
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulator-free approach towards missing-cluster defect formation in Zr-based UiO-66†
Patchanee Chammingkwan a,
Goji Yildun Shangkumab,
Le Thi Tuyet Maia,
Priyank Mohana,
Ashutosh Thakura,
Toru Wadaa and
Toshiaki Taniike*a
a,
Goji Yildun Shangkumab,
Le Thi Tuyet Maia,
Priyank Mohana,
Ashutosh Thakura,
Toru Wadaa and
Toshiaki Taniike*a
aGraduate School of Advanced Science and Technology, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: taniike@jaist.ac.jp
bDepartment of Chemistry, Faculty of Natural Sciences, University of Jos, P.M.B 2084, Jos Plateau State, Nigeria
First published on 28th July 2020
Abstract
By rigorous control of water, missing-cluster defects in Zr-based UiO-66 were generated to a remarkable extent without the need of acidic modulators. The presence of missing-cluster defects created hierarchical pore structures, which had a profound effect on the catalytic performance.
Introduction
Metal–organic frameworks (MOFs), constructed by three-dimensional assembly of metal ions/metal clusters and organic ligands, represent an emerging class of porous materials. The high degree of structural tunability, well-defined and controllable porosity, high surface areas, and high concentration of isolated metal ions have made MOFs promising materials in various fields such as photocatalysis, catalysis, separation and purification, gas/energy storage, and sensing.1–5 Recently, incorporation of defects in MOFs has attracted paramount attention. Removal of linkers widens a window for enhanced gas storage capacity and mass transportation.6–10 Meanwhile, the left under-coordinated metal atoms serve as catalytically active sites, otherwise as anchoring sites for other active elements.11–13 Here, the judicious control of defect formation is essential in achieving desired properties while maintaining well-defined and highly tunable structural features of the parent framework.Among a known variety of MOFs, defect engineering has been extensively investigated for Zr-based UiO-66,6–32 which is constructed by six-centred zirconium oxyhydroxide clusters linked via terephthalate linkers to form an fcu framework. In addition to its flexibility to tailor the functionality by the selection of the linker,18,19 the high degree of connectivity between metal clusters and linkers permits the formation of defects at a high concentration without the collapse of the overall structure.6,20–23 Two types of defects, missing-linker defects and missing-cluster defects, are known. The former is related to the irregular removal of linkers to generate point defects; the latter is created by the coupled removal of metal clusters and linkers connected to them in a concentrated manner to form a nano-domain of the reo topology. Though these two types of defects are likely similar in terms of chemical nature, they can significantly impact the mechanical and physical properties.24 Especially, the removal of clusters leaves meso-scale cavities to provide more open hierarchical pore structures that are beneficial for mass and proton transportation.25,26
Control over the missing-linker defect formation has been predominantly achieved through the addition of modulators.6–11,21,22,26,27 It is widely accepted that monocarboxylic acid with a lower pKa value than the original linker competitively coordinates with a metal cluster to facilitate the growth of crystals having a lower coordination number. On the other hand, limited modulators can promote the formation of the missing-cluster defects unless an excessive concentration is used.21 Based on energetic preference in DFT calculations, it was reported that capping species dominates the defect formation (i.e. type and concentration) through steric repulsion and hydrogen bonding patterns.28 So far, highly acidic modulators, particularly fluorinated carboxylic acids, were found to promote the formation of missing-cluster defects at a relatively small addition.21 However, the persistent aspect of fluorinated organic molecules in the environment raises a challenge in practical applications. A new approach to incorporate and manipulate the missing-cluster defects in a simple and more environmentally friendly fashion is highly desired. Herein, we report a novel modulator-free approach where missing-cluster defects can be generated and manipulated to a remarkable extent only by controlling “water” as one of the synthetic constituents without the need of acidic modulators. It was also proven that the missing-cluster defects produced based on the said method is effective for catalysis.
