
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(II)-catalyzed synthesis of multisubstituted indoles through sequential Chan–Lam and cross-dehydrogenative coupling reactions†
Xin Chenab,
Yunyun Biana,
Baichuan Moab,
Peng Sunab,
Chunxia Chen*ab and
Jinsong Peng *a
*a
aCollege of Chemistry, Chemical Engineering and Resource Utilization, Northeast Forestry University, No. 26 Hexing Road, Harbin, 150040, P. R. China
bMaterial Science and Engineering College, Northeast Forestry University, No. 26 Hexing Road, Harbin, 150040, P. R. China. E-mail: ccx1759@163.com; jspeng1998@nefu.edu.cn
First published on 30th June 2020
Abstract
Starting from arylboronic acids and ester (Z)-3-aminoacrylates, one-pot syntheses of diverse indole-3-carboxylic esters have been described through copper(II)-catalyzed sequential Chan–Lam N-arylation and cross-dehydrogenative coupling (CDC) reactions. The initial Chan–Lam arylation can proceed in DMF at 100 °C for 24 h to give ester (Z)-3-(arylamino)acrylate intermediates in the presence of Cu(OAc)2/tri-tert-butylphosphine tetrafluoroborate, a catalytic amount of myristic acid as the additive, KMnO4 and KHCO3. Sequentially, these in situ arylated intermediates can undergo an intramolecular oxidative cross-dehydrogenative coupling process in mixed solvents (DMF/DMSO = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 130 °C to give C3-functionalized multi-substituted indole derivatives.
:1) at 130 °C to give C3-functionalized multi-substituted indole derivatives.
1. Introduction
The indole unit is not only one of the most abundant structural motifs in natural products,1 but is also ubiquitous among agrochemicals,2 marketed medicines (such as dolasetron, tropisetron, indomethacin, proglumetacin, and ondansetron),3 and progressive functional materials.4 In particular, substituted indoles have been utilized as “privileged scaffolds” for drug discovery of anti-inflammatory, antihypertensive, anti-tumor, anti-HIV, and antimigraine agents.5 Considering the importance of indole scaffolds for pharmaceutical research, the development of practical syntheses of indole derivatives is of immense interest to synthetic chemists.Over the past century, a variety of approaches for indole preparation have been well established.6 While numerous classical procedures based on condensation and cyclization have been developed,6b there is still a great demand to explore new strategies and methodologies for the modular synthesis of functionalized indoles from easily available starting materials. Over the past decades, transition metal-mediated inter- and intramolecular C–C and C–N bond forming reactions have emerged as one of the most powerful and popular tools for indole syntheses.7 Following this tendency, an attractive C–H activation/cyclization strategy relying on the use of N-arylated enamine or imine intermediates have emerged in recent years (Scheme 1). In 2008, Glorius group8a reported the first Pd-catalyzed oxidative cyclization of N-aryl enaminones/esters for indole syntheses with Cu(OAc)2 as the oxidant. Shortly afterward, Cacchi9 demonstrated a copper-catalyzed aerobic version for the synthesis of indoles from N-aryl enaminones in DMF. Subsequent to these original works, different catalytic system (Pd,10 Cu,11 Fe,12 PIDA,13 I2,14 visible light,15 photoredox/metal,16 and electricity17) have been widely investigated to enlarge the substrate scope. Using simple and easily available substrates, domino one-pot processes combining the in situ formation of N-arylated enamines or imines with subsequent cyclization have also been developed for the synthesis of indoles. For instance, Jiao group18 pioneered studies to construct an indole backbone through an efficient Pd-catalyzed aerobic oxidative C–H functionalization approach from simple anilines and activated alkynes. Recently, Zhang and Cao19 reported a one-pot synthesis of 2-(perfluoroalkyl)indoles through sequential Michael-type addition and Pd(II)-catalyzed cross-dehydrogenative coupling reaction of anilines and methyl perfluoroalk-2-ynoates with oxygen as the sole oxidant. Yoshikai group20 developed a Pd-catalyzed method for the synthesis of indoles from N-aryl imines directly derived from simple and obtainable anilines and ketones. In addition, using simple anilines21 or aniline derivatives with directing groups,22–27 various transitional metal-catalyzed (such as Rh,22 Ru,23 Pd,24 Au,25 Ni,26 and Co27) protocols have been well established through group-directed oxidative C–H annulation of alkynes.
