
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetro Diels Alder protocol for regioselective synthesis of novel [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones†
Awad I.
Said
 ab,
Márta
Palkó
a,
Matti
Haukka
c and
Ferenc
Fülöp
*a
ab,
Márta
Palkó
a,
Matti
Haukka
c and
Ferenc
Fülöp
*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary. E-mail: awadsaid@aun.edu.eg; fulop@pharm.u-szeged.hu
bChemistry Department, Faculty of Science, Assiut University, Assiut, 71516, Egypt
cDepartment of Chemistry, University of Jyväskulä, FIN-40014, Jyväskulä, Finland
First published on 14th September 2020
Abstract
Reactions of diastereochemically varied norbornene-condensed 2-thioxopyrimidin-4-ones 6 and 10 with variously functionalized hydrazonoyl chlorides 2a–h gave regioselectively angular norbornene-based [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones 7a–h and 11a,c–e, respectively. Thermal retro Diels–Alder (RDA) reaction of 7a–h and 11a,c–e resulted in the target compounds 4a–h as single products. On the other hand, reactions of thiouracil 1 and hydrozonoyl chlorides 2a–e gave regioselectively [1,2,4]triazolo[4,3-a]pyrimidinone-5(1H)-ones 3a–e. The opposite regioselectivity of thiouracil 1 and norbornene-condensed 2-thioxopyrimidin-4-ones 6 and 10 was attributed to electronic factors according to DFT calculations. The angular structure of norbornene based [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones was confirmed by single crystal X-ray crystallography.
Introduction
Fused pyrimidines are important compounds, because of their wide spectrum of biological and pharmacological applications.1–4 Compounds bearing the [1,2,4]triazolo[4,3-a]pyrimidinone ring system were reported to possess a wide range of pharmacological activities including antitumor,5 antiallergic,6 antimicrobial,7 and 5α-reductase inhibitor properties.8Hydrazonoyl halides, known since 1930, have attracted the interest of synthetic chemists in designing and synthesizing different heterocycles.9 For example, [1,2,4]triazolo[4,3-a]pyrimidinones decorated with different functionalities can be incorporated into 2-thioxopyrimidin-4-ones by reacting the latter with the appropriate hydrazonoyl halide in the presence of a base.10 To the best of our knowledge, the reported reactions of thiouracil derivatives with hydrazonoyl chlorides gave substituted [1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones in a regioselective manner.11–15
Recently, synthetic chemists have utilized RDA protocols for designing and synthesizing novel heterocyclic scaffolds. This method is highly efficient to build a double bond into the heterocyclic system.16–23
In the present work, we report the formation of novel [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones 4a–h by reacting norbornene-condensed 2-thioxopyrimidin-4-ones 6 and 10 with hydrazonyl chlorides 2a–h with various functionalities followed by applying the RDA protocol. Note that 4a–h, cann't accessible significantly from the reactions of thiouracil with hydrazonoyl chlorides.
Results and discussion
The reaction of thiouracil 1 with varied functionalized hydrazonoyl chlorides 2a–e gave, as described earlier,11–15 [1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones 3a–e as the major and [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones 4a–e as the minor products. The ratio of 3a–e and 4a–e was 8–2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 1). The regioisomers could be easily separated by column chromatography via elution with EtOAc.
:1 (Scheme 1). The regioisomers could be easily separated by column chromatography via elution with EtOAc.
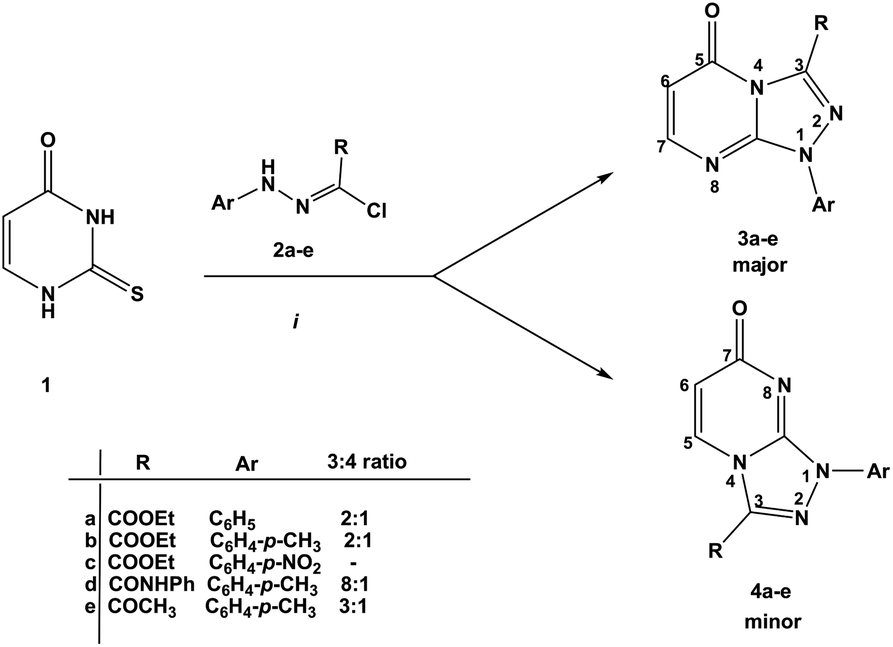 | ||
| Scheme 1 Solvents and conditions: dioxane, TEA, reflux 4–6 h. The ratio of compounds 3:4 was calculated from the 1H-NMR spectrum of the crude reaction mixture using the signals at around 6.1 ppm and 6.5 ppm for H-6 of regioisomers 3 and 4, respectively. Separation of the regioisomers by column chromatography, eluent: EtOAc, Rf = 3: 0.4; 4: 0.05. | ||
The challenge hence is how to have 4a–e as major products. Since 2-thioxopyrimidin-4-one 6 condensed to norbornene can easily be converted to thiouracil 1 by the RDA strategy,24 we used 6 instead of thiouracil 1 in the reaction with hydrazonoyl chlorides 2a–h (Scheme 2). Strikingly, the regioselectivity of the reaction was inversed. Only angular regioisomers, [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones 7a–h, were formed and isolated in moderate yields (Table 1).
