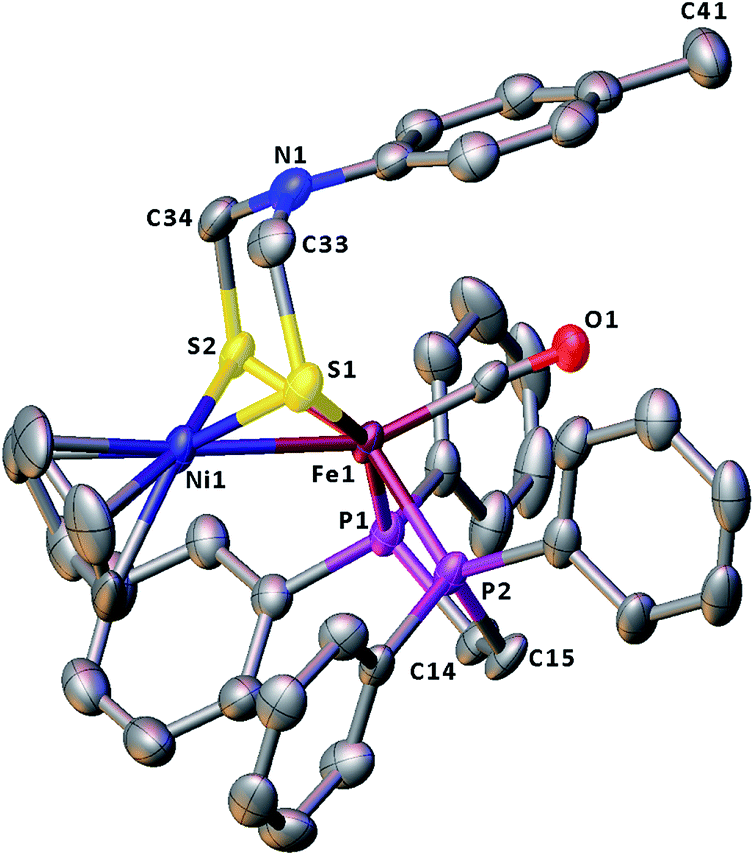
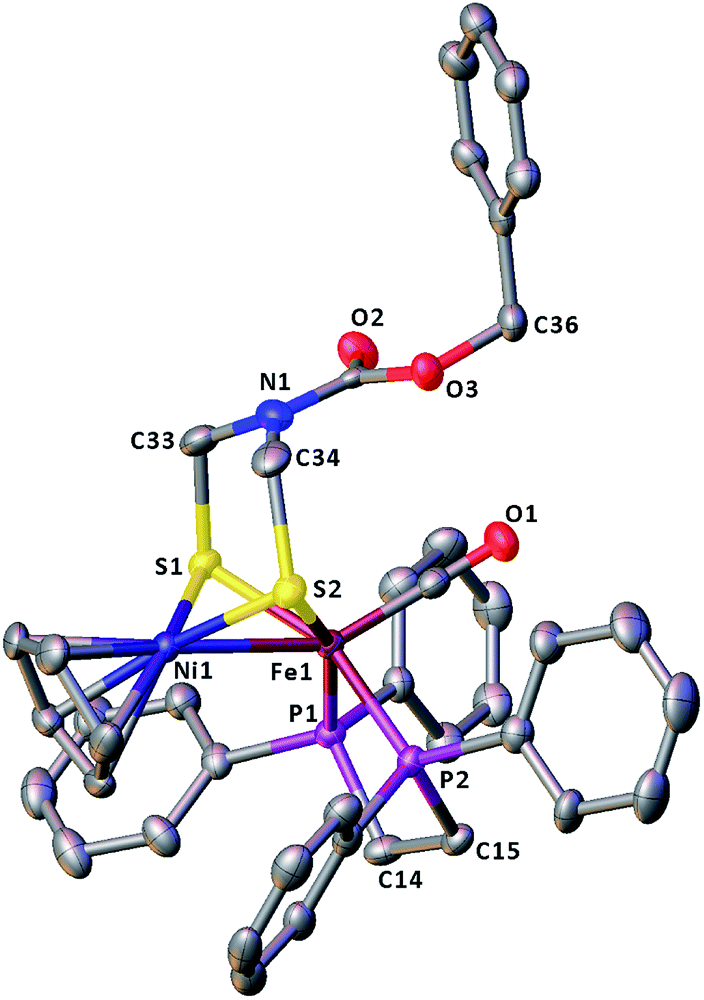
The molecular structures of the azadithiolato-bridged complexes 6–8 were confirmed by X-ray crystallographic analysis. While Fig. 3–5 show their molecular structures, Table 1 lists their selected bond lengths and angles. As shown in Fig. 3−5, the three biomimetic models are indeed the azadithiolato ligand-containing complexes that are composed of one monocation [CpNi{(μ-SCH2)2NC6H4Cl-4}Fe(CO)(dppv)]+, [CpNi{(μ-SCH2)2NC6H4Me-4}Fe(CO)(dppv)]+, or [CpNi{(μ-SCH2)2NCO2CH2Ph}Fe(CO)(dppe)]+ and one monoanion BF4−. Since the molecular structures of 6 and 7 are virtually the same as that of 8 (except that the 4-ClC6H4/dppv in 6 and 4-MeC6H4/dppv in 7 are replaced by the CO2CH2Ph/dppe in 8), we just discuss the structure of 8. In the monocation of 8, the Ni–Fe distance (2.5036 Å) is shorter than that (2.57 Å) of the [NiFe]-H2ase from D. Vulgaris Miyazaki F38 and the sum (2.56 Å) of the Ni and Fe covalent radii,39 indicative of the presence of a Ni–Fe metal–metal bond. In addition, the Ni–Fe distance of 8 is very close to that (2.5145 Å) of the reported 1,3-propanedithiolato(pdt)-bridged analogue [CpNi(pdt)Fe(dppe)(CO)]BF4.40 Owing to the presence of a Ni–Fe metal–metal bond in 8, the Ni center could be regarded as taking a “piano stool” geometry and the Fe center adopts a pseudo-octahedral geometry. In addition, the azadithiolato ligand in 8 is bridged between Ni and Fe atoms to construct two fused six-membered rings. The six-membered ring Fe1S1C33N1C34S2 shown in Fig. 3 possesses a boat conformation, whereas the six-membered ring Ni1S1C33N1C34S2 possesses a chair conformation. While the N-substituted CO2CH2Ph group is attached to the common N1 atom of the two fused six-membered rings with an axial type of bond, the terminal carbonyl ligand is bound to Fe1 atom in an axial position of the pseudo-octahedral geometry of the Fe center.