 | ||
| Scheme 1 Metal-catalyzed indole syntheses based on the N-arylated enamines and imines. | ||
We have been devoting our efforts to develop metal-catalyzed sequential one-pot processes containing a direct C–H functionalization step for the construction of heterocyclic frameworks.28 Over the past decades, oxidative cross-dehydrogenative coupling (CDC) reactions have emerged as one of the most powerful routes for C–C bond formations in organic synthesis.29 For indole syntheses, we recently developed a Cu(I)-catalyzed 2-fold arylation process through a tandem Ullmann-type C–N and cross-dehydrogenative coupling sequence from enamines and aryl iodides.30 More recently, a one-pot synthesis of 2-aryl indole-3-carboxylate derivatives has also been established by our group through stoichiometric copper salt-mediated sequential hydroamination and cross-dehydrogenative coupling reaction from simple anilines and ester arylpropiolates.31 Subsequent to our previous works, we became interested in developing a new entry to multi-substituted indoles from arylboronic acids and enamines through sequential copper(II)-catalyzed Chan–Lam oxidative N-arylation and cross-dehydrogenative coupling reaction (Scheme 2). Several potential issues need to be addressed: (1) the Chan–Lam arylation has been extended to numerous nucleophilic partners for carbon–heteroatom bonds formation,32 but not to enamines. (2) The Chan–Lam N-arylation involving arylboronic acids is known to occur in the presence of Cu(II) catalyst, while the Cu(I) catalyst has usually been employed in the C–H functionalization step.9,30 Merging fundamentally different copper catalysis into one-pot reaction is still challenging.
 | ||
| Scheme 2 Prospected sequence for the synthesis of indoles. | ||
2. Results and discussion
Ethyl (Z)-3-amino-3-phenylacrylate (2a) was selected as the substrate to react with phenylboronic acid (1a) in the presence of different combinations of copper catalysts, ligands, bases, solvents and oxidants (Table 1). The blank experiment (without copper catalyst and ligand) was examined in DMF at 70 °C for 24 h using KHCO3 as the base, and no desired product 3aa was obtained. A survey of copper catalysts showed that Cu(OAc)2 provided better results (20% yield) than CuI, CuBr, CuCl, Cu2O, Cu(OTf)2, CuCl2, CuBr2, CuSO4·5H2O, Cu(acac)2 and CuO (0–14% yields) with 2,2′-bipyridine (L1) as the ligand and air as the oxidant (entries 1–11). To increase the yield of 3aa, we then investigated the effect of various ligands (nitrogen-containing ligands L2–L6,33 N-heterocarbene ligand L7, mono-phosphorous ligands L8–L12, and Buchwald-type bulky biaryl phosphine ligands L13–L17, Scheme 3) on the reaction using Cu(OAc)2 as the catalyst (entry 7, Table 1). It turned out that tBu3P·HBF4 (L12) performed best, and the yield was improved to 25% with KHCO3 as the base under air atmosphere (Scheme 3). It is worth noting that ligand and oxidant are essential to this transformation and 3aa was not produced in the absence of ligand (entry 13) or oxidant (entry 14). The effect of other bases such as K2CO3, NaOH, Li2CO3, K3PO4, NaHCO3, and NaOAc on the reaction was next examined (entries 15–20), KHCO3 provided a better result with 25% isolated yield. A survey of reaction media showed that the use of DMF provided better results than those obtained in DMSO, THF, 1,4-dioxane, DMA, and tert-pentyl alcohol (entries 21–25). The binary mixed solvent composed of DMF and DMSO (DMF/DMSO, 2:1) gave the final product 3aa in 33% yield at 70 °C (entry 26). With increased reaction temperature and time, the yield of 3aa can been improved from 33% to 37% (entry 26). Among the range of oxidants (KMnO4, K2S2O8, TBHP, MnO2, and O2, entries 26–30) that were surveyed, KMnO4 appeared to be optimal and gave 3aa with an enhanced yield (48%, entry 27). Attempts have been made under higher air or O2 pressure (3 atm), however, the yields cannot obviously be improved. In 2001, Buchwald and co-workers demonstrated that aliphatic acid can increase the copper-catalyzed coupling reaction rate of arylboronic acids and amines through the coordination of aliphatic acid to the copper center to improve solubility of the copper catalyst in organic solvents.34 Given the importance of aliphatic acids in achieving a homogeneous catalyst system, various carboxylic acids such as myristic acid, palmitic acid, stearic acid, and trimethylacetic acid were explored as the additive. It was found that the addition of 1 equiv of myristic acid relative to Cu(OAc)2 improved the yield of 3aa to 55% (entry 31), while other carboxylic acids had little effect on the reaction (entries 32–34). In general, in the presence of combinations of Cu(OAc)2, tBu3P·HBF4, KHCO3, myristic acid and KMnO4, the synthesis of indoles 3 was conducted in a one-pot fashion using DMF/DMSO as mixed solvents.
