DOI:
10.1039/D0RA05159D
(Paper)
RSC Adv., 2020,
10, 30961-30974
A density functional theory study on silver and bis-silver complexes with lighter tetrylene: are silver and bis-silver carbenes candidates for SARS-CoV-2 inhibition? Insight from molecular docking simulation†
Received
11th June 2020
, Accepted 5th August 2020
First published on 21st August 2020
Abstract
Ribavirin and remdesivir have been preclinically reported as potential drugs for the treatment of SARS-CoV-2 infection, while light silver tetrylene complexes (NHEPh–AgCl and (NHEPh–AgCl)2 with E = C, Si, and Ge) have gained significant interest due to their promising applicability on the cytological scale. Firstly, the structures and bonding states of silver–tetrylene complexes (NHE–Ag) and bis-silver–tetrylene complexes (NHE–Ag-bis) were investigated using density functional theory (DFT) at the BP86 level with the def2-SVP and def2-TZVPP basis sets. Secondly, the inhibitory capabilities of the carbene complexes (NHC–Ag and NHC–Ag-bis) and the two potential drugs (ribavirin and remdesivir) on human-protein ACE2 and SARS-CoV-2 protease PDB6LU7 were evaluated using molecular docking simulation. The carbene ligand NHC bonds in a head-on configuration with AgCl and (AgCl)2, whereas, the other NHE (E = Si and Ge) tetrylene ligands bond in a side-on mode to the metal fragments. The bond dissociation energy (BDE) of the NHE–Ag bond in the complex families follows the order of NHC–Ag > NHSi–Ag > NHGe–Ag and NHSi–Ag-bis > NHGe–Ag-bis > NHC–Ag-bis. The natural bond orbital analysis implies that the [NHEPh→AgCl] and [(NHEPh)2→(AgCl)2] donations are derived mainly from the σ- and π-contributions of the ligands. The docking results indicate that both the ACE2 and PDB6LU7 proteins are strongly inhibited by silver–carbene NHC–Ag, bis-silver–carbene NHC–Ag-bis, ribavirin, and remdesivir with the docking score energy values varying from −17.5 to −16.5 kcal mol−1 and −16.9 to −16.6 kcal mol−1, respectively. The root-mean-square deviation values were recorded to be less than 2 Å in all the calculated systems. Thus, the present study suggests that silver–carbene NHC–Ag and bis-silver–carbene NHC–Ag-bis complexes are potential candidates to inhibit ACE2 and PDB6LU7, and thus potentially conducive to prevent infection caused by the SARS-CoV-2 virus.
Introduction
The interest in divalent carbon compounds has been revitalized by the discovery of highly stable nucleophilic N-heterocyclic carbenes (NHCs) by Arduengo in 1991.1,2 Due to their σ-donor capabilities, ease of preparation, functionalization,3,4 and strong metal–ligand interactions that allow stable multi-metallic arrangements,5–7 NHCs constitute a promising alternative to tertiary phosphines. Arduengo synthesized the first NHC–Ag(I) complex before applying the free carbene method two years later to successfully isolate free NHC.8 Besides, silver–NHC complexes9–11 have been reported to possess both antibiotic12–14 and anticancer properties15–18 and have untapped potential as drug candidates. Youngs et al. (2004) reported the first use of silver NHCs as antimicrobial agents.19 Also, recent studies have proposed a structure-anticancer activity correlation for NHC–silver(I) complexes.20,21 In particular, 1-methyl-3-(p-cyanobenzyl)-benzimidazol-2-ylidene silver acetate,21 methyl caffeine-derived silver acetate22,23 and their structural analogues have been studied. The results implied that there is a significant improvement in biological applicability, especially in clinical therapy, with the formation of metal–ligand coordination.24,25
Carbenes are electrophiles in their free state and become nucleophiles in an N-heterocyclic-coordinated system. N-heterocyclic carbenes (NHCs) are a well-known class of ligands with the capacity to form complexes with most main-group metals26 and transition metals,27 including rare earth elements.28 NHC–Ag(I) complexes have gained significant attention among the family of NHC metal complexes considering their synthesizability and applicability. In a previous study, two silver(I) complexes, silver(I)-2,6-bis(ethanolimidazolemethyl)pyridine hydroxide and silver(I)-2,6-bis(propanolimidazolemethyl)pyridine hydroxide were shown to exhibit stronger antimicrobial activity than AgNO3 against Escherichia coli, Staphylococcus aureus and Pseudomonas aeruginosa.19 Another important contribution was from Ghosh's research group for the synthesis and antimicrobial evaluation of NHC–silver complexes derived from 1-benzyl-3-tert-butylimidazole.17 Recently, Gurbuz and colleagues showed that new imidazolidin-2-ylidene silver complexes can exhibit effective antimicrobial activity against a variety of bacteria and fungi.29