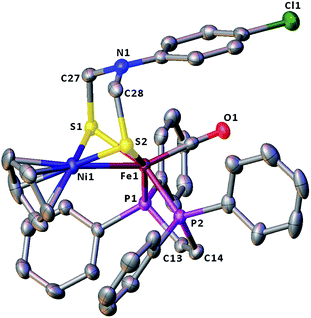 |
| | Fig. 3 Molecular structure of model complex 6 with ellipsoids drawn at a 50% probability level. All hydrogen atoms and the anion BF4− are omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for 6–8
| 6 |
| Ni(1)–Fe(1) |
2.5033(9) |
Fe(1)–S(1) |
2.2472(12) |
| Ni(1)–S(1) |
2.1783(13) |
Fe(1)–S(2) |
2.2361(11) |
| Ni(1)–S(2) |
2.1770(11) |
Fe(1)–P(1) |
2.2198(11) |
| N(1)–C(28) |
1.433(5) |
Fe(1)–P(2) |
2.2215(12) |
| Ni(1)–S(1)–Fe(1) |
68.87(4) |
S(2)–Fe(1)–S(1) |
87.09(4) |
| Ni(1)–S(2)–Fe(1) |
69.10(4) |
S(2)–Fe(1)–Ni(1) |
54.34(3) |
| S(2)–Ni(1)–S(1) |
90.34(4) |
S(1)–Fe(1)–Ni(1) |
54.26(3) |
| P(1)–Fe(1)–P(2) |
86.10(4) |
S(1)–Ni(1)–Fe(1) |
56.86(4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| 7 |
| Ni(1)–Fe(1) |
2.5071(9) |
Fe(1)–S(1) |
2.2497(14) |
| Ni(1)–S(1) |
2.1826(16) |
Fe(1)–S(2) |
2.2374(13) |
| Ni(1)–S(2) |
2.1795(14) |
Fe(1)–P(1) |
2.2268(15) |
| N(1)–C(33) |
1.429(7) |
Fe(1)–P(2) |
2.2249(14) |
| Ni(1)–S(1)–Fe(1) |
68.88(5) |
S(2)–Fe(1)–S(1) |
87.15(5) |
| Ni(1)–S(2)–Fe(1) |
69.15(4) |
S(2)–Fe(1)–Ni(1) |
54.33(4) |
| S(2)–Ni(1)–S(1) |
90.31(5) |
S(1)–Fe(1)–Ni(1) |
54.30(4) |
| P(1)–Fe(1)–P(2) |
86.13(5) |
S(1)–Ni(1)–Fe(1) |
56.83(4) |
|
| 8 |
| Ni(1)–Fe(1) |
2.5036(9) |
Fe(1)–S(1) |
2.2367(11) |
| Ni(1)–S(1) |
2.1807(13) |
Fe(1)–S(2) |
2.2306(12) |
| Ni(1)–S(2) |
2.1819(12) |
Fe(1)–P(1) |
2.2433(12) |
| N(1)–C(34) |
1.434(5) |
Fe(1)–P(2) |
2.2242(11) |
| Ni(1)–S(1)–Fe(1) |
69.04(4) |
S(2)–Fe(1)–S(1) |
87.28(4) |
| Ni(1)–S(2)–Fe(1) |
69.13(4) |
S(2)–Fe(1)–Ni(1) |
54.52(4) |
| S(1)–Ni(1)–S(2) |
89.93(4) |
S(1)–Fe(1)–Ni(1) |
54.43(3) |
| P(1)–Fe(1)–P(2) |
87.08(4) |
S(1)–Ni(1)–Fe(1) |
56.54(4) |
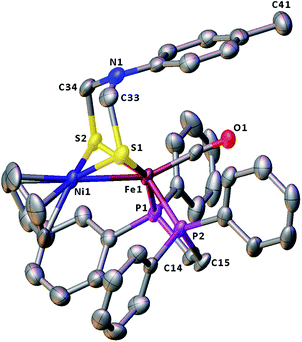 |
| | Fig. 4 Molecular structure of model complex 7 with ellipsoids drawn at a 50% probability level. All hydrogen atoms and the anion BF4− are omitted for clarity. | |
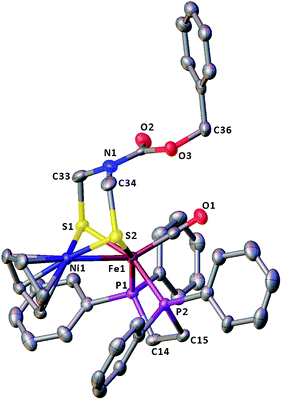 |
| | Fig. 5 Molecular structure of model complex 8 with ellipsoids drawn at a 50% probability level. All hydrogen atoms and the anion BF4− are omitted for clarity. | |
Electrochemical properties and electrocatalytic H2 production catalyzed by model complexes 7 and 9
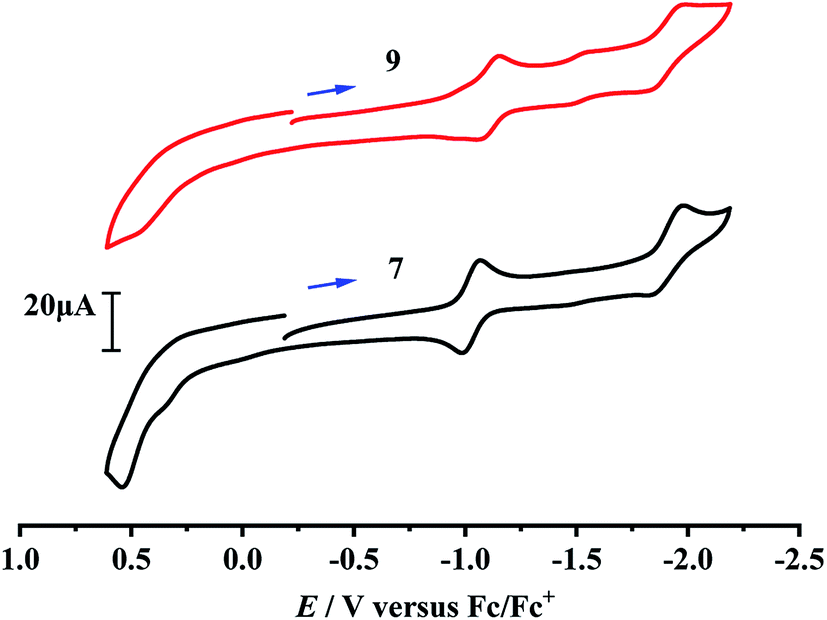
Although the electrochemical properties and electrocatalytic H2 production for some [NiFe]-H2ase models were previously studied by cyclic voltammetric techniques,40–46 there isn't any studied result regarding the electrochemical and electrocatalytic properties of the azadithiolato-bridged [NiFe]-H2ase models, so far. In order to see whether the prepared azadithiolato-bridged models 5–9 could have the electrocatalytic H2-producing ability, we chose models 7 and 9 (note that they have the same type of structures as 5, 6 and 8) as representatives to study their electrochemical properties by CV techniques using the supporting electrolyte n-Bu4NPF6 in MeCN at a scan rate of 100 mVs−1. As shown in Table 2 and Fig. 6, complex 7 displayed one quasi-reversible reduction wave at –1.07 V (due to its |Epc − EPa| = 82 mV and ipc/ipa = 1.05),47 one irreversible reduction wave at −1.98 V and one irreversible oxidation wave at 0.54 V, while complex 9 displayed one quasi-reversible reduction wave at −1.15 V (due to its |Epc − EPa| = 103 mV and ipc/ipa = 1.33),47 one irreversible reduction wave at −1.99 V, and one irreversible oxidation wave at 0.45 V, respectively. Previously, the electrochemical properties of the two pdt-bridged analogues of 7 and 9, namely complexes [CpNi(pdt)Fe(dppv)(CO)]BF4 (A) and [CpNi(pdt)Fe(dppe)(CO)]BF4 (B) (see Table 2) were studied by cyclic voltammetry.40 Similar to this case, the three redox events of 7 and 9 could be assigned to the NiIIFeII/NiIFeII, NiIFeII/NiIFeI and NiIIFeII/NiIII/FeII couples, respectively.