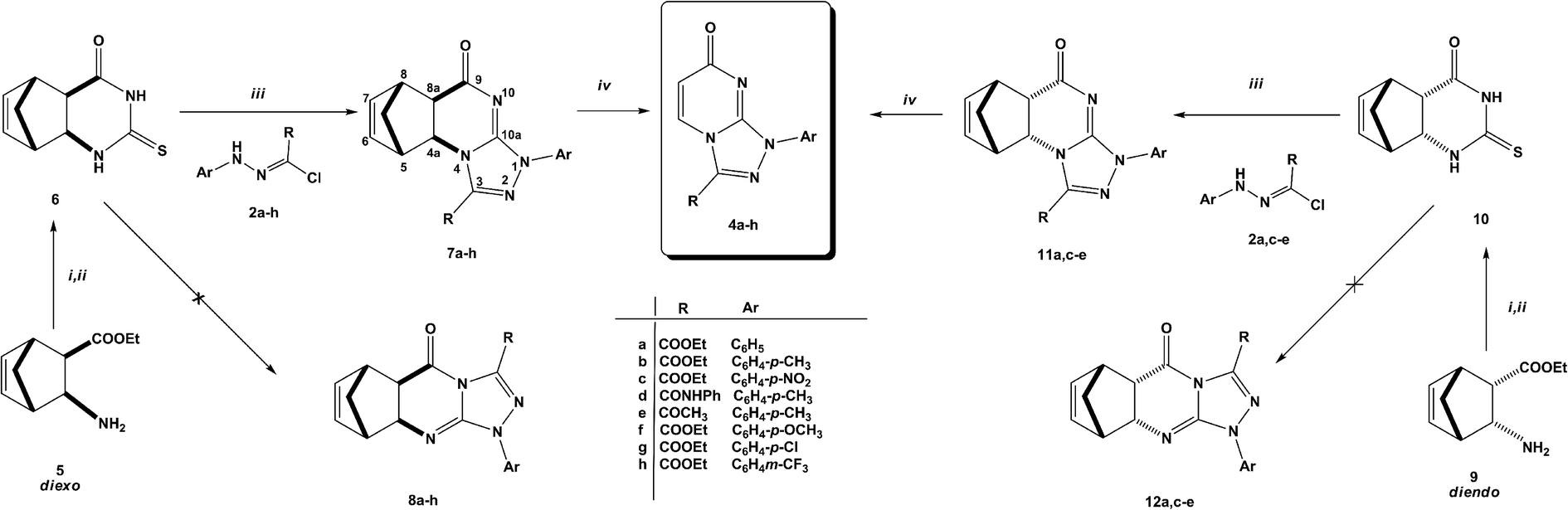 | ||
| Scheme 2 Solvents and conditions: (i) CSCl2, NaHCO3, CHCl3;27 (ii) NH3, MeOH;27 (iii) dioxane, TEA, reflux, 4–6 h; (iv) MW: in 1,2-DCM, 200 °C, 30–120 min, 200 W. | ||
| Entry | Energy difference ΔEa kJ mol−1 | Yieldb % | Entry | Energy difference ΔEa kJ mol−1 | Yieldb % |
|---|---|---|---|---|---|
| a Energy calculations were performed at the density functional theory (DFT) using the B3LYP functional. The 6-311G(d,p) basis sets were employed on all atoms. b The yield of isolated pure [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones by column chromatography. | |||||
| 7a | 50.75 | 66 | 7g | 50.7 | 68 |
| 8a | 8g | ||||
| 7b | 51.32 | 69 | 7h | 47.75 | 67 |
| 8b | 8h | ||||
| 7c | 47.48 | 60 | 11a | 51.13 | 72 |
| 8c | 12a | ||||
| 7d | 61.58 | 60 | 11c | 46.07 | 75 |
| 8d | 12c | ||||
| 7e | 38.84 | 45 | 11d | 58.16 | 70 |
| 8e | 12d | ||||
| 7f | 62.89 | 77 | 11e | 44.58 | 50 |
| 8f | 12e | ||||
In a similar manner, the reaction of diendo norbornene-condensed 2-thioxopyrimidin-4-one 10 with functionalized hydrazonoyl chlorides 2a,c–e was also found to be regioselective towards the formation of angular regioisomers, [1,2,4]triazolo[4,3-a]pyrimidin-7(1H)-ones 11a,c–e (Scheme 2) (Table 1). The effect of norbornene on the stereochemical outcome was attributed to electronic, rather, than steric factors. Specifically, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in thiouracil enforces the reaction towards the formation of [1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones 3a–e because (i) the nitrogen located closer to the CC double bond is more basic and (ii) tautomeric structure 1A is thermodynamically more stable than 1B (Scheme 3).