Coronaviruses (CoVs) are positive-sense, single-stranded RNA viruses infecting a wide range of animal hosts.30 A new virus first emerged in Wuhan, China, which has presently caused a global pandemic. It has been identified as a novel coronavirus (SARS-CoV-2), and is closely related to the severe acute respiratory syndrome from 2003 (SARS-CoV).31 Currently, no specific treatments have been found to be effective against this new virus. Thus, a practical approach is to determine whether or not existing antiviral drugs are reasonably effective for the treatment of SARS-CoV-2 infection. Several drugs, such as ribavirin, interferon, lopinavir–ritonavir, and corticosteroids, have been administered to patients with SARS or MERS, although the total efficacy of some drugs remains controversial.32 Wu Zhong et al. evaluated the antiviral effectiveness of five FAD-approved drugs, including ribavirin, penciclovir, nitazoxanide, nafamostat, chloroquine and two well-known broad-spectrum antiviral drugs, remdesivir (GS-5734) and favipiravir (T-705), against clinically isolated SARS-CoV-2 via in vitro experiments.33 In fact, ribavirin can be a potential drug for the suppression of 2019 SARS-CoV-2 considering its clinical efficacy demonstrated in 2003 SARS-CoV and 2012 MERS-CoV.34
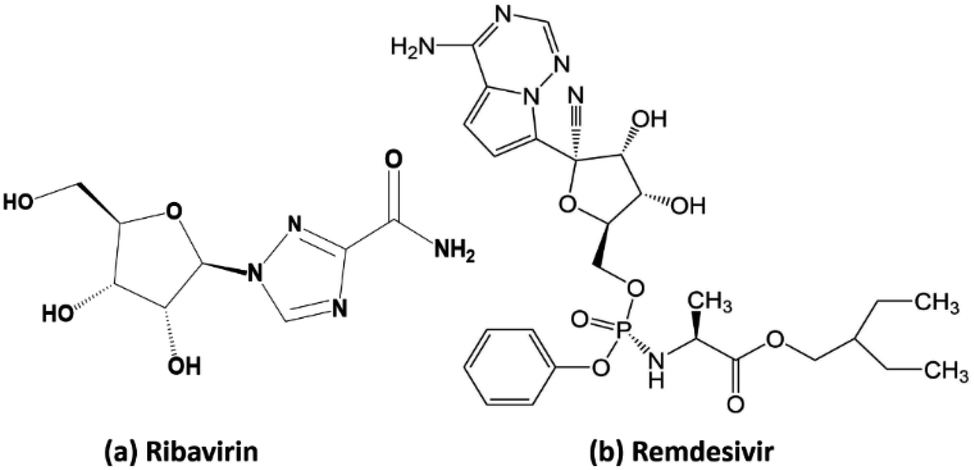
Ribavirin (i.e. 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide) is a guanosine analogue with a broad spectrum of antiviral activity against RNA and DNA viruses. This drug was patented in 1971 and approved for medical use in 1986.35 It is on the World Health Organization's list of essential medicines, the safest and most effective medicines needed in the healthcare system.36 In the treatment of hepatitis C, ribavirin is used in combination with other medications such as simeprevir, sofosbuvir, peginterferon alfa-2b and peginterferon alfa-2a. When administrated for the treatment of viral hemorrhagic fevers, it exhibits similar effectiveness for Lassa fever, Crimean–Congo hemorrhagic fever, and hantavirus infection. Remdesivir (GS-5734) is a nucleotide analog prodrug with broad-spectrum antiviral activity, which has been shown to inhibit filovirus, coronavirus, and paramyxovirus replication.37,38 In vitro, remdesivir shows potent antiviral activity against both the Malaysian and Bangladesh genotypes of Nipah virus. It reduces replication of Nipah virus Malaysia in primary human lung microvascular endothelial cells by more than four orders of magnitude.38 This indicates that the further testing of the efficacy of remdesivir against Nipah virus infection in vivo is appropriate.
Angiotensin-Converting Enzyme 2 (ACE2) is an integral membrane glycoprotein, which is known to be expressed in most tissues such as the kidney, endothelium, lungs, and heart.39,40 However, ACE2 is the host receptor of SARS-CoV-2 and SARS-CoV.40,48 Therefore, if the ACE2 protein can be inhibited, cells can be temporarily protected against infection from SARS-CoV-2. The structural database of ACE2 (https://www.uniprot.org/uniprot/Q9BYF1) can be referenced from UniProtKB (Scheme 1A).41 Moreover, protein PDB6LU7 is the main protease of SARS-CoV-2, functioning as a proteolytic enzyme that cuts polyproteins into functional pieces. Thus, if PDB6LU7 can be inhibited, the replication of SARS-CoV-2 can be restrained, temporizing its spread in order for human immunity to produce a sufficient number of antibodies to respond against this pathogen. The immune response can be neutralization, agglutination, or phagocytosis. The structure of PDB6LU7 (DOI: 10.2210/pdb6LU7/pdb) of SARS-CoV-2 has been archived recently by the Worldwide Protein Data Bank (Scheme 1B).41 In fact, Rohan et al. and Ran et al. independently carried out molecular docking simulation on the inhibitability of several potential candidates against ACE2 and PDB6LU7.42,43 These studies included two drugs, ribavirin and remdesivir, which exhibited significant inhibitory capability. Also, our preliminary investigation based on a similar virtual approach revealed that these two drugs interacted with the above two proteins mainly via their N-heterocyclic groups. This led to the implication of promising inhibitory efficacy of tetrylene complexes considering their resemblance of their molecular structure to N-heterocyclic rings.
 |
| | Scheme 1 (A) Angiotensin-converting enzyme 2 (DOI: 10.2210/pdbACE2/pdb) in the human body and (B) protein PDB6LU7 (DOI: 10.2210/pdb6LU7/pdb) in SARS-CoV-2. | |
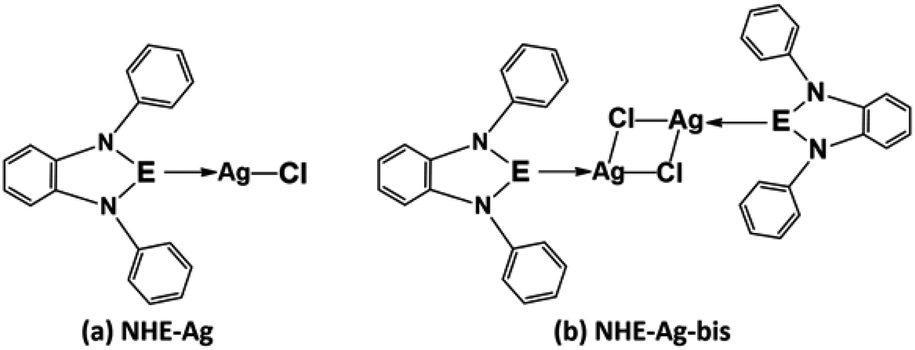
Our research included theoretical calculations of the quantum properties of the complexes using density functional theory (DFT) and further predictions on their inhibitability developed by molecular docking simulation (MDS). Firstly, the nature of bonding and the extent of σ and π interactions between the tetrylone ligands NHEPh and fragment AgCl in [NHEPh–AgCl] (NHE–Ag) and [(NHEPh–AgCl)2] (NHE–Ag-bis) (E = C, Si, and Ge) complexes were investigated (Scheme 2). The geometry and energy of the compounds NHE–Ag and NHE–Ag-bis and the free ligands NHE were optimized and calculated to reach the equilibrium structures of the complexes before their bond dissociation energies (BDEs) were examined with gradient-corrected density functional theory (DFT). The bonding states of the complexes were also examined by calculating the energetically low-lying occupied molecular orbitals (LOMO) for the σ- and π-orbitals in the complexes and free ligands using natural bond orbital (NBO) analysis. Then, the inhibitory effects of the considered NHC–Ag, NHC–Ag-bis, ribavirin and remdesivir compounds (Scheme 3) on the proteins ACE2 and PDB6LU7, which are crucial to tackle the infection of virus SARS-CoV-2 in the human physiological environment, were also investigated. To the best of our knowledge, to date, there is no experimental and theoretical information reported about the prevention of SARS-CoV-2 related to carbene complexes.