Table 2 Electrochemical data of 7/9a and A/Bb
| Complex |
Epc1/V |
Epc2/V |
Epa/V |
| All potentials are versus Fc/Fc+ in 0.1 M n-Bu4NPF6/MeCN at a scan rate of 0.1 V s−1. All potentials are versus Fc/Fc+ in 0.1 M n-Bu4NPF6/CH2Cl2 at a scan rate of 0.5 V s−1. |
| 7 |
−1.07 |
−1.98 |
0.54 |
| 9 |
−1.15 |
−1.99 |
0.45 |
| A40 |
−1.15 |
−2.18 |
0.72 |
| B40 |
−1.16 |
−2.15 |
0.70 |
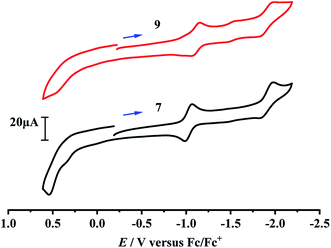 |
| | Fig. 6 Cyclic voltammograms of 7 and 9 (1.0 mM) in 0.1 M n-Bu4NPF6/MeCN at a scan rate of 0.1 V s−1. Arrows indicate the starting potential and scan direction. | |
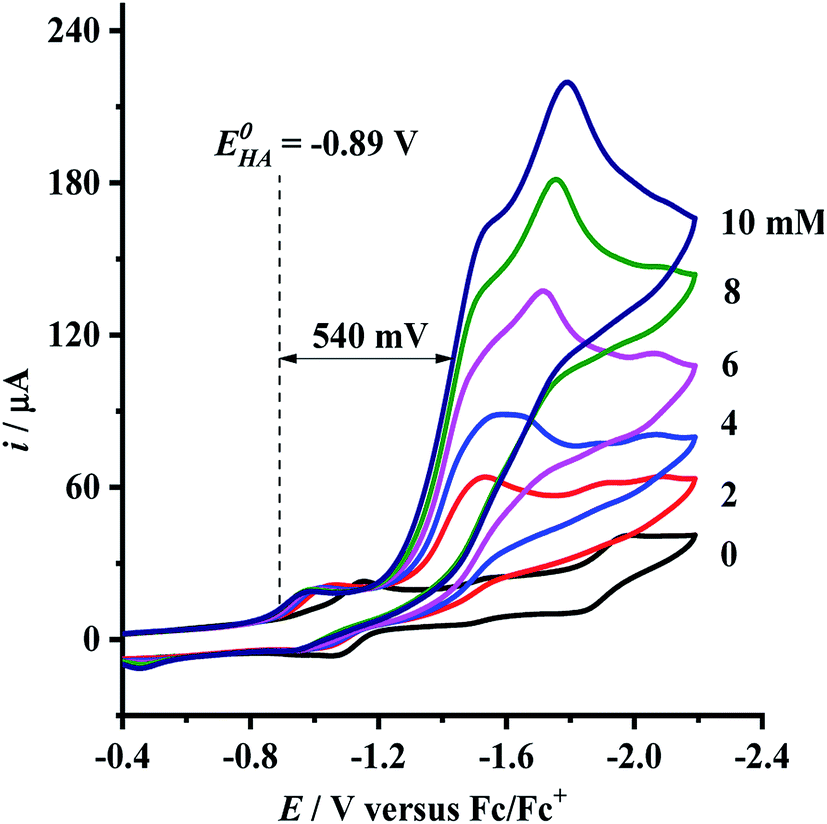
Having determined the electrochemical properties of 7 and 9, we further determined their cyclic voltammograms in the presence and absence (for comparison) of trifluoroacetic acid (TFA) (pKMeCNa = 12.7, 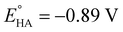 ),48,49 in order to see whether they could catalyze the proton reduction of TFA to give H2. As shown in Fig. 7 and 8, when TFA was sequentially added from 2 to 10 mM to MeCN solutions of 7 and 9, the current heights of their original first reduction waves were basically unchanged. However, in contrast to this, the current heights of their original second reduction waves were remarkably increased. Apparently, such an observation features an electrocatalytic proton reduction process from TFA to hydrogen.41,43 Note that through comparison of the reduction potentials Epc and peak currents ip in the CVs of 7/9 with those in the CVs of TFA without 7/9 (see Fig. S1 and Table S1 in the ESI†), models 7/9 were further proved to be catalysts for proton reduction. In addition, during the proton reductions catalyzed by 7 and 9, the original second reduction waves were positively shifted by ca. 15 and 45 mV, respectively; this is most likely due to protonation of the central N atoms in their bridged azadithiolato ligands.50
),48,49 in order to see whether they could catalyze the proton reduction of TFA to give H2. As shown in Fig. 7 and 8, when TFA was sequentially added from 2 to 10 mM to MeCN solutions of 7 and 9, the current heights of their original first reduction waves were basically unchanged. However, in contrast to this, the current heights of their original second reduction waves were remarkably increased. Apparently, such an observation features an electrocatalytic proton reduction process from TFA to hydrogen.41,43 Note that through comparison of the reduction potentials Epc and peak currents ip in the CVs of 7/9 with those in the CVs of TFA without 7/9 (see Fig. S1 and Table S1 in the ESI†), models 7/9 were further proved to be catalysts for proton reduction. In addition, during the proton reductions catalyzed by 7 and 9, the original second reduction waves were positively shifted by ca. 15 and 45 mV, respectively; this is most likely due to protonation of the central N atoms in their bridged azadithiolato ligands.50
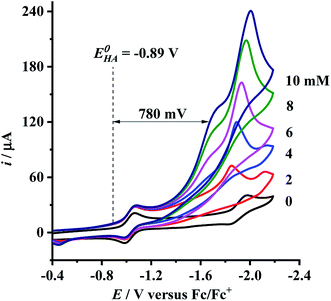 |
| | Fig. 7 Cyclic voltammograms of 7 (1.0 mM) with TFA (0–10 mM) in 0.1 M n-Bu4NPF6/MeCN at a scan rate of 0.1 V s−1. | |
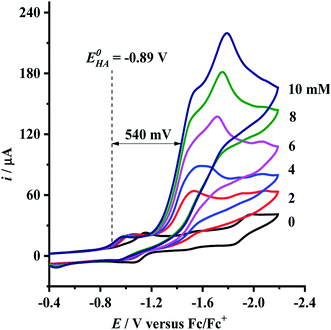 |
| | Fig. 8 Cyclic voltammograms of 9 (1.0 mM) with TFA (0–10 mM) in 0.1 M n-Bu4NPF6/MeCN at a scan rate of 0.1 V s−1. | |
Recently, we51 and others52 reported that some mononuclear Fe complexes can serve as catalysts for proton reduction to hydrogen. In order to observe whether the mononuclear Fe precursors of model complexes 5–8 (namely complexes 1–4) could catalyze the proton reductions, we chose complexes 3 and 4 as representatives to determine their cyclic voltammograms in the presence of TFA under the same conditions as those used for dertermination of models 7 and 9. We found that when TFA was sequentially added from 2 to 10 mM to MeCN solutions of 3 and 4, the current heights of the acid-induced reduction waves were considerably increased near their only one original reduction waves caused by their Fe(II)Fe(I) couples (see Fig. S2 and S3 in the ESI†). Obviously, such an observation demonstrates an electrocatalytic proton reduction process catalyzed by complexes 3 and 4. So, we might conclude that for mononuclear Fe complexes 3 and 4, only Fe makes contribution to their electrocatalytic proton reduction reactions caused by their Fe(II)/Fe(I) couples, while for dinuclear models 7 and 9, both Ni and Fe make contribution to the electrocatalytic proton reduction reactions occurred near their original second reduction waves caused by their Ni(I)Fe(II)/Ni(I)Fe(I) couples.