Experiment
Synthesis of UiO-66
A series of UiO-66 samples was prepared according to the previously reported procedure.29 1.63 mmol of ZrCl4 in 30 mL of dimethylformamide (DMF) and 2.28 mmol of terephthalic acid in 30 mL of DMF were mixed under N2 atmosphere. A specified amount of deionized water (0.2, 0.4, 0.8, 1.2 and 2.0 mL, corresponding to the molar ratio of 7, 14, 27, 41 and 68 with respect to ZrCl4, respectively) was added to the solution mixture prior to heating at 100 °C for 12 h under stirring. The solid product was collected by centrifugation, repetitively washed with DMF and methanol, and finally dried under vacuum at 90 °C for 24 h. The samples were named as UiO-X, in which X indicates the volume of added water. UiO-66-CH3 and UiO-66-NH2 samples (denoted as UiO-CH3-X and UiO-NH2-X, respectively) were prepared using the same procedure, except 2-methylterephthalic acid or 2-aminoterephthalic acid was used instead of terephthalic acid.Results and discussion
In this work, water was used to control the defect formation in UiO-66. A small amount of water was added to the mixture strictly under N2 to prevent water intrusion from the atmosphere due to the hygroscopic nature of ZrCl4. In the absence of water, only a trace amount of the solid product was formed, which is not unexpected as water is a requisite for the formation of the Zr4+ oxo/peroxo cluster.30 The addition of 0.2 mL of water (UiO-0.2) resulted in the formation of a white solid at a yield less than 50%, while the addition of water in the range of 0.4–2.0 mL (UiO-0.4, UiO-0.8, UiO-1.2 and UiO-2.0) led to similar product yields more than 80%. As the ZrCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O molar ratio of 1:8 is theoretically required for the formation of Zr6(OH)4O4(bdc)6, the low yield for UiO-0.2 was originated from an insufficient amount of water with respect to ZrCl4. The X-ray diffraction (XRD) patterns of all the samples exhibited the face-centered cubic lattice typical for the UiO-66 crystal (Fig. 1a and S1†). The crystallite size that was derived from the (111) reflection monotonously decreased by increasing the water amount, in agreement with the transmission electron microscopy observation (Fig. S4 and S7†). The reduction of the crystallite size was ascribed to accelerated nucleation of the oxoclusters by water, thus changing the crystallization process from nucleation-limited to growth-limited crystallization.27,30 From the XRD profiles, the broad diffraction peaks at 2θ = 5.9°, 9.6° and 10.6° were additionally observed for UiO-0.2, UiO-0.4 and UiO-0.8. These diffractions were similar to those reported by Goodwin and co-workers, when monocarboxylic acid was introduced as a modulator. They were assigned to the (110), (210) and (211) reflections of the reo topology.22 The reo phase existed as a nano-domain where the cluster is eight-connected instead of twelve to generate an octahedral cavity, so called a missing-cluster defect. In the case of UiO-1.2, only the peak at 5.9° was found in addition to a typical diffraction of UiO-66. Though this peak is very broad with a diffuse feature, it is sufficiently