C bond in thiouracil enforces the reaction towards the formation of [1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones 3a–e because (i) the nitrogen located closer to the CC double bond is more basic and (ii) tautomeric structure 1A is thermodynamically more stable than 1B (Scheme 3).
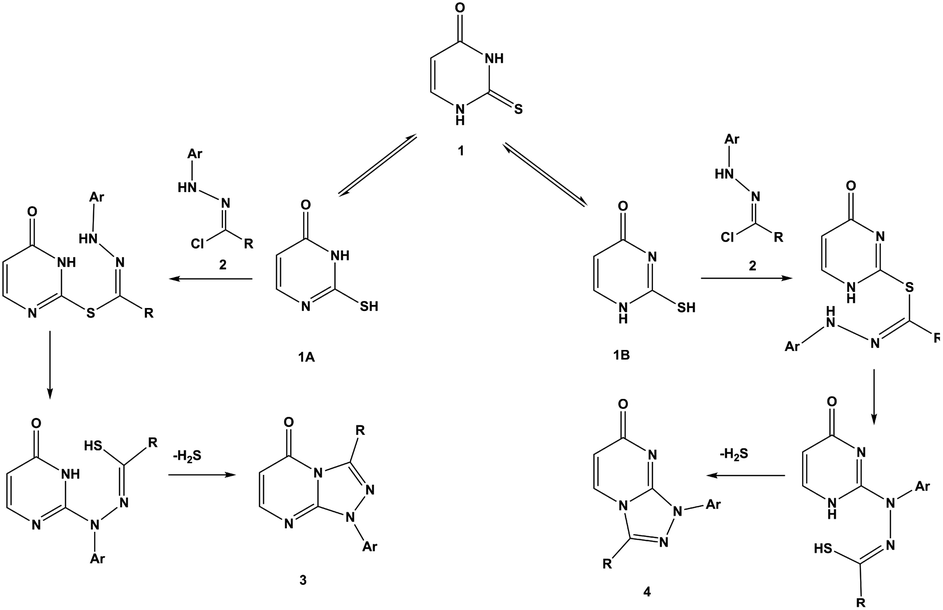 | ||
| Scheme 3 Reaction pathways of thiouracil 1 and hydrazonoyl chlorides 2 to form regioisomers 3 and 4. | ||
The lower yields of the reactions specially with hydrazonoyl chloride 2e was ascribed to the tendency of hydrazonoyl chlorides, whose configuration is Z,25 to isomerize to its E isomer under the reaction conditions as was deduced from the NMR spectra of the separated side product (ESI†). E isomer of hydrazonoyl chloride is more stable by hydrogen bonding and is hindered to be attacked by thione 6,10 to replace its chlorine atom (Scheme 4).
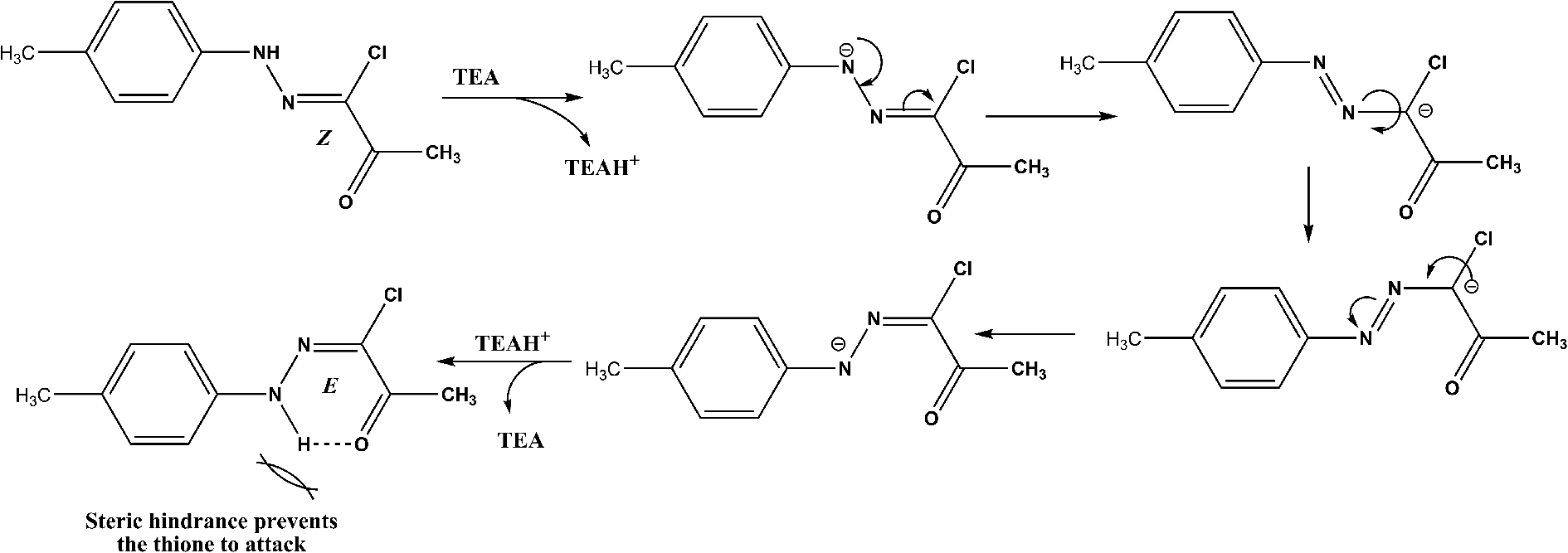 | ||
| Scheme 4 Isomerization of hydrazonoyl chlorides (2e as an example). | ||
After heating in 1,2-dichlorobenzene (1,2-DCB) at 200 °C under microwave conditions, norbornene-fused triazolo[4,3-a]pyrimidin-7(1H)-ones (7a–h and 11a,c–e) underwent RDA reaction producing triazolo[4,3-a]pyrimidin-7(1H)-ones 4a–h in moderate yields in a short time (30–90 minutes, Scheme 2 and Table 2). The RDA reaction was monitored by TLC and could also be noticed by the odor of cyclopentadiene. RDA reactions of diexo isomers are easier than their diendo counterparts (entries 6–9) due to higher stability of diendo isomers as was confirmed from DFT calculations (ESI†).
