
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of Ce addition in catalytic activity enhancement of TiO2-supported Ni for CO2 methanation reaction†
Ammarika Makdeea,
Kingkaew Chayakul Chanapattharapol *a,
Pinit Kidkhunthodb,
Yingyot Poo-arpornb and
Teruhisa Ohnoc
*a,
Pinit Kidkhunthodb,
Yingyot Poo-arpornb and
Teruhisa Ohnoc
aMaterials Chemistry Research Center, Department of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail: kingkaew@kku.ac.th; Fax: +66 432 02373; Tel: +66 430 09700 ext. 12371
bSynchrotron Light Research Institute (Public Organization), Nakhon Ratchasima 30000, Thailand
cDepartment of Applied Chemistry, Faculty of Engineering, Kyushu Institute of Technology, 1-1 Sensuicho, Tobata, Kitakyushu 804-8550, Japan
First published on 20th July 2020
Abstract
In this work, various amounts of Ce were added to TiO2 to form a mixed oxide support; CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) and then those synthesized supports were impregnated by 10 wt% Ni to produce a catalysts. The 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts were tested for CO2 methanation reaction by using a fixed-bed reactor in the temperature range of 100–500 °C. The sample was pretreated at 450 °C under H2 and then a mixed feed gas of CO2 and H2 was switched into the reactor to start the reaction. The results showed that 10 wt% Ni–Ce0.003Ti0.997O2 catalyst (the lowest Ce content) exhibited the highest CO2 conversion and CH4 yield. Moreover, 10 wt% Ni–Ce0.003Ti0.997O2 showed highly stable during the stability test (50 h.). The results indicated that upon addition of small amount of Ce into TiO2-supported Ni, the surface, structural, electrical and redox properties of the catalyst were improved to the extent that these properties can promote the catalytic activities for CO2 methanation. The Ce addition can improve the CO2 methanation catalytic activity by several ways. First, higher dispersion of Ni on catalysts surface upon addition of Ce was observed which resulted in higher adsorption rate of H2 on this metal active site. Second, formation of a larger amounts of oxygen vacancies as well as basicity improvement upon addition of Ce were occurred which can increase the CO2 adsorption on catalyst surface. Third, incorporation of Ce resulted in improving of a starting reduction temperature of Ni2+ to Ni0 for TiO2-supported Ni catalyst which can indicate that the reducibility of Ce-doped TiO2-supported Ni catalyst was enhanced and then alter its catalytic activity. However, increasing of Ce content led to lowering of CO2 methanation activities which resulted from increasing of basicity by Ce addition. The excess amounts of adsorbed CO2 would lead to competitive adsorption to H2 and then lead to a decrease of catalytic activity. Therefore, an appropriate amount of H2 and CO2 adsorption ability on catalyst surface was a prominent factor to dominate the catalytic activity.
1. Introduction
Carbon dioxide (CO2) is one of the greenhouse gases which is the most abundant in the atmosphere. It is the biggest contributor to the earth's temperature increases and climate change which are currently a main global problem. The concentration of CO2 in the atmosphere can be reduced by two methods (i) CO2 capture and storage (CCS) and (ii) CO2 conversion and utilization (CCU). However, while CO2 capture and storage is an important way to rapidly decrease CO2 levels in the atmosphere, the disadvantage of this method is the possible leakage of captured CO2.1 Therefore, CO2 conversion and utilization is a preferred method. This method not only reduces CO2 concentration but also generates useful products and fuels which respond to high energy demands. CO2 hydrogenation to form CH4 by using H2 as a reductant is one of the most common reactions which is used to decrease CO2 concentration and produce CH4. This reaction can be called CO2 methanation or Sabatier reaction according to eqn (1):| CO2 + 4H2 → CH4 + 2H2O, ΔH298 K = −165.12 kJ mol−1 | (1) |
One interesting benefit of CO2 methanation reaction is that the product, CH4, can be directly injected into natural gas pipelines, can be used as a starting material for the production of other chemicals and can be utilized as an energy carrier due to the transportation of H2 is limited. In addition, CO2 methanation can be performed under atmospheric pressure and low temperature due to its exothermic characteristics and it is also considerably faster than other reactions such as hydrogenation of CO2 to alcohol.2 However, this reaction needs eight electrons to convert CO2 to CH4 which is a significant kinetic barrier.3 Moreover, some side-reactions can occur at high temperature, such as reversed water gas shift (CO2 + H2 → CO + H2O) which can lead to lower CH4 selectivity.4 Therefore, catalysts must be employed in order to achieve a high reaction rate and high CH4 selectivity.5–8 In recent years, many research groups have been interested in CO2 methanation by using heterogeneous catalysts. It has been reported that supports and active species on catalyst surfaces affected the CO2 methanation catalytic activities of catalysts. For the active species, noble metals such as Ru and Rh provided the highest catalytic activities and CH4 selectivities.2,9 However, they are very expensive. Therefore, other metals with a lower price have been employed instead. Ni metal was widely used as an active species due to its inexpensive and high catalytic activity.9–16 The Ni metal is the most favorable active species for H2 dissociation to form atomic H which facilitates the CH4 formation and results in the enhancement of CO2 conversion and CH4 selectivity.17,18 The catalyst support also displays an important role in the catalytic activity of the catalyst. Recently, many metal oxides have been widely used as catalyst supports such as Al2O3, SiO2, ZrO2, TiO2, and CeO2.19–21 Among these supports, reducible supports (TiO2, CeO2, and ZrO2) were widely selected because of their excellent redox properties. Due to the CO2 methanation mechanism involved in the redox process, the reaction rate can be enhanced by facilitating this process. To increase the redox properties of metal oxide supports, some metals have been used to incorporate into the metal oxide lattice. These metals are called promoters. Several works have reported on the use of rare earth metals (La, Ce, Sm) as promoters in CO2 methanation reaction due to their functionality to catalyze the reaction rate by for example, modifying the surface properties, redox properties, electrical properties and surface basicity.22–29 Cerium oxide was usually used as a promoter due to its superior properties on improving the surface basicity and oxygen vacancies formation through valence change between Ce3+ and Ce4+.30,31 Bian et al.30 reported the effect of CeO2 addition as a promoter on Ni/Al2O3 catalysts. They found that the surface basicity and Ni dispersion on catalyst surfaces were improved upon the addition of CeO2 which resulted in high CO2 methanation activity in low-temperature ranges. Tada et al.31 reported that CeO2 plays an important role in promoting CO2 methanation and enhancing CH4 formation. CeO2 can improve the surface basicity in which CO2 can be easily adsorbed and reduced due to oxygen vacancies formation by addition of CeO2 into Ru/Al2O3. Moreover, they found that the specific surface area of the catalyst was enhanced upon the addition of CeO2.
In this work, we report on the catalytic activity of TiO2-supported Ni catalysts for CO2 methanation. Since, the reported on using TiO2 as a catalyst support for CO2 methanation is now still being developed, therefore, it is of interested and challenged to improve the TiO2 catalysts performance which can provide a better catalytic activity and can also produce the desired products. Ce was used as a promoter to modify the TiO2-supported Ni properties by incorporating it into TiO2 crystal structure in the form of CexTi1−xO2 mixed oxides. All of these catalysts were tested for catalytic activity for CO2 methanation and characterized by standard characterization methods such as X-ray diffraction (XRD), N2 adsorption–desorption, H2 adsorption, CO2 adsorption, transmission electron microscopy (TEM), scanning electron microscopy (SEM), inductively coupled plasma-optical emission spectroscopy (ICP-OES), X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS). All of these results were used to describe the role of Ce addition on enhancing the CO2 methanation reaction of TiO2-supported Ni.
2. Experimental
2.1 Materials
All chemicals in this experiment were analytical grade without further purification. Titanium(IV) isopropoxide (98+%) and cerium(III) nitrate hexahydrate (99.5%) were purchased from Acros Organics. Ethanol absolute anhydrous (99.9%) and nickel(II) nitrate hexahydrate (99%) were purchased from Carlo Erba. Nitric acid (65%) was provided by Acl Labscan. Deionized water was used for all preparations.2.2 Preparation of TiO2, CexTi1−x O2, TiO2-supported Ni and CexTi1−xO2-support Ni
TiO2 support was prepared by sol–gel method with titanium isopropoxide as a starting precursor. Titanium isopropoxide was first dissolved in ethanol, then DI water and HNO3 were added dropwise to Ti precursor solution and continuously stirred for 3 h. The mixture was dried at 100 °C for 24 h and calcined at 400 °C for 3 h. CexTi1−xO2 mixed oxide supports were also prepared by the same method as TiO2 support with Ce/Ti molar ratio of 1/200, 1/20, 1/10 and 1/5. The preparation procedure was the same as described above in which the aqueous solution of Ce(NO3)3·6H2O was co-added together with DI water and HNO3.10 wt% Ni doped TiO2 and CexTi1−xO2-supported were prepared by incipient wetness impregnation method. Ni(NO3)2·6H2O was used as a Ni precursor. An appropriate amount of precursor salt was dissolved in DI water. After that, the precursor solution was added to the prepared supports and then dried at 100 °C for 24 h and calcined at 400 °C for 3 h.
2.3 Catalysts characterization
X-ray diffraction (XRD) patterns of all catalysts were obtained from PANalytical Empyrean with Cu Kα radiation wavelength of 1.5406 Å. The obtained patterns were scanned in the 2θ range of 10° to 80°. The crystalline size (D) was calculated from Scherrer's equation as:
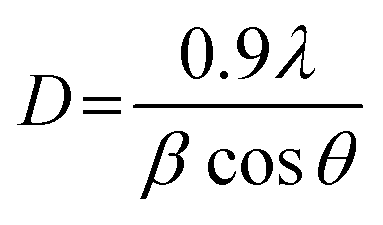 | (2) |
The specific surface area of the prepared catalysts was calculated from N2 adsorption–desorption isotherm at 77 K by Quantachrome NOVA 4200e instrument. Before the adsorption experiment, the catalysts were degassed at 100 °C for 1 h to clean the catalyst surface. The specific surface area was calculated by using the multipoint-BET method.
The morphology of the prepared catalysts was studied by transmission electron microscope (TEM) FEI/TECNAI G2 20. The catalysts were prepared by dispersing a catalyst powder in acetone with sonication and dropped on the copper grid before the scanning process.
Nickel dispersion mapping on catalysts surface was investigated by using a LEO 1450VP SEM-EDS instrument. Before scanning, the catalyst was dispersed on a carbon tape which was attached on a specimen stub and coated with gold to reduce the electron charge.
The metals content in the prepared catalysts can be determined by using inductively coupled plasma-optical emission spectroscopy (ICP-OES). Before measuring the metal content, the catalysts (0.020–0.025 g) were digested by using a mixture of 6 mL concentrated H2SO4 and 2 mL 30% H2O2 at high temperature. The standards and samples solution in the range of 0–10 ppm were analyzed by optima 100DVICP-OES (PerkinElmer). The detection wavelengths for Ce and Ni were 413.764 and 231.604 nm, respectively.
X-ray photoelectron spectroscopy (XPS) was used to investigate the surface components of the catalysts. Ti 2p XPS spectra of the catalysts were obtained by using a KRATOS AXIS-NOVA (Shimadzu Corporation) instrument with a monochromatic Al Kα as an X-ray source. The binding energy shift caused by relative surface charging was corrected by using the C 1s level (284.8 eV) as an internal standard. The XPS peaks were assumed to be Gaussian peaks and were resolved into each component by using XPSPEAK41 software.
2.4 CO2 and H2 adsorption experiments
In order to investigate the adsorption ability of both reactants in CO2 methanation reaction, the CO2 and H2 adsorption experiments were conducted at room temperature and under ambient pressure. For the CO2 adsorption experiment, the catalyst (0.13–0.14 g) was dispersed on a glass dish which was placed in a Tedlar bag. After that, 150 mL of mixed gas was injected into the Tedlar bag. The mixed gas consisted of 500 ppm CO2 and air (79% N2 and 21% O2). The concentration of CO2 was monitored every 30 min for 4 h by withdrawing the gas from the bag and detected by gas chromatograph (INFICON 3000 Micro GC) with a PLOT U column and TCD detector. The amount of adsorbed CO2 was calculated by subtracting the remaining CO2 concentration at interval times from the initial CO2 concentration. Finally, the relationship between adsorbed CO2 amounts as a function of time was constructed.The H2 adsorption experiment was conducted in a similar manner as CO2 adsorption experiment. The same amount of catalyst was spread on a glass dish and was placed in a Tedlar bag. The 150 mL of mixed gas (500 ppm H2 and air; 79% N2 and 21% O2) was injected into the bag and the adsorption experiment was left at room temperature for 4 h. After that, the remaining H2 amount was detected by gas chromatograph (Agilent Technologies 490 Micro GC) with a MS-5A column and TCD detector. The H2 uptake on the catalyst was calculated by using the same equation as the CO2 adsorption experiment. The data reported in this manuscript is an average of three reproducible experiments.