distinguishable from the base line and can be assigned to the (110) reflection of the reo phase with the domain size even smaller than the other three samples. The same approach was extended to UiO-66-CH3 and UiO-66-NH2 using water in the range of 0.3–0.9 mL. In XRD, they possess an isostructure to that of UiO-66 (Fig. 1b, c, S2 and S3†). The appearance of the diffraction peak at 2θ = 5.9° clearly evidenced the formation of the reo topology, emphasizing a crucial role of water on the formation of the missing-cluster defects regardless the type of the ligand. A similar tendency for the particle size reduction with the increase in the water amount was observed for both of the UiO-66-CH3 and UiO-66-NH2 series (Fig. S5–S7†). Among the employed linkers, UiO-66-NH2 exhibited the smallest particle size at the same water amount due to the crystallization acceleration effect of the amino group.31
:H2O molar ratio of 1:8 is theoretically required for the formation of Zr6(OH)4O4(bdc)6, the low yield for UiO-0.2 was originated from an insufficient amount of water with respect to ZrCl4. The X-ray diffraction (XRD) patterns of all the samples exhibited the face-centered cubic lattice typical for the UiO-66 crystal (Fig. 1a and S1†). The crystallite size that was derived from the (111) reflection monotonously decreased by increasing the water amount, in agreement with the transmission electron microscopy observation (Fig. S4 and S7†). The reduction of the crystallite size was ascribed to accelerated nucleation of the oxoclusters by water, thus changing the crystallization process from nucleation-limited to growth-limited crystallization.27,30 From the XRD profiles, the broad diffraction peaks at 2θ = 5.9°, 9.6° and 10.6° were additionally observed for UiO-0.2, UiO-0.4 and UiO-0.8. These diffractions were similar to those reported by Goodwin and co-workers, when monocarboxylic acid was introduced as a modulator. They were assigned to the (110), (210) and (211) reflections of the reo topology.22 The reo phase existed as a nano-domain where the cluster is eight-connected instead of twelve to generate an octahedral cavity, so called a missing-cluster defect. In the case of UiO-1.2, only the peak at 5.9° was found in addition to a typical diffraction of UiO-66. Though this peak is very broad with a diffuse feature, it is sufficiently distinguishable from the base line and can be assigned to the (110) reflection of the reo phase with the domain size even smaller than the other three samples. The same approach was extended to UiO-66-CH3 and UiO-66-NH2 using water in the range of 0.3–0.9 mL. In XRD, they possess an isostructure to that of UiO-66 (Fig. 1b, c, S2 and S3†). The appearance of the diffraction peak at 2θ = 5.9° clearly evidenced the formation of the reo topology, emphasizing a crucial role of water on the formation of the missing-cluster defects regardless the type of the ligand. A similar tendency for the particle size reduction with the increase in the water amount was observed for both of the UiO-66-CH3 and UiO-66-NH2 series (Fig. S5–S7†). Among the employed linkers, UiO-66-NH2 exhibited the smallest particle size at the same water amount due to the crystallization acceleration effect of the amino group.31
