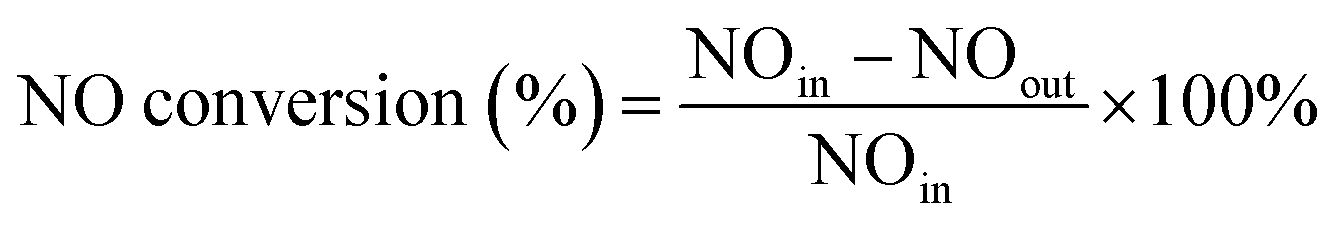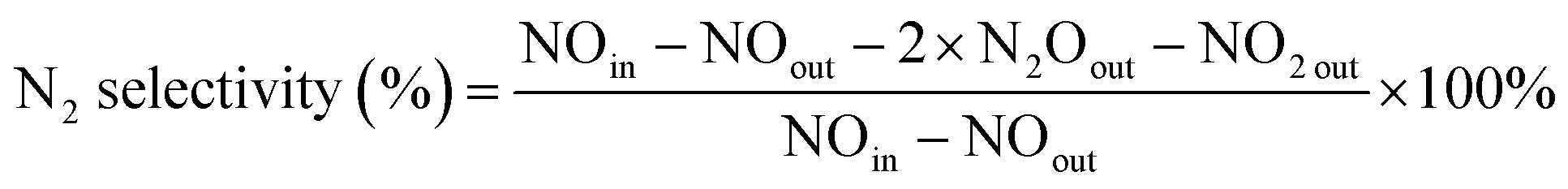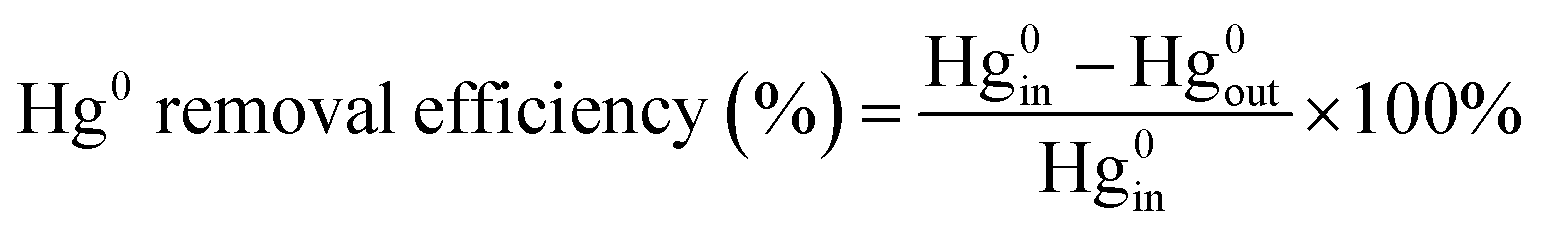DOI:
10.1039/D0RA04350H
(Paper)
RSC Adv., 2020,
10, 25325-25338
Enhancement of CeO2 modified commercial SCR catalyst for synergistic mercury removal from coal combustion flue gas
Received
15th May 2020
, Accepted 28th June 2020
First published on 3rd July 2020
Abstract
CeO2 modified commercial SCR (selective catalytic reduction) catalysts with different CeO2 content were prepared and researched for synergistic mercury removal from coal combustion flue gas in this study. The characterization analyses on the catalysts indicated that the introduction of CeO2 increased the surface area, the dispersity of the metal oxides on the TiO2 support and the redox behavior of the catalyst, which was beneficial to the catalytic activity. The experimental results confirmed that the CeO2 loading improved the catalytic efficiencies over the commercial SCR catalyst. The catalyst with a CeO2 content of 4% displayed the optimal performance for NO and synergistic Hg0 removal, of which the NO conversion and Hg0 removal efficiency reached 90.5% and 78.2%, respectively, at 300 °C in simulated coal-fired flue gas. The Hg0 removal activity, the independence of Hg0 removal from HCl concentration and the effects of SO2, NO and NH3 on Hg0 removal efficiency all became positive over the modified catalyst compared to over the raw one, which was mainly due to the sufficient chemisorbed oxygen derived from the synergy of V2O5 and CeO2 and the redox transformation between Ce3+ and Ce4+ on the catalyst surface. The CeO2 modification generated a significant enhancement on the catalytic performance and made the commercial SCR catalyst more suitable to be employed for NO and synergistic mercury removal in a coal combustion power plant.
1. Introduction
Mercury is a kind of extremely harmful pollutant in the ecological environment. It poses a serious threat to human health due to its hypertoxicity, persistence and bioaccumulation.1 According to the Global Mercury Assessment 2018 issued by the UN Environment Programme, the global anthropogenic mercury emission reached 2150 tons in 2015, which increased by 12% compared to that in 2010.2 Significant coal burning is one of the main reasons for the growth of mercury emissions. And coal combustion power plants are considered as the major anthropogenic source of mercury release.3 As the Minamata Convention on Mercury came into force in August 2017, the limit on mercury emission from coal-fired power plants will be more rigorous on the basis of the existing regulations.4 Therefore, it is urgent to pay extensive attention to mercury emission control of coal combustion power plants under the dual pressure of environmental protection and convention fulfillment.
Mercury in coal-fired flue gas exists mainly in the types of elemental Hg (Hg0), oxidized Hg (Hg2+) and particle bound Hg (HgP). Hg2+ and Hgp can be respectively captured by wet flue gas desulfurization (WFGD) and particulate matter control device (PMCD) of power plant because of their physical properties, while Hg0 is difficult to be controlled by the single pollutant control equipment due to its volatility and water insolubility.5,6 So the key to the control of mercury emission from coal combustion power plant is the removal of Hg0. Similarly with mercury, NOx is also a sort of hazardous contaminant with great harm to environment that coal burning releases, and NO occupies about 95% among NOx.7–9 Currently, the method of selective catalytic reduction (SCR) is generally used by coal-fired power plants for NO removal. Besides, the SCR catalyst has the capacity of oxidizing Hg0 to Hg2+ due to the existence of active oxygen on its surface, followed by Hg2+ being removed in the downstream WFGD.10,11 Compared with other Hg removal plans such as sorbent injection, utilizing SCR catalyst to synergistically remove Hg is remarkably cost-effective and meanwhile beneficial to the avoiding of secondary mercury pollution.12 Hence, it is promising for coal-fired power plant to adopt this approach to deal with the Hg removal from flue gas. And the research on the synergistic Hg0 oxidation with SCR catalyst has attracted more attention in recent years.
The commercial SCR catalyst that is currently used by coal combustion power plants is the TiO2-supported V2O5–WO3/TiO2 catalyst. A series of studies have been made on the Hg0 oxidation over the V2O5–WO3/TiO2 catalyst. The results indicated that the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on the catalyst surface could participate in Hg0 oxidation as the active sites. The Hg0 removal efficiency over the catalyst could reach 60–80% in general, and sometimes the efficiency was even higher than 90%.13,14 The increases of V2O5 loading, surface area and reaction temperature are in favor of the Hg0 oxidation activity.15 Especially, the existence of HCl in the flue gas had an obvious promotion on the Hg0 oxidation over the V2O5-based catalysts. Hg0 removal efficiency of V2O5–WO3/TiO2 was close to 100% at 380 °C with 4.5 mmol m−3 HCl contained in the reaction gas.16 The SiO2–TiO2–V2O5 catalyst likewise showed a Hg0 removal efficiency of nearly 100% in the co-presence of O2 and HCl.17 And the facilitation of HCl on the efficiency of commercial SCR catalyst was also testified by kinetic analysis.18 However, though the commercial V2O5–WO3/TiO2 catalyst displays certain Hg0 removal capacity under the appropriate conditions, it has apparent drawbacks such as the narrow working temperature range and the limited Hg0 removal efficiency at the SCR operating temperature.16,19 Meanwhile, the effectiveness of Hg0 removal depends heavily on the HCl concentration. The efficiency could be as high as 90% in the flue gas derived from burning high-rank coal, while in flue gas of burning low-rank coal only less than 30% was observed.17,20,21 This condition is distinctly disadvantageous to those power plants that combust sub-bituminous coal or lignite. So it is necessary to make modification on commercial SCR catalyst to improve its catalytic properties. In recent years, CeO2-based catalysts have gradually come into view of researchers due to its prominent catalytic activity. Related studies demonstrated that element Ce would help enhance the oxygen storage capacity of the catalyst, which led to the superior performance on NO and Hg0 removal. Illustratively, Gao et al.22 prepared CeO2/TiO2 catalyst by sol–gel method and found the NO conversion of the catalyst reached 93.4–98.6% in the wide temperature range of 250–450 °C; Li et al.23 investigated Hg0 removal activity of CeO2/TiO2 in simulated coal-fired flue gas and confirmed the optimal efficiency could attain 94%, and efficient Hg0 oxidation could be achieved even in the absence of HCl; Fan et al.24 acquired that the zeolite supported CeO2/HZSM-5 catalyst exhibited Hg0 removal efficiency of more than 95% among the range of 120–320 °C; Wang et al.25 loaded CeO2 on Ti-based pillared interlayered clays to examine the simultaneous NO and Hg0 removal efficiency over the catalyst, and the results showed that the NO conversion was almost 100% at 350 °C while Hg0 removal efficiency also reached higher than 50% in the same condition. In view of the advantage of the activity of catalyst containing CeO2, it is reasonable to speculate that using CeO2 to modify the V2O5–WO3/TiO2 catalyst will make a significant improvement on the catalytic properties of the catalyst. Zhao et al.19 has previously modified the TiO2 support with CeO2 and synthesized V2O5–WO3/TiO2–CeO2 catalyst, and the experimental study confirmed the enhancement of Hg0 removal performance of the catalyst, such as the efficiency and sulfur-resistance, resulted from the addition of CeO2. Some literatures also prepared the CeO2 modified V2O5–WO3(MoO3)/TiO2 to investigate the NO removal activity specifically, and the satisfactory NO conversions, sulfur-resistance and alkali metal resistance were obtained over the catalysts.26–28 Nevertheless, few literatures have made investigations on the effectiveness of employing CeO2 to directly modify the commercial SCR catalyst of power plant for synergistic Hg0 removal so far, which is of great value and close correlation to practical application. Moreover, the present commercial SCR catalyst is not replaceable in the short term, though some researched novel catalysts such as Mn-based, Cu-based, noble metal and perovskite structure catalysts displayed considerable Hg0 removal efficiency in the lab-scale tests.29–32 Thus, it can be seen that it is of great significance to examine the synergistic Hg0 removal performance of the CeO2 modified commercial V2O5–WO3/TiO2 catalyst.