To evaluate the electrocatalytic H2-producing ability of 7 and 9, we determined the ratio of the catalytic current (icat) to reductive peak current (ip) in the absence of the added acid (note that a higher icat/ip value indicates a faster catalysis).53 At a concentration of 10 mM TFA, the icat/ip values of 7 and 9 were calculated to be 6.0 and 5.3, respectively. It follows that the H2-producing ability catalyzed by complex 7 (with a 4-MeC6H4N group and a dppv ligand) is higher than that catalyzed by 9 (with an NH group and a dppe ligand). However, in order to compare the electrocatalytic H2-producing ability of the bridgehead N-substituted complex 8 with its parent complex 9, we further determined its icat/ip value by CV techniques to be 6.4 under the same conditions. Therefore, the H2-producing ability of complex 8 with a stronger electron-attracting PhCH2CO2N group is higher than that of complex 9 with a weaker electron-attracting NH group. In addition, at a 10 mM TFA concentration, the icat/ip values (5.3–6.4) of complexes 7–9 are higher than those (ca. 4) of the previously reported analogues [HNiFe(pdt)(dppe)(L)(CO)2]BF4 (L = P(OPh)3, PPh3).54
Since overpotential is also an important parameter for evaluating the electrocatalytic ability for a catalyst (note that a better catalyst has a lower overpotential), we used Evans' method to calculate the overpotentials of 7 and 9 according to the following two equations:49,55
| |
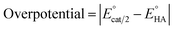 | (1) |
| |
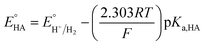 | (2) |
where
Ecat/2 is the catalytic half-wave potential and
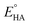
is the standard potential of the acid;
R is the ideal gas constant 8.314 J mol
−1 K
−1;
T is the absolute temperature 298 K;
F is the Faraday's constant 9.65 × 10
4 C mol
−1; in addition, the p
Ka value of TFA in MeCN and the standard redox potential
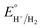
have been previously determined to be 12.7 (
ref. 48) and −0.14 V,
49 respectively. According to
eqn (2), the standard potential
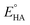
can be calculated to be −0.89 V and then according to
eqn (1), the overpotentials of
7 and
9 can be calculated to be 0.78 and 0.54 V, respectively (see
Fig. 7 and
8). At a 10 mM acid concentration, overpotentials of our azadithiolato-bridged dinuclear NiFe complexes
7 (0.78 V) and
9 (0.54 V) by using TFA as proton source are higher than those (
ca. 0.5 V) of the previously reported analogues [HNiFe(pdt)(dppe)(L)(CO)
2]BF
4 (L = P(OPh)
3, PPh
3) using TFA as proton source.
54 Particularly worth noting is that in terms of the studied results regarding the structure and catalytic function described above, complexes
7 and
9 might be regarded as not only the structural models but also the functional models for the active site of [NiFe]-H
2ases.
Summary and conclusion
We have synthesized the first azadithiolato-bridged dinuclear NiFe model complexes 5–8 and 9 by reactions of the N-substituted azadithiolato-chelated mononuclear Fe complexes 1–4 with Cp2Ni/HBF4·Et2O and by reaction of the N-substituted azadithiolato-bridged complex 8 with HBF4·Et2O, respectively. Particularly worth noting is that the spectroscopic characterization of all the new model complexes 5–9 and the X-ray crystallographic study on their three representative complexes 6–8 have proved that models 5–9 indeed contain an azadithiolato ligand that is bridged between their NiFe centers to form a butterfly Ni(II)S2Fe(II) cluster core with a Ni–Fe metal–metal bond. In addition, from the electrochemical and electrocatalytic studies, the two representative models 7 and 9 have been found to be catalysts for proton reduction to hydrogen using TFA as the proton source under electrochemical conditions. Therefore, according to the structural and functional similarity with the active site of [NiFe]-H2ases, complexes 5–9 might be regarded as the structural and functional models of [NiFe]-H2ases.
Experimental section
General comments
All reactions were carried out using standard Schlenk and vacuum-line techniques under an atmosphere of argon. While tetrahydrofuran (THF) and Et2O were distilled under Ar from sodium/benzophenone ketyl, CH2Cl2 was distilled from CaH2. t-BuONa, (η5-C5H5)2Ni, HBF4·Et2O (50–55% w/w in Et2O), anhydrous FeCl2 and other chemicals were commercially available and used as received. RN[CH2S(O)CMe]2 (R = Ph,56 4-ClC6H4,57 CO2CH2Ph,58 (dppv)Fe(CO)2Cl2 (dppv = 1,2-bis(diphenylphosphino)ethene)35 and (dppe)Fe(CO)2Cl2 (dppe = 1,2-bis(diphenylphosphino)ethane)36) were prepared according to the published procedures. While 1H, 13C{1H}, and 31P{1H} NMR spectra were obtained on a Bruker Avance 400 NMR spectrometer, IR spectra were recorded on a Bio-Rad FTS 135 infrared spectrophotometer. Elemental analyses were performed on an Elementar Vario EL analyzer. Melting points were determined on a SGW X-4 microscopic melting point apparatus and were uncorrected.
Preparation of [PhN(CH2S)2]Fe(CO)2(dppv) (1). A 250 mL Schlenk flask was charged with FeCl2 (0.634 g, 5.0 mmol) in 100 mL acetone and dppv (1.98 g, 5.0 mmol) in 20 mL of THF. The mixture was stirred at room temperature and bubbled with CO gas for 4 h to give a (dppv)Fe(CO)2Cl2-containing brown-red solution. Additionally, a 100 mL Schlenk flask was charged with thioester PhN[CH2S(O)CMe]2 (2.02 g, 7.5 mmol), sodium tert-butoxide (1.44 g, 15.0 mmol), and 40 mL of THF. The mixture was stirred at −78 °C for 4 h to give a PhN(CH2SNa)2-containing pale yellow solution. To the in situ prepared (dppv)Fe(CO)2Cl2-containing solution cooled to −78 °C was slowly added the prepared PhN(CH2SNa)2-containing solution and then the mixture was stirred at −78 °C for 5 h. Solvent was removed at reduced pressure and the residue was subjected to column chromatography (silica gel). Elution with CH2Cl2 developed a red band, from which 1 (0.622 g, 18%) was obtained as a brown-red solid, mp 93–94 °C. Anal. calcd for C36H31FeNO2P2S2: C, 62.52; H, 4.52; N, 2.03. Found: C, 62.35; H, 4.65; N, 2.00. IR (KBr disk): νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O 2018 (w), 1973 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 4.63 (s, 4H, CH2NCH2), 6.69–7.91 (m, 27H, CHCH, 5C6H5) ppm. 13C{1H} NMR (100 MHz, CDCl3): 48.6 (s, CH2NCH2), 114.7–134.7 (m, C6H5), 146.3–150.8 (m, CHCH), 211.8, 212.2 (2s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 79.9 (s, FeP) ppm.
O 2018 (w), 1973 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 4.63 (s, 4H, CH2NCH2), 6.69–7.91 (m, 27H, CHCH, 5C6H5) ppm. 13C{1H} NMR (100 MHz, CDCl3): 48.6 (s, CH2NCH2), 114.7–134.7 (m, C6H5), 146.3–150.8 (m, CHCH), 211.8, 212.2 (2s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 79.9 (s, FeP) ppm.
Preparation of [4-ClC6H4N(CH2S)2]Fe(CO)2(dppv) (2). The same procedure was followed as for the preparation of 1, except that 4-ClC6H4N[CH2S(O)CMe]2 (2.28 g, 7.5 mmol) was used instead of PhN[CH2S(O)CMe]2. 2 (0.835 g, 23%) was obtained as a brown-red solid, mp 145–146 °C. Anal. calcd for C36H30ClFeNO2P2S2: C, 59.56; H, 4.17; N, 1.93. Found: C, 59.47; H, 4.21; N, 1.69. IR (KBr disk): νCO 2020 (w), 1973 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 4.57 (s, 4H, CH2NCH2), 6.61–7.78 (m, 26H, CHCH, 4C6H5, C6H4) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): 48.0 (s, CH2NCH2), 115.5–144.1 (m, C6H5, C6H4), 150.2–150.6 (m, CHCH), 212.2 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 80.0 (s, FeP) ppm.