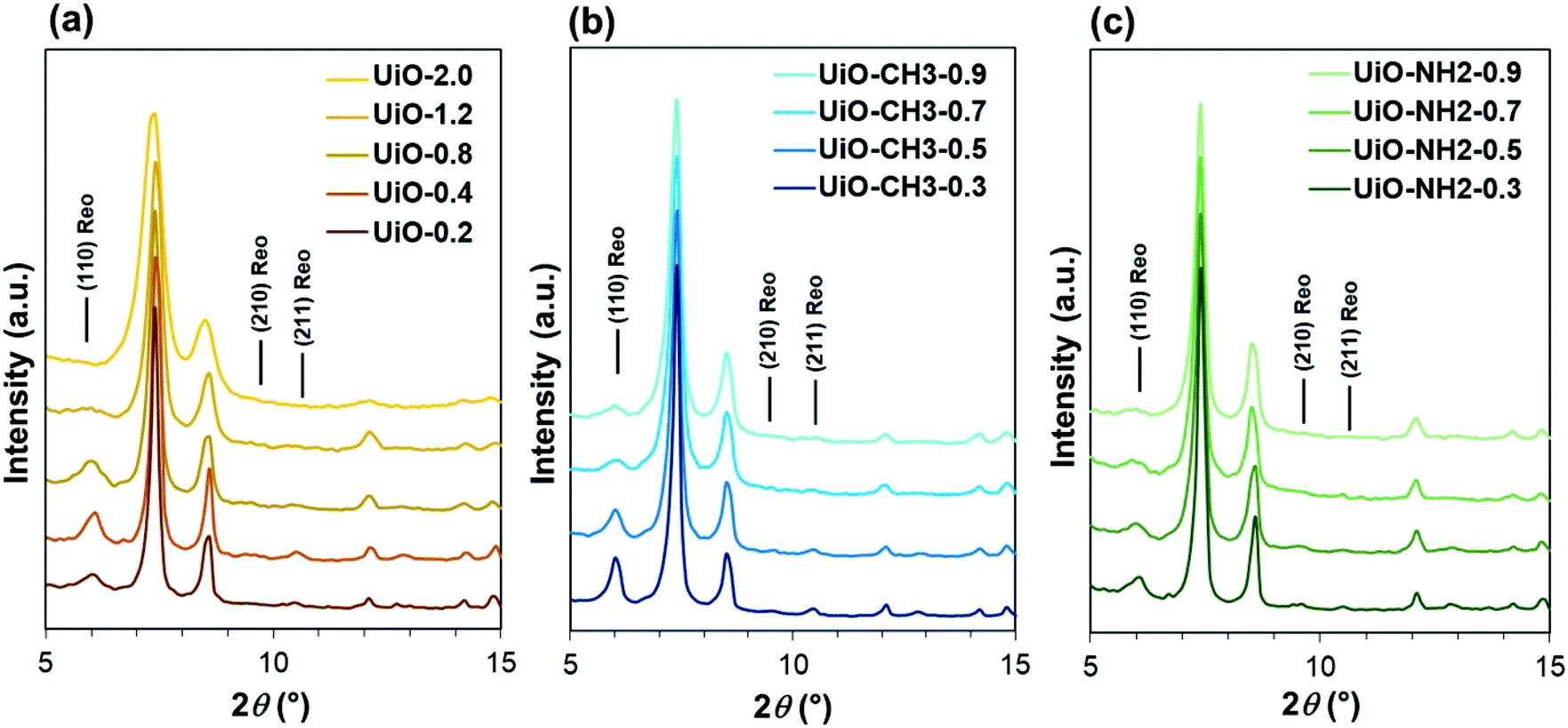 | ||
| Fig. 1 XRD patterns of (a) UiO-66, (b) UiO-66-CH3 and (c) UiO-66-NH2 samples. | ||
Based on the weight loss in the thermogravimetric (TG) analysis (Fig. S8 and Table S1†), all of the samples exhibited the number of the linker per cluster lower than 12. It must be noted that the TG method is known to be less accurate for the quantitative analysis of the organic content,12 especially for the amino-substituted UiO-66-NH2 that did not exhibit a clear plateau. However, the linker deficiency tended to be more pronounced for a smaller water amount for all the series. The microporous surface area and the mesoporous surface area were acquired from N2 adsorption/desorption experiments. In Table S2,† the microporous surface area of UiO-66 samples decreased with the increase in the water amount, while the tendency was opposite for the mesoporous surface area. The increase in the mesoporous surface area is originated from the reduction of the particle size, in which the intergrown morphology created the interparticle channel having the size in the mesopore region. As a result, the differences in microporous and mesoporous surface areas among the samples are hardly explained by the presence of defects due to the strong influences of the particle size. Internal pore structures in relation to the type of defects were interpreted from the N2 adsorption/desorption isotherms (Fig. S9–S21†). The pore size distributions constructed by the NLDFT method for UiO-2.0 and UiO-1.2 exhibited a broad peak with a small shoulder (Fig. 2a). The peak top is positioned at 10 Å, slightly larger than the pore size typically reported for defect-free UiO-66.11,32 Such an enlarged pore size has been reported for UiO-66 with a missing-linker defect where the shortage of the linkers from the metal cluster expanded the entire framework.6,11,12,32 On the other hand, the rest of the samples including UiO-66-CH3 and UiO-66-NH2 series equipped two modes of pores (Fig. 2b and c). The first mode centred at 10 Å similar to those of UiO-2.0 and UiO-1.2, while the second mode appeared at 16 Å. The appearance of the second mode was associated with the removal of the cluster to generate an octahedral cavity.22,32 These results agreed well with the observation of the reo topology by X-ray. Based on all the above observations, it was concluded that both of the missing-linker and missing-cluster defects co-existed in the samples, except UiO-2.0 that mainly contained the missing-linker defects. In the absence of modulators, the coordination vacancies can be filled by OH−/H2O and Cl− (generated in situ from the hydrolysis of ZrCl4). X-ray photoelectron spectroscopy analysis (Fig S22†) showed the absence of Cl, excluding Cl− as the compensating group. Indeed, the acid–base titration of UiO-66 revealed three reflection points (Fig. S23†), which are respectively attributed to μ3-OH, Zr–OH2 and Zr–OH.33 The latter two protons confirmed the charge compensation at the defective nodes by OH−/H2O.