O bond on the catalyst surface could participate in Hg0 oxidation as the active sites. The Hg0 removal efficiency over the catalyst could reach 60–80% in general, and sometimes the efficiency was even higher than 90%.13,14 The increases of V2O5 loading, surface area and reaction temperature are in favor of the Hg0 oxidation activity.15 Especially, the existence of HCl in the flue gas had an obvious promotion on the Hg0 oxidation over the V2O5-based catalysts. Hg0 removal efficiency of V2O5–WO3/TiO2 was close to 100% at 380 °C with 4.5 mmol m−3 HCl contained in the reaction gas.16 The SiO2–TiO2–V2O5 catalyst likewise showed a Hg0 removal efficiency of nearly 100% in the co-presence of O2 and HCl.17 And the facilitation of HCl on the efficiency of commercial SCR catalyst was also testified by kinetic analysis.18 However, though the commercial V2O5–WO3/TiO2 catalyst displays certain Hg0 removal capacity under the appropriate conditions, it has apparent drawbacks such as the narrow working temperature range and the limited Hg0 removal efficiency at the SCR operating temperature.16,19 Meanwhile, the effectiveness of Hg0 removal depends heavily on the HCl concentration. The efficiency could be as high as 90% in the flue gas derived from burning high-rank coal, while in flue gas of burning low-rank coal only less than 30% was observed.17,20,21 This condition is distinctly disadvantageous to those power plants that combust sub-bituminous coal or lignite. So it is necessary to make modification on commercial SCR catalyst to improve its catalytic properties. In recent years, CeO2-based catalysts have gradually come into view of researchers due to its prominent catalytic activity. Related studies demonstrated that element Ce would help enhance the oxygen storage capacity of the catalyst, which led to the superior performance on NO and Hg0 removal. Illustratively, Gao et al.22 prepared CeO2/TiO2 catalyst by sol–gel method and found the NO conversion of the catalyst reached 93.4–98.6% in the wide temperature range of 250–450 °C; Li et al.23 investigated Hg0 removal activity of CeO2/TiO2 in simulated coal-fired flue gas and confirmed the optimal efficiency could attain 94%, and efficient Hg0 oxidation could be achieved even in the absence of HCl; Fan et al.24 acquired that the zeolite supported CeO2/HZSM-5 catalyst exhibited Hg0 removal efficiency of more than 95% among the range of 120–320 °C; Wang et al.25 loaded CeO2 on Ti-based pillared interlayered clays to examine the simultaneous NO and Hg0 removal efficiency over the catalyst, and the results showed that the NO conversion was almost 100% at 350 °C while Hg0 removal efficiency also reached higher than 50% in the same condition. In view of the advantage of the activity of catalyst containing CeO2, it is reasonable to speculate that using CeO2 to modify the V2O5–WO3/TiO2 catalyst will make a significant improvement on the catalytic properties of the catalyst. Zhao et al.19 has previously modified the TiO2 support with CeO2 and synthesized V2O5–WO3/TiO2–CeO2 catalyst, and the experimental study confirmed the enhancement of Hg0 removal performance of the catalyst, such as the efficiency and sulfur-resistance, resulted from the addition of CeO2. Some literatures also prepared the CeO2 modified V2O5–WO3(MoO3)/TiO2 to investigate the NO removal activity specifically, and the satisfactory NO conversions, sulfur-resistance and alkali metal resistance were obtained over the catalysts.26–28 Nevertheless, few literatures have made investigations on the effectiveness of employing CeO2 to directly modify the commercial SCR catalyst of power plant for synergistic Hg0 removal so far, which is of great value and close correlation to practical application. Moreover, the present commercial SCR catalyst is not replaceable in the short term, though some researched novel catalysts such as Mn-based, Cu-based, noble metal and perovskite structure catalysts displayed considerable Hg0 removal efficiency in the lab-scale tests.29–32 Thus, it can be seen that it is of great significance to examine the synergistic Hg0 removal performance of the CeO2 modified commercial V2O5–WO3/TiO2 catalyst.
Based on the above presentations, this work takes CeO2 modified commercial SCR catalyst as the researching object, and conducts the experiments in simulated coal combustion flue gas (SFG). NO removal performance of the catalysts with different CeO2 loadings were first tested considering the primary purpose of SCR. Then the Hg0 removal activity of the catalysts was investigated in detail. Hg0 removal efficiencies over different CeO2-loading catalysts at different temperatures were evaluated, and the effects of individual flue gas components in SFG on the efficiency were detected as well. The characterization analyses of X-ray fluorescence (XRF), Brunauer–Emmett–Teller (BET), X-ray diffraction (XRD), H2-Temperature Programmed Reduction (H2-TPR) and X-ray photoelectron spectroscopy (XPS) were carried out to understand physical–chemical properties of the catalysts and explore the modification mechanism of CeO2. The study results of this work will present application prospect of the CeO2 modification on commercial SCR catalyst for improving the catalytic performance.
2. Materials and methods
2.1. Catalyst preparation
The honeycomb commercial SCR catalyst employed in this study was got from a catalyst corporation of China which professionally produces SCR catalyst of coal-fired power plant. The CeO2 modified catalysts were prepared by the solution impregnation method. The honeycomb catalyst was grinded to powder first and sieved with a 200 mesh sifter. Then a certain amount of the sieved fine catalyst powder was placed in a beaker, followed by the Ce(NO3)3 aqueous solution which contained the desired quantity of Ce(NO3)3 being filled into the beaker. The obtained slurry was stirred for 1 h and then exposed to an ultrasonic bath for 2 h. After the mixture was dried at 110 °C for 12 h and calcinated in air at 500 °C for 4 h sequentially, the final CeO2 modified commercial SCR catalyst was acquired. The mass fractions of CeO2 of 1%, 2%, 4% and 7% in the modified catalysts were designed. In the process of preparing the catalysts with different CeO2 loadings, the weight of the original catalyst powder was remained unchanged, and the CeO2 loading was controlled by the solvend amount of the added Ce(NO3)3 aqueous solution. The CeO2 modified catalysts were abbreviated as (x)CeO2-SCR (x represents the mass fraction of CeO2) in the later sections, and the catalyst without modification was designated as raw SCR. Additionally, the pure CeO2 catalyst was also prepared for comparison, which used Ce(NO3)3 as the precursor as well to maintain the consistency.
2.2. Catalyst characterizations
The characterization methods of XRF, BET, XRD, H2-TPR and XPS were carried out over the fresh and spent catalyst samples in order to understand the physical and chemical properties of the catalysts and analyze the CeO2 modification mechanism. The XRF analysis was conducted with an EAGLE III focusing fluorescence spectrograph which was operated at 38 kV. The measurement of the BET surface was accomplished on an ASAP 2020 porosimeter by means of N2 adsorption. The XRD analysis was performed using an X'Pert PRO diffractometer (Cu Kα radiation) of which the working voltage and emission current were 40 kV and 40 mA, respectively, with the scanning angle ranging from 10° to 80° (2θ). The test of H2-TPR was carried out on an Autochem 2920 analyzer with the operating temperature raised from 30 °C to 850 °C at a rate of 10 °C min−1, and the reaction gas was 50 mL min−1 10% H2/Ar. The XPS technique was implemented on a PerkinElmer PHI 5100 ESCA system with Al Kα X-ray source (hν = 1486.6 eV) to study the valence states of the elements and using the C 1s binding energy value of 284.6 eV for the spectra calibration.
2.3. Catalytic activity measurement
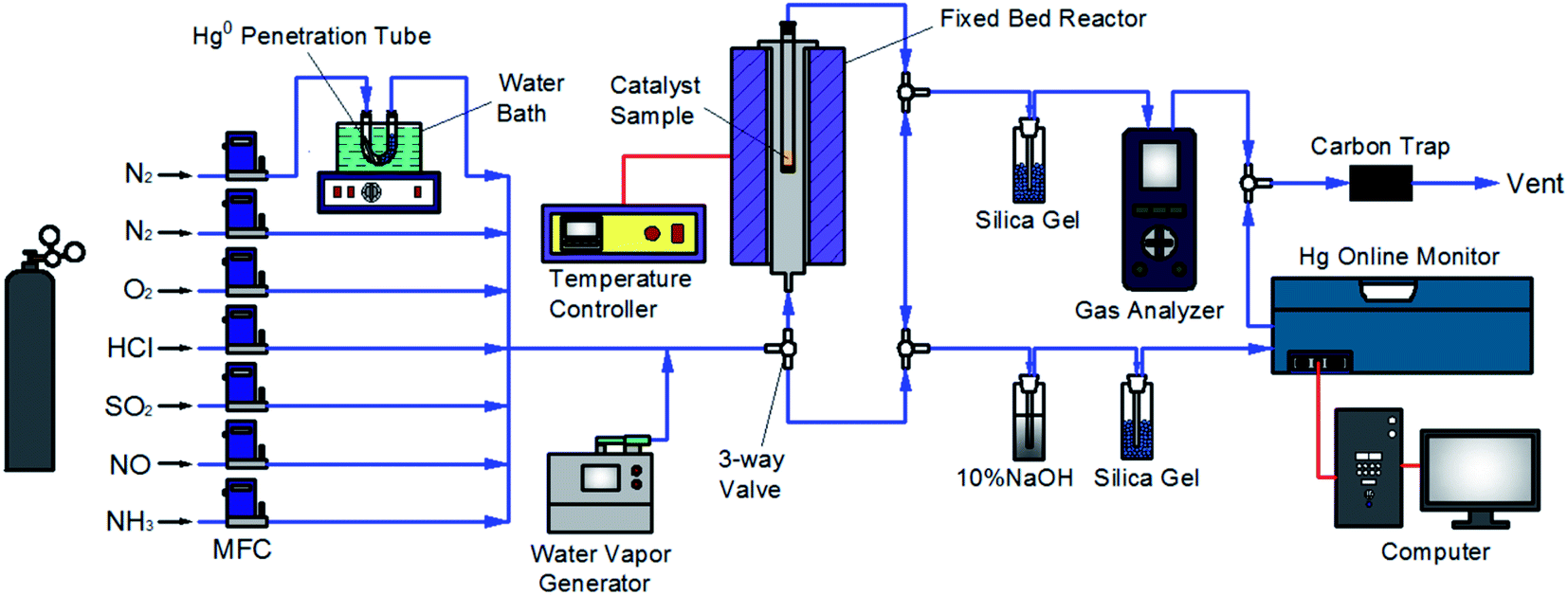
The experimental system used in this work was similar to that employed in our previous studies,33–35 as described in Fig. 1. Briefly, the flue gas components (N2, O2, HCl, SO2, NO, and NH3) came from standard cylinder gases and their gas flow was accurately controlled by the corresponding calibrated mass flowmeter, respectively. Water vapor (H2O) was produced by a steam generator. The continuous feed of Hg0 vapor of approximately 60 μg m−3 was generated from a Hg0 penetration tube (VICI, Metronics Inc., Santa Clara, CA) which was placed in a U-tube and heated by a water bath, with N2 carrying the Hg0 vapor into the flue gas. The catalytic reaction was made to occur in a fixed bed reactor with a temperature controller to set the reaction temperature. The NO and Hg0 concentrations in the flue gas were measured by a gas analyzer (AFRISO, Multilyzer STe, M60) and a Hg0 online monitor (Ohio Lumex, RA-915M), respectively. And the N2O and NO2 concentrations were monitored with a FTIR analyzer (Gasmet Dx4000). Several specific gas-washing bottles were added for eliminating the acid gas to prevent corrosion and interferences on the monitoring devices. The gas line of the system was heated by electric heating belt to avoid any possible adsorption of the gas components on the line before the measurement. The exhaust gas was purified by active carbon before discharged to atmosphere.
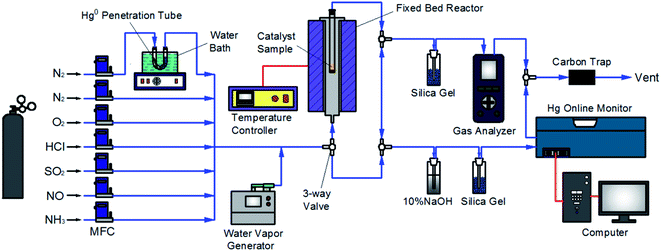 |
| | Fig. 1 Schematic diagram of the experimental system. | |
The experiments of this work were carried out under the condition of simulated coal-fired flue gas of which the composition was 4% O2, 10 ppm HCl, 800 ppm SO2, 400 ppm NO, 400 ppm NH3, 8% H2O and 60 μg m−3 Hg0 with N2 to balance unless otherwise noted. The total flow of the flue gas was controlled at 1 L min−1. The catalyst dosage was 0.5 g for each test, and the space velocity (GHSV) was correspondingly about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. In each test, the flue gas was first introduced to the bypass, and the concentrations of NO and Hg0 at the inlet of the reactor were acquired when the readings of the monitoring devices reached stability. Then the gas stream was switched to pass through the catalyst until the stable NO and Hg0 concentrations at the outlet of the reactor were obtained as well. The stability was defined as the fluctuation of the concentrations being no more than 5% for a period of at least 30 min. After each step of the experiment, the spent catalyst was replaced by fresh sample before starting the next test. The NO conversion, N2 selectivity and Hg0 removal efficiency adopted to evaluate the catalytic activity of the catalyst were respectively calculated by eqn (1)–(3) as follows.
000 h−1. In each test, the flue gas was first introduced to the bypass, and the concentrations of NO and Hg0 at the inlet of the reactor were acquired when the readings of the monitoring devices reached stability. Then the gas stream was switched to pass through the catalyst until the stable NO and Hg0 concentrations at the outlet of the reactor were obtained as well. The stability was defined as the fluctuation of the concentrations being no more than 5% for a period of at least 30 min. After each step of the experiment, the spent catalyst was replaced by fresh sample before starting the next test. The NO conversion, N2 selectivity and Hg0 removal efficiency adopted to evaluate the catalytic activity of the catalyst were respectively calculated by eqn (1)–(3) as follows.
| |
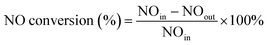 | (1) |
| |
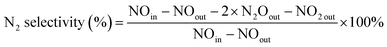 | (2) |
| |
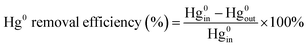 | (3) |
The subscript “in” and “out” in the equations represented the gas concentrations at the inlet and outlet of the reactor, respectively. As the outlet Hg0 concentration was read when it achieved a stable value, the catalyst was in the state of Hg saturated adsorption at this time and all the removed Hg was gaseous Hg2+. Additionally, the researched catalysts were verified to have almost no capacity for Hg0 removal at room temperature. So the physical adsorption of Hg0 was negligible, and the defined Hg0 removal efficiency here was equal to Hg0 oxidation efficiency.