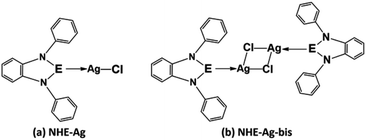 |
| | Scheme 2 Common molecular structure of the (a) NHE–Ag and (b) NHE–Ag-bis (E = C, Si, and Ge) complexes. | |
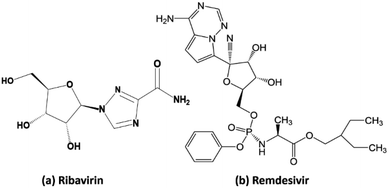 |
| | Scheme 3 Molecular structure of (a) ribavirin and (b) remdesivir. | |
Computational methods
Density functional theory calculation
Geometry optimization of the molecules was carried out without symmetry constraints using Turbomole 6.0 (ref. 44) and Gaussian 09 (ref. 45) at the BP86 (ref. 46 and 47)/def2-SVP48 level of theory. The former defined the coordinates of the molecules before the latter was utilised for optimizing the calculations. The molecules were also calculated by vibrational frequencies at the same functional to confirm that the structures were in global minimum on the potential energy surface (PES). For the silver atom, small-core quasi-relativistic effective core potentials (ECPs) were used.49 Single-point energies at the BP86/def2-SVP level optimized geometries were calculated with the application of the frozen-core approximation for non-valence-shell electrons. The same functional for geometry optimization was carried out at the larger def2-TZVPP50 basis set. RI approximation was used for all structure optimizations using the appropriate auxiliary basis sets. The bond dissociation energy (BDE), De (kcal mol−1) was determined at the BP86 level in conjunction with the def2-TZVPP basis set using the BP86/def2-SVP level optimized geometries. Considering information on the chemical bonding in the investigated compounds, the analysis on natural bond orbital (NBO)51 was performed to propose their intermolecular interactions and to imply donor–acceptor bonds. The NBO analysis was carried out at the BP86/def2-TZVPP//BP86/def2-SVP level of theory, which was proposed for the calculation of the Wiberg bond orders (WBI) and natural partial charges (NPA) as well as localized plotting molecular orbitals and orbital energies using NBO 5.1 available in Gaussian 09. Electron density distributions were revealed by bonding analysis. The HOMO energy, EHOMO, indicates the tendency of a molecule to donate electrons, whereas the ELUMO of a molecule refers to its electron accepting ability. The energy gap, ΔE = ELUMO − EHOMO, illustrates the tendency of inhibition efficiency of organic molecules towards the surface of metal elements. The ionization potential (I) and electron affinity (A) of the inhibitory molecules were calculated by applying Koopmans' theorem52 related to the HOMO and LUMO energy as: I = −EHOMO and A = −ELUMO. The obtained ionization potential and electron affinity values were used to yield the electronegativity (χ), global hardness (η), and global softness (S) of the molecules, which were calculated using the following three equations: χ = (I + A)/2; η = (I − A)/2; and S = 1/η, respectively.
Molecular docking simulation
All calculations for molecular docking simulation were investigated using the MOE 2015.10 software. The structural information of the proteins (ACE2 and PDB6LU7) and the structures of 6 potential drugs were required in order to implement the molecular docking. The structures were used to simulate the interactions between the drugs and proteins, and then to evaluate the bonding and docking score energy results. The results included the potential drug configurations, docking score (DS) energy, root-mean-square deviation (RMSD), types of interactions, and respective distances between the potential drug and proteins. A molecular docking simulation procedure involves 3 steps as follows.53–56
(a) Protein and ligand preparation. The structures of the ACE2 and PDB6LU7 proteins are available at UniProtKB41 and the Worldwide Protein Data Bank,57 respectively. The Quickprep tool was applied to prepare their structures and 3D protonation. The active zones of the proteins were confirmed based on the ligand position within a radius of 4.5 Å, and the presence of important amino acids and of the protein structures were saved in the *.pdb format. The six potential drugs were optimized via Conj Grad for the minimum energy; termination for energy change of 0.0001 kcal mol−1; max interactions of 1000; and using the Gasteiger–Huckel charge. Molecular dynamics on the MOE 2015.10 system were performed, and the structures of the proteins and potential drugs were saved in the *.sdf format.
(b) Docking investigation. The docking simulation parameters were set with the number of poses at 10 and kept for further analysis of the interaction, the maximum number of solutions per iteration at 1000, and the maximum number of solutions per fragmentation at 200.
(c) Docking results analysis. The docking score energy (DS) values demonstrated the binding ability between the potential drugs and the proteins (ACE2 and PDB6LU7). Evaluation of the docking score energy (DS) provided further insight into the interactions between the potential drugs and ACE2 and PDB6LU7 proteins. The performance of an interaction on the 2D and 3D planes of the drug–protein complexes was examined. The interactions formed between the potential drugs and important amino acids in the site–site distances of ACE2 and PDB6LU7 were analysed. Hydrogen bonds, ion bonds, π–π interactions, cation–π interactions, and van der Waals interactions were detected to gain information on the hydrophilic, hydrophobic, and solvent interactions. The interactions between the potential drugs and the proteins (ACE2 and PDB6LU7) were determined, allowing an inference to be reached on the inhibitory effects of the potential drugs on the host receptor ACE2 and SARS-CoV-2 main protease (PBD6LU7).