 | ||
| Fig. 2 Pore size distribution profiles derived by NLDFT of (a) UiO-66, (b) UiO-66-CH3 and (c) UiO-66-NH2 samples. | ||
In order to acquire the quantitative trend of the missing-cluster defect formation, the relative intensity of the (110) reo peak was calculated according to Shearer et al.21 As depicted in Fig. 3, the relative intensity of the (110) reo peak decreased along with the increase in the water amount, except UiO-0.2. To date, the mechanism for the formation of the reo phase in UiO-66 has not been well understood. As a modulator is not present, the coordination equilibrium shift due to competitive coordination can be ruled out. Recently, the partially deprotonated ligand model was proposed.32 In this model, the missing-cluster defects occur when ligands on the surface of adjacent clusters are partially deprotonated, thus being unable to accept the metal cluster between them. Herein, we did not observe the pendant coordinated-free carboxylate group as shown by the absence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and COH peaks in the Fourier transform infrared spectra (Fig. S24†), suggesting a different pathway for the missing-cluster defect formation. Considering that a small amount of water hardly alters the pKa value but greatly affects the defect formation, the origin of the missing-cluster defects is likely originated from the kinetic reason rather than the equilibrium in ligand deprotonation. On the basis of the metal cluster formation, the tetrameric species [Zr4(OH)8(H2O)16]8+ is known to be the most stable structure upon hydrolysis of ZrCl4 and ZrOCl2·8H2O in aqueous solution. In the presence of carboxylate ligands, the tetranuclear species rearranges into the thermodynamically stable hexanuclear building unit with 12-ligand coordination, plausibly through the breakage of the original structure and the formation of an intermediate species.34,35 At a limited accessibility to water molecules, it is anticipated that metastable species of the oxoclusters that allow a lower coordination with ligands might be formed and play a role of pre-nucleation species for the formation of the reo domain.
O and COH peaks in the Fourier transform infrared spectra (Fig. S24†), suggesting a different pathway for the missing-cluster defect formation. Considering that a small amount of water hardly alters the pKa value but greatly affects the defect formation, the origin of the missing-cluster defects is likely originated from the kinetic reason rather than the equilibrium in ligand deprotonation. On the basis of the metal cluster formation, the tetrameric species [Zr4(OH)8(H2O)16]8+ is known to be the most stable structure upon hydrolysis of ZrCl4 and ZrOCl2·8H2O in aqueous solution. In the presence of carboxylate ligands, the tetranuclear species rearranges into the thermodynamically stable hexanuclear building unit with 12-ligand coordination, plausibly through the breakage of the original structure and the formation of an intermediate species.34,35 At a limited accessibility to water molecules, it is anticipated that metastable species of the oxoclusters that allow a lower coordination with ligands might be formed and play a role of pre-nucleation species for the formation of the reo domain.
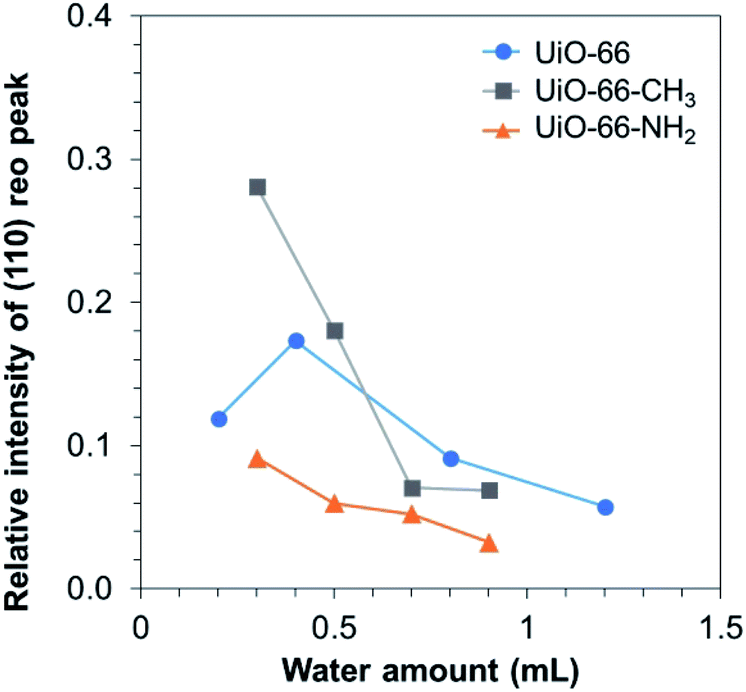 | ||
| Fig. 3 Relative intensity of (110) reo peak for UiO-66, UiO-66-CH3 and UiO-66-NH2. | ||