2.5 X-ray absorption spectroscopy (XAS)
The XANES experiments were carried out on Beamline 2.2 (time-resolved X-ray absorption spectroscopy) at the Synchrotron Light Research Institute (SLRI), Nakhon Ratchasima, Thailand. The bent crystal was used as an energy dispersive monochromator. A Si (111) crystal was used as a monochromator with an energy range of 2.4–10 keV. The linear image sensor was used as a detector. For the ex situ experiment, the sample was first mixed with boron nitride (BN) to obtain a homogeneous powder and pressed into a pellet and attached on the sample frame. The XANES spectra were recorded in the K (for Ni and Ti) and L3 (for Ce) edges energy in transmission mode. The ionization chamber was used as a detector and was placed in front of and behind the sample. For the in situ experiment, the sample was mixed with boron nitride, pressed into a pellet and then placed in an in situ cell. The sample was first pretreated by heating the sample cell from room temperature to 450 °C (ramp rate of 5 °C min−1) under H2 flowing at 24 mL min−1. The spectra were collected every 10 °C increment as the temperature increased. Next, the temperature was kept at 450 °C for 90 min, in order to reduce Ni ion species to metallic Ni, and the spectra were collected every 10 min. After the pretreatment, the temperature was reduced to 100 °C and the mixed feed gas composition of H2 (24 mL min−1) and CO2 (6 mL min−1) with the ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was switched into the in situ cell to start the CO2 methanation reaction. The reaction temperature was kept at 100 °C for 20 min and the spectra were collected every 10 min at this point. After that, the reaction temperature was increased to 150 °C (ramp rate of 5 °C min−1) and the spectra were collected at every 10 °C increment. When the reaction temperature reached 150 °C, the spectra were collected every 10 min for 20 min. Then, the reaction temperature was increased by 50 °C and the spectra were recorded during the increase and then holding the reaction temperature as described above. The reaction temperature range was 100–500 °C. The XANES spectra were obtained by subtracting I1 by I0 with the preprocessing software before being exported to Athena software.
:1 was switched into the in situ cell to start the CO2 methanation reaction. The reaction temperature was kept at 100 °C for 20 min and the spectra were collected every 10 min at this point. After that, the reaction temperature was increased to 150 °C (ramp rate of 5 °C min−1) and the spectra were collected at every 10 °C increment. When the reaction temperature reached 150 °C, the spectra were collected every 10 min for 20 min. Then, the reaction temperature was increased by 50 °C and the spectra were recorded during the increase and then holding the reaction temperature as described above. The reaction temperature range was 100–500 °C. The XANES spectra were obtained by subtracting I1 by I0 with the preprocessing software before being exported to Athena software.
2.6 CO2 methanation catalytic activity test
The catalytic activity of the prepared catalyst was tested for CO2 methanation reaction. The CO2 methanation reaction was studied in a flow reactor with the temperature in the range of 100–500 °C. The 50 mg of catalyst was sandwiched between two layers of quartz wool in the Pyrex tube. This Pyrex tube was placed inside the controllable temperature furnace. The K-type thermocouple was inserted at the top of the catalyst bed to monitor the reaction temperature. The feed gases flow rate was controlled by mass flow controllers. Before starting CO2 methanation, the catalyst was pretreated at 450 °C under H2 atmosphere with a flow rate of 50 mL min−1 for 1.5 h. The CO2 methanation was started by using mixed feed gas of CO2, H2 and He with a flow rate of 6, 24 and 10 mL min−1, respectively. The mixed feed gas was passed through the catalyst in the Pyrex tube and then the reactants and products were analyzed by on-line Agilent Technologies 6890N gas chromatograph with HEYSEP D packed column and TCD was used as a detector. The percentages of CO2 conversion, products yield, and selectivity were calculated by using the following equations;
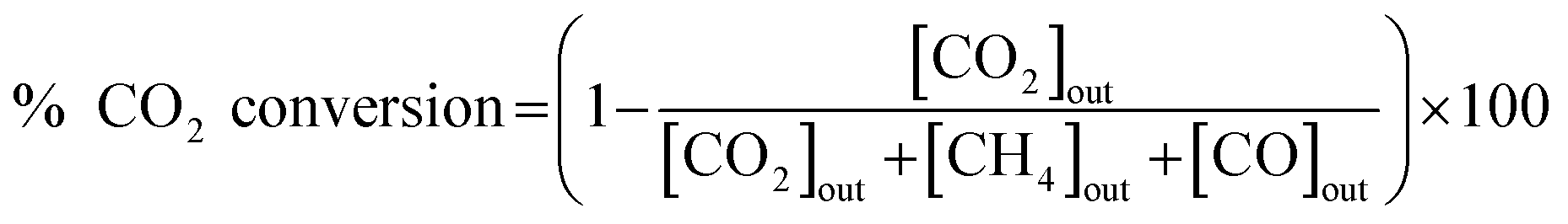 | (3) |
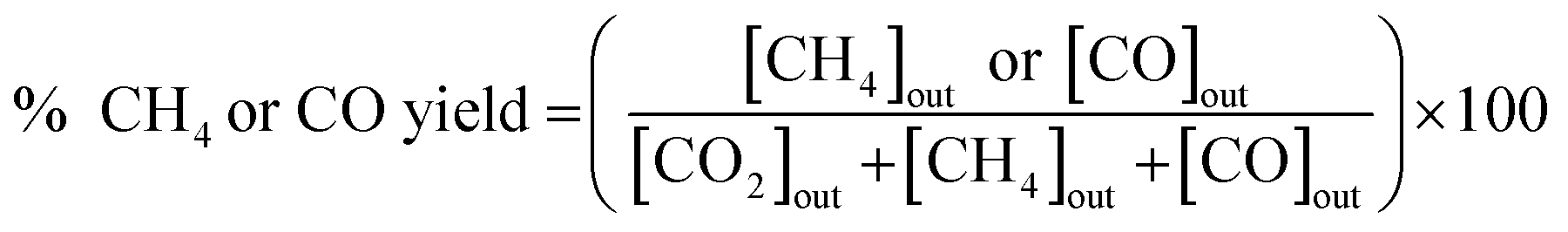 | (4) |
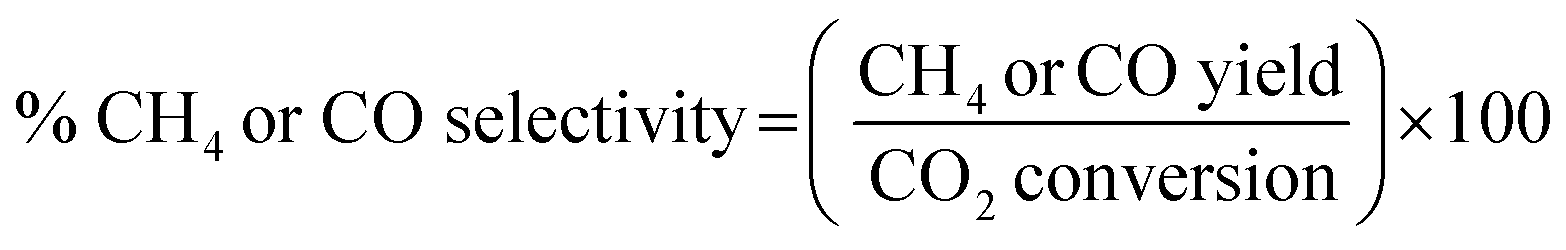 | (5) |
In order to exclude the heat and mass transfer under the kinetic reaction rate condition (reaction temperature range 230–280 °C), the separated experiments were conducted. The weight of catalyst (W) and total feed volume (Ftotal) were varied while the GHSV was kept constant.34 Two set of the same ratio between W/Ftotal; 0.05 g/40 mL min−1 and 0.1 g/80 mL min−1, were tested for CO2 methanation condition. The results showed that the CO2 conversion of 10 wt% Ni–Ce0.003Ti0.997O2 sample were similar for two sets (5.8% and 6.2%, respectively) at 250 °C.
3. Results and discussion
3.1 Standard characterization
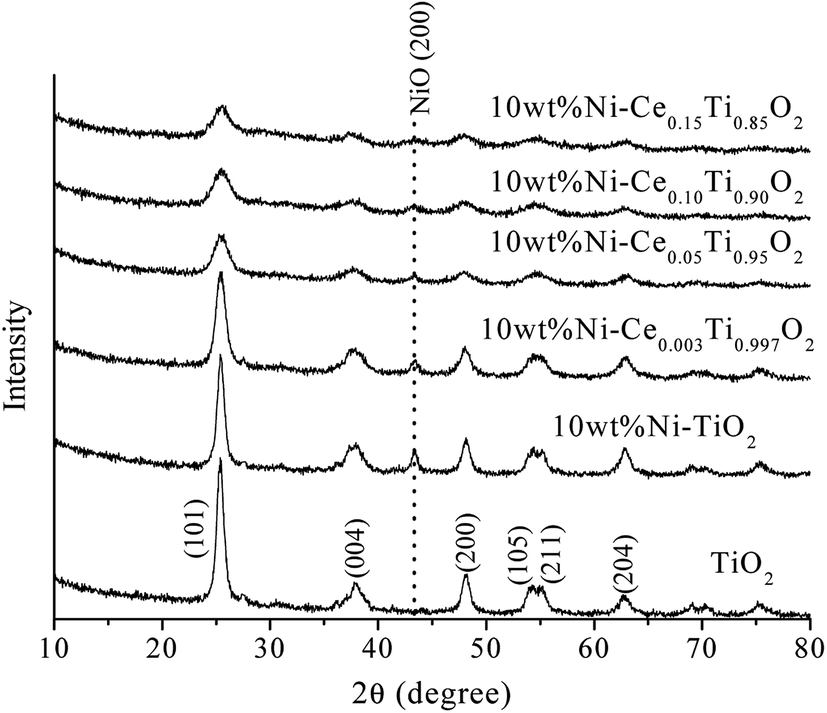 | ||
| Fig. 1 XRD patterns of TiO2 and 10 wt% NiCexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts. | ||
| Catalyst | Crystalline sizea (Å) | Lattice parametersb | Surface areac (m2 g−1) | Nid (wt%) | Ced (mol%) | ||
|---|---|---|---|---|---|---|---|
| a = b (Å) | c (Å) | Cell volume (Å3) | |||||
| a Calculated from (101) plane of TiO2 using Sherrer's equation.b Calculated from (101), (004), (200) planes of TiO2 using Bragg's equation.c Calculated by using multipoint BET method.d Calculated from ICP-OES results. | |||||||
| TiO2 | 103.9 | 3.786 | 9.503 | 136.2 | 138.02 | — | — |
| 10 wt% Ni–TiO2 | 108.6 | 3.793 | 9.568 | 137.7 | 67.63 | 11.01 | — |
| 10 wt% Ni–Ce0.003Ti0.997O2 | 76.8 | 3.802 | 9.581 | 138.5 | 82.16 | 10.78 | 0.24 |
| 10 wt% Ni–Ce0.05Ti0.95O2 | 62.6 | 3.833 | 9.676 | 142.1 | 123.48 | 10.87 | 4.11 |
| 10 wt% Ni–Ce0.10Ti0.90O2 | 54.2 | 3.834 | 9.795 | 143.9 | 136.87 | 11.44 | 9.40 |
| 10 wt% Ni–Ce0.15Ti0.85O2 | 51.5 | 3.850 | 9.935 | 147.3 | 150.03 | 12.61 | 15.30 |
In a comparison between different Ce loading catalysts, the characteristic peaks of cerium oxide were not observed, which indicated that Ce ions probably incorporated into TiO2 lattice by replacing Ti sites or interstitial sites to form a solid solution.42,43 These results could be confirmed by the expansion of the cell parameters with increased Ce content (Table 1). Additionally, the results of enlargement of TiO2 upon the addition of Ni and Ce indicated the incorporation of both larger Ni (Ni2+ = 0.69 pm) and Ce (Ce3+ = 1.01 pm) into the TiO2 (Ti4+ = 0.60 pm) lattice. It can be reported that the incorporation of added metal into the host lattice depended on the synthesis method. The sol–gel method usually produced the mixed oxide materials formed by replacing of host ions by added ions,42,44–47 while the impregnation method also produced the incorporation of added metal.48 To confirm the incorporation of added metals into TiO2, X-ray absorption spectroscopy in the region of extended X-ray absorption fine structure was studied and is further discussed in the next section.
Fig. 2 exhibits N2 adsorption–desorption isotherms of the prepared catalysts. The isotherm feature of TiO2 was similar to type IV isotherm with a hysteresis loop which indicated the existence of the mesoporous structure.37,49 When Ni and Ce metals were added into TiO2, the isotherm feature remained unchanged which indicated that the incorporation of Ni and Ce species into TiO2 did not alter the textural properties of TiO2. The calculated BET surface area of the prepared catalysts is presented in Table 1. The BET surface area of TiO2 was 138.02 m2 g−1 and decreased to 67.63 m2 g−1 for 10 wt% Ni–TiO2 which could be due to a partial blockage or collapse of small mesopores during the impregnation of Ni.12 In contrast, for Ce added samples, the specific surface area was increased with increased Ce content, which was due to the lowering of crystalline size of TiO2 crystal.