Results
Structures and energies of lighter tetrylene complexes
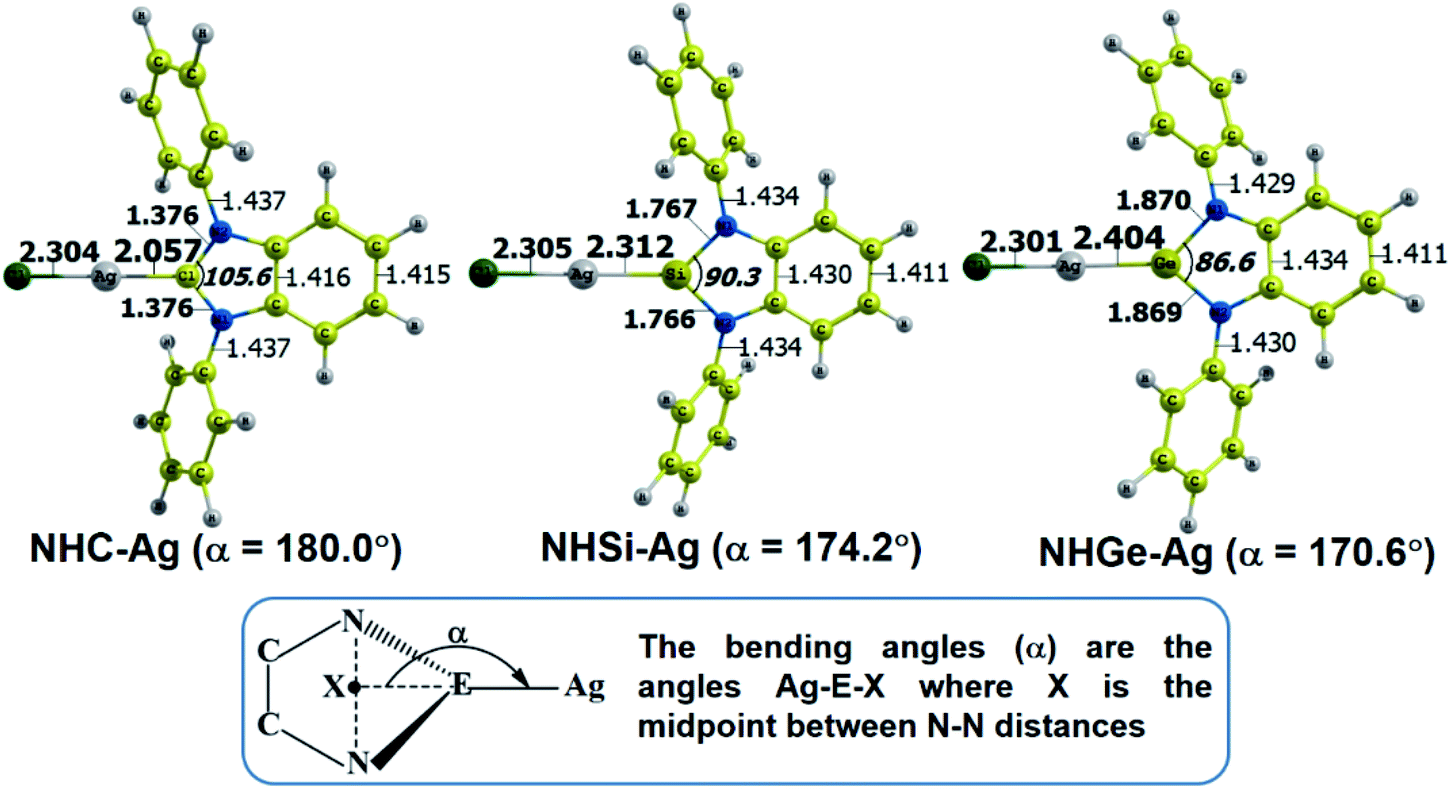
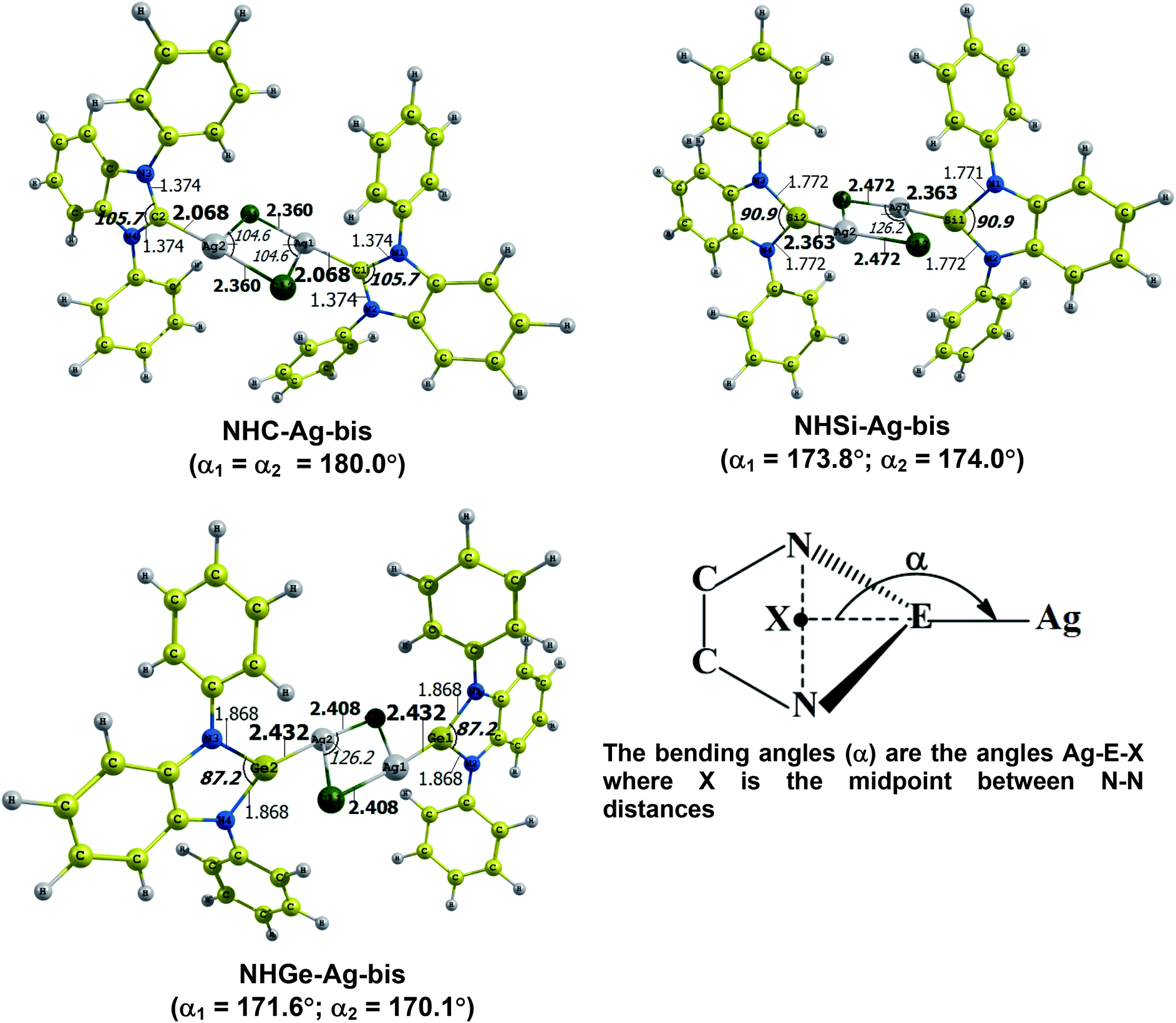
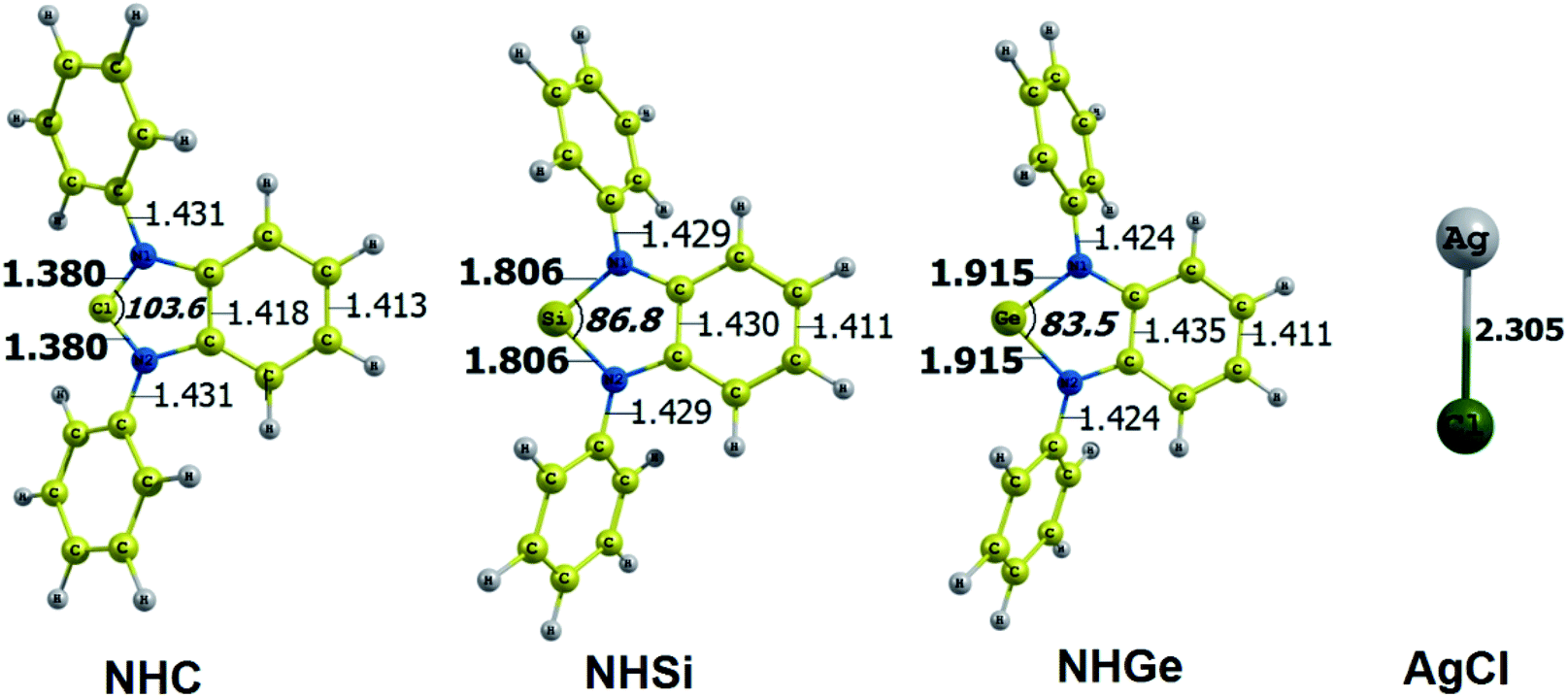
The optimized geometry with bond length, bond angle and bending angle of NHE–Ag, NHE–Ag-bis, and ligands NHE (E = C, Si, and Ge) are shown in Fig. 1, 2, and 3, respectively. The calculated Ag–E bond lengths of NHE–Ag (E = C, Si, and Ge), i.e. Ag–C, Ag–Si and Ag–Ge are 2.076 Å, 2.338 Å, and 2.448 Å, respectively. These results are consistent with the work on less-bulky N-heterocyclic carbene, silylene, and germylene complexes of AgCl investigated by Frenking et al.58 In detail, the calculated Ag–C bond length of NHC–Ag was the shortest (2.057 Å), which increased to 2.404 Å for the germylene complex. The equilibrium structure of the NHC–Ag complex in Fig. 1 shows that the lighter NHC ligands are bonded in a head-on manner to the metal fragment AgCl, i.e. a 180° alignment. However, the bending angle becomes much more noticeable if the E atom becomes heavier (bending angles, α, of NHSi–Ag = 174.2° and NHGe–Ag = 170.6°). Regarding the bis-complexes, the Ag1–C and Ag2–C bond lengths in the NHC–Ag-bis carbene complexes are both 2.068 Å (Fig. 2). These are also in good agreement with the results reported in another work by Frenking et