The catalytic performance of UiO-66 samples was evaluated in the Meerwein–Ponndorf–Verley (MPV) reduction of 4-tert-butylcyclohexanone with isopropanol (Table 1, entries 1–8) as well as in alcoholysis of styrene oxide with methanol (Table 1, entries 9–17). In the MPV reduction, UiO-66 samples were subjected to heat treatment at 150 °C under vacuum prior to the reaction. Note that the removal of the OH−/H2O compensating group to generate an open Lewis acidic site requires much lower temperature than that required for the removal of terminated carboxylate species11,36 or dehydroxylation of a perfect framework to form a sevenfold coordinated [Zr6O6]12+ clusters with a Lewis acidic character.23,37 In the blank experiment, the conversion reached 1.8% after 3 h, while it increased to 19–25% by the introduction of UiO-66. UiO-0.4, which exhibited the most intense reo peak, gave the highest activity among the samples. Prolonging the reaction time for UiO-0.4 to 24 h, the conversion increased up to 93.8%, surpassing those reported for missing-linker defect-type UiO-66.11 The achieved conversion was in a similar range to that obtained from highly active catalysts for the MPV reduction such as Zr- and Al-beta-zeolites.38,39 However, the time required to reach a similar conversion is longer for UiO-66. It is noted that the catalytic performance tended to follow the relative intensity of the (110) reo peak, but not exclusively. Particularly, a considerable activity was also obtained from UiO-2.0 that did not contain the reo domain. Instead, this sample possessed the smallest particle size and, hence, is expected to contain more under-coordinated Zr sites on the external surface. The presence of missing-cluster defects also affected the configurational isomerism of the product. The trans/cis ratio of the product tended to be lower for the samples containing the missing-cluster defects. It has been reported that the MPV reduction of 4-tert-butylcyclohexanone proceeds via the coordinative interaction of ketone and alcohol on a Lewis acidic site to form the cyclic six-membered transition state.39 In the beta-zeolite catalyst system, thermodynamically less stable cis-4-tert-butylcyclohexanol is favored as the transition state for cis-isomer is less bulky and can align along the restricted pore channels.38,39 On the other hand, trans-alcohol becomes the main product for mesoporous catalysts.39 In our case, the presence of missing-cluster defects generated the pore cavity close to mesopores. Hence, the increase in the selectivity towards cis-isomer is rather associated with the enhanced diffusion of the reactants to the active sites located in small pores. These results suggested that the accessibility to the active sites is rather important for the activation of bulky molecules, in which the missing-cluster defects provide not only the open Lewis sites, but also the hierarchical porous structure to facilitate the diffusion through interconnected mesopores. The catalytic recyclability and the structural retention were investigated for UiO-0.4. After 24 h of the MPV reduction, the nanoparticles were collected and re-used after repetitive washing with methanol. A slight reduction of the conversion was observed in the second run (Table 1, entry 8). The XRD patterns of UiO-0.4 after the first run and the second run were found to be identical to the original sample (Fig. S27†), indicating that the missing cluster defects remained intact after catalysis.