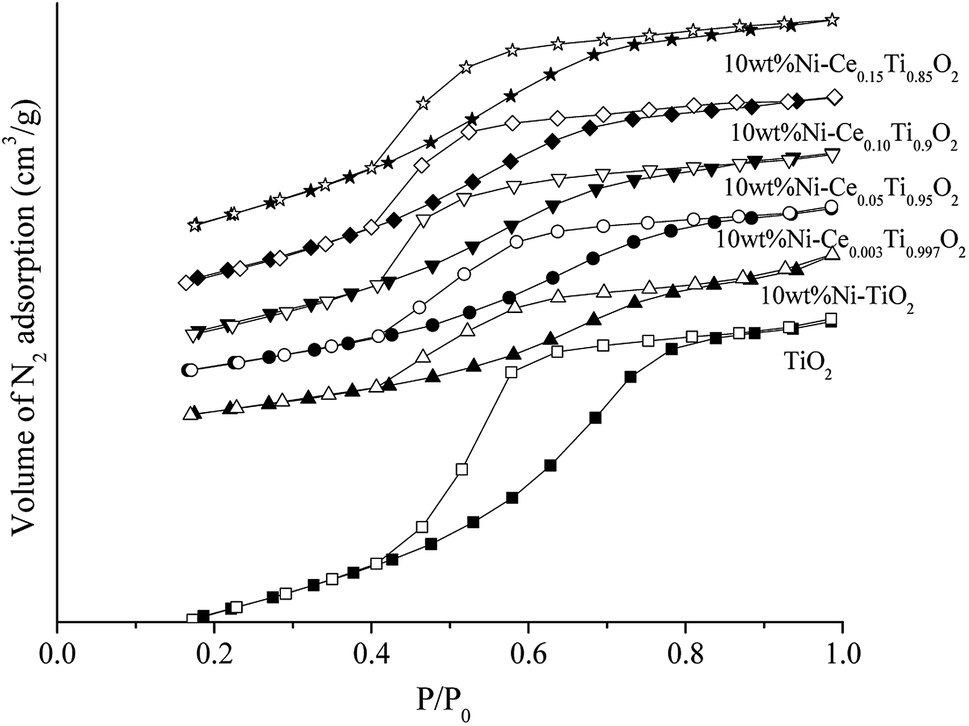 | ||
| Fig. 2 N2 adsorption–desorption isotherms of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts. A solid symbol is adsorption branch and open symbol is desorption branch. | ||
The Ni and Ce contents in 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts can be determined by using inductively coupled plasma-optical emission spectroscopy (ICP-OES), and the results are summarized in Table 1. The Ni amount of all catalysts exhibited a similar value at around 10 wt%. The calculated Ce content was 0.24, 4.11, 9.40 and 15.30 mol% which corresponded to the value of x for 10 wt% Ni–CexTi1−xO2 (x = 0.003, 0.05, 0.10 and 0.15). The morphologies of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003 and 0.10) catalysts were studied by using high-resolution TEM and the images are displayed in Fig. 3. All samples (for the left-hand side images) exhibit a spherical structure, compared to nanoaggregates assembled from many small nanocrystals, which led to the formation of a mesoporous structure. Moreover, upon incorporating Ce, the nanocrystal sizes were decreased, which corresponded to the XRD results. The right-hand side of Fig. 3 shows the selected area electron diffraction (SEAD) patterns of the corresponding samples. For TiO2, several diffraction planes were indexed to anatase TiO2 [(001), (004) and (200)], while for doped samples, the SEAD patterns exhibited several diffraction rings pattern which indicated a polycrystalline structure.46 Fig. 4 displays SEM and mapping images of Ni on catalysts surface for TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003 and 0.10). The loading amount of Ni was kept constant around 10 wt% for all samples, therefore, different Ni dispersion should be resulted from Ce addition. It is seen that 10 wt% Ni–Ce0.003Ti0.997O2 exhibited the highest dispersion of Ni on catalyst surface and that of Ni for 10 wt% Ni–Ce0.10Ti0.90O2 seemed to be lowered.
 | ||
| Fig. 3 TEM images and selected area of electron diffraction patterns of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003 and 0.10) catalysts. | ||
 | ||
| Fig. 4 SEM and mapping images of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003 and 0.10) catalysts. | ||
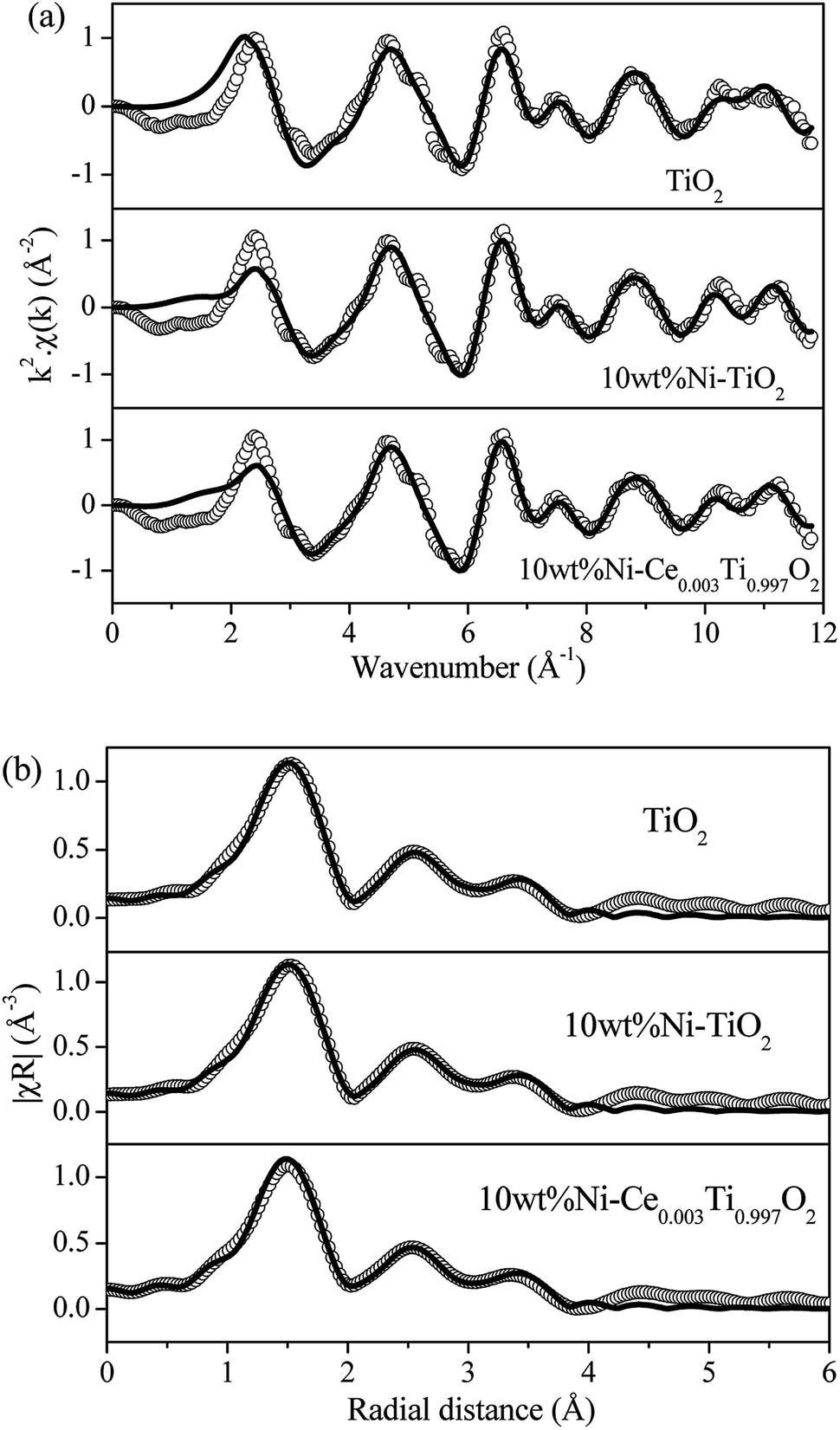 | ||
| Fig. 5 (a) Experimental (open circle) and calculated (black line) FT-EXAFS spectra in k-space for TiO2, 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.003Ti0.997O2. (b) Experimental (open circle) and calculated (black line) FT-EXAFS spectra in R-space for TiO2, 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.003Ti0.997O2. | ||
| Catalyst | Paths | EXAFS best fit parameters | R-Factor | |||
|---|---|---|---|---|---|---|
| N | S02 | σ2 | R (Å) | |||
| TiO2 | Ti–O | 4 | 0.95 | 0.0243 | 1.8249 | 0.0180 |
| 2 | 0.0002 | 1.9251 | ||||
| Ti–Ti | 4 | 0.0105 | 3.0337 | |||
| 4 | 0.0099 | 3.8523 | ||||
| 10 wt% Ni– TiO2 | Ti–O | 4 | 0.72 | 0.0040 | 1.9333 | 0.0064 |
| 2 | 0.0087 | 2.4159 | ||||
| Ti–Ni | 1 | 0.0002 | 2.9747 | |||
| Ti–Ti | 3 | 0.0227 | 3.0211 | |||
| 4 | 0.0084 | 3.8878 | ||||
| 10 wt% Ni–Ce0.003Ti0.997O2 | Ti–O | 4 | 0.65 | 0.0030 | 1.9295 | 0.0061 |
| 2 | 0.0048 | 2.4325 | ||||
| Ti–Ni | 1 | 0.0025 | 2.9674 | |||
| Ti–Ce | 1 | 0.0158 | 2.9892 | |||
| Ti–Ti | 2 | 0.0075 | 3.0550 | |||
| 4 | 0.00802 | 3.8954 | ||||
3.2 CO2 methanation catalytic activity study
The catalytic activity of all prepared catalysts was tested for the CO2 methanation reaction. Before starting the reaction, the catalyst was pretreated at 450 °C under H2 atmosphere with a flow rate of 50 mL min−1 for 1.5 h in order to reduce Ni2+ to Ni0.After that, the CO2 methanation reaction was started by switching the feed gas into the reactor. The catalytic activity of the prepared catalysts was studied in the temperature range of 100–500 °C. The percentages of CO2 conversion for all prepared catalysts are plotted as a function of reaction temperature, as shown in Fig. 6(a). It can be seen that the CO2 conversion were increased with increased reaction temperature. For TiO2 catalyst, the CO2 conversion was very low though the reaction temperature reached 500 °C. Upon doping of Ni onto TiO2, the CO2 conversion was dramatically enhanced. It is noteworthy that the incorporation of Ce into the TiO2 catalyst promoted the CO2 conversion ability. The results show that 10 wt% Ni–Ce0.003Ti0.997O2 exhibits the highest CO2 conversion with the maximum value of 65.29% at 450 °C, while increasing the Ce contents led to a decrease in catalytic activities. For the products (CH4 and CO) yield as illustrated in Fig. SI 1(a) and (b),† it was found that CH4 was the main product from CO2 methanation which produced by Ni-impregnated samples (10 wt% Ni–TiO2 and 10 wt% Ni–CexTi1−xO2, x = 0.003, 0.05, 0.10 and 0.15). On the other hand, CO which was produced by reversed water gas shift reaction (side reaction), was the main product for TiO2 catalyst. Moreover, the CO yield for 10 wt% Ni–TiO2 was higher than that of 10 wt% Ni–CexTi1−xO2 which could indicate that Ce incorporated probably suppressed the formation of undesired product (CO) of this reaction. From Fig. SI 1(b),† it is seen that the CO by-product was favorably enhanced when reaction temperature increase; therefore, the suitable reaction temperature for CO2 methanation for our reaction condition was 350 °C which exhibited low CO yield with high CO2 conversion and CH4 yield. Fig. 6(b) and (c) display the CH4 and CO selectivities for all synthesized samples. The results show that Ni-doped on TiO2 and CexTi1−xO2 catalysts were selectively toward CH4 while bare TiO2 catalyst was selectively toward CO. The calculated CO2 conversion, product yields and product selectivities of all catalysts for CO2 methanation at 350 °C are summarized in Table 3. In order to evidence that the enhancing of CO2 methanation rate resulted from Ni and Ce incorporated into TiO2, NiO and CeO2 catalysts were also prepared and tested for their catalytic activities. Fig. SI 2(a)–(c)† illustrate the CO2 conversion, CH4, and CO yields of NiO and CeO2 compared with TiO2 and 10 wt% Ni–Ce0.003Ti0.997O2. It can be seen that NiO and CeO2 exhibited relatively low CO2 conversion with low CH4 and CO yields when compared with the doped sample. Therefore, doped metals into TiO2 catalyst would play an important role in catalyzing the CO2 methanation.