al.,59 in which both Ag1–C and Ag2–C bonds are 2.084 Å in the [(NHCH)2–Ag] complex. Besides, the Ag1–E and Ag2–E bonds calculated for the heavier homologues, i.e. NHSi–Ag-bis (2.363 Å) and NHGe–Ag-bis (2.432 Å), are longer than that in the NHC–Ag-bis adduct. The equilibrium structure of NHC–Ag-bis shows that the NHC ligands bond in a head-on orientation to the silver atoms with an aligned angle α of 180.0°. The structures of NHE–Ag-bis contain a side-on bonded ligand, whose E = Si and Ge exhibit the bending angle of 174.0° in NHSi–Ag-bis and of 170.1° in NHGe–Ag-bis, respectively. The results are rather similar to the values previously obtained from the NHE–Ag complexes. Fig. 3 shows that the E–N bond lengths in the complexes positively correlate with the mass of their homologous free ligands. In detail, the E–N lengths in the NHE complexes are shorter than that in their NHE–bis counterparts. In addition, the optimized geometries of the silver-complexes NHE–Ag and NHE–Ag-bis (E = C, Si, and Ge) at the BP86/def2-SVP level with dispersive effect are presented in Fig. S1 and S2 (ESI†), respectively. The results reveal that the recorded deviation in the structural parameters, including bond length and bonding angles, is insignificant. Therefore, the geometries obtained without dispersive interactions are reliable.