3. Results and discussion
3.1. Characterization of the CeO2-SCR catalysts
3.1.1 XRF analysis. XRF analysis was adopted to investigate the element compositions and contents of the catalysts. The results were summarized in Table 1. Before the loading of CeO2, the content of V2O5 which was the active component and the content of WO3 using for improving the thermal stability and surface acidity in raw SCR catalyst were 0.98% and 6.63%, respectively. Both the values were among the ranges of the contents of V2O5 and WO3 in usual honeycomb commercial SCR catalyst, which were respectively 0.5–3% and 2–10%. The activity of SCR catalyst was generally in proportion to the content of V2O5. But exorbitant vanadium content would lead to the growing SO2/SO3 conversion.36 The V2O5 content of the raw SCR catalyst employed in this work was a moderate percent of about 1%, indicating this catalyst was well typical and representative. Small amount of SiO2 was also detected to contain in the catalyst, which was helpful for boosting the mechanical strength. For the CeO2 modified catalysts, the practical contents of CeO2 in the catalysts with different CeO2 loadings were very close to the corresponding designed values, which testified the accuracy of the preparation of the catalysts. Meanwhile, the addition of CeO2 did not cause apparent variations on the contents of V2O5, WO3 and SiO2 in the catalysts.
Table 1 Element compositions and contents of the CeO2 modified commercial SCR catalysts
| Catalyst |
Mass fraction (%) |
| CeO2 |
TiO2 |
V2O5 |
WO3 |
SiO2 |
| Raw SCR |
0 |
90.71 |
0.98 |
6.63 |
1.68 |
| 1% CeO2-SCR |
0.92 |
89.73 |
0.96 |
6.58 |
1.81 |
| 2% CeO2-SCR |
1.95 |
88.54 |
1.08 |
6.80 |
1.63 |
| 4% CeO2-SCR |
4.03 |
86.93 |
1.12 |
6.35 |
1.57 |
| 7% CeO2-SCR |
6.79 |
84.18 |
1.20 |
6.06 |
1.77 |
3.1.2 BET analysis. The surface structural properties of the CeO2 modified commercial SCR catalysts tested by BET analysis were listed in Table 2. According to the results, the surface area of the raw catalyst was at a relatively low level of 18.64 m2 g−1, which might result from the specific preparation process of the catalyst corporation. The introduction of CeO2 made a significant enhancement on the surface area and pore volume of the catalyst. The surface area increased dramatically from 18.64 m2 g−1 to 69.23 m2 g−1 with the loading of only 1% CeO2. The increase of surface area could raise the amount of the active sites available for Hg0 and other reactants on the catalyst surface, thereby it usually being beneficial to the catalytic activity.35,37 And the enlargement of pore volume was also in favor of the Hg0 removal capacity of the catalyst. The surface area showed a slight declined trend as the CeO2 loading augmented, which was probably due to the blockage of some surface micropores caused by the increasing CeO2 loading.38,39 It's worth noting that the surface area of the CeO2 modified catalysts was much closer to that of pure CeO2 than to the raw SCR catalyst, indicating that the surface area was obviously altered and controlled by CeO2 though it occupied only a minor proportion in the catalysts. By contrast, the pore size of the catalyst was not distinctly affected by the addition of CeO2, and the change was small.
Table 2 Surface structural properties of the CeO2 modified commercial SCR catalysts
| Catalyst |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
| Raw SCR |
18.64 |
0.069 |
19.579 |
| 1% CeO2-SCR |
69.23 |
0.287 |
16.555 |
| 2% CeO2-SCR |
66.39 |
0.279 |
17.081 |
| 4% CeO2-SCR |
64.92 |
0.285 |
17.248 |
| 7% CeO2-SCR |
61.83 |
0.233 |
15.553 |
| Pure CeO2 |
66.28 |
0.294 |
17.835 |
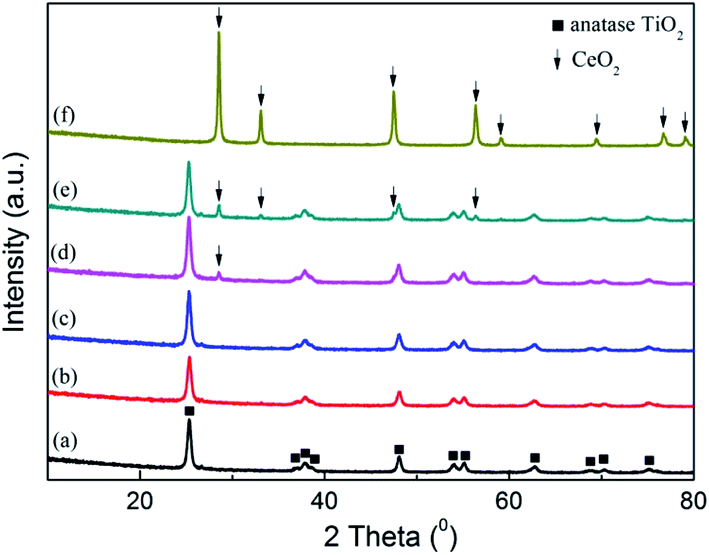
3.1.3 XRD analysis. The crystalline characteristic in the catalysts was investigated by XRD analysis, and the result was shown in Fig. 2. On the patterns of raw SCR catalyst and pure CeO2, only the peaks corresponding to anatase TiO2 and CeO2 were discovered respectively.23,30,40 With CeO2 doped into the commercial SCR catalyst, the peak intensity of TiO2 became weak gradually, and meanwhile the peak standing for CeO2 was not detected when the CeO2 content was lower than 4%. This phenomenon suggested that there existed some interaction between TiO2 and CeO2 in the catalysts.33,41,42 CeO2 was well dispersed and in the form of amorphous phase on the catalyst surface. As the CeO2 content reached 4%, a peak corresponding to CeO2 emerged on the pattern at 28.6°, indicating that the present load amount has made the dispersion of CeO2 on the catalyst reach the critical point of saturation. Further increasing the CeO2 loading would lead to the conversion of the doped CeO2 from amorphous phase to crystalline state. The emergence of distinct characteristic peaks corresponding to CeO2 on the profile of 7% CeO2-SCR confirmed this inference. In addition, the peaks of V2O5 and WO3 were not discovered on any catalyst pattern, displaying an amorphous distribution as well. More active substance existed in the amorphous phase was considered to be advantageous for the catalytic activity of the catalyst, while the appearance of the crystal of the active species was adverse to the catalytic performance.43,44
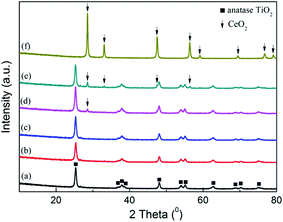 |
| | Fig. 2 XRD patterns of the catalysts ((a) raw SCR, (b) 1% CeO2-SCR, (c) 2% CeO2-SCR, (d) 4% CeO2-SCR, (e) 7% CeO2-SCR, (f) pure CeO2). | |
3.2. NO removal performance of the CeO2-SCR catalysts
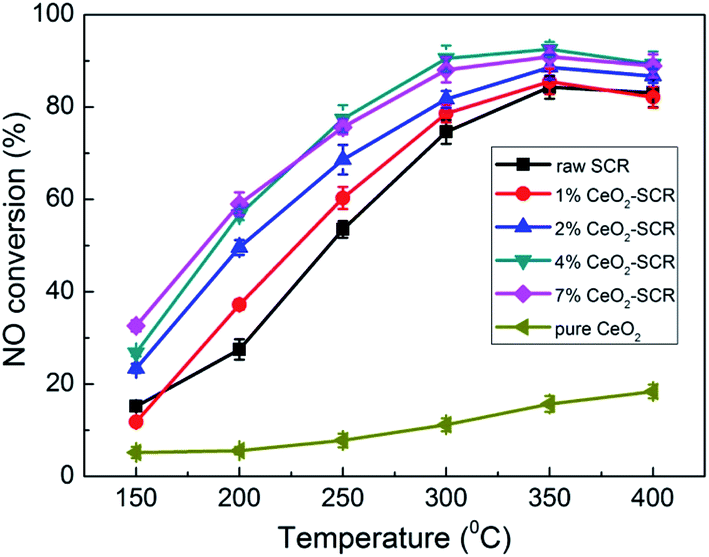
Considering the primary function of SCR catalyst was to remove NO for coal combustion power plant, NO removal activity of the CeO2 modified commercial SCR catalysts in simulated coal-fired flue gas was first examined prior to the investigation on Hg0 removal performance. The experimental results were shown in Fig. 3. The NO conversions of the catalysts showed a growing trend as the reaction temperature increased from 150 °C to 400 °C. The optimal temperature range was 300–400 °C which was consistent with that of literature report.41,45,46 NO conversion over the raw SCR catalyst in this range was 74.6–84.3%, which was a little lower than the efficiencies monitored in power plants. This might be attributed to the higher GHSV in the lab reactor than that under the practical conditions (2000–3000 h−1),47 which led to the shorter contact time between flue gas and catalyst. As CeO2 was added into the catalyst, NO conversion was apparently promoted. And the catalyst with the CeO2 loading of 4% exhibited the best activity for NO removal. The NO conversions were 90.5%, 92.5% and 89.3%, respectively, at the temperature points of 300–400 °C over 4% CeO2-SCR. Besides, the efficiency of 4% CeO2-SCR could also reach nearly 80% at 250 °C. Thus, the CeO2 modification not only improved NO conversion of commercial SCR catalyst, but also broadened the working temperature and enhanced the medium-low temperature activity of the catalyst. The superior NO removal performance of 4% CeO2-SCR was associated with the higher content of CeO2 dispersed in the amorphous phase, while the slightly decreased NO conversion over 7% CeO2-SCR compared to that over 4% CeO2-SCR might be due to the generation of CeO2 crystal in the catalyst. Additionally, the surface area was also a possible influence factor for the NO removal activity because the variation trend of the surface area was very close to that of the NO conversion among 4% CeO2-SCR, 7% CeO2-SCR and the raw catalyst. Therefore, the experimental acquirement was in good agreement with the characterization results. The efficiency of pure CeO2 was in a poor level among the testing temperature range, indicating that the element V was still responsible for the nice NO removal activity of CeO2-SCR though CeO2 generated modification effects on the catalysts. To sum up, the CeO2 modification led to an advancement upon the property of the commercial SCR catalyst and made it own prominent NO removal activity, which established a solid foundation on the utilization of the catalyst for synergistic Hg0 removal.
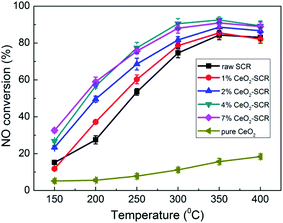 |
| | Fig. 3 NO conversion over the CeO2 modified commercial SCR catalysts under different reaction temperatures in simulated coal-fired flue gas. | |
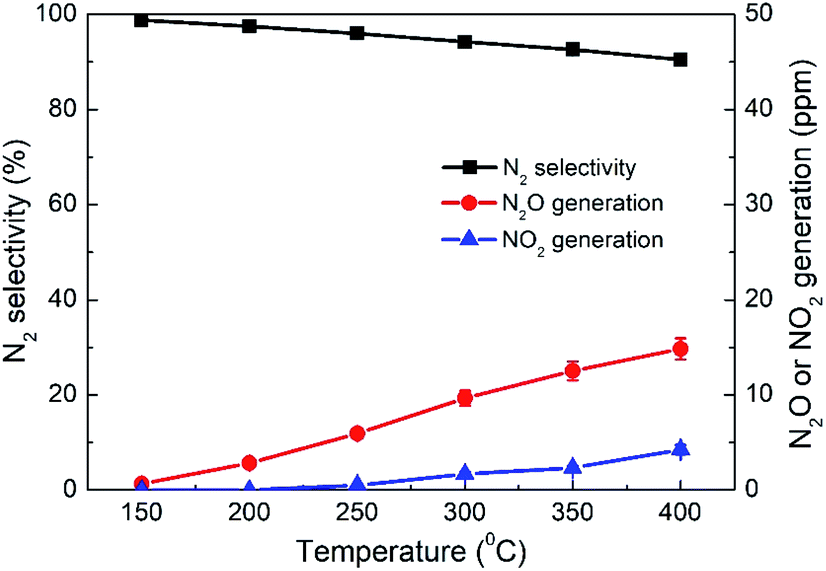
As another important evaluation index for NO removal performance, N2 selectivity was measured over the 4% CeO2-SCR catalyst which exhibited the highest NO conversion, and the results were shown in Fig. 4. Under SFG, the N2 selectivity over the catalyst reduced slightly with the increase of the reaction temperature, which was caused by the generation of a small amount of N2O and NO2 during the reaction. The detected concentrations of N2O were much higher than those of NO2. So the decrease of the N2 selectivity was mainly due to the N2O generation at the higher temperatures. Nevertheless, the N2O generation was lower than 15 ppm in the whole temperature range of 150–400 °C, and even the poorest N2 selectivity measured at 400 °C reached as high as 90.5%. Hence, the catalyst displayed great N2 selectivity in the NO removal process, further confirming the excellent NO removal performance of the CeO2-SCR catalyst in the simulated coal-fired flue gas.