| Entry | Starting material | Product | Reaction timea (min) | Isolated yieldb (%) |
|---|---|---|---|---|
| a T = 200 °C, Pmax = 200 W, in 1,2-DCB. b After column chromatography. | ||||
| 1 | 7a (exo) | 4a | 45 | 42 |
| 2 | 11a (endo) | 60 | 52 | |
| 3 | 7b (exo) | 4b | 60 | 54 |
| 4 | 7c (exo) | 4c | 90 | 41 |
| 5 | 11c (endo) | 30 | 45 | |
| 6 | 7d (exo) | 4d | 30 | 78 |
| 7 | 11d (endo) | 30 | 67 | |
| 8 | 7e (exo) | 4e | 120 | 90 |
| 9 | 11e (endo) | 30 | 57 | |
| 10 | 7f (exo) | 4f | 60 | 41 |
| 11 | 7g (exo) | 4g | 40 | 58 |
| 12 | 7h (exo) | 4h | 30 | 48 |
Geometry optimization and total energies calculations were performed at the density functional theory (DFT) using Becke's three-parameter Lee–Yang–Parr (B3LYP) exchange functional with 6-311G(d,p) basis sets, using Gaussian-09 program.26 Angular regioisomers 7a–h and 11a,c–e are thermodynamically more stable than their linear counterparts 8a–h and 12a,c–e, respectively (Table 1).
Fig. 1 shows the electron distribution and relative stability of tautomers A and B. The CC bond (such as in thiouracil) directs the tautomerism towards an excess of A. When it is absent (such as in 6,10), the tautomeric equilibrium is shifted towards B. Tautomer A is responsible for the formation of linear triazolo[4,3-a]pyrimidin-5(1H)-one regioisomers, while tautomer B leads to the formation of angular triazolo[4,3-a]pyrimidin-7(1H)-one regioisomers.
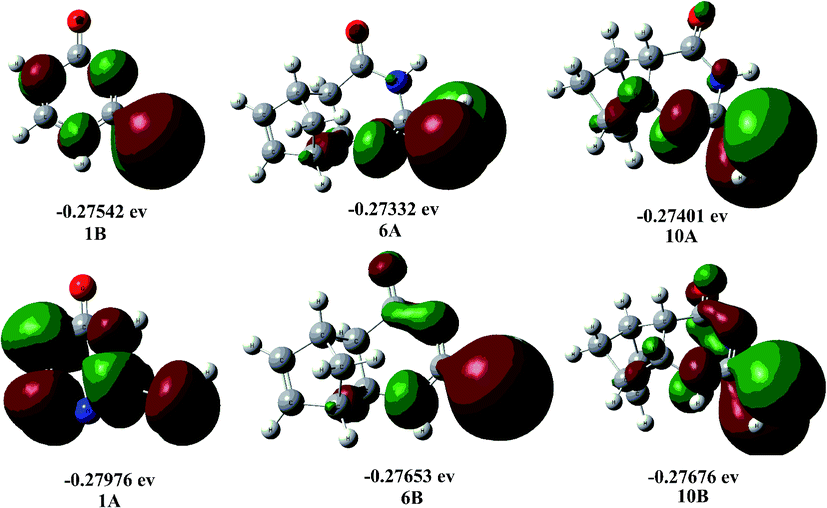 | ||
| Fig. 1 Optimized geometric structure and HOMO energy levels of tautomers A and B of compounds 1, 6 and 10. | ||
A conclusive evidence for the angular stereochemistry of the products of the reaction of norbornene-condensed 2-thioxopyrimidin-4-one 6,10 with hydrazonoyl chlorides 2a–h was obtained by X-ray crystallographic analysis of compounds 7e and 11d (Fig. 2 and 3).