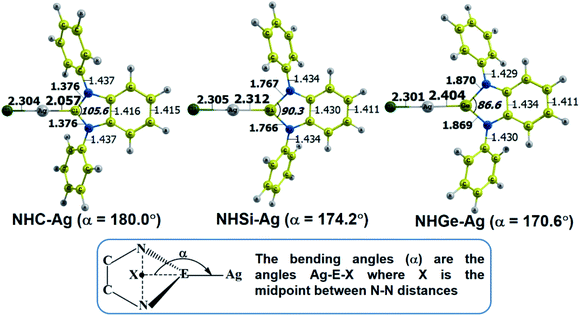 |
| | Fig. 1 Optimized geometries of NHE–Ag (E = C, Si, and Ge) silver-complexes at the BP86/def2-SVP level. Bond lengths are given in Å and angles in degrees. | |
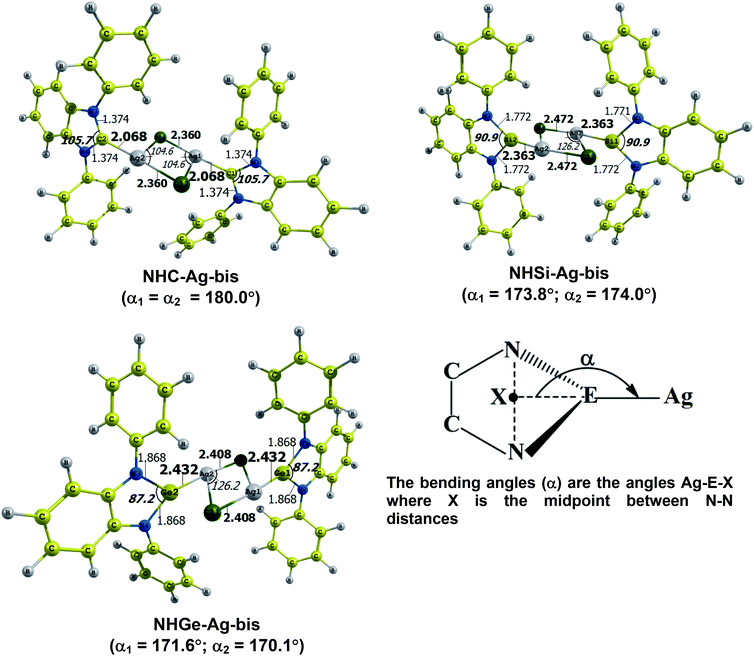 |
| | Fig. 2 Optimized geometries of NHE–Ag-bis (E = C, Si, and Ge) bis-silver-complexes at the BP86/def2-SVP level. Bond lengths are given in Å and angles in degrees. | |
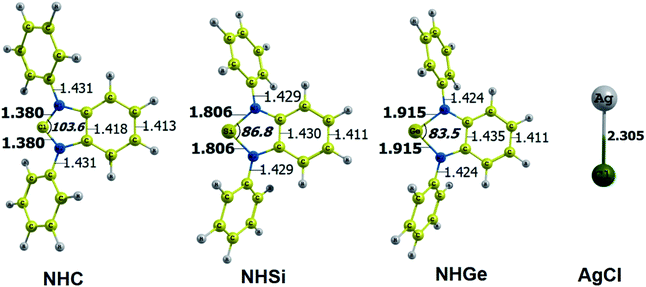 |
| | Fig. 3 Optimized geometries of tetrylene free ligands NHE (E = C, Si, and Ge) and metal fragment AgCl at the BP86/def2-SVP level of theory. Bond lengths are given in Å and angles in degrees. | |
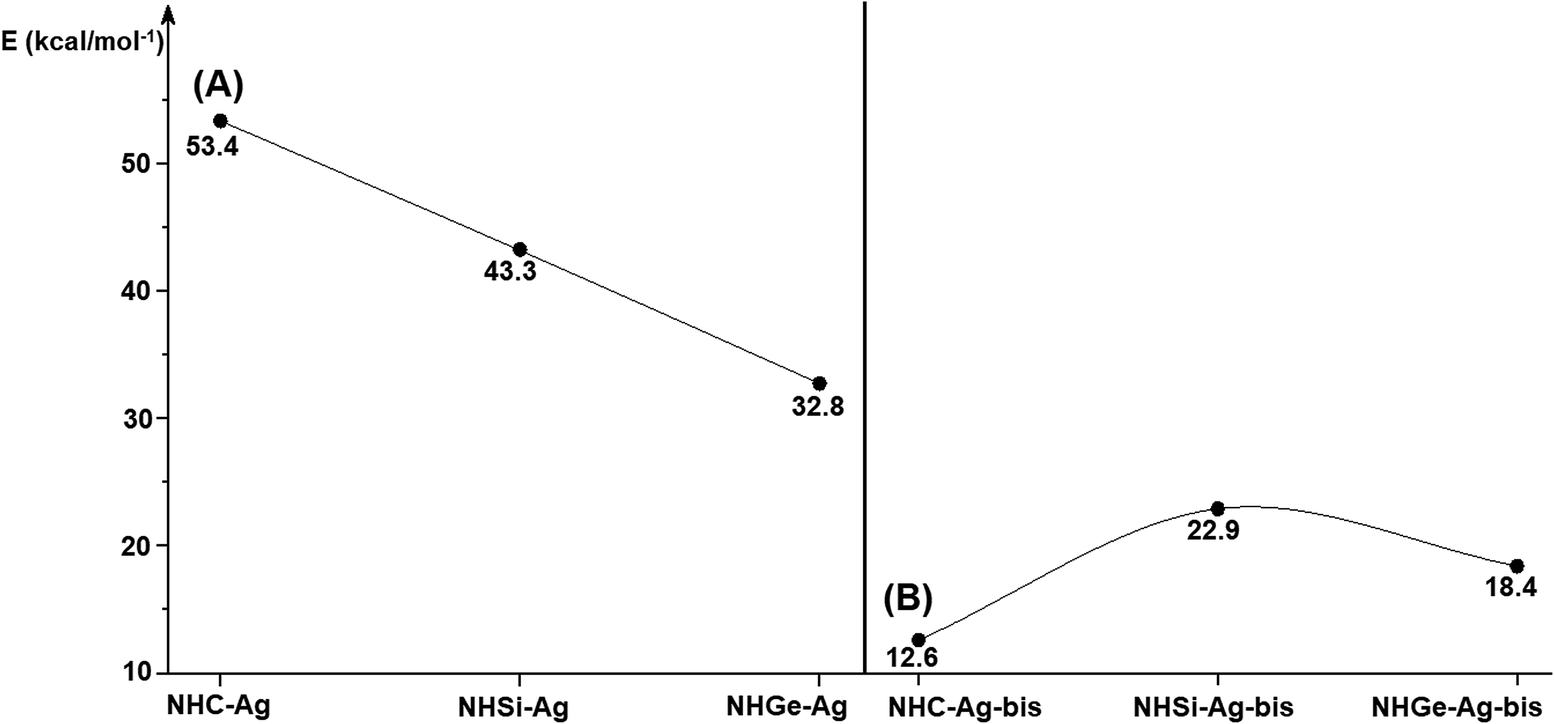
Fig. 4 shows the BDEs for the Ag–NHEMe bonds in NHE–Ag and NHE–Ag-bis. The energy exhibited a significant decrease from the carbene complex NHC–Ag (De = 53.4 kcal mol−1) to the germylene NHGe–Ag complex (De = 32.8 kcal mol−1), as shown in Fig. 4A. The calculations suggest that the bonding of the NHC ligand in NHC–Ag is the strongest and that in the heavier homologues NHE–Ag (E = Si and Ge) is weaker. These observations are in good agreement with the results reported in previous studies.60–62 The BDE tendency of AgCl–carbene and its homologous is also similar to other corresponding values for the AgCl–NHEH complexes (De = 56.5–29.9 kcal mol−1).58 Moreover, the BDEs calculated for the NHE–Ag-bis systems show a different pattern. Fig. 4B indicates that the BDEs exhibited the smallest value of De = 12.6 kcal mol−1 for NHC–Ag-bis, maximum for NHSi–Ag-bis (De = 22.9 kcal mol−1), and then smaller for NHGe–Ag-bis (De = 18.4 kcal mol−1). This observation is also in good agreement with a reported work on NHEMe–(AuCl)2 (E = C–Ge) transition metal tetrylene complexes.63 Although the BDEs consistently correlate with their associated values of either bond length or bending angle, α, obtained from the DFT analysis of the NHE–Ag complexes, the expected correlations were not observed when applied for the NHE–Ag-bis compounds. This inconsistency still requires further in-depth investigations in order to yield appropriate justification.