|
|||||
|---|---|---|---|---|---|
| Entry | Cu/ligand | Base | Solvent | Oxidant | Yieldb [%] |
| a Reaction conditions unless otherwise stated: 1a (0.5 mmol), 2a (0.2 mmol), base (0.6 mmol), catalyst (20 mol%), ligand (30 mol%), solvent (1.0 mL), air or oxidant, 70 °C, 24 h, air.b Isolated yield.c 1.5 equiv. of 1a.d 2.0 equiv. of 1a.e The effect of various ligands was investigated, see Scheme 1.f N2.g DMF (1.0 mL), 100 °C, 24 h; then DMSO (0.5 mL), 130 °C, 24 h.h 20 mol% of myristic acid was added.i 20 mol% of palmitic acid was added.j 20 mol% of stearic acid was added.k 20 mol% of trimethylacetic acid was added. | |||||
| 1 | CuI/L1 | KHCO3 | DMF | Air | 10/6c/7d |
| 2 | CuBr/L1 | KHCO3 | DMF | Air | Trace |
| 3 | CuCl/L1 | KHCO3 | DMF | Air | Trace |
| 4 | Cu2O/L1 | KHCO3 | DMF | Air | Trace |
| 5 | Cu(OTf)2/L1 | KHCO3 | DMF | Air | 5 |
| 6 | CuCl2/L1 | KHCO3 | DMF | Air | 8 |
| 7 | CuBr2/L1 | KHCO3 | DMF | Air | 7 |
| 8 | CuSO4·5H2O/L1 | KHCO3 | DMF | Air | 10 |
| 9 | Cu(OAc)2/L1 | KHCO3 | DMF | Air | 20 |
| 10 | Cu(acac)2/L1 | KHCO3 | DMF | Air | 14 |
| 11 | CuO/L1 | KHCO3 | DMF | Air | 0 |
| 12e | Cu(OAc)2/L2-L17 | KHCO3 | DMF | Air | 0–25 |
| 13 | Cu(OAc)2 | KHCO3 | DMF | Air | 0 |
| 14f | Cu(OAc)2/L12 | KHCO3 | DMF | — | 0 |
| 15 | Cu(OAc)2/L12 | K2CO3 | DMF | Air | 15 |
| 16 | Cu(OAc)2/L12 | NaOH | DMF | Air | 0 |
| 17 | Cu(OAc)2/L12 | Li2CO3 | DMF | Air | 0 |
| 18 | Cu(OAc)2/L12 | K3PO4 | DMF | Air | 20 |
| 19 | Cu(OAc)2/L12 | NaHCO3 | DMF | Air | 17 |
| 20 | Cu(OAc)2/L12 | NaOAc | DMF | Air | 18 |
| 21 | Cu(OAc)2/L12 | KHCO3 | DMSO | Air | 10 |
| 22 | Cu(OAc)2/L12 | KHCO3 | THF | Air | 15 |
| 23 | Cu(OAc)2/L12 | KHCO3 | 1,4-Dioxane | Air | Trace |
| 24 | Cu(OAc)2/L12 | KHCO3 | DMA | Air | 19 |
| 25 | Cu(OAc)2/L12 | KHCO3 | tert-Pentyl alcohol | Air | 20 |
| 26 | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | Air | 33 (37)g |
| 27g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | KMnO4 | 48 |
| 28g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | K2S2O8 | 21 |
| 29g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | TBHP | 20 |
| 30g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | MnO2 | 30 |
| 31g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | KMnO4 | 55h |
| 32g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | KMnO4 | 47i |
| 33g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | KMnO4 | 48j |
| 34g | Cu(OAc)2/L12 | KHCO3 | DMF/DMSO | KMnO4 | 45k |

 | ||
| Scheme 3 Effect of ligands on the copper-catalyzed annulations reaction. a Reaction conditions: 20 mol% Cu(OAc)2, KHCO3, DMF, air, 70 °C, 24 h; isolated yields. b PCy3 was directly used. | ||
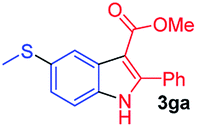
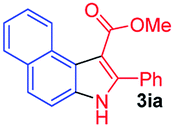
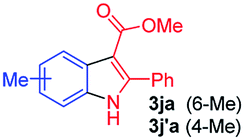
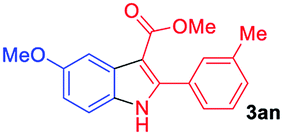
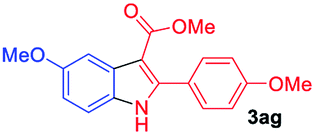
Having established the feasibility of indole synthesis via copper(II)-catalyzed sequential oxidative Chan–Lam arylation/CDC process, we then explored the generality of arylboronic acids using methyl or ethyl (Z)-3-amino-3-phenylacrylate as the coupling partner (Table 2). As shown in Table 2, diverse substituents (such as Me, OMe, F, Cl, ester, and methylthio) on the aromatic moiety of boronic acids were applicable, and the corresponding indole products 3aa–3ja can be obtained in 44–55% yields. Arylboronic acids containing electron-donating groups at the para (1b, 1c, and 1g), or meta (1j) position were generally more reactive than those bearing electron-withdrawing substituents (1d–1f) and provided higher yields (entries 2–7, and 10, Table 2). However, the incorporation of substituents in the ortho position of arylboronic acid seriously hampered copper-catalyzed oxidative annulation process, and the corresponding indoles cannot be obtained. In addition, α- or β-naphthyl boronic acids 1h and 1i can also be smoothly transformed into the corresponding products 3ha and 3ia in 49% and 50% yields, respectively (entries 8 and 9). For β-naphthyl boronic acid 1i, there are two possible C–H activation sites (α- and β-position), the CDC process occurred only at the α-site to give a single regioisomer 3ia (entry 9). The specificity of site selectivity (α/β) implies that the aromatic C–H alkenylation is probably an outcome of electrophilic aromatic substitutions. However, regioselectivity issues surfaced for meta-substituted arylboronic acid (1j), and a mixture of two regioisomers (3ja and 3j′a) was obtained in a nearly 2:1 ratio, indicating intramolecular CDC reaction occurred at the most sterically accessible site (entry 10).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | S-1 | P-3 | Yieldb | Entry | S-1 | P-3 | Yieldb |
| a Reaction conditions: 2 (0.2 mmol), arylboronic acids 1 (0.5 mmol), Cu(OAc)2 (20 mol%), tBu3P·HBF4 (30 mol%), myristic acid (0.04 mmol), KHCO3 (0.6 mmol), KMnO4 (0.1 mmol), DMF (1 mL), 100 °C, 24 h; then DMSO (0.5 mL), 130 °C, 24 h, air.b Yield of the isolated product.c The ratio of the regioisomers was determined by NMR analysis. | |||||||
| 1 |  |
 |
55 | 6 | 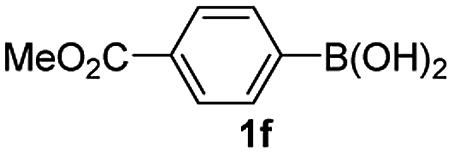 |
 |
44 |
| 2 | 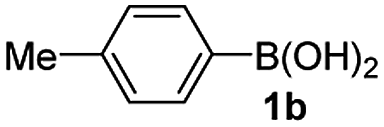 |
 |
54 | 7 | 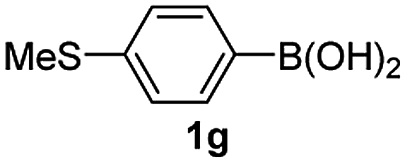 |
 |
50 |
| 3 |  |
 |
51 | 8 |  |
 |
49 |
| 4 |  |
 |
47 | 9 |  |
 |
50 |
| 5 |  |
 |
46 | 10c |  |
 |
52 (2:1) |

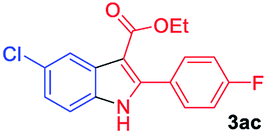
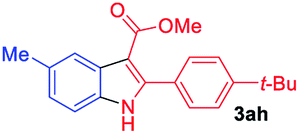
The scope and limitation of ester (Z)-3-aminoacrylate substrates were finally investigated in this one-pot sequential oxidative process. As shown in Table 3, several functional groups (such as Me, OMe, OEt, tBu, F, Cl, Br and CF3) are tolerated under the standard conditions. For ester (Z)-3-amino-3-arylpropiolates, the electronic nature of the aromatic motifs did not seem to affect the efficiency: both electron-donating (Me, MeO, EtO, and tBu) and electron-withdrawing substituents (F, Cl, Br, and CF3) can be incorporated at the para (2b–2j, entries 1–9) and meta (2k–2o, entries 10–14) position, providing indole derivatives 3ab–3ao in 42–53% yields. However, ester (Z)-3-amino-3-arylpropiolates (2p–2r, Fig. 1) with substituents in the ortho position of aromatic ring cannot proceed to give the corresponding products, probably because of the strong steric repulsion of the ortho substituent. In addition, when the scope of substrates was extended to aliphatic and carbocyclic enamines [for example, ethyl (Z)-3-aminobut-2-enoate 2s and 3-aminocyclohex-2-en-1-one 2u, Fig. 1], the reaction did not happen to give the corresponding products. Enamine substrates with an amide (2t), aromatic ketone (2v) or nitro group (2w) were also examined under the standard conditions; however, no product can be obtained. These unsuccessful results indicated that the electron density of olefin moiety was highly essential for CDC process.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | S-1 | P-3 | Yieldb | Entry | S-1 | P-3 | Yieldb |
| a Reaction conditions: 2 (0.2 mmol), arylboronic acids 1 (0.5 mmol), Cu(OAc)2 (20 mol%), tBu3P·HBF4 (30 mol%), myristic acid (0.04 mmol), KHCO3 (0.6 mmol), KMnO4 (0.1 mmol), DMF (1 mL), 100 °C, 24 h; then DMSO (0.5 mL), 130 °C, 24 h, air.b Yield of the isolated product. | |||||||
| 1 |  |
 |
47 | 9 | 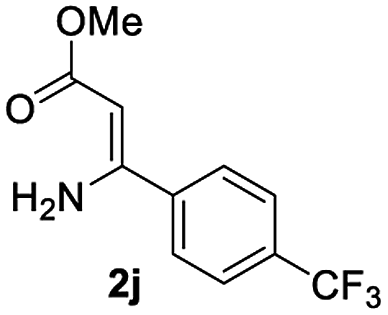 |
 |
49 |
| 2 |  |
 |
42 | 10 |  |
 |
47 |
| 3 |  |
 |
46 | 11 |  |
 |
46 |
| 4 |  |
 |
49 | 12 | 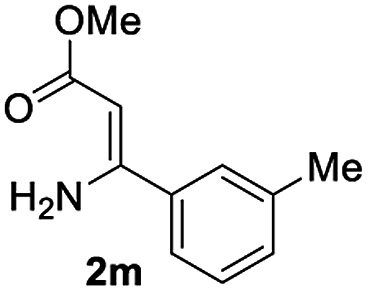 |
 |
49 |
| 5 |  |
 |
53 | 13 |  |
 |
49 |
| 6 |  |
 |
51 | 14 |  |
 |
48 |
| 7 |  |
 |
52 | 15 |  |
 |
51 |
| 8 |  |
 |
46 | ||||

 | ||
| Fig. 1 Unsuccessful enamine substrates. | ||