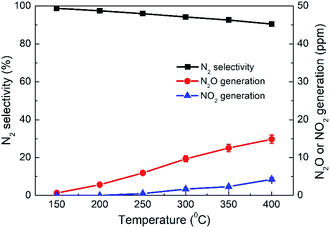 |
| | Fig. 4 N2 selectivity and N2O and NO2 generations over 4% CeO2-SCR under different reaction temperatures in simulated coal-fired flue gas. | |
3.3. Hg0 removal performance of the CeO2-SCR catalysts
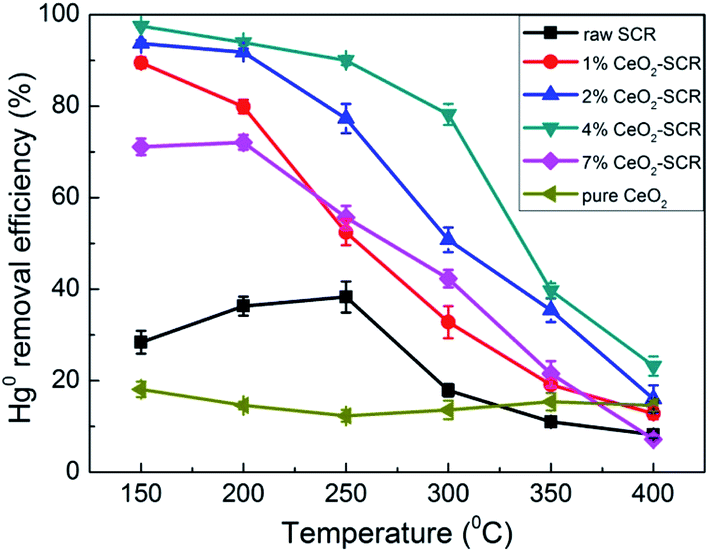
3.3.1 Hg0 removal efficiency under different temperatures in SFG. Hg0 removal performance of the CeO2 modified commercial SCR catalysts was then investigated as the emphasis. First, the Hg0 removal efficiencies of the catalysts in simulated coal-fired flue gas were measured under different reaction temperatures, and the results were shown in Fig. 5. As the temperature increased, the variation trend of the Hg0 removal efficiencies of the CeO2-SCR catalysts was opposite to that of the NO conversions, and it was a descending tendency. The possible reason for this phenomenon was that the lower temperature was beneficial to the Hg0 adsorption on the catalyst which was an essential procedure for Hg0 removal, and the Hg0 oxidation was realized mainly through the form of adsorbed Hg0 (Hg0ad).35,42,48 The introduction of CeO2 into the catalyst accelerated the Hg0 removal efficiency apparently. Analogously to the testing results of NO removal activity, the optimal sample for Hg0 removal was 4% CeO2-SCR as well, which corresponded to the characterization results again. Hg0 removal efficiency of 4% CeO2-SCR achieved more than 90% in the temperature range of 150–250 °C. Even at 300 °C which was among the conventional operating temperature of SCR catalyst (300–400 °C), 4% CeO2-SCR also exhibited the efficiency of as high as 78.2% on the basis of NO conversion guaranteed at 89.3%. So the catalyst showed remarkable activity for simultaneous NO and Hg0 removal. The prominent performance for synergistic Hg0 removal was mainly owed to the sufficient chemisorbed oxygen (Oad) of 4% CeO2-SCR led by the existence of Ce3+/Ce4+ ion pair and the oxygen transfer between them in the catalyst,38,49 which would be confirmed by the subsequent XPS analysis. The abundant Oad would facilitate Hg0 oxidation to generate HgO as the active species. The related reaction process was described by eqn (4)–(6). As the efficiencies of the raw catalyst and pure CeO2 were no more than 38.3%, the superior performance of the CeO2 modified commercial SCR catalyst was also primarily resulted from the synergy of V2O5 and CeO2 in the catalyst.50 In addition, considering the GHSV was much higher in the experimental condition than in actual flue gas of power plant, the catalytic efficiencies might be preferable in practical application. Hence, the SCR catalyst manifested to be more competent and promising for commercial use after the CeO2 modification.
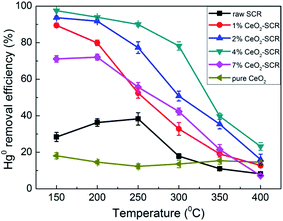 |
| | Fig. 5 Hg0 removal efficiency over the CeO2 modified commercial SCR catalysts under different reaction temperatures in simulated coal-fired flue gas. | |
3.3.2 Effects of the flue gas components on Hg0 removal efficiency. Effect of each flue gas component on the Hg0 removal efficiency of the CeO2-SCR catalyst was then investigated to reveal its role in Hg0 removal process. And the results were made comparison with those of the raw SCR catalyst to explore the reasons for the modification effect of CeO2 on the catalyst for Hg0 removal in simulated coal-fired flue gas. Because the optimum catalytic efficiencies were implemented at 300 °C over 4% CeO2-SCR with the NO conversion and synergistic Hg0 removal efficiency being 89.3% and 78.2%, respectively, the experiments of this part were carried out at 300 °C using 4% CeO2-SCR as the catalyst sample. The reaction atmosphere was SFG with the concentration of the investigated component changed and the others constant.
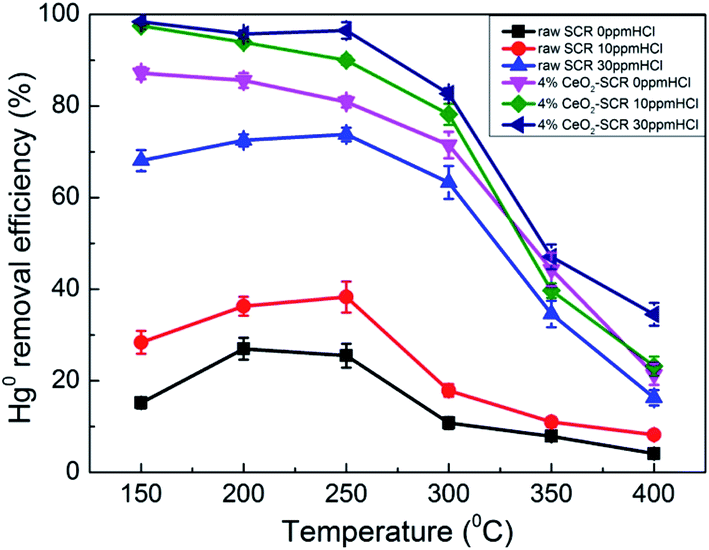
3.3.2.1. Effect of HCl. As the important oxidant for Hg0 oxidation in coal combustion flue gas, effect of HCl on the Hg0 removal efficiency of the catalysts was examined, and the results were shown in Fig. 6. For the raw catalyst, Hg0 removal efficiency was disadvantaged in the absence of HCl, and the highest value was only 27%. Even though 10 ppm HCl was added into the flue gas, the efficiency was still maintained at a low level since it was below 40% in the whole temperature range. Only when the HCl concentration increased from 10 ppm to 30 ppm did the Hg0 removal efficiency of the raw catalyst show a significant improvement. It increased by 35.5% and 45.4%, respectively, at 250 °C and 300 °C as the instances. The above results verified the viewpoint in the literatures that the commercial SCR catalyst was qualified to be utilized in the flue gas derived from burning bitumite with high HCl content while not appropriate to work under low HCl concentration caused by using low-rank coals for Hg0 removal.20,23 By contrast, after CeO2 modification, 