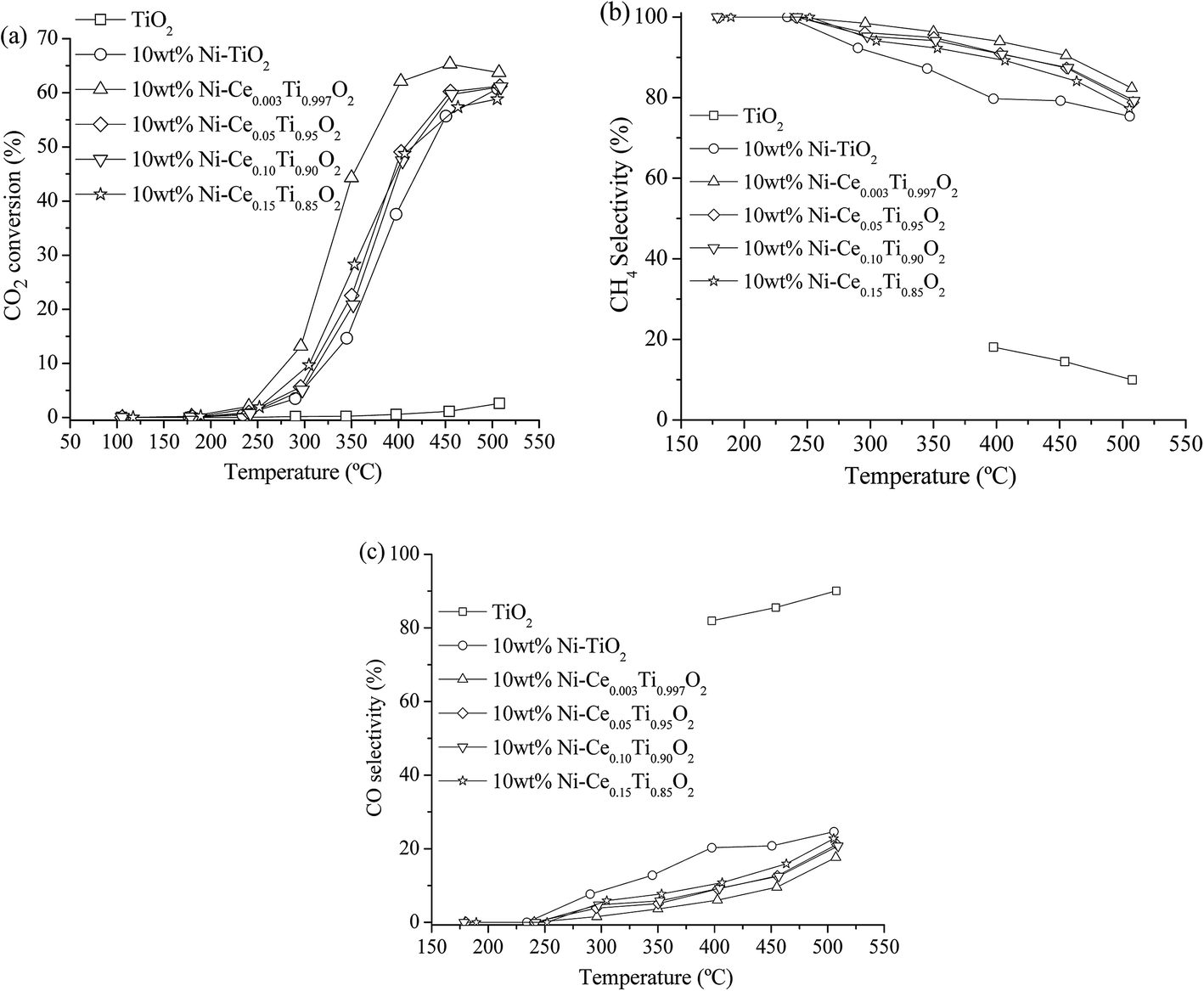 | ||
| Fig. 6 CO2 conversion (a), CH4 selectivity (b) and CO selectivity (c) of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts for CO2 methanation (CO2:H2 ratio = 1:4). | ||
| Catalyst | CO2 conversiona (%) | Yielda (%) | Selectivitya (%) | Eab (kJ mol−1) | ||
|---|---|---|---|---|---|---|
| CO | CH4 | CO | CH4 | |||
| a Reaction temperature at 350 °C.b Calculated by using the temperature range of 230–280 °C (CO2 conversion less than 15%). | ||||||
| TiO2 | 0.22 | 0.22 | 0.00 | 100 | — | — |
| 10 wt% Ni–TiO2 | 14.66 | 1.87 | 12.78 | 12.78 | 87.22 | 79.35 |
| 10 wt% Ni–Ce0.003Ti0.997O2 | 44.28 | 1.62 | 42.66 | 3.66 | 96.34 | 74.86 |
| 10 wt% Ni–Ce0.05Ti0.95O2 | 22.55 | 1.14 | 21.41 | 5.08 | 94.92 | 78.79 |
| 10 wt% Ni–Ce0.10Ti0.90O2 | 20.83 | 1.22 | 19.61 | 5.84 | 94.16 | 78.57 |
| 10 wt% Ni–Ce0.15Ti0.85O2 | 28.28 | 2.18 | 26.09 | 7.72 | 92.28 | 78.98 |
In order to illustrate the role of the catalysts in enhancing the reaction rate at low temperature, the kinetic study in a term of apparent activation energy (Ea) was carried out. The absence of heat and mass transfer were evidenced as mentioned in experimental part. To determine the apparent activation energy, an Arrhenius plot was constructed. This plot is a relationship between ln(rate) and the reciprocal of absolute temperature corresponding to the Arrhenius equation as presented in eqn (6)
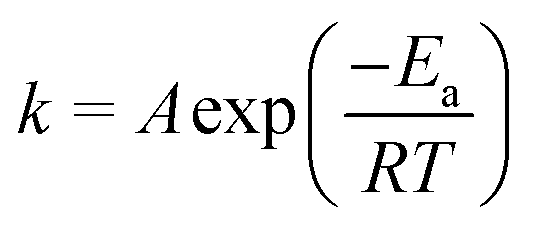 | (6) |
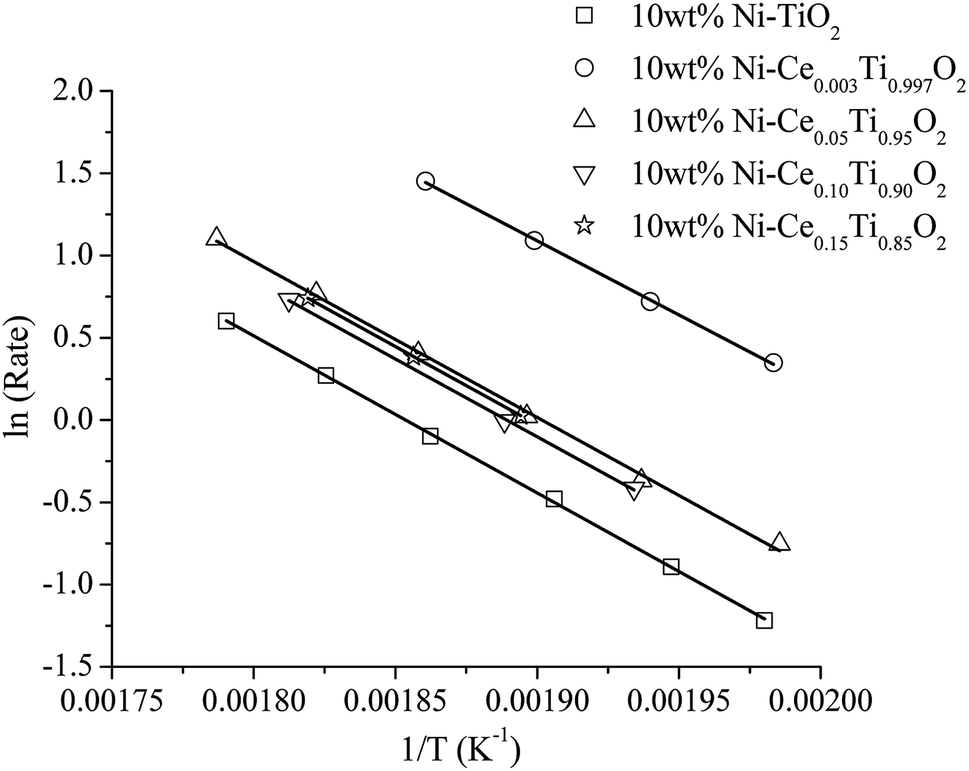 | ||
| Fig. 7 The Arrhenius plots of 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts. | ||
It can be seen that the activation energy of TiO2 could not be determined at low temperature because CO2 conversion was very low. The activation energies of the prepared catalysts appeared in the range of 75–80 kJ mol−1. The apparent activation energies were decreased with the incorporation of Ce into TiO2 and 10 wt% Ni–Ce0.003Ti0.997O2 exhibits the lowest apparent activation energy which is in agreement with the highest catalytic activity for CO2 methanation.
The long-term stability of the catalyst is also an important property for CO2 methanation because metallic Ni can be sintered under exothermic reaction conditions and carbon deposition can also formed on the catalysts surface which can lead to fast deactivation. In this work, a long-term stability test of the best catalyst (10 wt% Ni–Ce0.003Ti0.997O2) was investigated at 350 °C for 50 h. Fig. 8 displays the percentages of CO2 conversion, CH4 yield, and selectivity for the reaction time of 50 h. It can be seen that the catalyst exhibited greatly stable CO2 conversion, CH4 yield, and selectivity within the whole range of stability test (50 h) without an obvious decrease of those values.
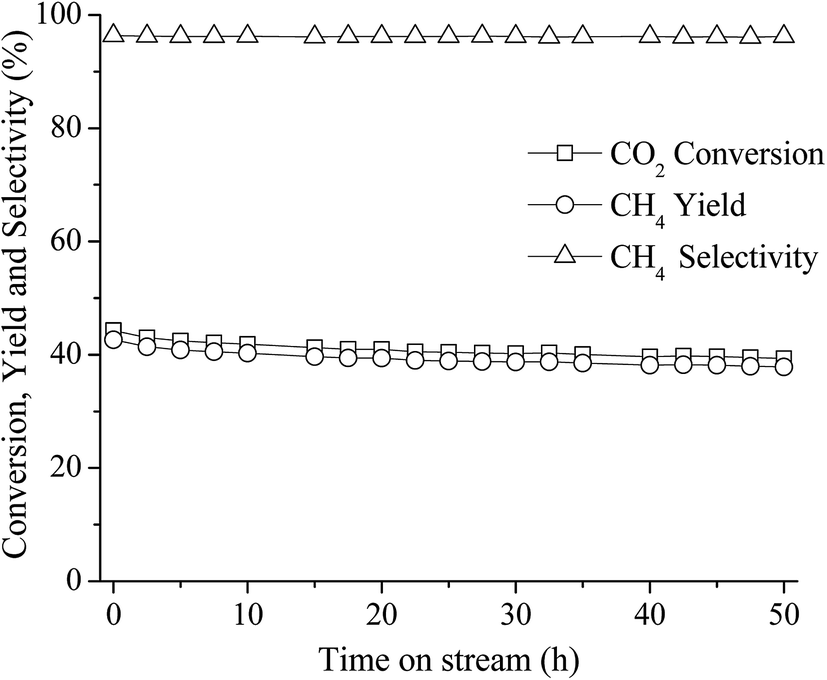 | ||
| Fig. 8 50 h long-term stability test over 10 wt% Ni–Ce0.003Ti0.997O2 at 350 °C (CO2:H2 ratio = 1:4). | ||
To explain the excellent stability under feed stream of this catalyst at this reaction temperature (350 °C), TEM images of used catalysts at interval reaction temperatures (250 °C, 350 °C and 450 °C) were compared. Fig. 9(a) displays TEM images of 10 wt% Ni–Ce0.003Ti0.997O2 catalyst after reaction at 250 °C, 350 °C and 450 °C. The results show that the mean particle sizes of this catalyst after all reaction temperatures were in the rage of 10–12 nm. Agglomeration of particles upon increased of reaction temperature was not observed which indicated that the thermal sintering of the metallic Ni was inhibited.50 Moreover, carbon deposition on catalyst surface was not also observed (form EDX results). Therefore, from this behaviour, this catalyst exhibited an excellent stability under feed stream with high CH4 yield and selectivity.
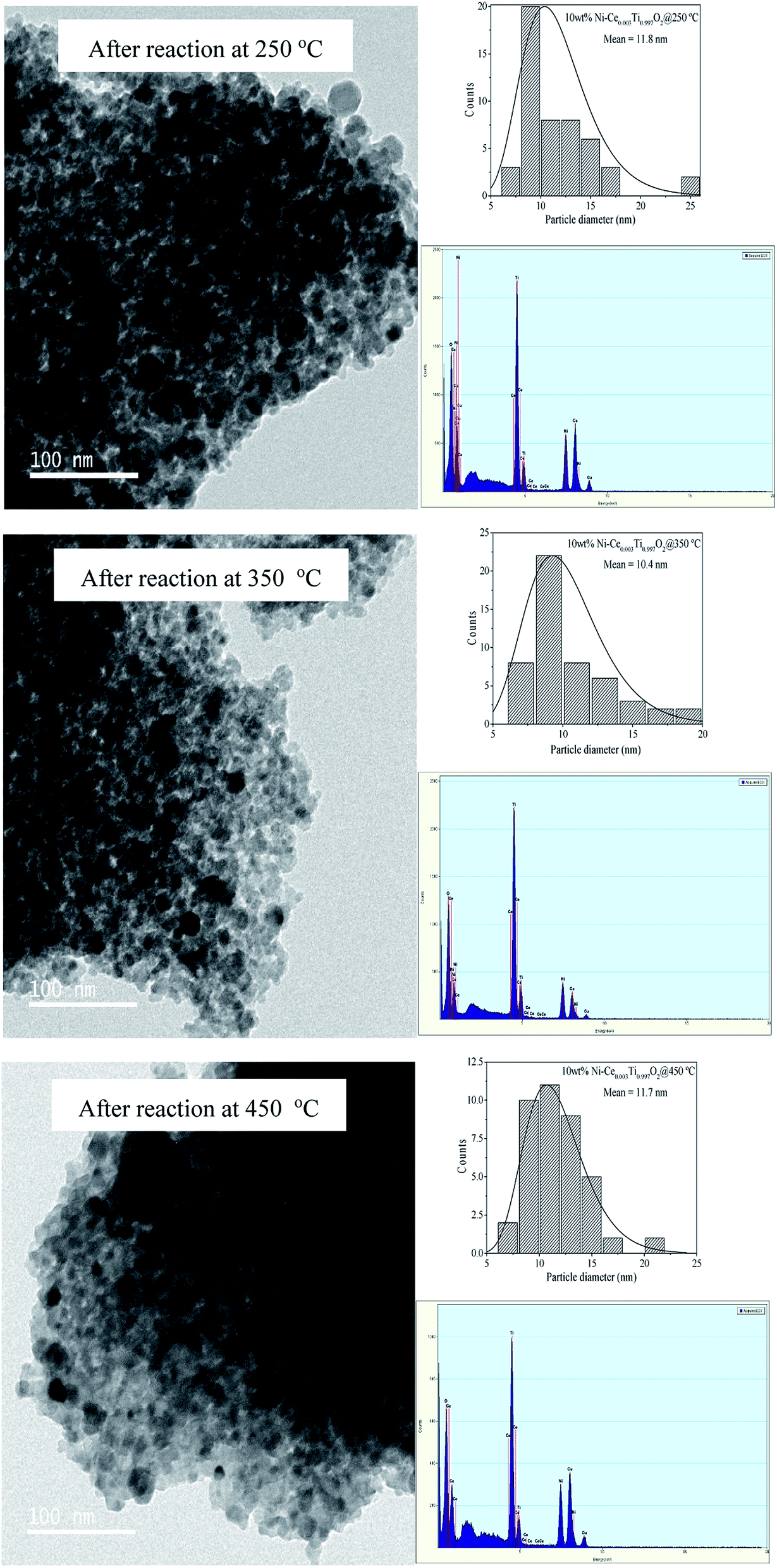 | ||
| Fig. 9 TEM images of 10 wt% Ni–Ce0.003Ti0.997O2 after reaction at 250, 350 and 450 °C. | ||
3.3 X-ray absorption spectroscopy (XAS)
X-ray absorption spectroscopy (XAS) technique is used to investigate the electronic states of probe elements in the catalyst. X-ray absorption is the process in which X-ray radiation excites an electron in a core level of an absorbing atom to empty level or is ejected from the atom as a photoelectron. The absorption coefficient of probe atom as a function of excited photon energy is recorded and the plot between these two parameters is called XAS spectrum. The XAS spectrum can be divided into two regions; X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). Information on the oxidation state is obtained from the XANES region while the information about types of neighboring atom, coordination number and bond length are obtained from the EXAFS region. In this work, we used XAS technique for (i) evidence of the oxidation state and local structure of the catalyst and (ii) monitoring the catalyst changing during CO2 methanation reaction.In this part, the freshly prepared, pretreated and used catalysts were employed to test for XANES experiment in order to investigate the oxidation states of Ti, Ni and Ce probe atoms in different states of catalysts. Data analysis from XANES spectra can provide a good deal of useful information such as electronic states, electronic structure and electron density. It is known that the XANES spectra feature in the pre-edge region are sensitive to the electronic structure and crystal structure changing in the materials.51,52 These changes were monitored to study the influence of metal substitution into TiO2 by Ni and Ce. Fig. 10(a) illustrates the Ti K edge in the pre-edge region for freshly prepared TiO2, 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.05Ti0.95O2 catalysts. There are three main peaks in pre-edge region; A1, A2 and A3. Feature A1 is attributed from transition of 1s → 3d-t2g state. Feature A2 and A3 resulted from excitation of 1s electron to hybridized p–d (t2g) and p–d (eg) on the Ti central atom, respectively.53,54 These three pre-edge peaks intensities were related to the symmetry of the crystal structure. A distortion and oxygen vacancies can cause an increase in the pre-edge intensity.55,56 From Fig. 10(a), the pre-edge intensity was increased upon the addition of 10 wt% Ni onto TiO2 which was due to an incorporation of Ni into TiO2 lattice. In addition, these pre-edge peaks intensities were further increased when Ce addition for 10 wt% Ni–Ce0.05Ti0.95O2 catalyst. This indicated that Ce was further incorporated into TiO2 lattice and led to more defects and oxygen vacancies in the doped catalyst. Fig. 10(b) and (c) show a comparison between Ni0 and Ni2+ standards with three different catalysts for 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.05Ti0.95O2, respectively. For NiO standard, the white line peak at 8350 eV was observed which is attributed to the transition from 1s to the unoccupied 4p state and the other peak at 8366 eV was a signature of Ni in 6-fold coordinated by O atoms.57–59 The Ni foil spectrum exhibits a shoulder at around 8334 eV which is assigned to the electron transition from 1s to 3d orbital.60 Comparison between the XANES spectra of standards and samples can initially indicate the oxidation states of the probe atom. However, from our result, the XANES spectra of all samples were not completely identical to both standards. Therefore, the oxidation state of samples cannot be estimated by only comparison with standard spectra. For this aspect, the apparent oxidation state of Ni should be determined. To confirm the apparent oxidation states of the probe atom (Ni), the relationship between the Ni K edge energy of different samples relative to the edge energy of Ni metal (ΔE) and oxidation state of Ni was constructed as shown in Fig. SI 3(a) and (b).† For both 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.05Ti0.95O2, the Ni oxidation state of freshly prepared catalyst was Ni2+ while pretreated and used samples were close to Ni0. It is seen that the reduction of Ni2+ was not completed to Ni0 which was probably due to the oxidation of some Ni species on the catalyst when exposed in air, since these catalysts (pretreated and used catalysts) were obtained after those steps in the catalytic activity process.