|
||||
|---|---|---|---|---|
| Entry | Sample | Time (h) | Temp. (°C) | Conv.c (%) |
| a 4-tert-Butylcyclohexanone (0.5 mmol), isopropanol (2.0 mL), a catalyst (10 mg), and n-decane (20 μL) as an internal standard were heated at 80 °C.b Styrene oxide (1 mmol), methanol (1.0 mL), a catalyst (5.5 mg), and n-decane (20 μL) as an internal standard were heated at 55 °C.c Determined by gas chromatography. The number in the parenthesis indicated the trans/cis ratio of 4-tert-butylcyclohexanol products.d Nanoparticles were recovered after 24 h of the reaction and re-used after repetitive washing with methanol. | ||||
| 1 | Blank | 3 | 80 | 1.8 |
| 2 | UiO-0.2 | 3 | 80 | 20.9 (4.2) |
| 3 | UiO-0.4 | 3 | 80 | 25.6 (3.9) |
| 4 | UiO-0.4 | 24 | 80 | 93.8 (3.9) |
| 5 | UiO-0.8 | 3 | 80 | 22.8 (3.7) |
| 6 | UiO-1.2 | 3 | 80 | 19.6 (4.4) |
| 7 | UiO-2.0 | 3 | 80 | 20.9 (4.5) |
| 8 | Re-UiO-0.4d | 24 | 80 | 86.6 (3.5) |

|
||||
|---|---|---|---|---|
| Entry | Sample | Time (h) | Temp. (°C) | Conv.c (%) |
| 9 | Blank | 0.5 | 55 | 0 |
| 10 | UiO-0.2 | 0.5 | 55 | 9.8 |
| 11 | UiO-0.4 | 0.5 | 55 | 13.2 |
| 12 | UiO-0.4 | 1 | 55 | 17.0 |
| 13 | UiO-0.4 | 2 | 55 | 22.0 |
| 14 | UiO-0.4 | 24 | 55 | 72.1 |
| 15 | UiO-0.8 | 0.5 | 55 | 12.9 |
| 16 | UiO-1.2 | 0.5 | 55 | 8.1 |
| 17 | UiO-2.0 | 0.5 | 55 | 7.6 |
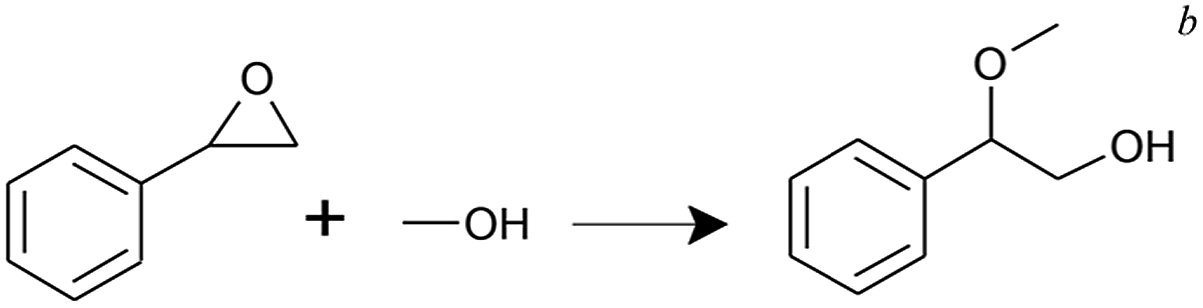
In alcoholysis of styrene oxide, the as-synthesized UiO-66 samples were used without the heat treatment. The conversion reached 7–13% in the presence of UiO-66 samples after 0.5 h with 100% selectivity towards 2-methoxy-2-phenylethanol. Similar to the case of the MPV reduction, UiO-0.4 gave the highest conversion among the samples. As the Lewis acidic character of the Zr atom is shielded by OH−/H2O in the absence of heat treatment, it is likely that the reaction is achieved by the Brønsted acid sites via the addition of proton to the epoxide oxygen.40 A higher activity of UiO-0.4 is believed to be associated with additional Brønsted sites at the Zr–OH/Zr–OH2 defective nodes. As well, the presence of missing-cluster defects might indirectly facilitate the reaction through facile proton mobility in the hydrated environment and larger pore cavities.
Conclusion
In summary, defect engineering in UiO-66 and its isoreticular derivatives was achieved by the rigorous control of water addition during the synthesis. Especially, we report for the first time that the missing-cluster defects can be generated without the addition of acidic modulators. This approach provides a method of fine-tuning missing-cluster defects in a simple and environmentally friendly fashion. The presence of the missing-cluster defects give rise to beneficial influences on the catalytic performance of UiO-66, where the missing-cluster defects provide additional Brønsted/Lewis acidic sites and facilitate diffusion of the reagents due to the hierarchical pore structure.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Chen, X. Zhang, F. Bi, X. Zhang, Y. Yang and Y. Wang, J. Colloid Interface Sci., 2020, 571, 275–284 CrossRef CAS PubMed.