Preparation of [4-MeC6H4N(CH2S)2]Fe(CO)2(dppv) (3). In order to prepare 3, we should first prepare its precursor, the new bis(thioester) 4-MeC6H4N[CH2S(O)CMe]2. (i) Preparation of 4-MeC6H4N[CH2S(O)CMe]2: a 250 mL three-necked flask was charged with 4-MeC6H4NH2 (10.7 g, 100 mmol), 37% aqueous formaldehyde (24.2 mL, 320 mmol), thioacetic acid (14.6 mL, 200 mmol), and ethanol (100 mL). While stirring, the mixture was heated to about 60 °C and stirred at this temperature for 3 h. To the resulting mixture was added 100 mL of ice-water to give some precipitates. The precipitates were collected by filtration, washed with some cold ethanol, and finally dried under vacuum to give 4-MeC6H4N[CH2S(O)CMe]2 (25.6 g, 90%) as a white solid, mp 66–67 °C. Anal. calcd for C13H17NO2S2: C, 55.10; H, 6.05; N, 4.95. Found: C, 55.35; H, 6.30; N, 4.66. IR (KBr disk): νCO 1688 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 2.27 (s, 3H, CH3C6H4), 2.36 (s, 6H, 2CH3CO), 5.10 (s, 4H, CH2NCH2), 6.68–7.10 (m, 4H, C6H4) ppm. 13C{1H} NMR (100 MHz, CDCl3): 20.5 (s, CH3C6H4), 31.4 (s, CH3CO), 53.1 (s, CH2NCH2), 114.7–141.6 (m, C6H4), 196.1 (s, CO) ppm. (ii) Preparation of 3: the same procedure was followed as for the preparation of 1, except that 4-MeC6H4N[CH2S(O)CMe]2 (2.13 g, 7.5 mmol) was employed in place of PhN[CH2S(O)CMe]2. 3 (0.741 g, 21%) was obtained as a brown-red solid, mp 113–114 °C. Anal. calcd for C37H33FeNO2P2S2: C, 62.98; H, 4.71; N, 1.99. Found: C, 62.72; H, 4.95; N, 2.05. IR (KBr disk): νCO 2035 (w), 1973 (vs.) cm−1. 1H NMR (400 MHz, acetone-d6): 2.18 (s, 3H, CH3), 4.56 (s, 4H, CH2NCH2), 6.92–7.80 (m, 24H, 4C6H5, C6H4), 8.17–8.31 (m, 2H, CHCH) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): 17.7 (s, CH3), 45.9 (s, CH2NCH2), 113.1–131.6 (m, C6H5, C6H4), 144.7–145.8 (m, CHCH), 200.1 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 77.9 (s, FeP) ppm.
Preparation of [PhCH2O2CN(CH2S)2]Fe(CO)2(dppe) (4). The same procedure was followed as for the preparation of 1, except that PhCH2O2CN[CH2S(O)CMe]2 (2.46 g, 7.5 mmol) was used in place of PhN[CH2S(O)CMe]2. 4 (1.32 g, 35%) was obtained as a brown-red solid, mp 91–92 °C. Anal. calcd for C38H35FeNO4P2S2: C, 60.73; H, 4.69; N, 1.86. Found: C, 60.42; H, 4.94; N, 1.66. IR (KBr disk): νCO 2013 (vs.), 1968 (vs.); νCO 1700 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 3.02–4.56 (m, 6H, PCH2CH2P, OCH2); 4.89–5.30 (m, 4H, CH2NCH2); 7.30–7.76 (m, 25H, 5C6H5) ppm. 13C{1H} NMR (100 MHz, CDCl3): 24.6–57.0 (m, PCH2CH2P, OCH2), 66.7–68.0 (m, CH2NCH2), 127.2–154.7 (m, C6H5), 210.8 (s, CO), 213.0, 213.3 (2s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 44.3 (s, FeP) ppm.
Preparation of [CpNi{(μ-SCH2)2NPh}Fe(CO)(dppv)]BF4 (5). While stirring, nickelocene (0.038 g, 0.2 mmol) was dissolved in diethyl ether (5 mL) and then HBF4·Et2O (27.2 μL, 0.2 mmol) was added. After the mixture was stirred at room temperature for 15 min, a brown-red suspension was formed. To this suspension was added a solution of 1 (0.138 g, 0.2 mmol) in CH2Cl2 (5 mL). The new mixture was stirred at room temperature for 2 h, solvent was removed at reduced pressure to give a residue, which was subjected to column chromatography (silica gel), elution with CH2Cl2/acetone (v/v = 10:1) developed a major red band, from which 5 (0.052 g, 30%) was obtained as brown-red solid. Mp 220–221 °C. Anal. calcd for C40H36BF4FeNNiOP2S2: C, 54.96; H, 4.15; N, 1.60. Found: C, 54.86; H, 3.98; N, 1.62. IR (KBr disk): νCO 1956 (m) cm−1. 1H NMR (400 MHz, acetone-d6): 4.43 (s, 5H, C5H5), 4.68, 5.47 (dd, J = 14.6 Hz, 4H, CH2NCH2), 6.77–8.13 (m, 25H, 5C6H5), 8.71, 8.86 (2s, 2H, CHCH) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): 54.4 (s, CH2NCH2), 94.5, 95.9 (2s, C5H5), 115.4–142.2 (m, C6H5), 149.3–149.9 (m, CHCH), 205.9 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 82.7 (s, FeP) ppm.
Preparation of [CpNi{(μ-SCH2)2NC6H4Cl-4}Fe(CO)(dppv)]BF4 (6). The same procedure was followed as for the preparation of 5, except that 2 (0.145 g, 0.2 mmol) was utilized instead of 1. Complex 6 (0.113 g 62%) was obtained as a brown-red solid, mp 197–198 °C. Anal. calcd for C40H35BClF4FeNNiOP2S2: C, 52.88; H, 3.88; N, 1.54. Found: C, 52.84; H, 4.01; N, 1.62. IR (KBr disk): νCO 1952 (m) cm−1. 1H NMR (400 MHz, acetone-d6): 4.47 (s, 5H, C5H5), 4.64, 5.49 (dd, 4H, J = 15.4 Hz, CH2NCH2), 7.32–8.13 (m, 24H, 4C6H5, C6H4), 8.73, 8.88 (2s, 2H, CHCH) ppm. 13C{1H} NMR (100 MHz, CD2Cl2): 52.6, 54.1 (2s, CH2NCH2), 94.8, 96.5 (2s, C5H5), 117.0–142.4 (m, C6H5, C6H4), 149.4–151.3 (m, CHCH), 210.9 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 83.9 (s, FeP) ppm.
Preparation of [CpNi{(μ-SCH2)2NC6H4Me-4}Fe(CO)(dppv)]BF4 (7). The same procedure was followed as for the preparation of 5, except that 3 (0.141 g, 0.2 mmol) was employed in place of 1. Complex 7 (0.038 g, 21%) was obtained as a brown-red solid, mp 210–211 °C. Anal. calcd for C41H38BF4FeNNiOP2S2: C, 55.45; H, 4.31; N, 1.58. Found: C, 55.23; H, 4.28; N, 1.34. IR (KBr disk): νCO 1955 (s) cm−1. 1H NMR (400 MHz, acetone-d6): 2.12 (s, 3H, CH3), 4.42 (s, 5H, C5H5), 4.67, 5.41 (dd, 4H, J = 13.0 Hz, CH2NCH2), 7.14–8.13 (m, 24H, 4C6H5, C6H4), 8.69–8.84 (m, 2H, CHCH) ppm. 13C{1H} NMR (100 MHz, CD3CN): 20.1 (s, CH3), 54.8 (s, CH2NCH2), 96.1 (s, C5H5), 117.0–141.5 (m, C6H5, C6H4), 150.5–151.2 (m, CHCH), 219.5 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 83.7 (s, FeP) ppm.