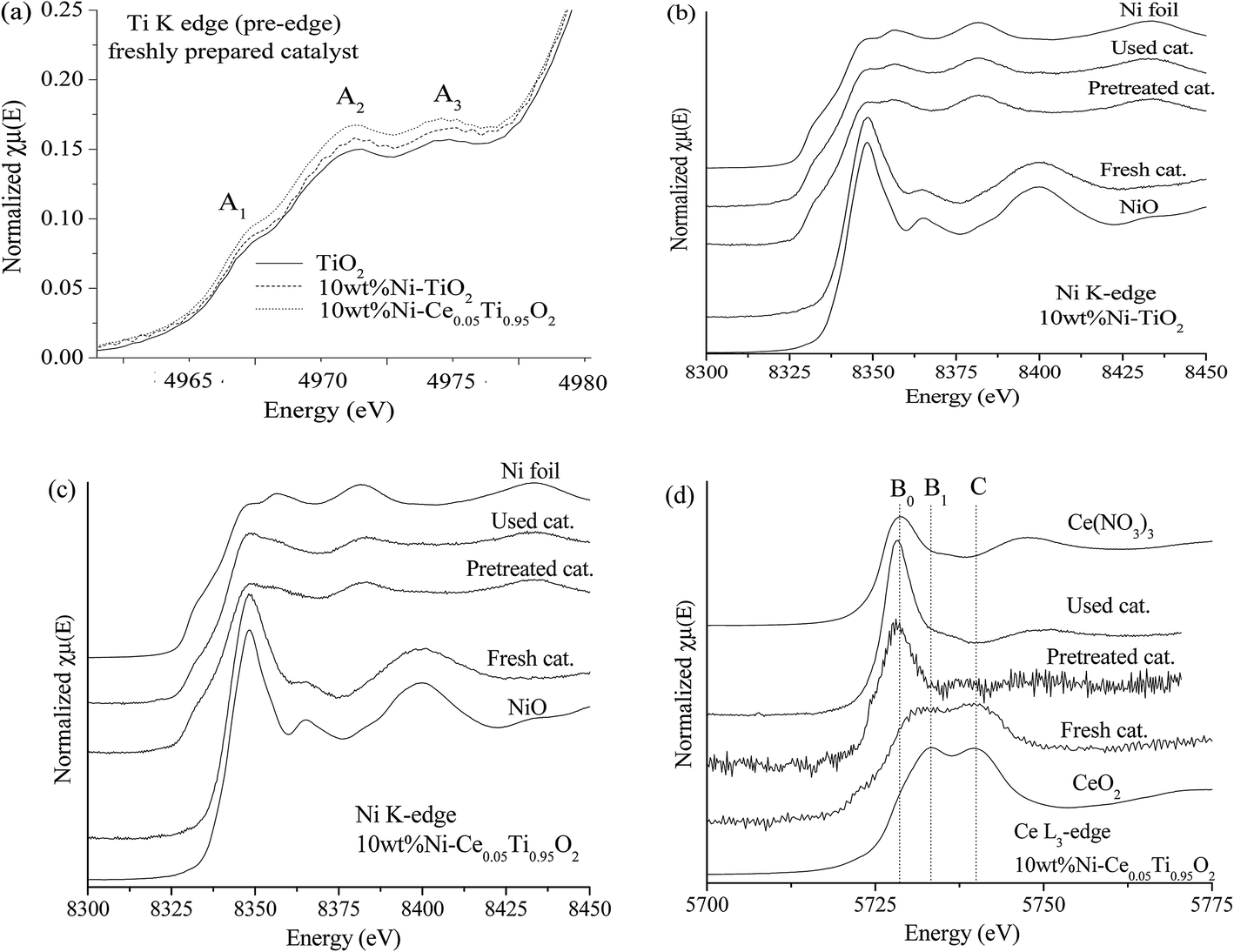 | ||
| Fig. 10 (a) The Ti K edge XANES spectra in the pre-edge region of fresh TiO2, 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.05Ti0.95O2 catalysts. The Ni K edge XANES spectra of fresh, pretreated and used 10 wt% Ni–TiO2 (b) and 10 wt% Ni–Ce0.05Ti0.95O2 (c) catalysts. (d) The Ce L3 edge XANES spectra of fresh, pretreated and used 10 wt% Ni–Ce0.05Ti0.95O2 catalyst. | ||
Fig. 10(d) illustrates Ce L3 edge XANES spectra of three different types of 10 wt% Ni–Ce0.05Ti0.95O2 comparing with Ce3+ and Ce4+ standards. For Ce(NO3)3·6H2O (Ce3+) standard, a single white line, labeled as B0, is assigned as the transition of an electron from Ce core 2p3/2 state to 4f15d state. For CeO2 (Ce4+) standard, two peaks were observed. These two peaks (labeled as B1 and C) are attributed to electron transition from Ce 2p3/2 to a final state with the configuration of 4f15d and 4f05d.61,62 For Ce3+, the transition of an electron from Ce 2p3/2 to an empty 5d state in which the 4f valence state of Ce is occupied by a single electron occurred, while for Ce4+, the 4f state is formally unoccupied, but it can borrow an electron from its oxygen neighbors; thus Ce4+ exhibits a mixed-valence state during the exchange of electrons. For freshly prepared catalysts, the XANES spectrum feature was similar to that of CeO2. Upon being pretreated by H2 and after the reaction, the XANES spectra were quite similar to Ce3+ standard. Fig. SI 3(c)† shows that the apparent oxidation state of freshly prepared 10 wt% Ni–Ce0.05Ti0.95O2 was lower than 4+ which indicated a mixing between some Ce3+and Ce4+ in the sample. While for pretreated and used catalysts, the oxidation state of Ce was lower than that of freshly prepared catalyst and quite close to Ce3+ which was due to the reduction of Ce4+ to Ce3+ under H2 atmosphere.
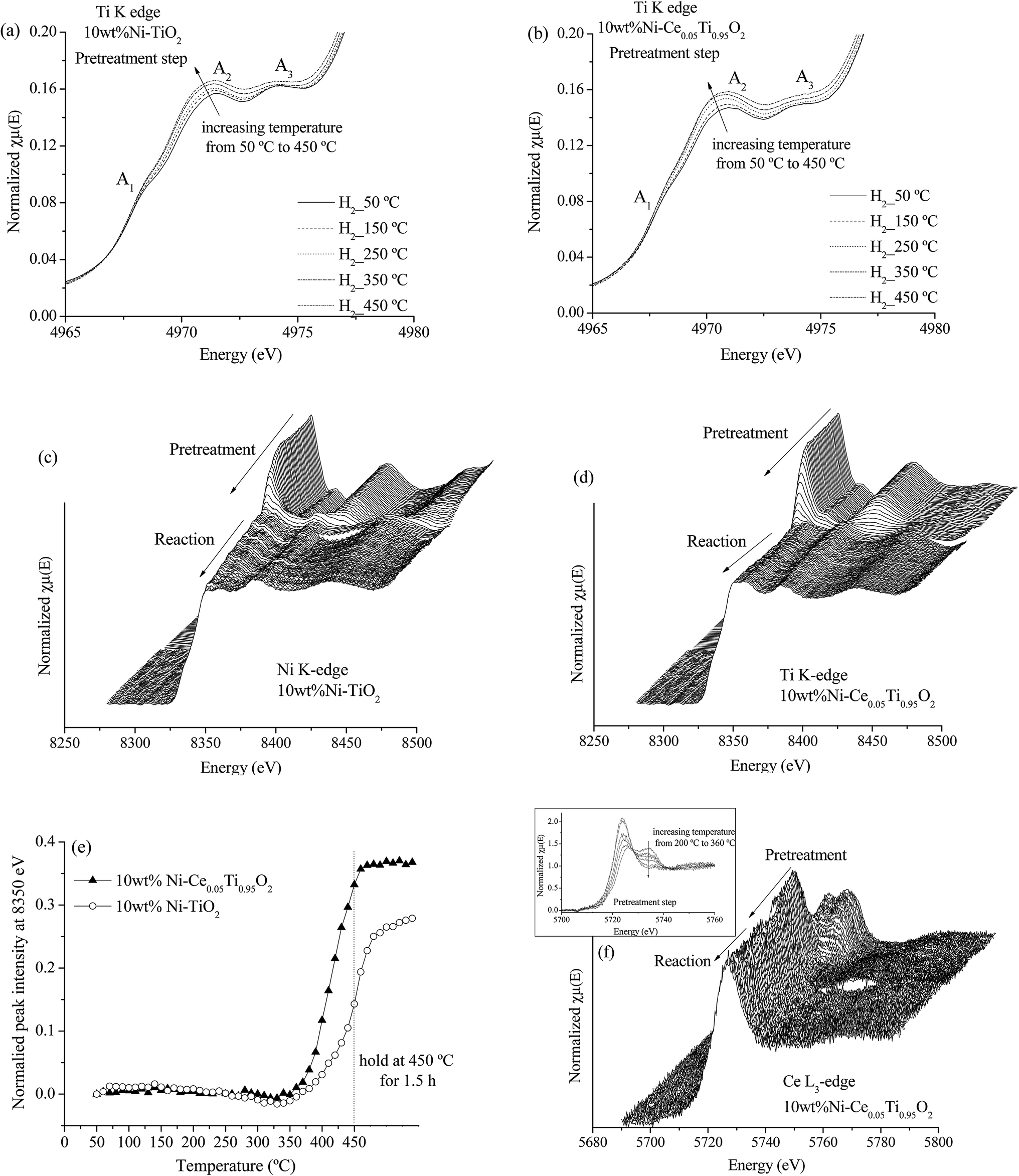 | ||
| Fig. 11 Ti K edge XANES in the pre-edge region during pretreatment step for 10 wt% Ni–TiO2 (a) and 10 wt% Ni–Ce0.05Ti0.95O2 (b). Ni K edge XANES spectra during pretreatment and CO2 methanation reaction for 10 wt% Ni–TiO2 (c) and 10 wt% Ni–Ce0.05Ti0.95O2 (d). (e) The normalized peak intensity at 8350 eV from Ni K edge XANES spectra as a function of pretreated temperature for 10 wt% Ni–TiO2 and 10 wt% Ni–Ce0.05Ti0.95O2. (f) Ce L3 edge XANES spectra during pretreatment and CO2 methanation reaction for 10 wt% Ni–Ce0.05Ti0.95O2 (inset; Ce L3 edge XANES spectra during pretreatment step). | ||
Fig. 11(c) and (d) illustrate the Ni K edge XANES spectra of Ni-impregnated on TiO2 and Ce0.05Ti0.95O2 catalysts. These two catalysts exhibit the same changing of XANES spectra during two steps. For the pretreatment step, the oxidation state of Ni was changed from Ni2+ (initial state for pretreated catalyst) to Ni0 with increased temperature from 50–450 °C. Note that, in the in situ experiment, Ni2+ was completely reduced to Ni0 as shown in Fig. SI 4,† since the sample was performed under H2 atmosphere. However, the starting reduction temperature of these two samples were different. Fig. 11(e) shows the different starting reduction temperatures of both samples. The plot was expressed as normalized peak intensity at 8350 eV (white line position of Ni2+) as a function of temperature. The normalized peak intensity was calculated from the difference between peak intensity at 8350 eV of the initial state and that of each temperature divided by peak intensity at the initial state. Therefore, an increase of this value indicated a lowering of the white line intensity of Ni2+, i.e., Ni2+ was reduced to Ni0. From Fig. 11(e), the starting reduction temperature of 10 wt% Ni–TiO2 was higher than that of 10 wt% Ni–Ce0.05Ti0.95O2. This result can indicate an improvement of reducibility of Ce-doped catalyst which could alter its catalytic activity. In addition, under CO2 methanation conditions, the oxidation states of Ni for both catalysts remained unchanged from the initial state (Ni0 after pretreatment step). Fig. 11(f) shows Ce L3 edge XANES spectra of 10 wt% Ni–Ce0.05Ti0.95O2 during pretreatment and CO2 methanation reaction. It can be seen that the oxidation state changing of Ce4+ to Ce3+ during the pretreatment process occurred. The Ce4+ was started to reduce at the reduction temperature around 220 °C and completely reduced to Ce3+ at around 360 °C (as shown in the inset). After that, the oxidation state of Ce was not changed during the reaction. From the above results, it can be summarized that (1) the changing of environmental structure around Ti during the pretreatment process was found which in accordance with the changing of Ce oxidation state from Ce4+ to Ce3+. Due to the incorporation of Ce into TiO2 lattice, the oxygen vacancies or defect sites existent upon Ce modification was observed, (2) for Ni oxidation state change, the reduction of Ni2+ to Ni0 at lower temperature upon the addition of Ce into the catalyst could be used to describe the enhancing of CO2 methanation activity for the Ce-doped sample.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) from the databases with experimental EXAFS data by using the Artemis program. The structural parameters from EXAFS fitting are summarized in Table 4 which presented the coordination number (N), radial distance (R) between absorbing Ni atoms with neighboring Ni atoms, Debye–Waller factor (σ2) and R-factor. The fitting results exhibited that a first strong peak at around 2.0 Å (2.48 Å from fitting) indicated a single scattering path of absorbing Ni atom with the nearest neighboring Ni atoms (Ni–Ni). For Ni foil standard, the calculated coordination number was 12 which indicated that the absorbing Ni atom was surrounded by 12 neighboring Ni atoms. However, the calculated coordination number of 10 wt% Ni–Ce0.003Ti0.997O2 catalyst at 100, 350 and 550 °C decreased to 9.3, 8.4 and 5.3, respectively. The lowering of Ni–Ni coordination numbers at higher temperatures, i.e., Ni–Ni coordination number at 100 °C = 9.3 and at 550 °C = 5.3; indicated a higher unsaturated Ni–Ni coordination number. High unsaturated numbers of Ni species can lead to existing more active site/defect and can accelerate the dissociative adsorption for H2 to involve in the reaction.14,63 Moreover, the Debye–Waller factor of Ni–Ni pairs increased when the reaction temperature increased. This indicated the deviation of the Ni atom position from the cubic close-packed (ccp) Ni structure due to the more vibration of atoms at high temperature. This result is in agreement with Liu et al.64