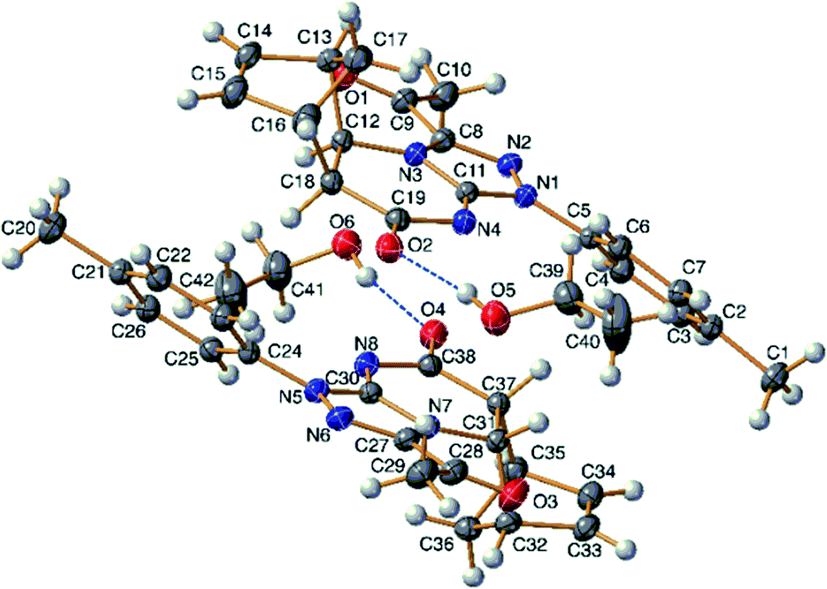 | ||
| Fig. 2 TELP image of 7e at 50% probability level. There are two independent molecules in the asymmetric unit. | ||
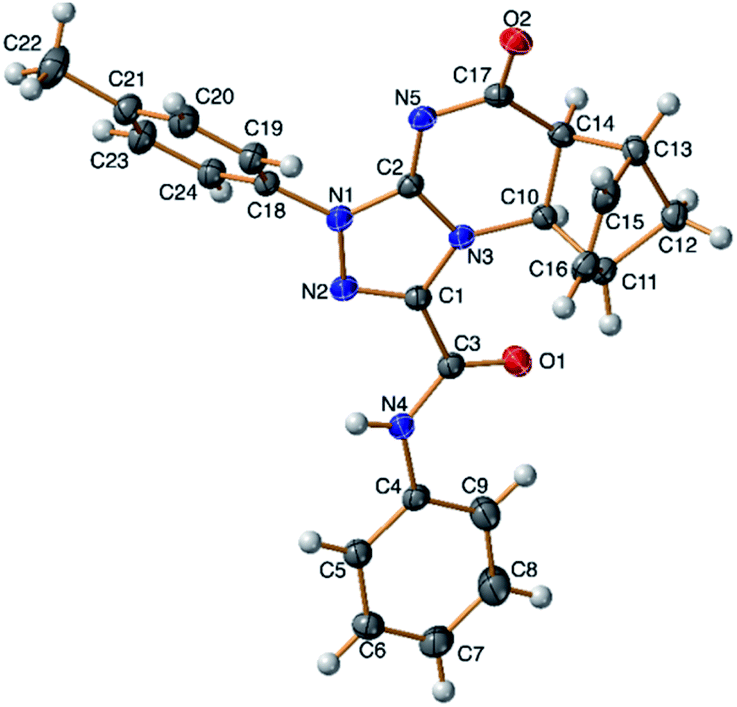 | ||
| Fig. 3 TELP image of 11d at 50% probability level. | ||
Experimental section
Materials and methods
NMR spectra were recorded at 500.20 MHz for 1H-NMR and at 125.62 MHz for 13C-NMR in CDCl3 at room temperature, using a Bruker AV NEO Ascend 500 spectrometer (Bruker Biospin, Karlsruhe, Germany) with Double Resonance Broad Band Probe (BBO). Tetramethylsilane (TMS) was used as internal standard. Microwave-promoted reactions were performed using sealed reaction vials (10 mL) in a microwave (CEM, Discover, SP) cavity (CEM Corporation, Matthews, NC, USA). Reactions were monitored by thin layer chromatography (TLC) using aluminum sheets coated with silica gel (POLYGRAM®SIL G/UV254, Merck). TLC plates were inspected under UV light. Melting points were measured with a Hinotek-X4 micro melting point apparatus (Hinotek, Ningbo, China) and are uncorrected. All theoretical calculations were performed using the Gaussian 09 package. The geometrical optimization was performed at the density functional theory (DFT) using the B3LYP. The 6-311G(d, p) basis sets were employed by all atoms.Diexo 6 and diendo10 norbornene-fused 2-thioxopyrimidin-4-ones were prepared from the corresponding norbornene amino ester 5 and 9, respectively, according to the reported procedures.27 Hydrazonoyl chlorides 2a–h were synthesized according to the reported procedures.28–30
X-ray diffraction data of 7e and 11d were collected on a Rigaku Oxford Diffraction Supernova diffractometer using Mo Kα (7e) or Cu Kα (11d) radiation. The CrysAlisProX software package31 was used for cell refinements and data reductions. The structures were solved by intrinsic phasing method using the SHELXX software.32 The data was corrected with empirical absorption correction based of equivalent reflections (7e) or analytical absorption correction (11d) CrysAlisProX structural refinements31 were carried out using SHELXLX software.32 The NH hydrogen atoms were located from the difference Fourier map and refined isotropically. Other hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–1.00 Å and Uiso = 1.2–1.5Ueq (parent atom). The crystallographic details are summarized in Table S7.†
General procedure for the synthesis of [1,2,4]triazolo-[4,3-a]pyrimidin-5(1H)-ones 3a–e and 5,8-methano[1,2,4]-triazolo[4,3-a]quinazolinones 7a–h and 11a,c–e
To a mixture of 0.5 mmol of 2-thioxopyrimidin-4-one (1,6,10) and 0.5 mmol of hydrazonyl chloride 2a–h in dioxane (10 mL), 100 μL triethyl amine (TEA) were added. The mixture was heated under reflux conditions with stirring until completion of the reaction as confirmed by the disappearance of H2S odor and TLC monitoring (n-hexane/EtOAC = 2:1 as the eluent). The solvent was evaporated under reduced pressure, the residue was dissolved in CHCl3 (50 mL) and extracted with water (3 × 10 mL). Then the CHCl3 layer was dried on Na2SO4, the solvent was evaporated, and the residue was dissolved in 5 mL EtOAc and purified by column chromatography on silica gel eluted by EtOAc. In the reaction of thiouracil 1 with hydrazonoyl chloride 2a–e, the major 3a–e and minor 4a–e regioisomers were separated using TLC on silica gel (eluent EtOAc): 3a–e, Rf 0.4, 4a–eRf 0.05. The products were crystallized from Et2O to produce white or light yellow crystals.