Preparation of [CpNi{(μ-SCH2)2NCO2CH2Ph}Fe(CO)(dppe)]BF4 (8). The same procedure was followed as for the preparation of 5, except that 4 (0.150 g, 0.2 mmol) was used instead of 1. Complex 8 (0.052 g, 28%) was obtained as a brown-red solid, mp 121–122 °C. Anal. calcd for C42H40BF4FeNNiO3P2S2: C, 54.00; H, 4.32; N, 1.50. Found: C, 54.27; H, 4.43; N, 1.58. IR (KBr disk): νCO 1940 (vs.); νCO 1704 (vs.) cm−1. 1H NMR (400 MHz, CDCl3): 3.03–4.05 (m, 6H, PCH2CH2P, OCH2), 4.54 (s, 5H, C5H5), 4.98–5.31 (m, 4H, CH2NCH2), 7.12–7.86 (m, 25H, 5C6H5) ppm. 13C{1H} NMR (100 MHz, CD3CN): 29.5, 29.8 (2s, PCH2CH2P), 50.7 (s, CH2NCH2), 69.5 (s, OCH2), 96.5 (s, C5H5), 128.9–154.1 (m, C6H5), 211.0 (s, CO), 216.4 (s, CO) ppm. 31P{1H} NMR (162 MHz, CDCl3): 78.3, 79.1 (2s, FeP) ppm.
Preparation of [CpNi{(μ-SCH2)2NH}Fe(CO)(dppe)]BF4 (9). To a solution of 8 (0.093 g, 0.1 mmol) in CH2Cl2 (5 mL) were added PhOMe (0.5 mL) and HBF4·Et2O (408 μL, 3.0 mmol). The mixture was stirred at room temperature for 4 h to give a brown-red solution. Solvents were removed to give a residue, which was subjected to column chromatography (silica gel). Elution with CH2Cl2/acetone developed a major red band, from which 9 (0.034 g, 42%) was obtained as a brown-red solid, mp 170 °C (dec). Anal. calcd for C34H34BF4FeNNiOP2S2: C, 51.04; H, 4.28; N, 1.75. Found: C, 50.81; H, 4.56; N, 1.70. IR (KBr disk): νCO 1939 (vs.) cm−1. 1H NMR (400 MHz, CD3CN): 2.26 (br.s, 1H, NH), 3.04–4.42 (m, 8H, PCH2CH2P, CH2NCH2), 4.61 (s, 5H, C5H5), 7.43–7.95 (m, 20H, 4C6H5) ppm. 13C{1H} NMR (100 MHz, CD3CN): 29.2, 29.6 (2s, PCH2CH2P), 52.4 (s, CH2NCH2), 95.7 (s, C5H5), 129.2–133.8 (m, C6H5), 218.3 (s, CO) ppm. 31P{1H} NMR (162 MHz, acetone-d6): 78.1 (s, FeP) ppm.
Electrochemical and electrocatalytic experiments
Acetonitrile (Amethyst Chemicals, HPLC grade) for electrochemical and electrocatalytic experiments was used directly without further purification. A solution of 0.1 M n-Bu4NPF6 in MeCN was used as the electrolyte in all CV experiments. The n-Bu4NPF6 electrolyte was dried in an oven at 110 °C for at least 24 h. Dry N2 was purged through the solutions for at least 10 min before the measurements were taken. The measurements were made by using a BAS Epsilon potentiostat. All voltammograms were obtained in a three-electrode cell with a 3 mm diameter glass carbon working electrode, a platinum counter electrode, and a Ag/Ag+ (0.01 M AgNO3/0.1 M n-Bu4NPF6 in MeCN) reference electrode under an atmosphere of nitrogen. The working electrode was polished with 0.05 μm alumina paste and sonicated in water for about 10 min prior to use. All potentials are quoted against Fc/Fc+ potential.
X-ray crystal structure determinations of 6−8
While single crystals of 6–8 for X-ray diffraction analysis were grown by slow diffusion of hexane into their CH2Cl2 solutions at −5 °C. A single crystal of 6–8 was mounted on a Rigaku MM-007 (rotating anode) diffractometer equipped with a Bruker P4 accessory. While the data of 6 and 8 were collected using a confocal monochromator with Mo Kα radiation (λ = 0.71073 Å) in the ω scanning mode at 113 K, data of 7 were collected using a confocal monochromator with Mo Kα radiation (λ = 0.71073 Å) in the ω scanning mode at 293 K, respectively. Data collection, reduction, and absorption correction were performed by the CRYSTALCLEAR program.59 The structures were solved by direct methods using the SHELXS program60 and refined by full-matrix least-squares techniques (SHELXL)61 on F2. Hydrogen atoms were located by using the geometric method. Details of crystal data, data collections, and structure refinements are summarized in Table 3.
Table 3 Crystal data and structure refinement details for 6–8
| |
6 |
7 |
8 |
| Formula |
C40H35BClF4FeNNiO |
C41H38BF4FeNNiO |
C42H40BF4FeNNiO3 |
| P2S2 |
P2S2·CH2Cl2 |
P2S2·CH2Cl2 |
| Mw |
908.57 |
973.08 |
1019.10 |
| Cryst syst |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P121/c1 |
P121/c1 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
17.861(4) |
17.8793(11) |
10.113(2) |
| b (Å) |
11.063(2) |
11.0429(5) |
12.365(3) |
| c (Å) |
22.252(5) |
22.4895(10) |
18.383(4) |
| α (°) |
90 |
90 |
75.55(3) |
| β (°) |
108.15(3) |
108.615(6) |
87.96(3) |
| γ (°) |
90 |
90 |
77.76(3) |
| V (Å3) |
4178.2(16) |
4208.0(4) |
2175.1(8) |
| Z |
4 |
4 |
2 |
| Crystal size (mm) |
0.05 × 0.04 × 0.03 |
0.06 × 0.05 × 0.03 |
0.20 × 0.18 × 0.12 |
| Dc (g cm−3) |
1.444 |
1.536 |
1.556 |
| μ (mm−1) |
1.088 |
1.148 |
1.117 |
| F(000) |
1856 |
1992 |
1044 |
| Reflns collected |
49278 |
43090 |
26271 |
| Reflns unique |
9958 |
8586 |
10291 |
| θmin/max (°) |
2.078/27.863 |
1.905/26.372 |
1.144/27.834 |
| Final R |
0.0693 |
0.0712 |
0.0567 |
| Final RW |
0.1648 |
0.1987 |
0.1575 |
| GOF on F2 |
1.127 |
1.061 |
1.060 |
| Δρmax/min (e Å−3) |
0.536/−0.576 |
1.000/−1.039 |
0.738/−1.255 |
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (21772106) and the Ministry of Science and Technology of China (973 program 2014CB845604) for financial support.
References
- R. Cammack, Nature, 1999, 397, 214–215 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg Chem., 2005, 10, 239–249 CrossRef CAS PubMed.
- R. Cammack, M. Frey and R. Robson, Hydrogen as a fuel: Learning from nature, Taylor & Francis, London, 2001 Search PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- S. Ogo, Coord. Chem. Rev., 2017, 334, 43–53 CrossRef CAS.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- N. J. Lindenmaier, S. Wahlefeld, E. Bill, T. Szilvási, C. Eberle, S. Yao, P. Hildebrandt, M. Horch, I. Zebger and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 2208–2211 CrossRef CAS PubMed.
- C. U. Perotto, G. Marshall, G. J. Jones, E. S. Davies, W. Lewis, J. McMaster and M. Schröder, Chem. Commun., 2015, 51, 16988–16991 RSC.