m) from the databases with experimental EXAFS data by using the Artemis program. The structural parameters from EXAFS fitting are summarized in Table 4 which presented the coordination number (N), radial distance (R) between absorbing Ni atoms with neighboring Ni atoms, Debye–Waller factor (σ2) and R-factor. The fitting results exhibited that a first strong peak at around 2.0 Å (2.48 Å from fitting) indicated a single scattering path of absorbing Ni atom with the nearest neighboring Ni atoms (Ni–Ni). For Ni foil standard, the calculated coordination number was 12 which indicated that the absorbing Ni atom was surrounded by 12 neighboring Ni atoms. However, the calculated coordination number of 10 wt% Ni–Ce0.003Ti0.997O2 catalyst at 100, 350 and 550 °C decreased to 9.3, 8.4 and 5.3, respectively. The lowering of Ni–Ni coordination numbers at higher temperatures, i.e., Ni–Ni coordination number at 100 °C = 9.3 and at 550 °C = 5.3; indicated a higher unsaturated Ni–Ni coordination number. High unsaturated numbers of Ni species can lead to existing more active site/defect and can accelerate the dissociative adsorption for H2 to involve in the reaction.14,63 Moreover, the Debye–Waller factor of Ni–Ni pairs increased when the reaction temperature increased. This indicated the deviation of the Ni atom position from the cubic close-packed (ccp) Ni structure due to the more vibration of atoms at high temperature. This result is in agreement with Liu et al.64
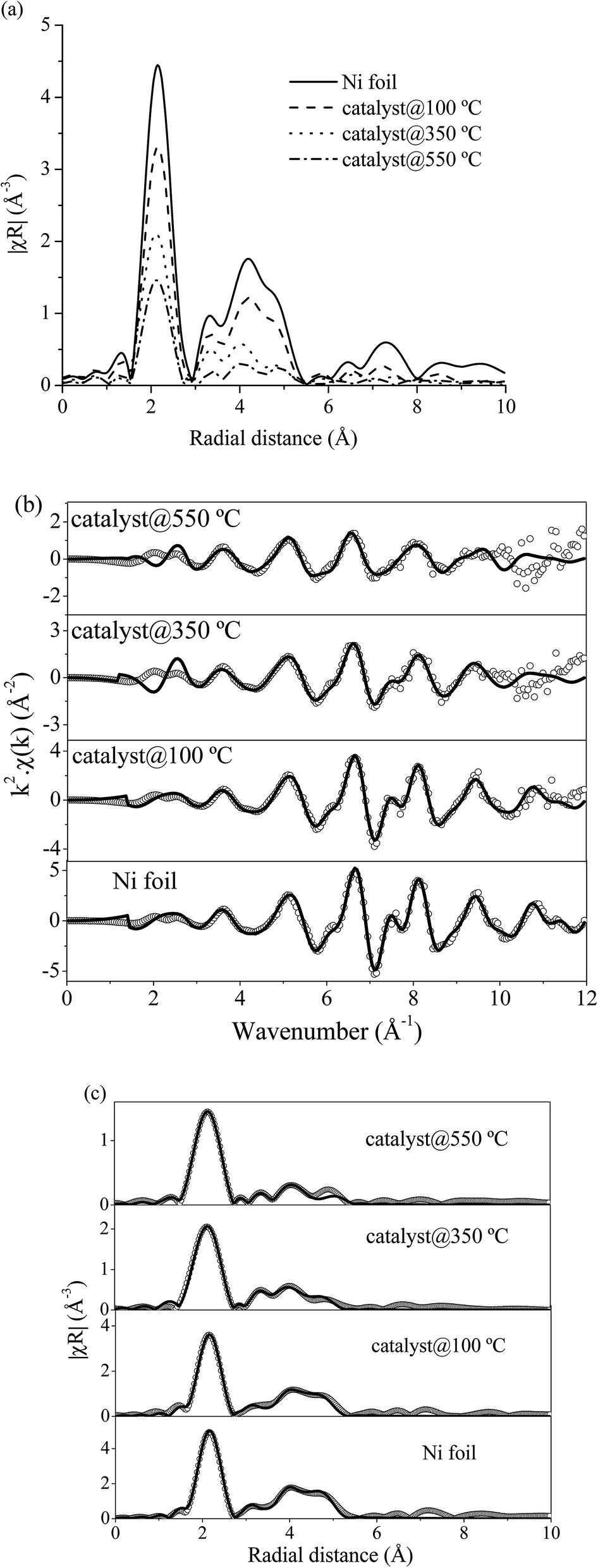 | ||
| Fig. 12 (a) Normalized Ni K-edge EXAFS spectra in R space of Ni foil and 10 wt% Ni–Ce0.003Ti0.997O2 catalyst at reaction temperature of 100, 350 and 550 °C. (b) Experimental (open circle) and calculated (black line) FT-EXAFS spectra in k-space of Ni foil and 10 wt% Ni–Ce0.003Ti0.997O2 catalyst at reaction temperature of 100, 350 and 550 °C. (c) Experimental (open circle) and calculated (black line) FT-EXAFS spectra in R-space of Ni foil and 10 wt% Ni–Ce0.003Ti0.997O2 catalysts at reaction temperature of 100, 350 and 550 °C. | ||
| Catalyst | Path | EXAFS best fit parameters | R-Factor | ||
|---|---|---|---|---|---|
| N | σ2 | R (Å) | |||
| Ni foil | Ni–Ni | 12.0 | 0.0071 | 2.48 | 0.0055 |
| 10 wt% Ni–Ce0.003Ti0.997O2 at 100 °C | Ni–Ni | 9.3 | 0.0073 | 2.48 | 0.0080 |
| 10 wt% Ni–Ce0.003Ti0.997O2 at 350 °C | Ni–Ni | 8.4 | 0.0105 | 2.48 | 0.0074 |
| 10 wt% Ni–Ce0.003Ti0.997O2 at 550 °C | Ni–Ni | 5.3 | 0.0106 | 2.48 | 0.0028 |
However, the H2 adsorption ability on an active site was one of the main factor to describe the catalytic performance. Therefore, to explain the catalytic activities enhancement, understanding on the adsorption behavior of two reactants should be investigated.
3.4 The role of Ce addition on TiO2-supported Ni catalyst on enhancing CO2 methanation
It is known that many properties such as surface properties (surface area, pore size and volume), electrical, structural and redox properties can dominate the catalytic activities of the catalysts. In this work, Ce was added into TiO2-supported Ni to improve the catalytic activity for CO2 methanation reaction. The results showed that the catalytic activity of TiO2-supported Ni was significantly increased upon addition of small amount of Ce (0.003 mol%). It is noted that Ni species can facilitate the dissociative-adsorption of H2 molecules and, in this work, the amounts of impregnated Ni were similar for all doped catalysts (10 wt% Ni); thus, the difference of catalytic activities of samples with different amounts of Ce should be resulted from the effects of Ce addition. Therefore, it is implied that the addition of Ce would lead to improvement of some catalyst properties which were already characterized and investigated by several techniques.In this part, the effects of Ce addition on TiO2-supported Ni catalytic activity for CO2 methanation are summarized.
(1) Addition of Ce improved the dispersion of Ni on catalyst surface. The results from SEM and mapping images in characterization section indicated that the dispersion of Ni on catalysts surface was improved upon addition of Ce when compared with that of Ni on bare TiO2 without Ce addition. Moreover, the highest Ni dispersion was obtained by addition of small amount of Ce (0.003 mol%). It is known that H2 molecules favor to adsorb on metallic surface (Ni0) in dissociative paths, i.e., H2 dissociative adsorption occurs in H atoms formed on metallic surface. Then the dissociated H atoms were further involved in the reaction of CO2 methanation. Therefore, larger area of Ni active metal would lead to larger amount of dissociated H atoms to be involved in the reaction pathways. In order to evidence the role of Ni active metal area on enhancing the catalytic activity, the H2 adsorption experiment on all synthesized catalysts was conducted. Fig. 13 illustrates the H2 adsorption capacity at room temperature for 4 h of all prepared catalysts. From Fig. 13, it can be seen that the lowest H2 adsorption capacity can be observed on the TiO2 surface while the H2 adsorption capacity increased when Ni was added. The role of Ni sites on H2 adsorption has been reported by many research groups.2,65,66 It is due to Ni on the catalyst surface acting as the active species for H2 adsorption by dissociation of H2 to form atomic H, in which atomic H facilitates the methane formation. From this results, the highest H2 adsorption capacity was observed for 10 wt% Ni–Ce0.003Ti0.997O2 which exhibited the highest Ni dispersion among all Ce doped catalysts. Therefore, the catalyst with the lowest Ce amount trended to exhibit the highest CO2 methanation activity. Moreover, Ce addition can also promote the stability of catalyst by prevent the sintering of catalyst and also exhibited coking deposition inhibition.