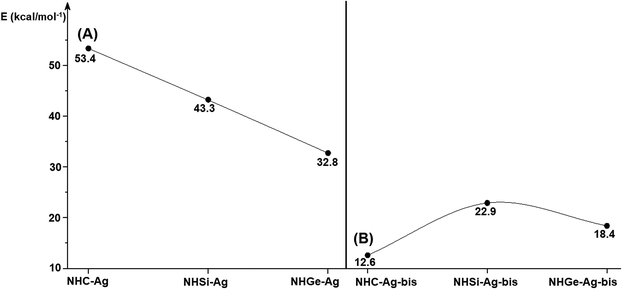 |
| | Fig. 4 Bond dissociation energies of NHE–AgCl bond in (A) NHE–Ag silver tetrylene complexes, (B) and NHE–Ag-bis (E = C, Si, and Ge) bis-silver tetrylene complexes calculated at the BP86/def2-TZVPP//BP86/def2-SVP level. | |
Bonding analysis of lighter tetrylene complexes
Table 1 shows the results of the NBO partitioning analysis, including Wiberg Bond Indices (WBI) and natural partial charge (NPA) of the NHE–Ag and NHE–Ag-bis complexes. The calculated partial charges show that the metal fragments AgCl and (AgCl)2 in the complexes always carry a negative charge from NHC–Ag (−0.23 e) and NHC–Ag-bis (−0.44 e). The magnitude of charge donation to the AgCl fragment in the NHE–Ag and NHE–Ag-bis complexes is lower than the corresponding donation observed from other transition metal moieties such as W(CO)5 and Mo(CO)4 in similar NHEMe ligands.60,62 In fact, the more negative charges of −0,47 e and −0.77 e have been reported in the transition metal fragments W(CO)5 and Mo(CO)4, respectively. The WBI for the Ag–E bond in the tetrylene complexes is 0.22 for NHC–Ag, 0.15 for NHSi–Ag and 0.20 for NHGe–Ag. However, the WBI of the E–N1 and E–N2 bonds remained almost similar for NHC–Ag-bis (0.33), NHSi–Ag-bis (0.45), and NHGe–Ag-bis (0.40). The natural population analysis reveals that the electrostatic charges of the carbon atoms in the fragment of the NHE–Ag and NHE–Ag-bis complexes are nearly neutral, while Si and Ge carry relatively larger positive charge values. The NPA values for NHSi–Ag, NHSi–Ag-bis, NHGe–Ag, and NHGe–Ag-bis are 1.15 e, 1.18 e, 1.07 e, and 1.10 e, respectively.
Table 1 NBO analysis on NHE–Ag and NHE–Ag-bis tetrylene complexes with Wiberg Bond Indices (WBI) and natural partial charges (NPA) at the BP86/def2-TZVPP//BP86/def2-SVP level. The partial charges, q, are given in electrons [e]
| Molecule |
Bond |
WBI |
qAgCl [e] |
Atom |
NPA [e] |
| NHC–Ag |
C1–Ag |
0.22 |
|
Ag |
0.36 |
| C1–N1 |