- C. U. Perotto, C. L. Sodipo, G. J. Jones, J. P. Tidey, A. J. Blake, W. Lewis, E. S. Davies, J. McMaster and M. Schröder, Inorg. Chem., 2018, 57, 2558–2569 CrossRef CAS PubMed.
- F. Osterloh, W. Saak, D. Haase and S. Pohl, Chem. Commun., 1997, 979–980 RSC.
- L.-C. Song, Y. Lu, L. Zhu and Q.-L. Li, Organometallics, 2017, 36, 750–760 CrossRef CAS.
- J. Jiang, M. Maruani, J. Solaimanzadeh, W. Lo, S. A. Koch and M. Millar, Inorg. Chem., 2009, 48, 6359–6361 CrossRef CAS PubMed.
- D. Sellmann, F. Geipel, F. Lauderbach and F. W. Heinemann, Angew. Chem., Int. Ed., 2002, 41, 632–634 CrossRef CAS.
- L.-C. Song, X.-Y. Gao, W.-B. Liu, H.-T. Zhang and M. Cao, Organometallics, 2018, 37, 1050–1061 CrossRef CAS.
- M. C. Smith, J. E. Barclay, S. P. Cramer, S. C. Davies, W.-W. Gu, D. L. Hughes, S. Longhurst and D. J. Evans, J. Chem. Soc., Dalton Trans., 2002, 2641–2647 RSC.
- J. A. W. Verhagen, M. Lutz, A. L. Spek and E. Bouwman, Eur. J. Inorg. Chem., 2003, 3968–3974 CrossRef CAS.
- Z. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950–8951 CrossRef CAS PubMed.
- D. Sellmann, F. Lauderbach and F. W. Heinemann, Eur. J. Inorg. Chem., 2005, 371–377 CrossRef CAS.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- Y. Ohki, K. Yasumura, M. Ando, S. Shimokata and K. Tatsumi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3994–3997 CrossRef CAS PubMed.
- D. Brazzolotto, L. Wang, H. Tang, M. Gennari, N. Queyriaux, C. Philouze, S. Demeshko, F. Meyer, M. Orio, V. Artero, M. B. Hall and C. Duboc, ACS Catal., 2018, 8, 10658–10667 CrossRef CAS.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pécaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
-
(a) L.-C. Song, X.-Y. Yang, M. Cao, X.-Y. Gao, B.-B. Liu, L. Zhu and F. Jiang, Chem. Commun., 2017, 53, 3818–3821 RSC;
(b) L.-C. Song, X.-Y. Yang, X.-Y. Gao and M. Cao, Inorg. Chem., 2019, 58, 39–42 CrossRef CAS PubMed;
(c) L.-C. Song, X.-J. Sun, P.-H. Zhao, J.-P. Li and H.-B. Song, Dalton Trans., 2012, 41, 8941–8950 RSC.
- H.-J. Fan and M. B. Hall, J. Am. Chem. Soc., 2001, 123, 3828–3829 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J.-M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- L. P. Battaglia, G. P. Chiusoli, D. Delledonne, M. Nardelli, C. Pelizzi and G. Predieri, Gazz. Chim. Ital., 1989, 119, 345–348 CAS.
- T. A. Manuel, Inorg. Chem., 1963, 2, 854–858 CrossRef CAS.
- A. Salzer and H. Werner, Angew. Chem., Int. Ed., 1972, 11, 930–932 CrossRef CAS.
- C. Elschenbroich, Organometallics. Third, Completely Revised and Extended Edition, WILEY-VCH Verlag GMBH & Co. KGdt, Weinheim Germany, 2006, p. 506 Search PubMed.
- C. Fichtner, C. Laurich, E. Bothe and W. Lubitz, Biochemistry, 2006, 45, 9706–9716 CrossRef CAS PubMed.
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- G. M. Chambers, M. T. Huynh, Y. Li, S. Hammes-Schiffer, T. B. Rauchfuss, E. Reijerse and W. Lubitz, Inorg. Chem., 2016, 55, 419–431 CrossRef CAS PubMed.
- L.-C. Song, X.-F. Han, W. Chen, J.-P. Li and X.-Y. Wang, Dalton Trans., 2017, 46, 10003–10013 RSC.
- M. E. Carroll, B. E. Barton, D. L. Gray, A. E. Mack and T. B. Rauchfuss, Inorg. Chem., 2011, 50, 9554–9563 CrossRef CAS PubMed.
- L.-C. Song, M. Cao and Y.-X. Wang, Dalton Trans., 2015, 44, 6797–6808 RSC.
- S. Canaguier, L. Vaccaro, V. Artero, R. Ostermann, J. Pécaut, M. J. Field and M. Fontecave, Chem.-Eur. J., 2009, 15, 9350–9364 CrossRef CAS PubMed.
- M. Y. Darensbourg, I. Font, D. K. Mills, M. Pala and J. H. Reibenspies, Inorg. Chem., 1992, 31, 4965–4971 CrossRef CAS.
- B. Adhikary, S. Liu and C. R. Lucas, Inorg. Chem., 1993, 32, 5957–5962 CrossRef CAS.
- P. Zanello, Inorganic Electrochemistry. Theory, Practice and Application, Thomas Graham House, Cambridge, UK, 2003 Search PubMed.
- K. Izutsu, Acid-Base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell Scientific Publications, Oxford, UK, 1990 Search PubMed.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- W. Dong, M. Wang, T. Liu, X. Liu, K. Jin and L. Sun, J. Inorg. Biochem., 2007, 101, 506–513 CrossRef CAS PubMed.
-
(a) L.-C. Song, L. Zhu, F.-Q. Hu and Y.-X. Wang, Inorg. Chem., 2017, 56, 15216–15230 CrossRef CAS PubMed;
(b) L.-C. Song, F.-Q. Hu, M.-M. Wang, Z.-J. Xie, K.-K. Xu and H.-B. Song, Dalton Trans., 2014, 43, 8062–8071 RSC.
-
(a) S. C. Eady, T. Breault, L. Thompson and N. Lehnert, Dalton Trans., 2016, 45, 1138–1151 RSC;
(b) S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033–8036 CrossRef CAS PubMed.
- G. A. N. Felton, C. A. Mebi, B. J. Petro, A. K. Vannucci, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Organomet. Chem., 2009, 694, 2681–2699 CrossRef CAS.
- B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877–14885 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- L.-C. Song, W.-B. Liu and B.-B. Liu, Organometallics, 2020, 39, 1431–1439 CrossRef CAS.
- T. Izawa, Y. Terao and K. Suzuki, Tetrahedron: Asymmetry, 1997, 8, 2645–2648 CrossRef CAS.
- C. Sommer, C. P. Richers, W. Lubitz, T. B. Rauchfuss and E. J. Reijerse, Angew. Chem., Int. Ed., 2018, 57, 5429–5432 CrossRef CAS PubMed.
- Rigaku Americas and Rigaku, CrystalClear and CrystalStructure, Rigaku and Rigaku Americas, The Woodlands, TX, 2007 Search PubMed.