4% CeO2-SCR exhibited much more prominent Hg0 removal efficiency than raw SCR catalyst under the same HCl concentration. The performance over 4% CeO2-SCR was even better without HCl than that over the raw catalyst in the presence of 30 ppm HCl. A limited increase of the efficiency of 4% CeO2-SCR was observed as the HCl concentration raised. Nevertheless, the catalyst displayed satisfactory Hg0 removal activity when exposed to 10 ppm HCl. The Hg0 removal efficiencies were excellent at 150–300 °C. Therefore, the CeO2 modification weakened the dependence of Hg0 removal activity of the catalyst on HCl content of the flue gas. This was really good news for power plants combusting sub-bituminous coal and lignite which occupied the majority of all items. The reason for the superior Hg0 removal efficiency of 4% CeO2-SCR under low HCl concentration was also due to the improved content of Oad on the catalyst surface. More HCl could be converted by the abundant Oad to form active Cl (Cl*) which had strong oxidation, followed by Hg0 being oxidized to HgCl2 by Cl*.51,52 Through this way, the introduced CeO2 enhanced the HCl utilization of the catalyst. The involved reactions could be described by eqn (7) and (8). Meanwhile, this was also one of the main reasons for the higher Hg0 removal efficiency over 4% CeO2-SCR compared to that over raw SCR in the simulated coal-fired flue gas besides the direct oxidation by Oad.| | |
2HCl + Oad → 2Cl* + H2O
| (7) |
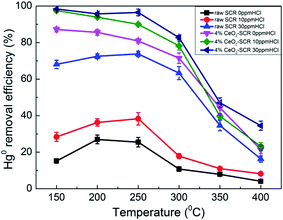 |
| | Fig. 6 Effect of HCl on Hg0 removal efficiency of raw SCR catalyst and 4% CeO2-SCR in simulated coal-fired flue gas (reaction gas: SFG with 0, 10, 30 ppm HCl). | |
| | |
Hg0 + SO3 + Oad → HgSO4
| (10) |
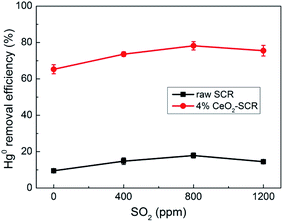 |
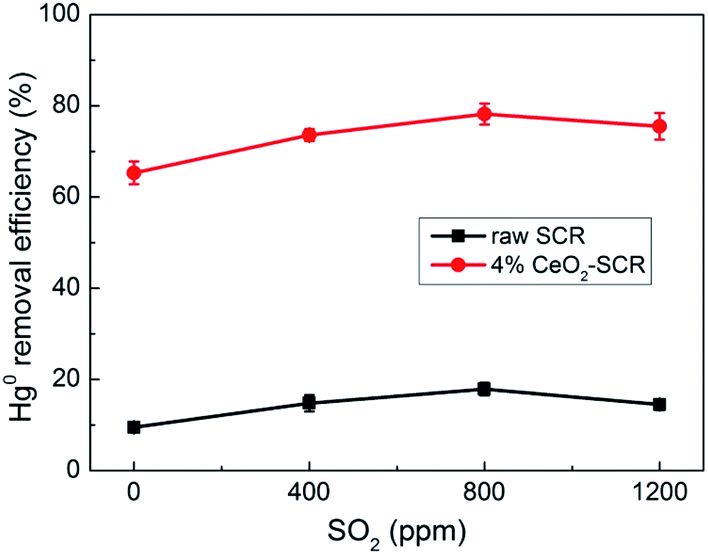
| | Fig. 7 Effect of SO2 on Hg0 removal efficiency of raw SCR catalyst and 4% CeO2-SCR in simulated coal-fired flue gas (reaction gas: SFG with 0, 400, 800, 1200 ppm SO2). | |
| | |
Hg0 + 2NO2 + 2Oad → Hg(NO3)2
| (12) |
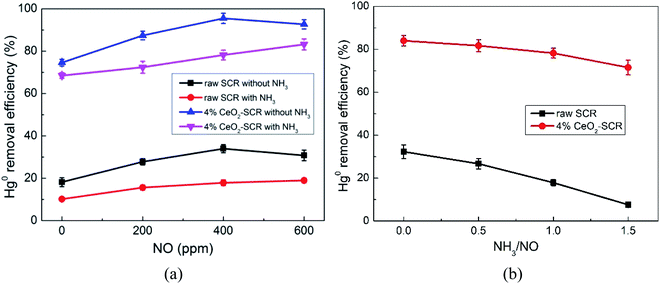 |
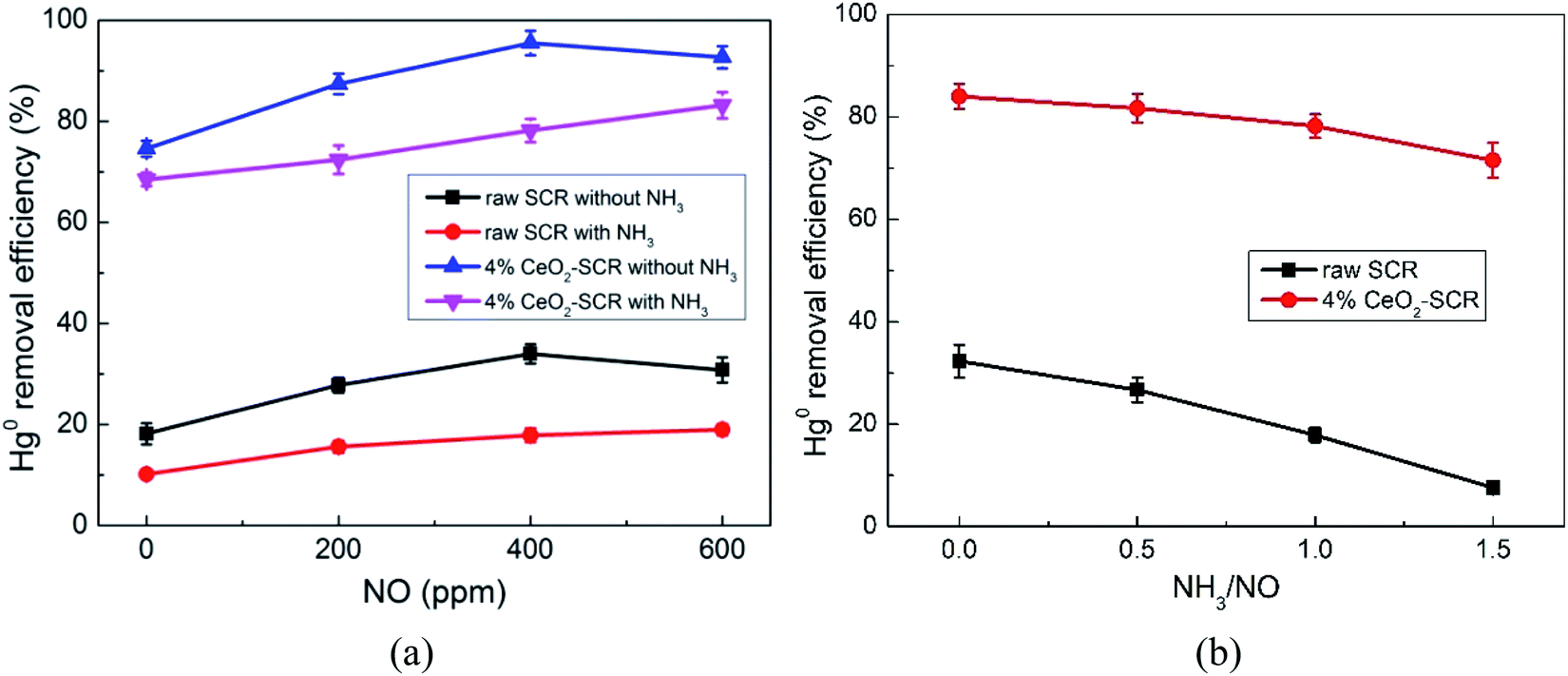
| | Fig. 8 Effects of NO and NH3 on Hg0 removal efficiency of raw SCR catalyst and 4% CeO2-SCR in simulated coal-fired flue gas ((a) effect of NO, reaction gas: SFG with 0, 200, 400, 600 ppm NO in the presence or absence of NH3; (b) effect of NH3, reaction gas: SFG with 0, 200, 400, 600 ppm NH3). | |
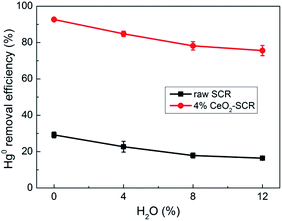 |
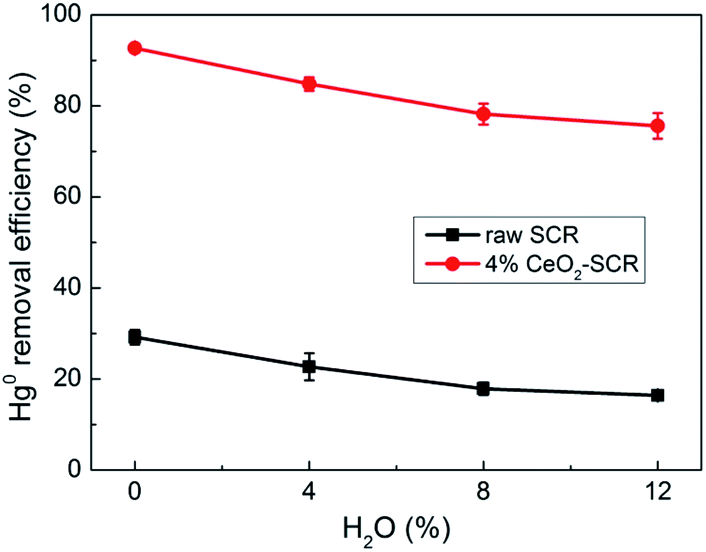
| | Fig. 9 Effect of H2O on Hg0 removal efficiency of raw SCR catalyst and 4% CeO2-SCR in simulated coal-fired flue gas (reaction gas: SFG with 0, 4, 8, 12% H2O). | |
3.4. Modification mechanism of CeO2 explored by XPS analysis
According to the above experimental results, the CeO2 modification generated excellent results on the NO and Hg0 removal performance of commercial SCR catalyst. The characterization results of BET and XRD could present the related reasons for the modification effects in a certain degree. In order to further explore the modification mechanism of CeO2 on the catalyst, H2-TPR and XPS analyses were carried out to detect the redox behavior and valence states (or types) of the elements in the raw and modified catalysts.
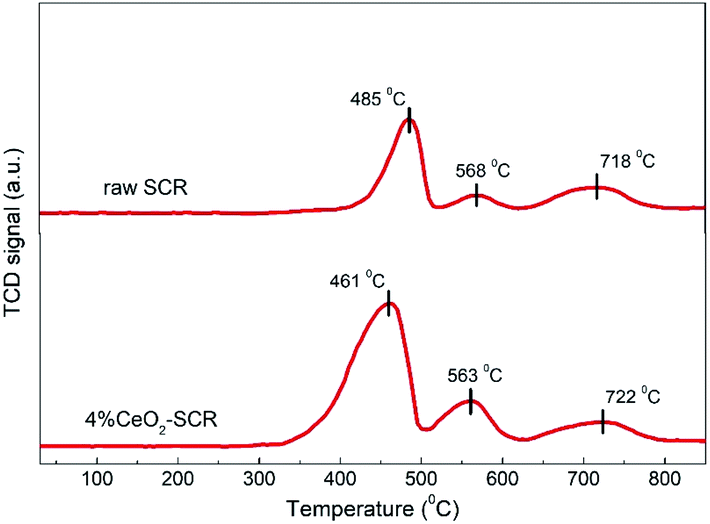
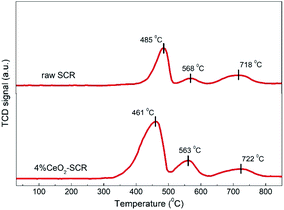
3.4.1 H2-TPR analysis. H2-TPR analysis was implemented over the raw SCR and 4% CeO2-SCR catalysts, and the results were shown in Fig. 10. On the profile of the raw catalyst, the peaks emerged at 485 °C and 568 °C could be attributed to the reduction of V5+ and surface oxygen, respectively, and the broad shoulder peak at around 720 °C was corresponded to the overlap of the reduction of W6+ and lattice oxygen.44,60,61 By contrast, a reduction peak was observed at 461 °C on the profile of 4% CeO2-SCR. As Ce4+ was reported to reduce at about 470 °C, this peak was considered to be the overlapped reduction peak of V5+ and Ce4+.62 It was evident that the temperature of this peak was lowered and the intensity was strengthened dramatically compared to the peak of the raw catalyst at 485 °C, which indicated that the synergy of element V and Ce reinforced the reactivity of the catalyst. In addition, the reduction peak of surface oxygen of 4% CeO2-SCR at 563 °C was much stronger than that of the raw catalyst, so it demonstrated the existence of Ce enhanced the oxygen storage capacity of the catalyst. Combining the above factors, the integral area of the reduction profile was obviously larger over 4% CeO2-SCR than over the raw catalyst, suggesting the improved redox behavior of the catalyst led by the CeO2 modification. The superior redox behavior was favorable to the NO and Hg0 removal performance, which was one of main reasons for the prominent catalytic efficiencies of the CeO2 modified commercial SCR catalyst.
 |