O), 155.9(CH), 155.3(CO), 147.9(C), 136.2(C), 135.9(C), 129.5(CH), 128.1(CH), 121.1(CH), 103.2(CH), 64.1(CH2), 13.9(CH3).
O), 155.9(CH), 155.4(CO), 147.8(C), 138.3(C), 135.7(C), 133.7(C), 129.9(CH), 121.2(CH), 102.9(CH), 64.0(CH2), 21.1(CH3, p-tolyl), 13.9(CH3).
O), 155.4(CH), 154.9(C), 147.9(C), 146.1(C), 141.1(C), 136.7(C), 125.1(CH), 120.3(CH), 104.8(CH), 64.4(CH2), 13.9(CH3).
O), 156.8(CH), 150.8(CO), 148.6(C), 139.7(C), 139.0(C), 137.6(C), 133.3(C), 129.9(CH), 129.1(CH), 125.2(CH), 122.0(CH), 120.4(CH), 102.8(CH), 21.2(CH3, p-tolyl).
O), 155.6(CO), 155.5(CH), 148.4(C), 141.4(C), 138.5(C), 133.6(C), 130.0(CH), 121.3(CH), 103.5(CH), 29.8(COCH3), 21.1(CH3, p-tolyl).
O), 156.3(CO), 154.1(C), 140.3(CH), 137.7(C), 136.1(C), 134.3(CH), 129.1(CH), 128.2(CH), 122.2(CH), 63.3(CH2), 57.3(CH), 52.8(CH), 49.7(CH), 43.9(CH2), 40.5(CH), 14.2(CH3).
O), 156.4(CO), 153.9(C), 140.5(CH), 138.1(C), 137.5(C), 134.3(CH), 133.7(C), 130.1(CH), 122.2(CH), 63.5(CH2), 57.3(CH), 52.9(CH), 49.5(CH), 43.7(CH2), 40.5(CH), 21.0(CH3, p-tolyl), 14.0(CH3).
O), 156.0(CO), 154.4(C), 146.1(C), 141.2(C), 140.2(CH), 138.5(C), 134.4(CH), 124.7(CH), 121.6(CH), 63.8(CH2), 57.4(CH), 53.0(CH), 49.8(CH), 44.1(CH2), 40.6(CH), 14.2(CH3).
O), 154.0(CO), 153.0(CO), 139.9(CH), 139.4(C), 138.3(C), 136.1(C), 134.8(CH), 133.5(C), 129.7(CH), 129.4(CH), 125.7(CH), 122.0(CH), 120.4(CH), 57.4(CH), 52.9(CH), 49.8(CH), 43.9(CH2), 40.5(CH), 21.1(CH3).
O), 176.7(CO), 154.1(C), 142.1(C), 140.0(CH), 138.1(C), 134.7(CH), 133.8(C), 129.7(CH), 122.1(CH), 57.4(CH), 52.7(CH), 49.8(CH), 44.4(CH2), 40.6(CH), 26.5(COCH3), 21.1(CH3, p-tolyl).
O), 159.0(CO), 156.3(C), 153.7(C), 140.4(CH), 137.4(C), 134.3(CH), 129.1(C), 123.9(CH), 114.2(CH), 63.3(CH2), 57.3(CH), 55.5(CH), 52.8(CH), 49.7(CH), 43.9(CH2), 40.5(OCH3), 14.1(CH3).
O), 156.1(CO), 154.0(C), 140.3(CH), 137.8(C), 134.8(C), 134.3(CH), 133.6(C), 129.2(CH), 123.1(CH), 63.5(OCH2), 57.3(CH), 52.8(CH), 49.7(CH), 43.9(CH2), 40.4(CH), 14.1(CH3).
O), 156.1(CO), 154.3(C), 140.4(CH), 138.1(C), 136.7(C), 134.5(CH), 131.8(C), 131.7(C), 130.0(CH), 125.4(CH), 124.5(CH), 118.6(CH), 63.7(CH2), 57.3(CH), 53.0(CH), 49.8(CH), 44.0(CH2), 40.3(CH), 14.2(CH3).
O), 156.3(CO), 154.1(C), 140.3(CH), 137.8(C), 136.1(C), 132.1(CH), 129.1(CH), 127.9(CH), 122.2(CH), 63.4(CH2), 57.1(CH), 49.5(CH), 49.4(CH), 46.4(CH2), 41.2(CH), 14.1(CH3).