To gain insight into the mechanism of the reaction, some designed control experiments were conducted. When the reaction of 1a and 2a was conducted in DMF for 24 h, the Chan–Lam N-arylated intermediate 9 and 3aa can be isolated in 68% and 10% yields, respectively. Then, the intermediate 9 was carried out under the optimized reaction conditions, 65% of the desired product 3aa was obtained. Based on the above results and previous literature reports from our group30,31 and others,35 a reaction mechanism for the copper(II)-catalyzed oxidative annulation of enamines with arylboronic acids was proposed. The Scheme 4 showed a simplified sequence of events beginning with the Cu(OAc)2. First, a soluble active cupric tetradecanoate species was formed through the anion exchange reaction of Cu(OAc)2 and myristic acid in the presence of base.34 The coordination of (Z)-enamine 2 to the Cu(II) center to form 4, which then reacted with a base to form a Cu–N bond and afforded an intermediate complex 5. Engagement of arylboronic acid 1 led to transmetalation via 4-membered transition state35a and delivered aryl-Cu(II) species 6. The intermediate 6 was then oxidized by Cu(II) to form an Cu(III) species 8, and the subsequent reductive elimination gave N-arylated intermediate 9 and a Cu(I)OAc species 7. Completion of the catalytic cycle was achieved via oxidation to Cu(II) in the presence of KMnO4 (Chan–Lam C–N coupling process). The coordination of N-arylated intermediate 9 to Cu(II) gave a six-membered chelate ring complex 10. A new C–Cu(II) complex 12 was then formed through sequential a base-promoted deprotonation of N–H, dissociation of acetate anion and complexation of the resulting cationic Cu(II) species at α-carbon of 11. A deprotonation/re-protonation process of alkyl-Cu(II) complex 12 afforded an alkenyl copper 13 under basic conditions. Ortho-cupration of phenyl ring of 13 provided a six-membered copper-cycle intermediate 14,9 which was then transformed into the product 3 and Cu(0) species 15 through reductive elimination process. Finally, Cu(0) was oxidized to the active Cu(II) catalyst by KMnO4 for the next Chan–Lam arylation reaction.
 | ||
| Scheme 4 Proposed catalytic pathway for the formation of indole 3. | ||
3. Conclusions
In summary, we have developed a new one-pot approach to diverse multi-substituted indoles through copper-catalyzed oxidative annulations of enamines with readily accessible arylboronic acids. The accomplished reaction comprises an intermolecular Chan–Lam arylation followed by an intramolecular cross-dehydrogenative coupling reaction promoted by the same copper catalyst. The success of the reaction heavily relies on the careful selection of proper additive and oxidant. The combination of myristic acid and KMnO4 was found to be essential for the formation of C3-functionalized multi-substituted indoles. Considering a broad substrate scope and considerable valance of the products for medicinal science, this novel synthetic method could be of utility for the discovery of drugs.4. Experimental section
4.1. General information
Chemicals were all purchased from commercial supplies and used without further purification unless otherwise stated. Solvents were dried and purified according to the standard procedures before use. Reactions were monitored by analytical thin-layer chromatography (TLC). All reactions were conducted in dried glassware. Purification of reaction products was done by flash chromatography with 230–400 mesh silica gel. Ester (Z)-3-aryl-3-aminoacrylate substrates were prepared according to the literature methods.36 Melting points were determined on a melting point apparatus in open capillaries and are uncorrected. Infrared spectra of samples were recorded from 4000 to 500 cm−1 in ATR (attenuated total reflectance) mode using an FT-IR instrument. 1H NMR spectra were recorded on a 400 or 500 MHz spectrometer, and 13C NMR spectra were recorded at 100, 125 or 150 MHz. Unless otherwise stated, deuterochloroform (CDCl3) was used as a solvent. Chemical shifts (δ) are given in parts per million downfield relative to tetramethylsilane (TMS). Chemical shifts for carbon resonances are reported in parts per million and are referenced to the carbon resonance of the solvent CHCl3 (δ = 77.16 ppm). The splitting patterns are reported as s (singlet), d (doublet), dd (double doublet), td (triplet of doublet), t (triplet), q (quartet), br (broad), and m (multiplet). Coupling constants are given in hertz. High-resolution mass spectra were recorded on a BIO TOF Q mass spectrometer equipped with an electrospray ion source (ESI), operated in the positive mode.4.2. General procedure for synthesis of indole-3-carboxylate derivatives