| | Fig. 10 H2-TPR profiles of the raw SCR and 4% CeO2-SCR catalysts. | |
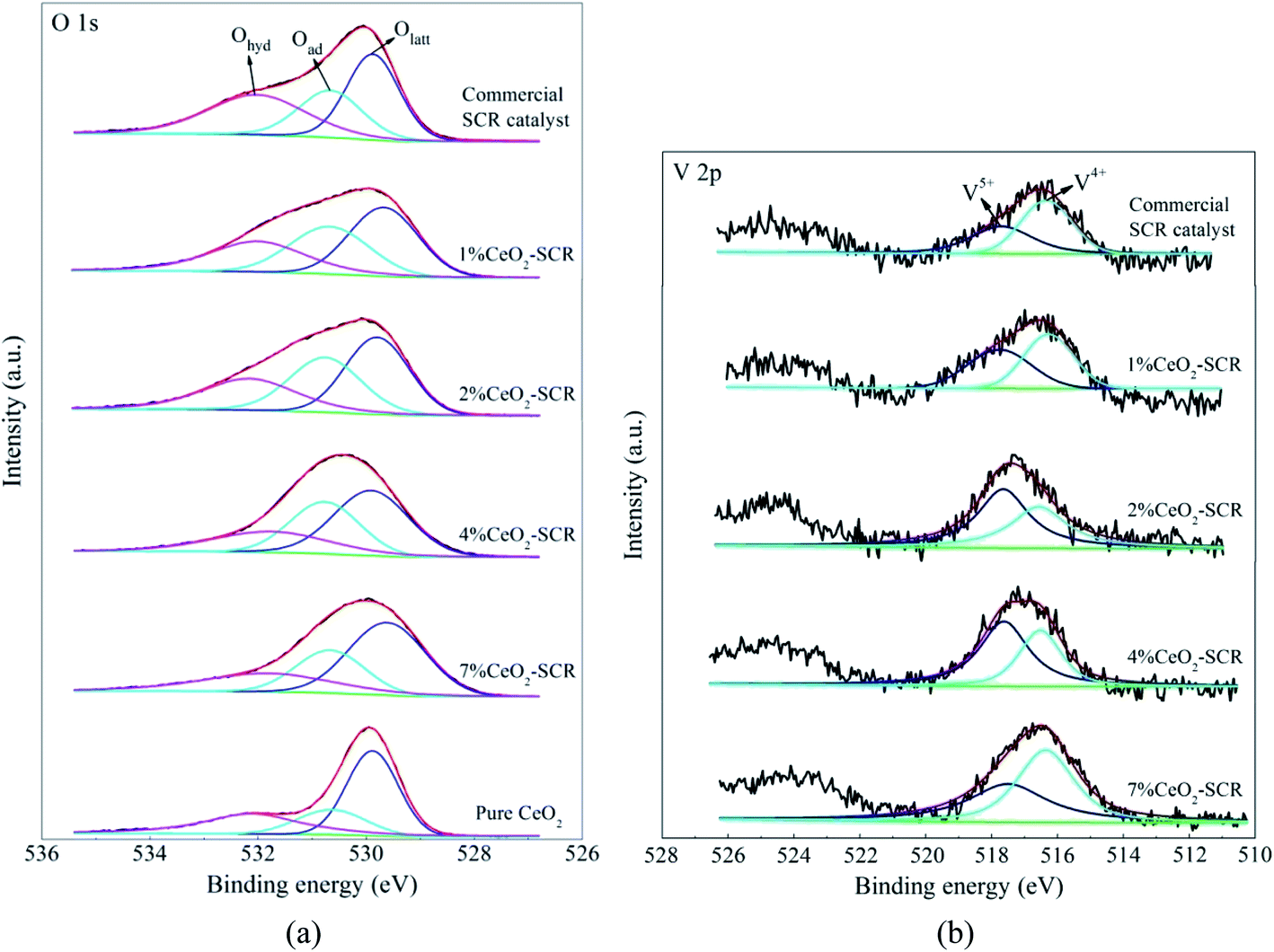
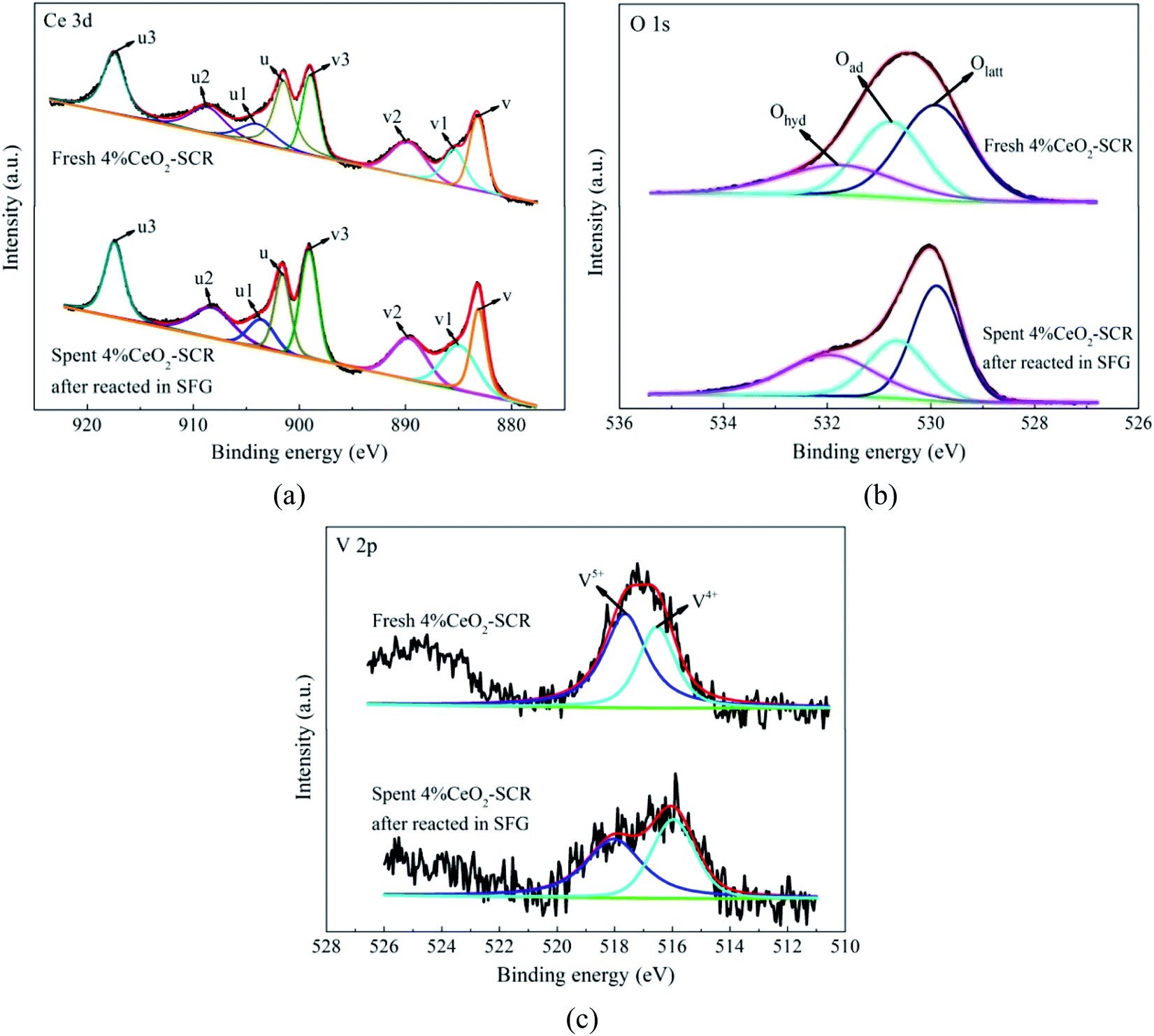
3.4.2 XPS analysis. The XPS spectra of the elements for the fresh catalysts, together with the fitting results of the curves, were shown in Fig. 11. For the spectra of O 1s, the fitting peaks were assigned to lattice oxygen (Olatt), chemisorbed oxygen (Oad) and oxygen of hydroxyl and free water (Ohyd) in sequence at the binding energies from small to large,25,63 as shown in Fig. 11(a). And the fitting peaks of V 2p at the binding energies of approximately 516.4 eV and 517.6 eV could be distributed to V4+ and V5+, respectively,64,65 which was shown in Fig. 11(b). In addition, the analysis on the spent catalyst sample of 4% CeO2-SCR after reacted in simulated coal-fired flue gas was conducted as well. The obtained curves of Ce 3d, O 1s and V 2p were made comparisons with those of the fresh catalyst, and the results were shown in Fig. 12. On the curves of the element Ce as shown in Fig. 12(a), the fitting peaks of u, u2, u3, v, v2 and v3 were attributed to Ce4+, while the peaks of u1 and v1 were corresponded to Ce3+.38,66 And the spectra of O and V for the spent catalyst were shown respectively in Fig. 12(b) and (c). The ratios of each elemental type or valence state in the corresponding elements of the catalysts were acquired through integrating the fitting peaks and calculating the peak area. The calculation results for the elements of the fresh and spent catalysts were summarized in Tables 3 and 4, respectively.
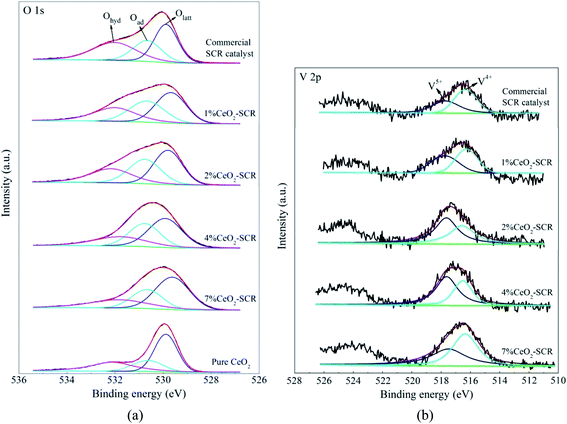 |
| | Fig. 11 XPS spectra of O 1s and V 2p for the fresh raw and CeO2 modified commercial SCR catalysts ((a) O 1s; (b) V 2p). | |
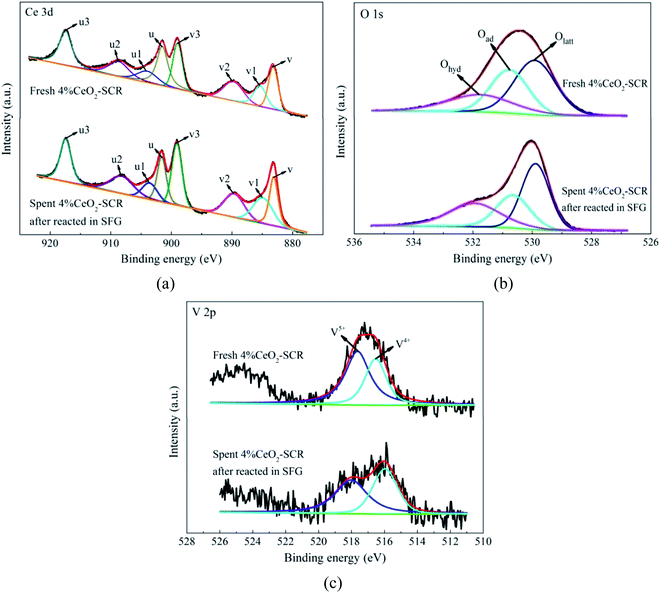 |
| | Fig. 12 XPS spectra of Ce 3d, O 1s and V 2p for the fresh 4% CeO2-SCR catalyst and the spent 4% CeO2-SCR catalyst after reacted in SFG ((a) Ce 3d; (b) O 1s; (c) V 2p). | |
Table 3 The surface atomic contents of O and the ratios of Oad and V5+ in the corresponding elements on the catalysts determined by XPS
| Catalyst |
Content of O (%) |
Oad/(Olatt + Oad + Ohyd) (%) |
Content of Oad (%) |
V5+/(V4+ + V5+) (%) |
| Raw SCR |
46.0 |
26.1 |
12.0 |
43.5 |
| 1% CeO2-SCR |
64.9 |
31.0 |
20.1 |
48.0 |
| 2% CeO2-SCR |
64.6 |
31.4 |
20.3 |
56.4 |
| 4% CeO2-SCR |
62.6 |
32.5 |
20.4 |
60.8 |
| 7% CeO2-SCR |
57.4 |
25.8 |
14.8 |
46.1 |
| Pure CeO2 |
46.2 |
19.8 |
9.1 |
— |
Table 4 The ratios of Ce3+, Oad and V5+ in the corresponding elements on the fresh and spent 4% CeO2-SCR catalysts determined by XPS
| Catalyst |
Ce3+/(Ce4+ + Ce3+) (%) |
Oad/(Olatt + Oad + Ohyd) (%) |
Ohyd/(Olatt + Oad + Ohyd) (%) |
V5+/(V4+ + V5+) (%) |
| Fresh 4% CeO2-SCR |
16.6 |
32.5 |
22.8 |
60.8 |
| Spent 4% CeO2-SCR after reacted in SFG |
20.5 |
27.0 |
30.2 |
54.7 |
According to the testing results, the addition of CeO2 into the catalyst improved both the surface atomic content of O and the proportion of Oad, which led to the increase of the content of Oad on the catalyst, as the data listed in Table 3. It could be judged from the results of Ce 3d of 4% CeO2-SCR shown in Fig. 12(a) and Table 4 that Ce3+ and Ce4+ coexisted in the modified catalysts. The presence of Ce3+ with a proportion of 16.6% could create charge imbalance and unsaturated chemical bonds on the surface, which was favorable for the generation of chemisorbed oxygen, thereby raising the content of Oad and boosting the oxygen storage capacity of the catalyst.38,61,67 Oad was the active oxygen species that could participate in the catalytic reactions. 4% CeO2-SCR owned the highest content of Oad among the catalysts, which was another important reason for its optimal NO and Hg0 removal performance. As the CeO2 loading increased from 4% to 7%, the Oad content on the catalyst declined and it was even lower than that of the raw catalyst. This result could be associated with the conversion of CeO2 to the crystalline phase in 7% CeO2-SCR according to the XRD results, which made it disadvantaged for the forming of Oad from the loaded CeO2, and meanwhile the forming of crystalline CeO2 might consume a number of the intrinsic Oad on the surface. Besides Oad, the intensity of the V5+ peak and the ratio of V5+ were also enlarged with the introduction of CeO2. The increase of the V5+ proportion might be attributed to part of V4+ being oxidized by the abundant Oad to V5+ on the modified catalysts. V5+ was the active species in V-based catalyst as well, which had good oxidation and was beneficial to Hg0 removal activity. So the adequate Oad was also presented in the form of V2O5. As the content of Oad on the surface of pure CeO2 did not show an advantage, it further demonstrated the superior oxygen storage capacity was the result of the synergy of CeO2 and V2O5 in the CeO2-SCR catalysts.