O), 156.0(CO), 154.5(C), 146.0(C), 141.2(C), 140.4(CH), 138.5(C), 132.2(CH), 124.6(CH), 121.6(CH), 63.8(CH2), 57.2(CH), 49.6(CH), 49.4(CH), 46.5(CH2), 41.2(CH), 14.2(CH3).
O), 154.0(C), 153.0(C), 140.1(CH), 139.6(C), 138.1(C), 136.3(C), 133.6(C), 132.4(CH), 129.6(CH), 129.3(CH), 125.7(CH), 121.9(CH), 120.2(CH), 57.2(CH), 49.6(CH), 49.5(CH), 46.4(CH2), 41.2(CH), 21.1(CH3).
O), 177.0(CO), 154.1(C), 142.3(C), 140.2(CH), 138.1(C), 133.7(C), 132.2(CH), 129.7(CH), 121.9(CH), 57.1(CH), 49.5(CH), 49.3(CH), 46.3(CH2), 41.2(CH), 26.6(COCH3), 21.1(CH3, p-tolyl).
RDA protocol for the synthesis of [1,2,4]triazolo[4,3-a]pyrimidines 4a–h
Norbornene-fused [1,2,4]triazolo[4,3-a]pyrimidinones 7a–h or 11a,c–e (0.1 mmol) was dissolved in 1,2-DCM (2 mL) in a 10 mL sealed reaction vial. The solution was stirred at 200 °C for 30–120 min. at max. 200 W microwave irradiation. After completing the reaction, monitored by TLC, the solvent was evaporated, the residue was dissolved in EtOAc and purified by column chromatography on silica gel eluting with EtOAc.O), 156.2(CO), 149.2(C), 135.9(C), 132.4(C), 130.6(CH), 129.4(CH), 128.2(CH), 121.4(CH), 114.9(CH), 63.9(OCH2), 14.2(CH3).
O), 156.2(CO), 149.1(C), 138.3(C), 133.4(C), 132.2(C), 130.5(CH), 129.8(CH), 121.3(CH), 114.9(CH), 63.8(OCH2), 21.1(CH3), 14.1(CH3).
O), 155.8(CO), 149.3(C), 146.3(C), 140.7(C), 133.2(C), 130.8(CH), 125.0(CH), 120.9(CH), 114.9(CH), 64.2(OCH2), 14.1(CH3).
O), 152.9(CO), 138.4(C), 135.8(C), 134.0(C), 133.3(C), 131.1(CH), 130.5(CH), 129.9(CH), 129.4(CH), 127.7(CH), 126.0(CH), 121.1(CH), 120.4(CH), 114.7(CH), 21.1(CH3).
O), 168.5(CO), 149.1(C), 138.5(C), 136.8(C), 133.5(C), 131.1(CH), 129.9(CH), 121.1(CH), 114.9(CH), 26.4(CH3), 21.1(CH3).
O), 159.3(CO), 156.2(C), 149.0(C), 132.1(C), 130.5(CH), 128.8(C), 123.2(CH), 114.9(CH), 114.4(CH), 63.7(OCH2), 55.6(OCH3), 14.1(CH3).
O), 156.1(CO), 149.1(C), 134.5(C), 133.9(C), 132.5(C), 130.6(CH), 129.5(CH), 122.3(CH), 115.0(CH), 64.0(OCH2), 14.1(CH3).
O), 156.0(CO), 149.2(C), 136.4(C), 132.8(C), 130.2(C), 124.7(CH), 124.7(CH), 124.5(CH), 117.8(CH), 117.8(CH), 115.0(CH), 64.1(CH2), 14.1(CH3).
Conclusions
In the present work, we prepared triazolo[4,3-a]pyrimidin-7(1H)-ones 4a–h. These compounds cannot be prepared by the reactions of thiouracil 1 and hydrazonoyl chloride 2a–h, because these reactions are regioselective towards [1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones 3a–d. For overcoming this obstacle, we used diexo6 or diendo10 norbornene-condensed 2-thioxopyrimidin-4-ones characterized by two structural features: (i) the absence of the CC bond in the pyrimidinone moiety that ensures the direction of the regioselectivity towards triazolo[4,3-a]pyrimidin-7(1H)-ones and (ii) the easy removal of cyclopentadiene by the RDA reaction and the concomitant rebuilding the CC bond in the pyrimidinone moiety to obtain triazolo[4,3-a]pyrimidin-7(1H)-ones 4a–h as the final products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Hungarian Research Foundation (OTKA No. K 115731). The financial support of the GINOP-2.3.2-15-2016-00038 project is acknowledged. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is acknowledged. In addition, Tempus Public Foundation is also acknowledged for its financial support (No. AK-00167-002/2019).Notes and references
- H. Dong, Z. Gao, R. Li, Y. Hu, H. Dong and Z. Xie, RSC Adv., 2014, 4, 55827 RSC.
- B. Jafari, N. Yelibayeva, M. Ospanov, S. A. Ejaz, S. Afzal, S. U. Khan, Z. A. Abilov, M. Z. Turmukhanova, S. N. Kalugin, S. Safarov, J. Lecka, J. Sévigny, Q. Rahman, P. Ehlers, J. Iqbal and P. Langer, RSC Adv., 2016, 6, 107556 RSC.