A 10 mL Schlenk tube or standard vial equipped with a magnetic stirring bar was charged with ester (Z)-3-aryl-3-aminoacrylates (0.2 mmol, 1.0 equiv.), aryl boronic acid (0.5 mmol, 2.5 equiv.), KMnO4 (0.1 mmol, 15.8 mg), and KHCO3 (60 mg, 0.6 mmol, 3.0 equiv.), and then Cu(OAc)2 (0.04 mmol, 8.0 mg), tBu3P·HBF4 (0.06 mmol, 17.4 mg) and myristic acid (0.04 mmol, 9.1 mg) were added. Finally, DMF (1.0 mL) was added to the mixture via syringe at room temperature under air. The vial was sealed and put into a preheated oil bath at 100 °C for 24 h. The mixture was cooled to room temperature, dimethyl sulfoxide (0.5 mL) was then added via syringe, and the reaction mixture was heated to 130 °C for another 24 h. Finally, the mixture was cooled to room temperature, quenched with water (3 mL), and diluted with ethyl acetate (5 mL). The layers were separated, and the aqueous layer was extracted with 3 × 5 mL of ethyl acetate. The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The crude product was then purified by a chromatography silica gel (H), eluting with ethyl acetate/petroleum ether (10–15%).:3j′a = 2:1) is determined by NMR; IR (KBr, cm−1): 3297, 2949, 1682, 1452, 1214, 1127, 1048, 813, 768, 696; 1H NMR (400 MHz, CDCl3) δ 8.50 (br, 1.5Hoverlap), 8.07 (d, J = 8.2 Hz, 1H3ja), 7.65–7.63 (m, 2H3ja), 7.55–7.51 (m, 1H3j′a), 7.45–7.40 (m, 4.50Hoverlap), 7.22 (d, J = 8.0 Hz, 0.55H3j′a), 7.17–7.10 (m, 2.54Hoverlap), 7.01 (d, J = 7.1 Hz, 0.53H3j′a), 3.83 (s, 3H3ja), 3.76 (s, 1.56H3j′a), 2.65 (s, 1.50H3j′a), 2.47 (s, 3H3ja).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from Special Project for Double First-Class-Cultivation of Innovative Talents (201815), the Fundamental Research Funds for the Central Universities (2572017DB07), and Natural Science Foundation of Heilongjiang Province (B2017002, LC2018003).Notes and references
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed.
- C.-L. Do-Thanh, J. J. Vargas, J. W. Thomas, G. R. Armel and M. D. Best, J. Agric. Food Chem., 2016, 64, 3533 CrossRef CAS PubMed.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159 CrossRef CAS PubMed.
- (a) Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057 CrossRef CAS PubMed; (b) M. Vlasselaer and W. Dehaen, Molecules, 2016, 21, 785 CrossRef PubMed.
- (a) F. R. de Sa Alves, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2009, 9, 782 CrossRef PubMed; (b) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1 CrossRef CAS PubMed; (c) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620 CrossRef CAS PubMed; (d) T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9 CrossRef CAS PubMed.
- (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (b) F. T. Douglass and K. T. Pavan, Tetrahedron, 2011, 67, 7195 CrossRef PubMed; (c) R. Vicente, Org. Biomol. Chem., 2011, 9, 6469 RSC; (d) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29 RSC; (e) G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728 RSC; (f) M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7, 52852 RSC.
- (a) G. Battistuzzi, S. Cacchi and G. Fabrizi, Eur. J. Org. Chem., 2002, 2671 CrossRef CAS; (b) K. Kruger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef; (c) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed; (d) M. Platon, R. Amardeil, L. Djakovitch and J. C. Hierso, Chem. Soc. Rev., 2012, 41, 3929 RSC; (e) T. L. Guo, F. Huang, L. Yu and Z. Yu, Tetrahedron Lett., 2015, 56, 296 CrossRef CAS; (f) S. W. Youn and T. Y. Ko, Asian J. Org. Chem., 2018, 7, 1467 CrossRef CAS.