- Y. Yang, Z. Zheng, W. Ji, J. Xu and X. Zhang, J. Hazard. Mater., 2020, 395, 122686 CrossRef CAS PubMed.
- Y. Wang, Y. Yang, N. Liu, Y. Wang and X. Zhang, RSC Adv., 2018, 8, 33096–33102 RSC.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- Y. Liu, X.-Y. Xie, C. Cheng, Z.-S. Shao and H.-S. Wang, J. Mater. Chem. C, 2019, 35, 10743–10763 RSC.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, J. Hazard. Mater., 2019, 365, 597–605 CrossRef CAS PubMed.
- X. Zhang, Y. Yang, X. Lv, Y. Wang, N. Liu, D. Chen and L. Cui, J. Hazard. Mater., 2019, 366, 140–150 CrossRef CAS PubMed.
- X. Zhang, X. Shi, J. Chen, Y. Yang and G. Lu, J. Environ. Chem. Eng., 2019, 7, 103405 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, ACS Catal., 2016, 6, 235–247 CrossRef CAS.
- D. Yang, C. A. Gaggioli, D. Ray, M. Babucci, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2020, 142, 8044–8056 CrossRef CAS PubMed.
- X. Chen, Y. Lyu, Z. Wang, X. Qiao, B. C. Gates and D. Yang, ACS Catal., 2020, 10, 2906–2914 CrossRef CAS.
- J. Wang, L. Liu, C. Chen, X. Dong, Q. Wang, L. Alfilfil, M. R. AlAlouni, K. Yao, J. Huang, D. Zhang and Y. Han, J. Mater. Chem. A, 2020, 8, 4464–4472 RSC.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. K. P. Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. J. Hoffman, V. Meynen, C. Marquez, D. E. De Vos, V. Van Speybroeck, K. Leus and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 3174–3183 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- B. Bueken, N. Van Velthoven, A. Krajnc, S. Smolders, F. Taulelle, C. Mellot-Draznieks, G. Mali, T. D. Bennett and D. De Vos, Chem. Mater., 2017, 29, 10478–10486 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F.-X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC.
- S. Yuan, L. Zou, J.-S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme, T. Cagin and H.-C. Zhou, Nat. Commun., 2017, 8, 15356 CrossRef CAS.
- J. M. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Chem. Mater., 2015, 27, 2286–2289 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS.
- K. L. Svane, J. K. Bristow, J. D. Gale and A. Walsh, J. Mater. Chem. A, 2018, 6, 8507–8513 RSC.
- G. Y. Shangkum, P. Chammingkwan, D. X. Trinh and T. Taniike, Membranes, 2018, 8, 129 CrossRef PubMed.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U.-H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491–2500 CrossRef CAS PubMed.
- F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. De Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- B. Shan, S. M. McIntyre, M. R. Armstrong, Y. Shen and B. Mu, Ind. Eng. Chem. Res., 2018, 57, 14233–14241 CrossRef CAS.
- R. C. Klet, Y. Liu, T. C. Wang, J. P. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- C. Hennig, S. Weiss, W. Kraus, J. Kretzschmar and A. C. Scheinost, Inorg. Chem., 2017, 56, 2473–2480 CrossRef CAS PubMed.
- M. G. Goesten, M. F. De Lange, A. I. Olivos-Suarez, A. V. Bavykina, P. Serra-Crespo, C. Krywka, F. M. Bickelhaupt, F. Kapteijn and J. Gascon, Nat. Commun., 2016, 7, 11832 CrossRef CAS PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- J. Wang, K. Okumura, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2015, 493, 112–120 CrossRef CAS.
- J. C. Van der Waal, E. J. Creyghton, P. J. Kunkeler, K. Tan and H. Van Bekkum, Top. Catal., 1997, 4, 261–268 CrossRef.
- S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction, transmission electron microscopy, thermogravimetric analysis, N2 adsorption/desorption measurement, X-ray photoelectron spectroscopy, acid–base titration, Fourier-transform infrared spectroscopy, analysis of reaction products, catalyst recyclability and structural retention. See DOI: 10.1039/d0ra04812g |
| This journal is © The Royal Society of Chemistry 2020 |