-
(a) G. M. Sheldrick, SHELXS97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Some electrocatalytic plots/data (Fig. S1–S3/Table S1) and some IR/NMR spectra (Fig. S4–S23). CCDC 2001290–2001292. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04344c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Bei-Bei Liua,
Wen-Bo Liua and
Zheng-Lei Tana
*ab,
Bei-Bei Liua,
Wen-Bo Liua and
Zheng-Lei Tana
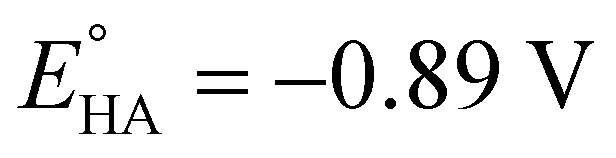 ),48,49 in order to see whether they could catalyze the proton reduction of TFA to give H2. As shown in Fig. 7 and 8, when TFA was sequentially added from 2 to 10 mM to MeCN solutions of 7 and 9, the current heights of their original first reduction waves were basically unchanged. However, in contrast to this, the current heights of their original second reduction waves were remarkably increased. Apparently, such an observation features an electrocatalytic proton reduction process from TFA to hydrogen.41,43 Note that through comparison of the reduction potentials Epc and peak currents ip in the CVs of 7/9 with those in the CVs of TFA without 7/9 (see Fig. S1 and Table S1 in the ESI†), models 7/9 were further proved to be catalysts for proton reduction. In addition, during the proton reductions catalyzed by 7 and 9, the original second reduction waves were positively shifted by ca. 15 and 45 mV, respectively; this is most likely due to protonation of the central N atoms in their bridged azadithiolato ligands.50
),48,49 in order to see whether they could catalyze the proton reduction of TFA to give H2. As shown in Fig. 7 and 8, when TFA was sequentially added from 2 to 10 mM to MeCN solutions of 7 and 9, the current heights of their original first reduction waves were basically unchanged. However, in contrast to this, the current heights of their original second reduction waves were remarkably increased. Apparently, such an observation features an electrocatalytic proton reduction process from TFA to hydrogen.41,43 Note that through comparison of the reduction potentials Epc and peak currents ip in the CVs of 7/9 with those in the CVs of TFA without 7/9 (see Fig. S1 and Table S1 in the ESI†), models 7/9 were further proved to be catalysts for proton reduction. In addition, during the proton reductions catalyzed by 7 and 9, the original second reduction waves were positively shifted by ca. 15 and 45 mV, respectively; this is most likely due to protonation of the central N atoms in their bridged azadithiolato ligands.50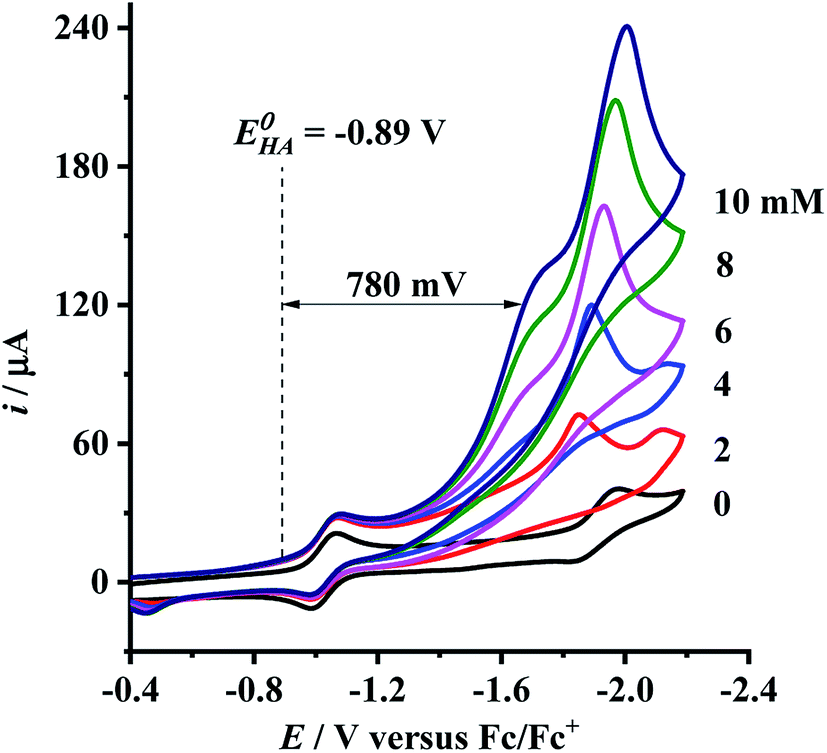
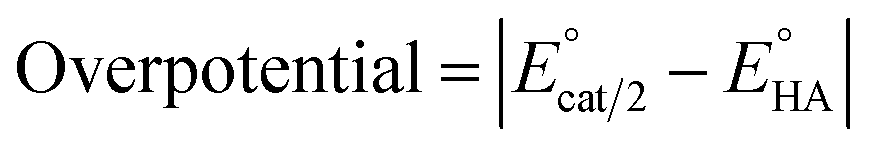
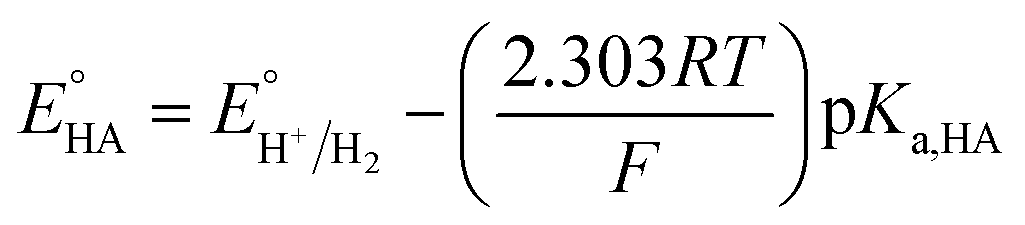
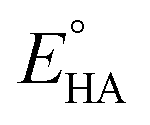 is the standard potential of the acid; R is the ideal gas constant 8.314 J mol−1 K−1; T is the absolute temperature 298 K; F is the Faraday's constant 9.65 × 104 C mol−1; in addition, the pKa value of TFA in MeCN and the standard redox potential
is the standard potential of the acid; R is the ideal gas constant 8.314 J mol−1 K−1; T is the absolute temperature 298 K; F is the Faraday's constant 9.65 × 104 C mol−1; in addition, the pKa value of TFA in MeCN and the standard redox potential 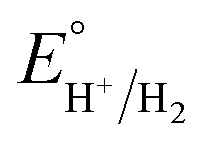 have been previously determined to be 12.7 (ref. 48) and −0.14 V,49 respectively. According to eqn (2), the standard potential
have been previously determined to be 12.7 (ref. 48) and −0.14 V,49 respectively. According to eqn (2), the standard potential 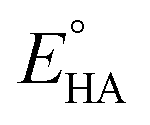 can be calculated to be −0.89 V and then according to eqn (1), the overpotentials of 7 and 9 can be calculated to be 0.78 and 0.54 V, respectively (see Fig. 7 and 8). At a 10 mM acid concentration, overpotentials of our azadithiolato-bridged dinuclear NiFe complexes 7 (0.78 V) and 9 (0.54 V) by using TFA as proton source are higher than those (ca. 0.5 V) of the previously reported analogues [HNiFe(pdt)(dppe)(L)(CO)2]BF4 (L = P(OPh)3, PPh3) using TFA as proton source.54 Particularly worth noting is that in terms of the studied results regarding the structure and catalytic function described above, complexes 7 and 9 might be regarded as not only the structural models but also the functional models for the active site of [NiFe]-H2ases.
can be calculated to be −0.89 V and then according to eqn (1), the overpotentials of 7 and 9 can be calculated to be 0.78 and 0.54 V, respectively (see Fig. 7 and 8). At a 10 mM acid concentration, overpotentials of our azadithiolato-bridged dinuclear NiFe complexes 7 (0.78 V) and 9 (0.54 V) by using TFA as proton source are higher than those (ca. 0.5 V) of the previously reported analogues [HNiFe(pdt)(dppe)(L)(CO)2]BF4 (L = P(OPh)3, PPh3) using TFA as proton source.54 Particularly worth noting is that in terms of the studied results regarding the structure and catalytic function described above, complexes 7 and 9 might be regarded as not only the structural models but also the functional models for the active site of [NiFe]-H2ases.