After the 4% CeO2-SCR catalyst was reacted in SFG, the XPS spectra of O 1s and V 2p for the spent catalyst were compared with those for the fresh one. The results indicated that the intensity of both the Oad and V5+ peaks reduced apparently after the reaction, as shown in Fig. 12(b) and (c). The variation could be observed more intuitively by the results in Table 4 that the ratios of Oad and V5+ in the corresponding elements decreased from 32.5% to 27% and from 60.8% to 54.7%, respectively, in the reaction process, while the ratio of Ce3+ increased from 16.6% to 20.5%. The variation trends of the ratios of Oad and Ce3+ on the catalyst were in accordance with those in the related literatures after the catalysts were spent.19,68 The decline of the ratio of Oad demonstrated it indeed participated in the catalytic reactions as the active substance. And the decrease of the proportion of V5+ suggested the redox behavior between element V and Ce on the catalyst during the reactions, which could be expressed by eqn (13). Combining the eqn (13) with the previous eqn (5), it could be seen that it occurred the process of the redox transformation between Ce3+ and Ce4+ on the surface, in which the chemisorbed oxygen was generated. The formed Oad then involved in the catalytic reactions such as eqn (6), (7) and (9)–(12) so that the performance of the catalyst for synergistic Hg0 removal in SFG was improved. Besides Oad, the ratio of Ohyd increased by 7.4% in the spent catalyst. On one hand, H2O contained in the flue gas adsorbed on the catalyst and formed hydroxyl during the reaction, which caused the competitive adsorption with Hg0 and led to the inhibition of H2O on Hg0 removal efficiency; on the other hand, the increased proportion of Ohyd might also be due to the generated H2O of eqn (7), thereby further demonstrating the occurrence of this reaction.
| | |
V5+ + Ce3+ → V4+ + Ce4+
| (13) |
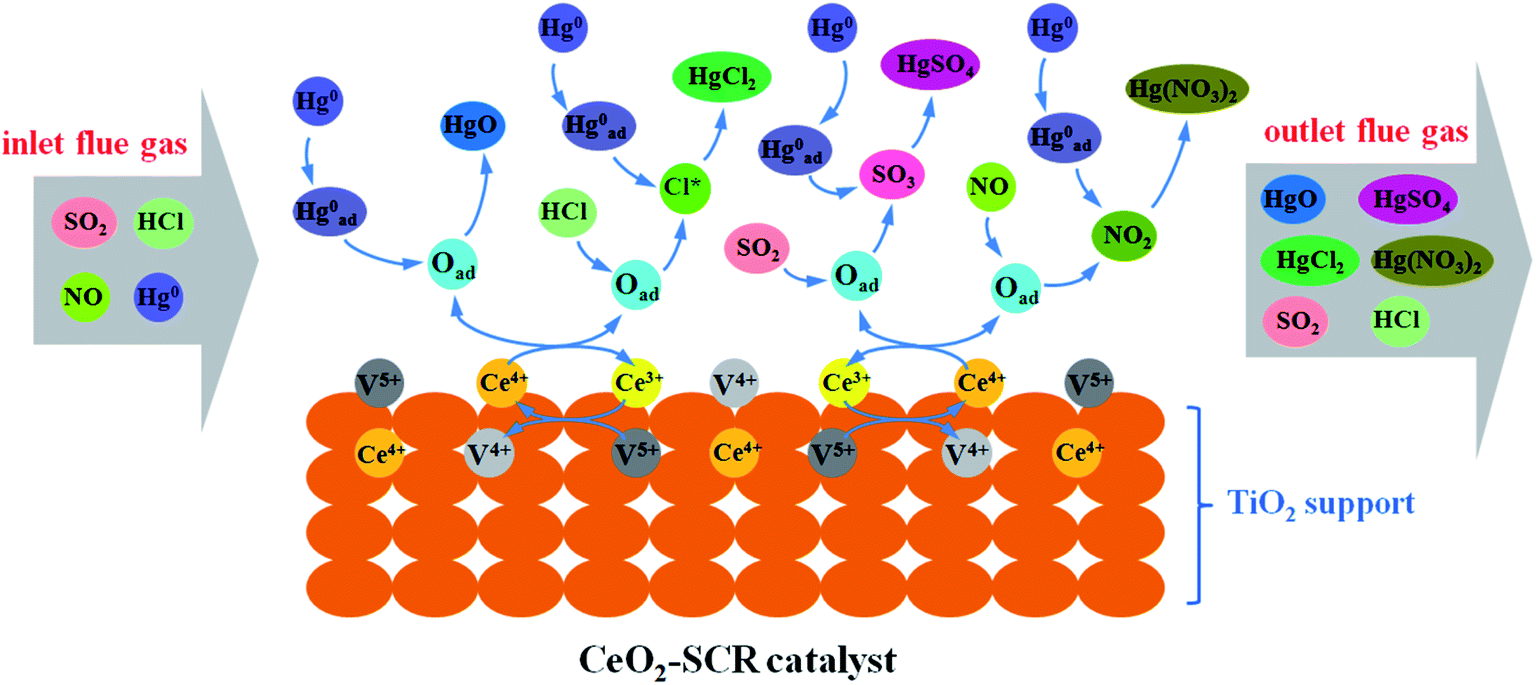
Combining the experimental results and the XPS analysis conclusions, the modification effects of CeO2 on commercial SCR catalyst was mainly owed to the more sufficient chemisorbed oxygen which derived from the interaction between element V and Ce and the redox transformation between Ce3+ and Ce4+ on the catalyst surface. The abundant Oad improved the catalytic activity of the catalyst and the promotion of related flue gas components such as HCl on the Hg0 removal efficiency. Integrating these factors, the catalytic property for synergistic Hg0 removal was enhanced by the CeO2 modification. The modification process was described more vividly and specifically by the illustration shown in Fig. 13.
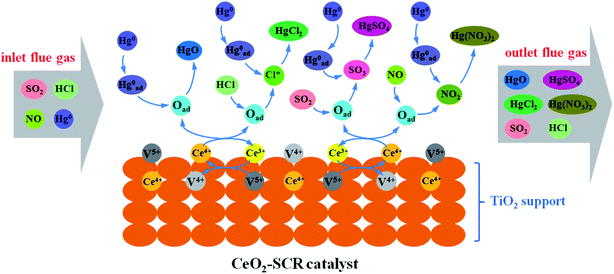 |
| | Fig. 13 Description of the mechanism of CeO2 modification on the synergistic Hg0 removal performance of commercial SCR catalyst. | |
4. Conclusions
CeO2 modified commercial SCR catalyst was prepared and investigated for NO and synergistic Hg0 removal. The research results indicated that the CeO2 loading improved a series of properties of the catalyst. Concretely, the BET surface area, the dispersity of the metal oxides on TiO2 support and the redox behavior were increased with the introduction of CeO2 into the catalyst, which was favorable to the catalytic activity. The catalyst with the CeO2 content of 4% exhibited the optimal performance for simultaneous NO and Hg0 removal. The NO conversion of 4% CeO2-SCR was as high as 90.5% at 300 °C in SFG with excellent N2 selectivity, while the synergistic Hg0 removal efficiency could reach 78.2% under the same condition. Owing to the abundant chemisorbed oxygen generated from the synergy of V2O5 and CeO2 and the redox transformation between Ce3+ and Ce4+, the Hg0 removal activity, the HCl utilization and NH3-resistance in Hg0 removal process and the promotion of SO2 and NO on Hg0 removal efficiency were improved over 4% CeO2-SCR compared to over the raw catalyst. On account of these factors, the CeO2 modification made an enhancement on the synergistic Hg0 removal performance of the commercial SCR catalyst in simulated coal-fired flue gas, especially under low HCl concentration. Therefore, the CeO2 modified commercial SCR catalyst was a potential candidate to be practically applied in coal combustion power plant for NO and synergistic mercury removal.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Fundamental Research Fund of Shandong University (No. 2020GN008), the National Key Research and Development Program of China (No. 2016YFB0600604), the National Nature Science Foundation of China (NSFC) (No. 41672148) and the Foundation of State Key Laboratory of Coal Combustion (No. FSKLCCA1910). The authors would like to thank the Analysis and Test Center of Huazhong University of Science & Technology for the characterization work.
References
- P. J. Landrigan, R. O. Wright and L. S. Birnbaum, Mercury toxicity in children, Science, 2013, 342, 1447 CrossRef CAS PubMed
 .
. - UNEP, Global mercury assessment 2018, UNEP Chemicals and Health Branch, Geneva, Switzerland, 2019 Search PubMed .
- M. Rumayor, M. Díaz-Somoano, M. A. Lopez-Anton and M. R. Martínez-Tarazona, Application of thermal desorption for the identification of mercury species in solids derived from coal utilization, Chemosphere, 2015, 119, 459–465 CrossRef CAS PubMed .
- Q. Wu, S. Wang, K. Liu, G. Li and J. Hao, Emission-limit-oriented strategy to control atmospheric mercury emissions in coal-fired power plants toward the implementation of the Minamata Convention, Environ. Sci. Technol., 2008, 52, 11087–11093 CrossRef PubMed .
- Q. Shi, Y. Wang, X. Zhang, B. Shen, F. Wang and Y. Zhang, Hierarchically porous biochar synthesized with CaCO3 template for efficient Hg0 adsorption from flue gas, Fuel Process. Technol., 2020, 199, 106247 CrossRef CAS .
- X. Geng, Y. Duan, S. Zhao, Y. Xu, T. Huang, J. Hu and S. Ren, Study of mercury-removal performance of mechanical-chemical-brominated coal-fired fly ash, Energy Fuels, 2019, 33, 6670–6677 CrossRef CAS .
- G. Busca, M. A. Larrubia, L. Arrighi and G. Ramis, Catalytic abatement of NOx: chemical and mechanistic aspects, Catal. Today, 2005, 107–108, 139–148 CrossRef CAS .
- P. G. Smirniotis, P. M. Sreecanth, D. A. Peña and R. G. Jenkins, Manganese oxide catalysts supported on TiO2, Al2O3, and SiO2: a comparison for low-temperature SCR of NO with NH3, Ind. Eng. Chem. Res., 2006, 45, 6436–6443 CrossRef CAS .
- C. Peng, J. Liang, H. Peng, R. Yan, W. Liu, Z. Wang, P. Wu and X. Wang, Design and synthesis of Cu/ZSM-5 catalyst via a facile one-pot dual-template strategy with controllable Cu content for removal of NOx, Ind. Eng. Chem. Res., 2018, 57, 14967–14976 CrossRef CAS .
- S. Eswaran and H. G. Stenger, Understanding mercury conversion in selective catalytic reduction (SCR) catalysts, Energy Fuels, 2005, 19, 2328–2334 CrossRef CAS .
- A. Suarez Negreira and J. Wilcox, Role of WO3 in the Hg oxidation across the V2O5–WO3–TiO2 SCR catalyst: a DFT study, J. Phys. Chem. C, 2013, 117, 24397–24406 CrossRef CAS .
- J. Yang, Q. Li, M. Li, W. Zhu, Z. Yang, W. Qu, Y. Hu and H. Li, In situ decoration of selenide on copper foam for the efficient immobilization of gaseous elemental mercury, Environ. Sci. Technol., 2020, 54, 2022–2030 CrossRef CAS PubMed .
- H. Kamata, S. Ueno, T. Naito, A. Yamaguchi and S. Ito, Mercury oxidation by hydrochloric acid over a VOx/TiO2 catalyst, Catal. Commun., 2008, 9, 2441–2444 CrossRef CAS .
- C. L. Senior, Oxidation of mercury across selective catalytic reduction catalysts in coal-fired power plants, J. Air Waste Manage. Assoc., 2006, 56, 23–31 CrossRef CAS PubMed .
- W. Lee and G.-N. Bae, Removal of elemental mercury (Hg(0)) by nanosized V2O5/TiO2 catalysts, Environ. Sci. Technol., 2009, 43, 1522–1527 CrossRef CAS PubMed .
- H. Kamata, S. Ueno, T. Naito and A. Yukimura, Mercury oxidation over the V2O5(WO3)/TiO2 commercial SCR catalyst, Ind. Eng. Chem. Res., 2008, 47, 8136–8141 CrossRef CAS .
- H. Li, Y. Li, C.-Y. Wu and J. Zhang, Oxidation and capture of elemental mercury over SiO2–TiO2–V2O5 catalysts in simulated low-rank coal combustion flue gas, Chem. Eng. J., 2011, 169, 186–193 CrossRef CAS .
- W. Gao, Q. Liu, C.-Y. Wu, H. Li, Y. Li, J. Yang and G. Wu, Kinetics of
mercury oxidation in the presence of hydrochloric acid and oxygen over a commercial SCR catalyst, Chem. Eng. J., 2013, 220, 53–60 CrossRef CAS .
- L. Zhao, C. Li, J. Zhang, X. Zhang, F. Zhan, J. Ma, Y. Xie and G. Zeng, Promotional effect of CeO2 modified support on V2O5–WO3/TiO2 catalyst for elemental mercury oxidation in simulated coal-fired flue gas, Fuel, 2015, 153, 361–369 CrossRef CAS .
- Y. Cao, Z. Gao, J. Zhu, Q. Wang, Y. Huang, C. Chiu, B. Parker, P. Chu and W. Pan, Impacts of halogen additions on mercury oxidation, in a slipstream selective catalyst reduction (SCR), reactor when burning sub-bituminous coal, Environ. Sci. Technol., 2008, 42, 256–261 CrossRef CAS PubMed .
- C. W. Lee, R. K. Srivastava, S. B. Ghorishi, J. Karwowski, T. W. Hastings and J. C. Hirschi, Pilot-scale study of the effect of selective catalytic reduction catalyst on mercury speciation in illinois and powder river basin coal combustion flue gases, J. Air Waste Manage. Assoc., 2006, 56, 643–649 CrossRef CAS PubMed .
- X. Gao, Y. Jiang, Y. Zhong, Z. Luo and K. Cen, The activity and characterization of CeO2–TiO2 catalysts prepared by the sol-gel method for selective catalytic reduction of NO with NH3, J. Hazard. Mater., 2010, 174, 734–739 CrossRef CAS PubMed .
- H. Li, C.-Y. Wu, Y. Li and J. Zhang, CeO2–TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas, Environ. Sci. Technol., 2011, 45, 7394–7400 CrossRef CAS PubMed .