- (a) S. Würtz, S. Rakshit, J. J. Neumann, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 7230 CrossRef PubMed; (b) J. J. Neumann, S. Rakshit, T. Dröge, S. Würtz and F. Glorius, Chem. - Eur. J., 2011, 17, 7298 CrossRef CAS PubMed.
- R. Bernini, G. Fabrizi, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2009, 48, 8078 CrossRef CAS PubMed.
- (a) X.-L. Lian, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 3360 CrossRef CAS PubMed; (b) A. Mishra and I. Deb, Adv. Synth. Catal., 2016, 358, 2267 CrossRef CAS.
- (a) F. Huang, P. Wu, L. Wang, J. Chen, C. Sun and Z. Yu, J. Org. Chem., 2014, 79, 10553 CrossRef CAS PubMed; (b) P. Drouhin and R. J. K. Taylor, Eur. J. Org. Chem., 2015, 2333 CrossRef CAS; (c) J. Liu, W. Wei, T. Zhao, X. Liu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2016, 81, 9326 CrossRef CAS PubMed; (d) F.-Z. Hu, S.-H. Zhao, H. Chen, S.-W. Yu, X.-Y. Xu, W.-C. Yuan and X.-M. Zhang, ChemistrySelect, 2017, 2, 1409 CrossRef CAS.
- Z.-H. Guan, Z.-Y. Yan, Z.-H. Ren, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2010, 2823 RSC.
- W. Yu, Y. Du and K. Zhao, Org. Lett., 2009, 11, 2417 CrossRef CAS PubMed.
- (a) Z. He, W. Liu and Z. Li, Chem.–Asian J., 2011, 6, 1340 CrossRef CAS PubMed; (b) Z. Jia, T. Nagano, X. Li and A. S. C. Chan, Eur. J. Org. Chem., 2013, 858 CrossRef CAS.
- (a) C.-J. Wu, Q.-Y. Meng, T. Lei, J.-J. Zhong, W.-Q. Liu, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Catal., 2016, 6, 4635 CrossRef CAS; (b) W.-Q. Liu, T. Lei, Z.-Q. Song, X.-L. Yang, C.-J. Wu, X. Jiang, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2017, 19, 3251 CrossRef CAS PubMed.
- J. Zoller, D. C. Fabry, M. A. Ronge and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13264 CrossRef CAS PubMed.
- S. Tang, X. Gao and A. Lei, Chem. Commun., 2017, 53, 3354 RSC.
- (a) Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572 CrossRef CAS PubMed; (b) L. Ren, Z. Shi and N. Jiao, Tetrahedron, 2013, 69, 4408 CrossRef CAS.
- D. Shen, J. Han, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Org. Lett., 2015, 17, 3283 CrossRef CAS PubMed.
- Y. Wei, I. Deb and N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098 CrossRef CAS PubMed.
- (a) S. Kramer, K. Dooleweerdt, A. T. Lindhardt, M. Rottländer and T. Skrydstrup, Org. Lett., 2009, 11, 4208 CrossRef CAS PubMed; (b) G. Zhang, H. Yu, G. Qin and H.-M. Huang, Chem. Commun., 2014, 50, 4331 RSC; (c) D. Shen, J. Han, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Org. Lett., 2015, 17, 3283 CrossRef CAS PubMed.
- (a) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed; (b) J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426 CrossRef CAS PubMed; (c) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (d) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed; (e) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294 CrossRef CAS PubMed; (f) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (g) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed; (h) T. Matsuda and Y. Tomaru, Tetrahedron Lett., 2014, 55, 3302 CrossRef CAS; (i) L. Zheng and R. Hua, Chem. - Eur. J., 2014, 20, 2352 CrossRef CAS PubMed; (j) K. Muralirajan and C. H. Cheng, Adv. Synth. Catal., 2014, 356, 1571 CrossRef CAS; (k) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS; (l) P.-Y. Tao and Y.-X. Jia, Chem. Commun., 2014, 50, 7367 RSC; (m) S. Kathiravan and I. A. Nicholls, Chem. Commun., 2014, 50, 14964 RSC; (n) B. Zhou, Y. Yang, H. Tang, J. Du, H. Feng and Y. Li, Org. Lett., 2014, 16, 3900 CrossRef CAS PubMed; (o) Z.-L. Fan, S.-S. Song, W. Li and A. Zhang, Org. Lett., 2015, 17, 310 CrossRef CAS PubMed; (p) L. Kong, F. Xie, S. Yu, Z. Qi and X. Li, Chin. J. Catal., 2015, 36, 925 CrossRef CAS; (q) Z. Zhou, G.-X. Liu, Y. Chen and X.-Y. Lu, Adv. Synth. Catal., 2015, 357, 2944 CrossRef CAS; (r) Y. Kim and S. Hong, Chem. Commun., 2015, 51, 11202 RSC; (s) H. Yan, H. Wang, X. Li, X. Xin, C. Wang and B. Wan, Angew. Chem., Int. Ed., 2015, 54, 10613 CrossRef CAS PubMed; (t) H. J. Kim, D. C. Fabry, S. Madera and M. Rueping, Org. Chem. Front., 2019, 6, 2319 RSC.