- V. S. Dinakaran, B. Bomma and K. K. Srinivasan, Der Pharma Chem., 2012, 4, 255 CAS.
- R. Mishra and I. Tomar, Int. J. Pharm. Sci. Res., 2011, 2, 758 CAS.
- M. Fares, S. M. Abou-Seri, H. A. Abdel-Aziz, S. E. S. Abbas, M. M. Youssef and R. A. Eladwy, Eur. J. Med. Chem., 2014, 83, 155 CrossRef CAS; A. O. Abdelhamid, S. M. Gomha, N. A. Abdelriheem and S. M. Kandeel, Molecules, 2016, 21, 929 CrossRef PubMed.
- B. Loev, J. H. Musser, R. E. Brown, H. Jones, R. Kahen, F. C. Huang, A. Khandwala, P. Sonnino-Goldman and M. J. Leibowitz, J. Med. Chem., 1985, 28, 363 CrossRef CAS PubMed; B. A. El-Gazzar, M. M. El-Enanyb and M. N. Mahmoud, Bioorg. Med. Chem., 2008, 16, 3261 CrossRef PubMed.
- M. S. M. Ahmed and T. A. Farghaly, Lett. Org. Chem., 2018, 15, 183 Search PubMed; S. M. Riyadh, J. Chin. Chem. Soc., 2005, 52, 545 CrossRef CAS.
- T. A. Farghaly, S. M. Gomha, E. M. H. Abbas and M. M. Abdalla, Arch. Pharm., 2012, 345, 117 CrossRef CAS PubMed.
- A. S. Shawali, Chem. Rev., 1993, 2731 CrossRef CAS; A. S. Shawali, J. Adv. Res., 2016, 7, 873 CrossRef PubMed.
- S. M. Riyadh, Molecules, 2011, 16, 1834 CrossRef CAS PubMed.
- H. M. Hassaneen, H. A. Abdelhadi and T. A. Abdallah, Tetrahedron, 2001, 57, 10133 CrossRef CAS.
- H. M. Hassneen and T. A. Abdallah, Molecules, 2003, 8, 333 CrossRef CAS; N. A. Abdel Hafez, T. A. Farghaly, M. A. Al-Omar and M. M. Abdall, Eur. J. Med. Chem., 2010, 45, 4838 CrossRef PubMed.
- S. M. Riyadh, Molecules, 2011, 16, 1834 CrossRef CAS PubMed; S. M. Gomha, S. A. Ahmed and A. O. Abdelhamid, Molecules, 2015, 20, 1357 CrossRef.
- T. A. Abdallah, M. A. Darwish and H. M. Hassaneen, Molecules, 2002, 7, 494 CrossRef CAS.
- G. S. Masaret and T. A. Farghaly, Curr. Org. Synth., 2018, 15, 126 CrossRef CAS.
- K. Suzuki, K. Inomata and Y. Endo, Org. Lett., 2004, 6, 409 CrossRef CAS PubMed.
- I. Nekkaa, M. Palkó, I. M. Mándity, F. Miklós and F. Fülöp, Eur. J. Org. Chem., 2018, 32, 4456 CrossRef.
- F. Miklós, Z. Tóth, M. M. Hänninen, R. Sillanpää, E. Forró and F. Fülöp, Eur. J. Org. Chem., 2013, 22, 4887 CrossRef.
- B. Fekete, M. Palkó, S. Mándity, M. Haukka and F. Fülöp, Eur. J. Org. Chem., 2016, 21, 3519 CrossRef.
- S. H. Frayne, R. M. Stolz and B. H. Northrop, Org. Biomol. Chem., 2019, 17, 7878 RSC.
- B. Zhang, Y. Li, Z. Zhang, Y. An, Y. Wen, X. Gou, S. Quan, X. Wang and Y. Liang, J. Am. Chem. Soc., 2019, 141, 9731 CrossRef CAS.
- F. Csende, G. Stájer and F. Fülöp, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, 2014, vol. 5, p. 518 Search PubMed.
- S. Kotha and S. Banerjee, RSC Adv., 2013, 3, 7642 RSC.
- M. Palkó, M. El Haimer, Z. Kormányos and F. Fülöp, Molecules, 2019, 24, 772 CrossRef.
- A. M. Asiri, A. O. Al-Youbi, M. E. M. Zayed and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1964 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev., 1988, 37, 785 Search PubMed; A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- G. Stajer, A. E. Szabo and P. Sohar, Heterocycles, 1999, 51, 1849 CrossRef CAS.
- V. S. Matiychuk, M. A. Potopnyk, R. Luboradzki and M. D. Obushak, Synthesis, 2011, 11, 1799 Search PubMed.
- R. Silvestri, M. G. Cascio, G. La Regina, F. Piscitelli, A. Lavecchia, A. Brizzi, S. Pasquini, M. Botta, E. Novellino, V. Di Marzo and F. Corell, J. Med. Chem., 2008, 51, 1560 CrossRef CAS.
- J. Liu, M. Nie, Y. Wang, J. Hu, F. Zhang, Y. Gao, Y. Liu and P. Gong, Eur. J. Med. Chem., 2016, 123, 431 CrossRef CAS.
- Rikagu Oxford Diffraction, CrysAlisPro, Rikagu Oxford Diffraction inc., Yarnton, Oxfordshire, England, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980577 and 1980578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04345a |
| This journal is © The Royal Society of Chemistry 2020 |