- X. Fan, C. Li, G. Zeng, X. Zhang, S. Tao, P. Lu, Y. Tan and D. Luo, Hg0 removal from simulated flue gas over CeO2/HZSM-5, Energy Fuels, 2012, 26, 2082–2089 CrossRef CAS .
- Y. Wang, B. Shen, C. He, S. Yue and F. Wang, Simultaneous removal of NO and Hg0 from flue gas over Mn-Ce/Ti-PILCs, Environ. Sci. Technol., 2015, 49, 9355–9363 CrossRef CAS PubMed .
- L. Chen, J. Li and M. Ge, Promotional effect of Ce-doped V2O5–WO3/TiO2 with low vanadium loadings for selective catalytic reduction of NOx by NH3, J. Phys. Chem. C, 2009, 113, 21177–21184 CrossRef CAS .
- L. Song, J. Chao, Y. Fang, H. He, J. Li, W. Qiu and G. Zhang, Promotion of ceria for decomposition of ammonia bisulfate over V2O5–MoO3/TiO2 catalyst for selective catalytic reduction, Chem. Eng. J., 2016, 303, 275–281 CrossRef CAS .
- H. Li, J. Miao, Q. Su, Y. Yu, Y. Chen, J. Chen and J. Wang, Improvement in alkali metal resistance of commercial V2O5–WO3/TiO2 SCR catalysts modified by Ce and Cu, J. Mater. Sci., 2019, 54, 14707–14719 CrossRef CAS .
- A. Zhang, W. Zheng, J. Song, S. Hu, Z. Liu and J. Xiang, Cobalt manganese oxides modified titania catalysts for oxidation of elemental mercury at low flue gas temperature, Chem. Eng. J., 2014, 236, 29–38 CrossRef CAS .
- W. Xu, H. Wang, X. Zhou and T. Zhu, CuO/TiO2 catalysts for gas-phase Hg0 catalytic oxidation, Chem. Eng. J., 2014, 243, 380–385 CrossRef CAS .
- C. Antuna-Nieto, E. Rodríguez, M. A. Lopez-Anton, R. García and M. R. Martínez-Tarazona, A candidate material for mercury control in energy production processes: carbon foams loaded with gold, Energy, 2018, 159, 630–637 CrossRef CAS .
- S. Zhang, Y. Zhao, M. Díaz-Somoano, J. Yang and J. Zhang, Synergistic mercury removal over the CeMnO3 perovskite structure oxide as a selective catalytic reduction catalyst from coal combustion flue gas, Energy Fuels, 2018, 32, 11785–11795 CrossRef CAS .
- S. Zhang, Y. Zhao, Z. Wang, J. Zhang, L. Wang and C. Zheng, Integrated removal of NO and mercury from coal combustion flue gas using manganese oxides supported on TiO2, J. Environ. Sci., 2017, 53, 141–150 CrossRef PubMed .
- S. Zhang, Y. Zhao, J. Yang, Y. Zhang, P. Sun, X. Yu, J. Zhang and C. Zheng, Simultaneous NO and mercury removal over MnOx/TiO2 catalyst in different atmospheres, Fuel Process. Technol., 2017, 166, 282–290 CrossRef CAS .
- S. Zhang, Y. Zhao, J. Yang, J. Zhang and C. Zheng, Fe-modified MnOx/TiO2 as the SCR catalyst for simultaneous removal of NO and mercury from coal combustion flue gas, Chem. Eng. J., 2018, 348, 618–629 CrossRef CAS .
- C. Orsenigo, L. Lietti, E. Tronconi, P. Forzatti and F. Bregani, Dynamic investigation of the role of the surface sulfates in NOx reduction and SO2 oxidation over V2O5–WO3/TiO2 catalysts, Ind. Eng. Chem. Res., 1998, 37, 2350–2359 CrossRef CAS .
- H. Xu, Y. Ma, S. Zhao, W. Huang, Z. Qu and N. Yan, Enhancement of Ce1−xSnxO2 support in LaMnO3 for the catalytic oxidation and adsorption of elemental mercury, RSC Adv., 2016, 6, 63559–63567 RSC .
- N. Yang, R. Guo, Y. Tian, W. Pan, Q. Chen, Q. Wang, C. Lu and S. Wang, The enhanced performance of ceria by HF treatment for selective catalytic reduction of NO with NH3, Fuel, 2016, 179, 305–311 CrossRef CAS .
- P. Wang, Q. Wang, X. Ma, R. Guo and W. Pan, The influence of F and Cl on Mn/TiO2 catalyst for selective catalytic reduction of NO with NH3: a comparative study, Catal. Commun., 2015, 71, 84–87 CrossRef CAS .
- X. Gao, Y. Jiang, Y. Fu, Y. Zhong, Z. Luo and K. Cen, Preparation and characterization of CeO2/TiO2 catalysts for selective catalytic reduction of NO with NH3, Catal. Commun., 2010, 11, 465–469 CrossRef CAS .
- Z. Wu, B. Jiang, Y. Liu, W. Zhao and B. Guan, Experimental study on a low-temperature SCR catalyst based on MnOx/TiO2 prepared by sol-gel method, J. Hazard. Mater., 2007, 145, 488–494 CrossRef CAS PubMed .
- H. Li, C.-Y. Wu, Y. Li and J. Zhang, Superior activity of MnOx–CeO2/TiO2 catalyst for catalytic oxidation of elemental mercury at low flue gas temperatures, Appl. Catal., B, 2012, 111–112, 381–388 CrossRef CAS .
- D. A. Peña, B. S. Uphade and P. G. Smirniotis, TiO2-supported metal oxide catalysts for low-temperature selective catalytic reduction of NO with NH3: I. Evaluation and characterization of first row transition metals, J. Catal., 2004, 221, 421–431 CrossRef .
- L. Zhao, C. Li, S. Li, Y. Wang, J. Zhang, T. Wang and G. Zeng, Simultaneous removal of elemental mercury and NO in simulated flue gas over V2O5/ZrO2–CeO2 catalyst, Appl. Catal., B, 2016, 198, 420–430 CrossRef CAS .
- S. A. Benson, J. D. Laumb, C. R. Crocker and J. H. Pavlish, SCR catalyst performance in flue gases derived from subbituminous and lignite coals, Fuel Process. Technol., 2005, 86, 577–613 CrossRef CAS .
- J. Zhang, F. Liu, J. Liang, H. Yu, W. Liu, X. Wang, H. Peng and P. Wu, Exploring the nanosize effect of mordenite zeolites on their performance in the removal of NOx, Ind. Eng. Chem. Res., 2019, 58, 8625–8635 CAS .
- Y. Li, P. D. Murphy, C.-Y. Wu, K. W. Powers and J.-C. J. Bonzongo, Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas, Environ. Sci. Technol., 2008, 42, 5304–5309 CrossRef CAS PubMed .
- F. Scala and S. Cimino, Elemental mercury capture and oxidation by a regenerable manganese-based sorbent: the effect of gas composition, Chem. Eng. J., 2015, 278, 134–139 CrossRef CAS .
- S. Zhang, M. Díaz-Somoano, Y. Zhao, J. Yang and J. Zhang, Research on the mechanism of elemental mercury removal over Mn-based SCR catalysts by a developed Hg-TPD method, Energy Fuels, 2019, 33, 2467–2476 CrossRef CAS .
- H. Li, L. Zhu, S. Wu, Y. Liu and K. Shih, Synergy of CuO and CeO2 combination for mercury oxidation under low-temperature selective catalytic reduction atmosphere, Int. J. Coal Geol., 2017, 170, 69–76 CrossRef CAS .
- H. Li, C.-Y. Wu, Y. Li, L. Li, Y. Zhao and J. Zhang, Role of flue gas components in mercury oxidation over TiO2 supported MnOx–CeO2 mixed-oxide at low temperature, J. Hazard. Mater., 2012, 243, 117–123 CrossRef CAS PubMed .
- Y. Zhuang, J. Laumb, R. Liggett, M. Holmes and J. Pavlish, Impacts of acid gases on mercury oxidation across SCR catalyst, Fuel Process. Technol., 2007, 88, 929–934 CrossRef CAS .
- W. Xu, H. He and Y. Yu, Deactivation of a Ce/TiO2 catalyst by SO2 in the selective catalytic reduction of NO by NH3, J. Phys. Chem. C, 2009, 113, 4426–4432 CrossRef CAS .
- S. Gao, P. Wang, F. Yu, H. Wang and Z. Wu, Dual resistance to alkali metals and SO2: vanadium and cerium supported on sulfated zirconia as an efficient catalyst for NH3-SCR, Catal. Sci. Technol., 2016, 6, 8148–8156 RSC .
- H. Xu, Y. Ma, B. Mu, W. Huang, Q. Hong, Y. Liao, Z. Qu and N. Yan, Enhancing the catalytic oxidation of elemental mercury and suppressing sulfur-toxic adsorption sites from SO2-containing gas in Mn-SnS2, J. Hazard. Mater., 2020, 392, 122230 CrossRef CAS PubMed .
- G. Busca, L. Lietti, G. Ramis and F. Berti, Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: a review, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS .
- P. Wang, S. Su, J. Xiang, H. You, F. Cao, L. Sun, S. Hu and Y. Zhang, Catalytic oxidation of Hg0 by MnOx–CeO2/γ-Al2O3 catalyst at low temperatures, Chemosphere, 2014, 101, 49–54 CrossRef CAS PubMed .
- G. Qi, R. T. Yang and R. Chang, MnOx–CeO2 mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3 at low temperatures, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS .
- J. Yang, Y. Zhao, J. Zhang and C. Zheng, Regenerable cobalt oxide loaded magnetosphere catalyst from fly ash for mercury removal in coal combustion flue gas, Environ. Sci. Technol., 2014, 48, 14837–14843 CrossRef CAS PubMed .
- Q. Wan, L. Duan, J. Li, L. Chen, K. He and J. Hao, Deactivation performance and mechanism of alkali (earth) metals on V2O5–WO3/TiO2 catalyst for oxidation of gaseous elemental mercury in simulated coal-fired flue gas, Catal. Today, 2011, 175, 189–195 CrossRef CAS .
- Y. Jiang, Z. Xing, X. Wang, S. Huang, X. Wang and Q. Liu, Activity and characterization of a Ce–W–Ti oxide catalyst prepared by a single step sol-gel method for selective catalytic reduction of NO with NH3, Fuel, 2015, 151, 124–129 CrossRef CAS .
- W. Yao, Y. Liu, X. Wang, X. Weng, H. Wang and Z. Wu, The superior performance of sol-gel made Ce–O–P catalyst for selective catalytic reduction of NO with NH3, J. Phys. Chem. C, 2016, 120, 221–229 CrossRef CAS .
- L. Li, L. Chen, M. Kong, Q. Liu and S. Ren, New insights into the
deactivation mechanism of V2O5–WO3/TiO2 catalyst during selective catalytic reduction of NO with NH3: synergies between arsenic and potassium species, RSC Adv., 2019, 9, 37724–37732 RSC .
- S. He, J. Zhou, Y. Zhu, Z. Luo, M. Ni and K. Cen, Mercury oxidation over a vanadia-based selective catalytic reduction catalyst, Energy Fuels, 2009, 23, 253–259 CrossRef CAS .
- Y. Li, W. Liu, R. Yan, J. Liang, T. Dong, Y. Mi, P. Wu, Z. Wang, H. Peng and T. An, Hierarchical three-dimensionally ordered macroporous Fe–V binary metal oxide catalyst for low temperature selective catalytic reduction of NOx from marine diesel engine exhaust, Appl. Catal., B, 2020, 268, 118455 CrossRef .
- R. Yan, S. Lin, Y. Li, W. Liu, Y. Mi, C. Tang, L. Wang, P. Wu and H. Peng, Novel shielding and synergy effects of Mn–Ce oxides confined in mesoporous zeolite for low temperature selective catalytic reduction of NOx with enhanced SO2/H2O tolerance, J. Hazard. Mater., 2020, 396, 122592 CrossRef CAS PubMed .
- R. Fan, Z. Li, Y. Wang, C. Zhang, Y. Wang, Z. Ding, X. Guo and R. Wang, Effects of WO3 and SiO2 doping on CeO2–TiO2 catalysts for selective catalytic reduction of NO with ammonia, RSC Adv., 2020, 10, 5845–5852 RSC .
- J. Yang, M. Zhang, H. Li, W. Qu, Y. Zhao and J. Zhang, Simultaneous NO reduction and Hg0 oxidation over La0.8Ce0.2MnO3 perovskite catalysts at low temperature, Ind. Eng. Chem. Res., 2018, 57, 9374–9385 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Yongchun Zhao
*a,
Yongchun Zhao