- (a) L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764 CrossRef CAS PubMed; (b) Z. Zhang, H. Jiang and Y. Huang, Org. Lett., 2014, 16, 5976 CrossRef CAS PubMed; (c) S. Allu and K. C. Kumara Swamy, Adv. Synth. Catal., 2015, 357, 2665 CrossRef CAS.
- (a) F. Zhou, X.-L. Han and X.-Y. Lu, Tetrahedron Lett., 2011, 52, 4681 CrossRef CAS; (b) J.-L. Chen, Q. Y. Pang, Y. B. Sun and X. Li, J. Org. Chem., 2011, 76, 3523 CrossRef CAS PubMed.
- S. Kramer, K. Dooleweerdt, A. T. Lindhardt, M. Rottländer and T. Skrydstrup, Org. Lett., 2009, 11, 4208 CrossRef CAS PubMed.
- W. Song and L. Ackermann, Chem. Commun., 2013, 49, 6638 RSC.
- (a) Z.-Z. Zhang, B. Liu, J.-W. Xu, S.-Y. Yan and B.-F. Shi, Org. Lett., 2016, 18, 1776 CrossRef CAS PubMed; (b) H. Wang, M. Moselage, M. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705 CrossRef CAS; (c) Q. Lu, S. Vásquez-Céspedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352 CrossRef CAS; (d) A. Lerchen, S. Vasquez-Cespedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208 CrossRef CAS PubMed; (e) Y. J. Liang and N. Jiao, Angew. Chem., Int. Ed., 2016, 55, 4035 CrossRef CAS PubMed.
- (a) C. Chen, G. Shang, J. Zhou, Y. Yu, B. Li and J. Peng, Org. Lett., 2014, 16, 1872 CrossRef CAS PubMed; (b) G. Zhao, C. Chen, Y. Yue, Y. Yu and J. Peng, J. Org. Chem., 2015, 80, 2827 CrossRef CAS PubMed; (c) Y. Yue, J. Peng, D. Wang, Y. Bian, P. Sun and C. Chen, J. Org. Chem., 2017, 82, 5481 CrossRef CAS PubMed; (d) X. Li, Y. Bian, X. Chen, H. Zhang, W. Wang, S. Ren, X. Yang, C. Lu, C. Chen and J. Peng, Org. Biomol. Chem., 2019, 17, 321 RSC; (e) X. Li, X. Chen, H. Wang, C. Chen, P. Sun, B. Mo and J. Peng, Org. Biomol. Chem., 2019, 17, 4014 RSC.
- (a) C. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) C. Liu, J.-W. Yuan, M. Gao, S. Tang, W. Li, R.-Y. Shi and A.-W. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed.
- Y. Li, J. Peng, X. Chen, B. Mo, X. Li, P. Sun and C. Chen, J. Org. Chem., 2018, 83, 5288 CrossRef CAS PubMed.
- P. Sun, J. Yang, Z. Song, Y. Cai, Y. Liu, C. Chen, X. Chen and J. Peng, Synthesis, 2020, 52, 75 CrossRef CAS.
- Selected reviews, see: (a) J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 829 CrossRef CAS; (b) K. S. Rao and T.-S. Wu, Tetrahedron, 2012, 68, 7735 CrossRef; (c) X. Ma, F. Liu and D. Mo, Chinese J. Org. Chem., 2017, 37, 1069 CrossRef CAS; (d) X. Duan, N. Liu, J. Wang and J. Ma, Chinese J. Org. Chem., 2019, 39, 661 CrossRef.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742 CrossRef CAS.
- J. C. Antilla and S. L. Buchwald, Org. Lett., 2001, 3, 2077 CrossRef CAS PubMed.
- (a) J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, J. Am. Chem. Soc., 2017, 139, 4769 CrossRef CAS PubMed; (b) A. E. King, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 5044 CrossRef CAS PubMed; (c) J. P. Collman and M. Zhong, Org. Lett., 2000, 2, 1233 CrossRef CAS PubMed.
- G. Hou, W. Li, M. Ma, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12844 CrossRef CAS PubMed.
- X. Guo, J. Han, Y. Liu, M. Qin, X. Zhang and B. Chen, J. Org. Chem., 2017, 82, 11505 CrossRef CAS PubMed.
- X. Zhang, D. Zhang-Negrerie, J. Deng, Y. Du and K. Zhao, J. Org. Chem., 2013, 78, 12750 CrossRef CAS PubMed.
- V. A. Bodunov, E. E. Galenko, A. V. Galenko, M. S. Novikov and A. F. Khlebnikov, Synthesis, 2018, 50, 2784 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C NMR, HRMS and IR spectra for new compounds. See DOI: 10.1039/d0ra04592f |
| This journal is © The Royal Society of Chemistry 2020 |