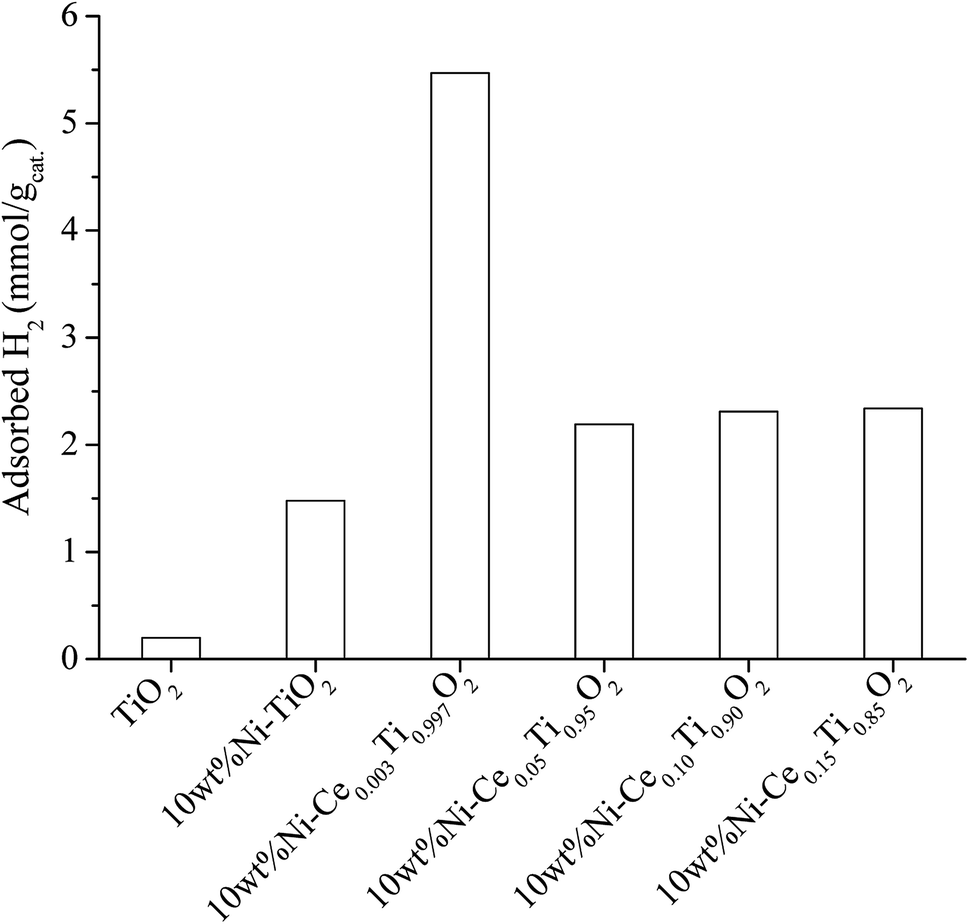 | ||
| Fig. 13 H2 adsorption result of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts. | ||
(2) Ce addition can produce the oxygen vacancies within TiO2 lattice. The results from XRD analysis, addition of Ni led to an enlargement of TiO2 unit cell and further increased of unit cell of TiO2 was observed upon increased of Ce loading. Therefore, addition of these two metals led to producing defects or oxygen vacancies within TiO2 lattice. These structural changes were due to the incorporation of larger added metal ions into TiO2 lattice (perhaps interstitial site or by replacing Ti4+ site). Moreover, another result that confirmed this structural modification was an XANES and EXAFS of Ti K edge which indicated the distortion of environmental neighboring atoms around the Ti probe atom upon the addition of Ni and Ce. However, the structural change of Ti obtained from XAS was too small to observe, which could be due to the lower amount of added metals when compared with the amount of bulk Ti in the catalyst, where the XAS technique was a bulk technique. Therefore, in order to evidence the incorporation of added metals into TiO2 lattice which can produce the Ti3+ (oxygen vacancies) on a catalyst, X-ray photoelectron spectroscopy (XPS), which was a surface sensitive technique was used. Fig. 14(a)–(c) reveal Ti 2p XPS spectra of TiO2, 10 wt% Ni–TiO2, and 10 wt% Ni–Ce0.003Ti0.997O2, respectively. The obtained XPS peak of the catalyst was analyzed by the fitting of XPS peaks with Gaussian–Lorentzian peaks after the subtraction of a Shirley background using the XPSPEAK41 program. The information from fitting include the peak position and peak area. The oxidation state of Ti in the catalysts can be obtained by comparing the peak position from fitting with the peak position of references. It can be seen that Ti 2p XPS spectra of TiO2 in Fig. 14(a) exhibits two peaks at around 458 and 464 eV corresponding to the electron transition of Ti4+ from 2p3/2 and 2p1/2 to form a photoelectron. For XPS spectra of 10 wt% Ni–TiO2 (Fig. 14(b)) and 10 wt% Ni–Ce0.003Ti0.997O2 (Fig. 14(c)) catalysts, four XPS peaks can be observed. The two XPS peaks at around 458 and 464 eV were the characteristic of Ti4+ species while another two peaks at around 456 and 462 eV were the electron transition from 2p3/2 and 2p1/2 for Ti3+ species. Therefore, Ti composition of Ni and Ce co-added catalysts contained the mixing of Ti4+and Ti3+ species on the surface. The presence of Ti3+ in the catalyst structure indicated the formation of oxygen vacancies on the catalyst surface.67 However, the Ti3+/Ti4+ XPS peak area ratio of 10 wt% Ni–Ce0.003Ti0.997O2 was higher than that of 10 wt% Ni–TiO2 which indicated that higher oxygen vacancies sites than that of 10 wt% Ni–TiO2. The oxygen vacancies are the active species for CO2 adsorption which interacts with the oxygen of CO2 molecules by strong interaction and weakened the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.50,65,68 The oxygen vacancies for CO2 adsorption can be generated by some Ti4+ being substituted by metal with a lower oxidation state (Ce3+ and Ni2+) and some Ti4+ being transformed to Ti3+ and then oxygen atoms were lost from the catalyst structure.
O bond.50,65,68 The oxygen vacancies for CO2 adsorption can be generated by some Ti4+ being substituted by metal with a lower oxidation state (Ce3+ and Ni2+) and some Ti4+ being transformed to Ti3+ and then oxygen atoms were lost from the catalyst structure.
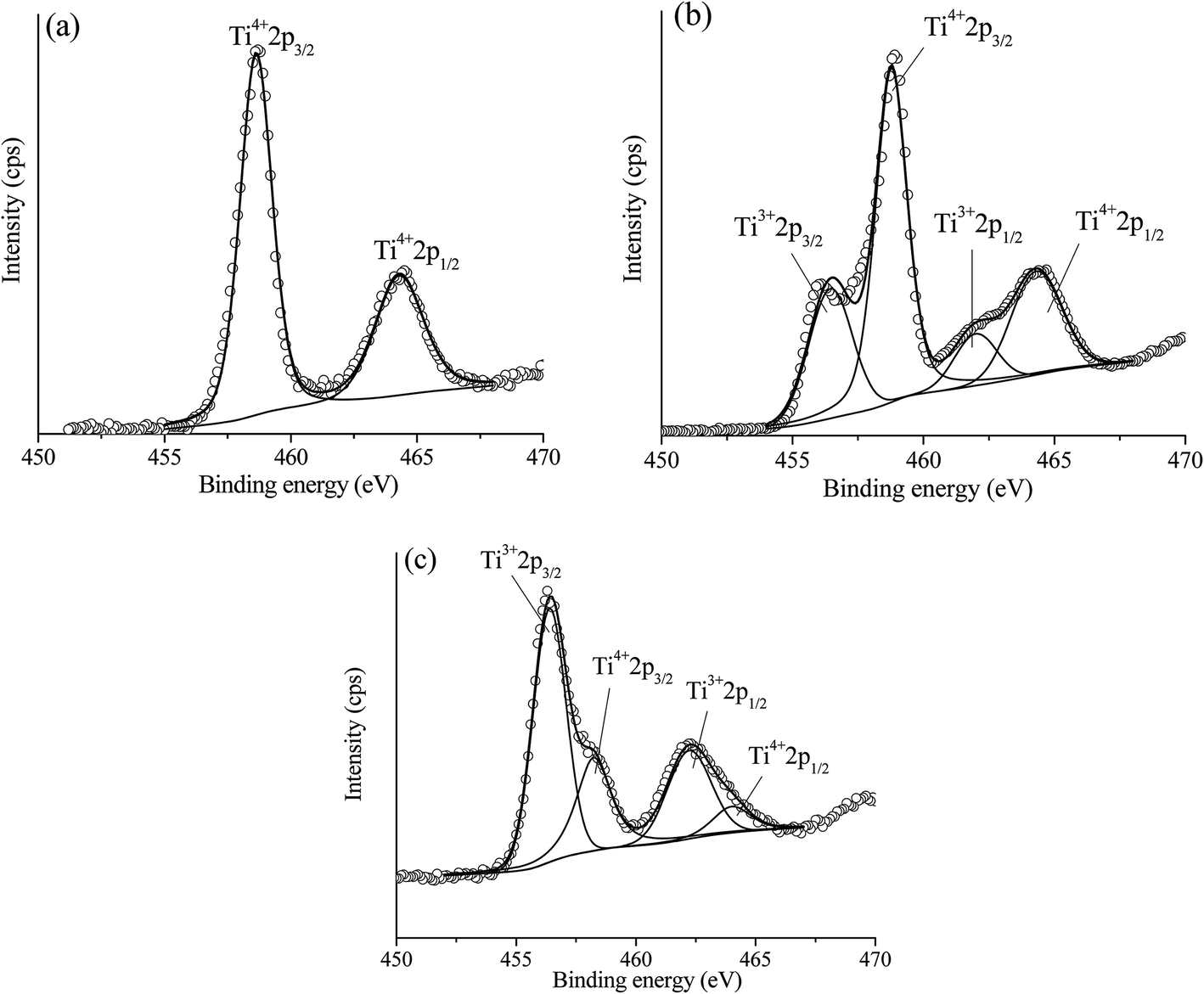 | ||
| Fig. 14 Experimental (open circle) and calculated (black line) Ti 2p XPS spectra of TiO2 (a), 10 wt% Ni–TiO2 (b) and 10 wt% Ni–Ce0.003Ti0.997O2 (c). | ||
Beside the existence of oxygen vacancies that can affect CO2 adsorption, the addition of Ce into TiO2 also increased CO2 uptake by creating a basicity site. Due to weak acidic properties of CO2, increasing the basicity of the catalyst can also improve the adsorption of CO2. Many works have reported on increasing catalyst basicity by adding some metal oxide, such as La2O3 and MgO.69,70 Therefore, in order to evidence the improvement of basicity of catalysts, a CO2 adsorption experiment under ambient pressure and temperature was carried out. To investigate the CO2 adsorption efficiency of the prepared catalysts, diluted CO2 was injected to the catalysts and kept at room temperature (298 K) for 4 h. After that, the amount of unadsorbed CO2 was detected by Micro GC every 30 min. Fig. 15 reveals time-dependent CO2 adsorption of all the prepared catalysts. It can be seen that CO2 adsorption of all prepared catalysts reached the equilibrium point within 1 h. The CO2 adsorption capacity (mmol of CO2 per g of catalyst) of TiO2 showed the lowest capacity, while the CO2 adsorption capacity increased upon adding Ni metal in TiO2 and further increased with an increase of Ce content. The catalysts with the highest Ce amount exhibited the maximum CO2 adsorption capacity, which is probably due to the highest basicity. However, the trend of increasing CO2 uptake with increased Ce content was not related to the catalytic activities results. Therefore, it indicated that CO2 adsorption ability was not the main factor to enhance the reaction rate.
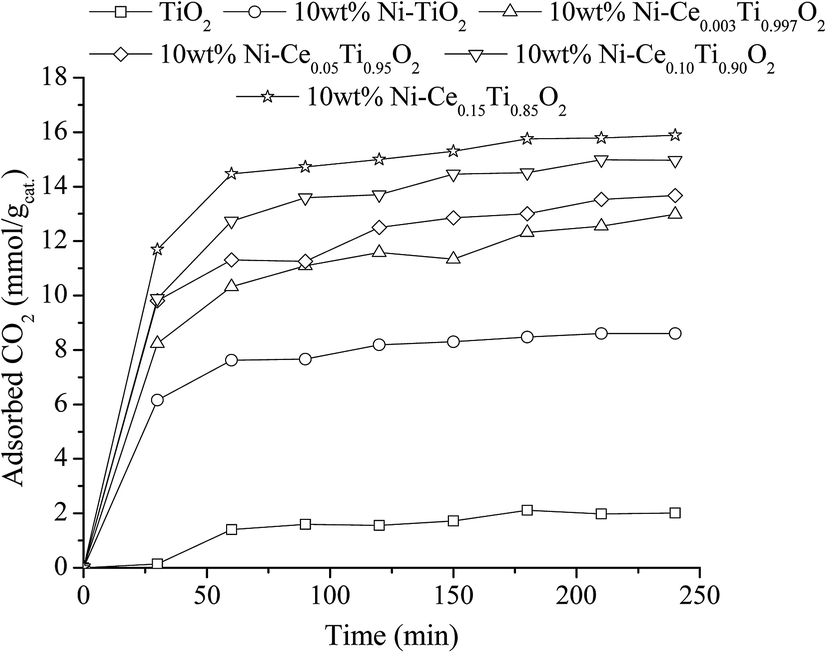 | ||
| Fig. 15 CO2 adsorption result of TiO2 and 10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts. | ||
(3) Ce addition can alter the reducibility of catalysts. Higher reducibility of Ce-doped catalysts can be evidenced by in situ XANES results for Ni K edge, which revealed that the starting reduction temperature of 10 wt% Ni–Ce0.05Ti0.95O2 exhibited a lower reduction temperature of Ni2+ → Ni0. This lowering of reduction temperature can indicate that the reduction of Ni2+ was facilitated by the addition of Ce to improve the reducibility of the catalyst which can lead to more effectively catalyzation of the reaction, as mentioned by other works.26,71,72
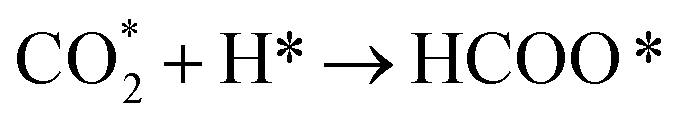
The CO2 methanation reaction involved the interaction between CO2 and H2 molecule on the catalyst surface. Several pathways of the CO2 methanation mechanism on the catalyst surface have been proposed by many research groups. Ren et al.73 proposed one of the mechanisms for CO2 methanation as follows:
| H2 + 2* → 2H* | (7) |
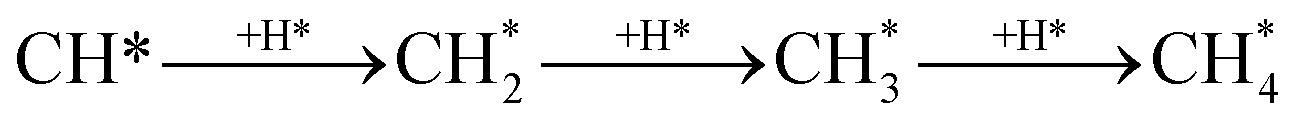 | (8) |
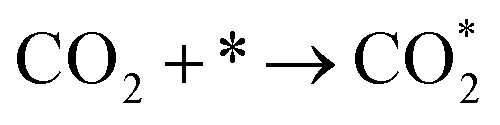 | (9) |
| HCOO* → CO* + OH* | (10) |
| CO* + 2H* → CH* +OH* | (11) |
 | (12) |
| OH* + H* → H2O* → H2O + * | (13) |
4. Conclusion
10 wt% Ni–CexTi1−xO2 (x = 0, 0.003, 0.05, 0.10 and 0.15) catalysts were prepared and characterized by several techniques. The catalytic activity of the prepared catalysts was studied for the CO2 methanation reaction. The 10 wt% Ni–Ce0.003Ti0.997O2 catalyst displayed the best CO2 methanation catalytic activity which showed the highest CO2 conversion and CH4 yield with low CO yield entire the reaction temperature range. The role of Ce addition on TiO2-supported Ni in enhancing CO2 methanation catalytic activities were discussed in term of surface, structural, electrical and redox properties changes. The electronic state of Ni species during pretreatment and CO2 methanation showed that the active state of Ni species was Ni0 which can be activated by being pretreated under H2 flowing at 450 °C and this active state remained unchanged during the reaction. The role of Ce in enhancing the CO2 methanation catalytic activity of TiO2-supported Ni was to modify the catalyst properties as follows;(i) Ce can promote the dispersion of Ni on catalysts surface and also prevent the sintering of Ni species during the reaction which can lead to high catalytic activity and high stability.
(ii) Upon the incorporation of Ni into TiO2 lattice, the expansion of unit cell and oxygen vacancies were observed and further increased of Ce loading led to higher degree of unit cell enlargement and larger amount of oxygen vacancies formation. Moreover, Ce can increase the basicity of catalysts, which can be evidenced by CO2 adsorption results. It can be seen that higher amounts of Ce led to high CO2 adsorption. However, excess amounts of CO2 adsorption can also inhibit the H2 adsorption on the catalyst surface leading to lowering of catalytic activities.
(iii) Ce can increase the reducibility of the catalyst by facilitating reduction of Ni2+ to Ni0 at lower temperature.
Therefore, it can be explained that the CO2 methanation reaction on the prepared catalysts depends on the adsorbed H2 amount on the catalyst surface, or that the adsorbed H2 on the catalyst surface was the limiting reagent and the main factor that improved the CO2 methanation catalytic activity of the prepared catalyst. Furthermore, the catalytic stability of the 10 wt% Ni–Ce0.003Ti0.997O2 catalyst was investigated at 350 °C for 50 h. The result exhibited that the 10 wt% Ni–Ce0.003Ti0.997O2 catalyst was greatly stable CO2 conversion, CH4 yield and CH4 selectivity entire 50 h of stability test.
Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
The authors would like to thank the Department of Chemistry, Faculty of Science, Khon Kaen University, Thailand and Applied Chemistry Section, Department of Materials Science, Faculty of Engineering, Kyushu Institute of Technology, Japan for providing research facilities. Thanks to the Synchrotron Light Research Institute (Public Organization) for XAS analysis and generous beamtime. The financial support from the Science Achievement Scholarship of Thailand (SAST) and the scholarship for research overseas which was supported by the Graduate School, Khon Kaen University, are gratefully acknowledged.References
- I. Dimitriou, P. Garcıa-Gutierrez, R. H. Elder, R. M. Cuéllar-Franca, A. Azapagicb and R. W. K. Allena, Energy Environ. Sci., 2018, 8, 1775–1789 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- J. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- J. R. Rostrup-Nielsen, K. Pedersen and J. Sehested, Appl. Catal., 2007, 330, 134–138 CrossRef CAS.
- T. T. M. Nguyen, L. Wissing and M. S. Skjøth-Rasmussen, Catal. Today, 2013, 215, 233–238 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- I. Fechete and J. C. Vedrine, Molecules, 2015, 20, 5638–5666 CrossRef CAS PubMed.
- P. Riani, G. Garbarinob, M. A. Lucchini, F. Canepa and G. Busca, J. Mol. Catal. A: Chem., 2014, 383–384, 10–16 CrossRef CAS.
- D. Pandey, K. Ray, R. Bhardwaj, S. Bojja, K. V. R. Chary and G. Deo, Int. J. Hydrogen Energy, 2018, 43, 4987–5000 CrossRef CAS.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma and J. Wang, Appl. Catal., B, 2019, 243, 262–272 CrossRef CAS.
- R. Ye, W. Gong, Z. Sun, Q. Sheng, X. Shi, T. Wang, Y. Yao, J. J. Razink, L. Lin, Z. Zhou, H. Adidharma, J. Tang, M. Fan and Y. Yao, Energy, 2019, 188, 116059 CrossRef CAS.
- M. Wolf, L. Hui Wong, C. Schüler and O. Hinrichsen, J. CO2 Util., 2020, 36, 276–287 CrossRef CAS.
- J. Guilera, J. d. Valle, A. Alarcón, J. A. Díaz and T. Andreu, J. CO2 Util., 2019, 30, 11–17 CrossRef CAS.
- S. Tada, S. Ikeda, N. Shimoda, T. Honma, M. Takahashi, A. Nariyuki and S. Satokawa, Int. J. Hydrogen Energy, 2017, 42, 30126–30134 CrossRef CAS.
- B. Lu and K. Kawamoto, Catal. Sci. Technol., 2014, 4, 4313–4321 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Muktid, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- T. A. Le, M. S. Ki, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- M. Yamasaki, M. Komori, E. Akiyama, H. Habazaki, A. Kawashima, K. Asami and K. Hashimoto, Mater. Sci. Eng., 1999, A267, 220–226 CrossRef CAS.
- H. Takano, K. Izumiya, N. Kumagai and K. Hashimoto, Appl. Surf. Sci., 2011, 257, 8171–8176 CrossRef CAS.
- Ni. Perkas, G. Amirian, Z. Zhong, J. Teo, Y. Gofer and A. Gedanken, Catal. Lett., 2009, 130, 455–462 CrossRef CAS.
- G. Zhi, X. Guo, Y. Wang, G. Jin and X. Guo, Catal. Commun., 2011, 16, 56–59 CrossRef CAS.
- H. Liu, X. Zou, X. Wang, X. Lu and W. Ding, J. Nat. Gas Chem., 2012, 21, 703–707 CrossRef CAS.
- C. Gaudillere, L. Navarrete and J. M. Serra, Int. J. Hydrogen Energy, 2017, 42, 895–905 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- L. Bian, L. Zhang, R. Xia and Z. Li, J. Nat. Gas Sci. Eng., 2015, 27, 1189–1194 CrossRef CAS.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Int. J. Hydrogen Energy, 2014, 39, 10090–10100 CrossRef CAS.
- P. Kidkhunthod, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2017, 8, 035007 Search PubMed.
- W. Klysubun, P. Kidkhunthod, P. Tarawarakarn, P. Sombunchoo, C. Kongmark, S. Limpijumnong, S. Rujirawat, R. Yimnirun, G. Tumcharern and K. Faungnawakij, J. Synchrotron Radiat., 2017, 24, 707–716 CrossRef CAS PubMed.
- C. M. Kalamaras, S. Americanou and A. M. Efstathiou, J. Catal., 2011, 279, 287–300 CrossRef CAS.
- N. Li, X. Zou, M. Liu, L. Wei, Q. Shen, R. Bibi, C. Xu, Q. Ma and J. Zhou, J. Phys. Chem. C, 2017, 121, 25795–25804 CrossRef CAS.
- Y. Jiang, Z. Jin, C. Chen, W. Duan, B. Liu and X. Chen, RSC Adv., 2017, 7, 12856–12870 RSC.
- M. Tahir and N. A. S. Amin, Appl. Catal., B, 2015, 162, 98–109 CrossRef CAS.
- S. Rajendran, D. Manojb, K. Raju, D. D. Dionysioud, M. Naushade, F. Graciaf, L. Cornejo, M. A. Gracia-Pinillag and T. Ahamad, Sens. Actuators, B, 2018, 264, 27–37 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- D. R. Shinde, P. S. Tambade, M. G. Chaskar and K. M. Gadave, Drinking Water Eng. Sci., 2017, 10, 109–117 CrossRef CAS.
- H. Yan, D. Zhang, J. Xu, Y. Lu, Y. Liu, K. Qiu, Y. Zhang and Y. Luo, Nanoscale Res. Lett., 2014, 9, 1–7 CrossRef CAS PubMed.
- L. Li, F. Chen, J. Q. Lu and M. F. Luo, J. Phys. Chem. A, 2011, 115, 7972–7977 CrossRef CAS PubMed.
- S. L. Reis, E. C. C. Souza and E. N. S. Muccillo, Solid State Ionics, 2011, 192, 172–175 CrossRef CAS.
- B. Choudhury, B. Borah and A. Choudhury, Photochem. Photobiol., 2012, 88, 257–264 CrossRef CAS PubMed.
- S. Esposito, Materials, 2019, 12, 668 CrossRef CAS PubMed.
- V. R. Akshay, B. Arun, G. Mandal and M. Vasundhar, Phys. Chem. Chem. Phys., 2019, 21, 2519–2532 RSC.
- F. Ocampo, B. Louis and A. Roger, Appl. Catal., A, 2009, 369, 90–96 CrossRef CAS.
- K. C. Chanapattharapol, S. Krachuamram, A. Makdee, P. Unwiset and S. Srikwanjai, J. Rare Earths, 2017, 35, 1197–1205 CrossRef CAS.
- Z. Fan, F. Meng, J. Gong, H. Li, Z. Ding and B. Ding, J. Mater. Sci.: Mater. Electron., 2016, 27, 11866–11872 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, D. Nie, X. Lian, Z. Lu, H. Chen, K. Zhang and P. Ge, Int. J. Hydrogen Energy, 2017, 42, 15523–15539 CrossRef CAS.
- E. R. Aluri and A. P. Grosvenor, Phys. Chem. Chem. Phys., 2013, 15, 10477–10486 RSC.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- C. Wattanawikkam, W. Pecharapa and K. N. Ishihara, Ceram. Int., 2017, 43, S397–S402 CrossRef CAS.
- M. Sahoo, A. K. Yadav, S. N. Jha, D. Bhattacharyya, T. Mathews, N. K. Sahoo, S. Dash and A. K. Tyagi, J. Phys. Chem. C, 2015, 119, 17640–17647 CrossRef CAS.
- B. Hsieh, M. Tsai, C. Pan, W. Su, J. Rick, J. Lee, Y. Yang and B. Hwang, NPG Asia Mater., 2017, 9, e403 CrossRef CAS.
- K. Huang, K. Sasaki, R. R. Adzic and Y. Xing, J. Mater. Chem., 2012, 22, 16824–16832 RSC.
- T. Tangcharoen, W. Klysubun and C. Kongmark, J. Mol. Struct., 2018, 1156, 524–533 CrossRef CAS.
- J. K. Kesavan, I. Luisetto, S. Tuti, C. Meneghini, G. Iucci, C. Battocchio, S. Mobilio, S. Casciardi and R. Sisto, J. CO2 Util., 2018, 23, 200–211 CrossRef CAS.
- T. Tangcharoen, W. Klysubun and C. Kongmark, J. Mol. Struct., 2019, 1182, 219–229 CrossRef CAS.
- X. Meng, C. Wan, Y. Wang and X. Ju, J. Alloys Compd., 2018, 735, 1637–1647 CrossRef CAS.
- A. Worayingyong, S. Sang-urai, M. F. Smith, S. Maensiri and S. Seraphin, Appl. Phys. A: Mater. Sci. Process., 2014, 117, 1191–1201 CrossRef CAS.
- K. C. Chanapattharapol, S. Krachuamram, P. Kidkhunthod and Y. Pooarporn, Solid State Sci., 2020, 99, 106066 CrossRef CAS.
- P. Helden, J. Berga and I. M. Ciobîcă, Catal. Sci. Technol., 2012, 2, 491–494 RSC.
- J. Liu, C. Li, F. Wang, S. He, H. Chen, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Catal. Sci. Technol., 2013, 3, 2627–2633 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Appl. Catal., A, 2014, 486, 115–122 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, J. Zhang, K. Yuan, L. Wang, K. Wu, G. Xu and W. Chen, RSC Adv., 2016, 6, 28489–28499 RSC.
- B. Bharti, S. Kumar, H. Lee and R. Kumar, Sci. Rep., 2016, 6, 32355 CrossRef CAS PubMed.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Appl. Catal., B, 2018, 238, 393–403 CrossRef CAS.
- H. Y. Kim, H. M. Lee and J. Park, J. Phys. Chem. C, 2010, 114, 7128–7131 CrossRef CAS.
- M. Ding, J. Tu, Q. Zhang, M. Wang, N. Tsubaki, T. Wang and L. Ma, Biomass Bioenergy, 2016, 85, 12–17 CrossRef CAS.
- F. B. Noronha, E. C. Fendley, R. R. Soares, W. E. Alvarez and D. E. Resasco, Chem. Eng. J., 2001, 82, 21–31 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04934d |
| This journal is © The Royal Society of Chemistry 2020 |