0.32 |
−0.23 |
C1 |
0.22 |
| C1–N2 |
0.32 |
|
N1; N2 |
−0.33 |
| Ag–Cl |
0.31 |
|
Cl |
−0.59 |
| NHSi–Ag |
Si–Ag |
0.15 |
|
Ag |
0.24 |
| Si–N1 |
0.44 |
−0.33 |
Si |
1.15 |
| Si–N2 |
0.44 |
|
N1; N2 |
−0.72 |
| Ag–Cl |
0.33 |
|
Cl |
−0.57 |
| NHGe–Ag |
Ge–Ag |
0.20 |
|
Ag |
0.29 |
| Ge-N1 |
0.37 |
−0.30 |
Ge |
1.07 |
| Ge-N2 |
0.37 |
|
N1; N2 |
−0.68 |
| Ag–Cl |
0.31 |
|
Cl |
−0.57 |
| NHC–Ag-bis |
(C1–Ag)bis |
0.23 |
|
Ag-bis |
0.40 |
| (C1–N1)bis |
0.33 |
−0.44 |
(C1)bis |
0.20 |
| (C1–N2)bis |
0.33 |
|
N1; N2 |
−0.34 |
| (Ag–Cl)bis |
0.30 |
|
Cl |
−0.62 |
| NHSi–Ag-bis |
(Si–Ag)bis |
0.20 |
|
Ag |
0.30 |
| (Si–N1)bis |
0.45 |
−0.33 |
Si/Si |
1.18 |
| (Si–N2)bis |
0.45 |
|
N1; N2 |
−0.75 |
| (Ag–Cl)bis |
0.32 |
|
Cl |
−0.50 |
| NHGe–Ag-bis |
(Ge–Ag)bis |
0.22 |
|
Ag |
0.35 |
| (Ge-N1)bis |
0.40 |
−0.31 |
Ge/Ge |
1.10 |
| (Ge-N2)bis |
0.40 |
|
N1; N2 |
−0.70 |
| (Ag–Cl)bis |
0.32 |
|
Cl |
−0.51 |
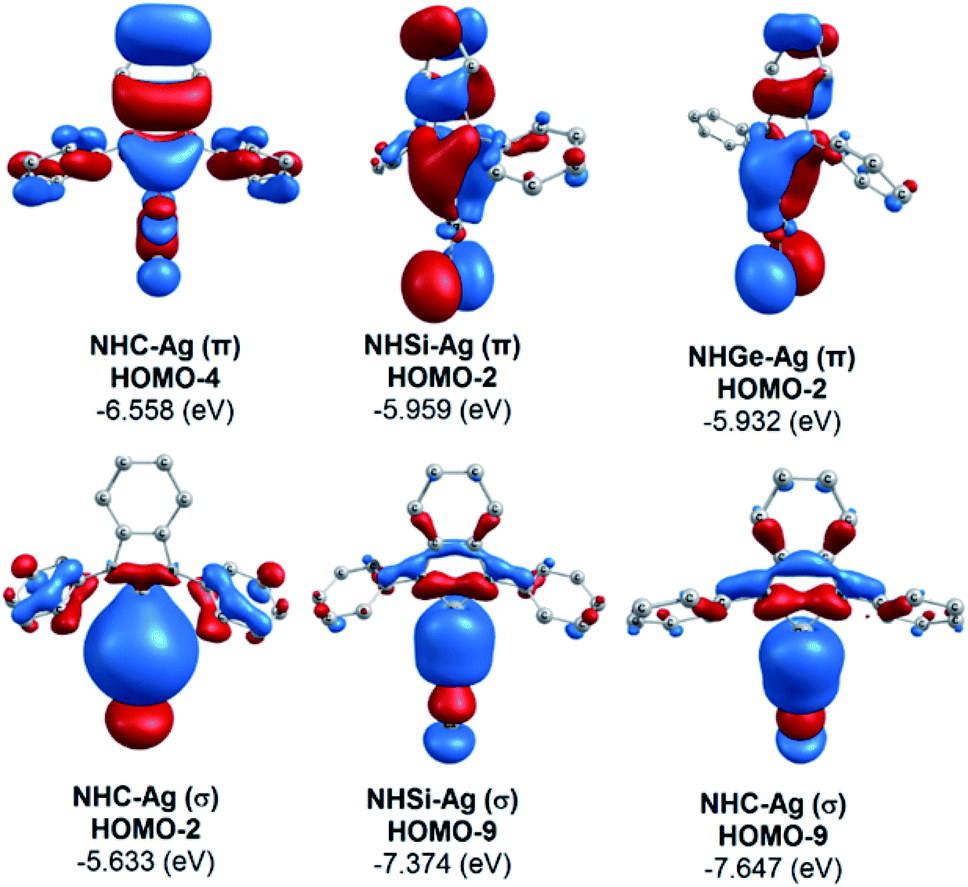
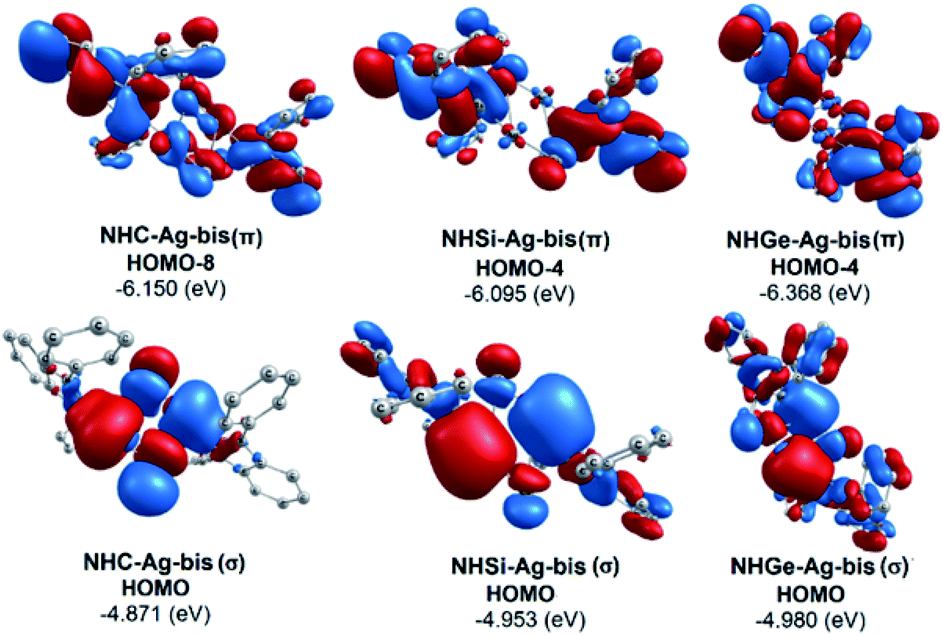
The bonding analysis of the NHE–Ag and NHE–Ag-bis complexes also displayed the strength of the σ- and π-donation in ClAg←NHEPh and (ClAg)2←(NHEPh)2 (E = C to Ge). Fig. 5 and 6 show the occupied molecular orbitals and orbital energies of the σ-type and π-type MOs calculated derived from the two types of NHE–Ag and NHE–Ag-bis complexes at the BP86/TZVPP level. This reveals that the energy levels of the π-donor orbitals of the NHE–Ag complexes are higher than that of their σ-donor orbitals, except for NHC–Ag. In contrast, the NHE–Ag-bis complexes present the opposite tendency, i.e. the energy levels of their π-donor orbitals are lower than their σ-donor counterparts. Also, the extensively occupied shape of the molecular orbitals indicates that NHEPh→AgCl and (NHEPh)2→(AgCl)2 not only perform significant σ donation, but also exhibit noticeable π donation in the tetrylene complexes.
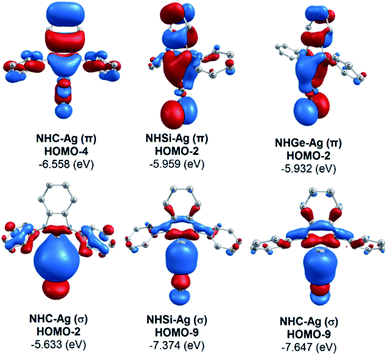 |
| | Fig. 5 Molecular orbitals and orbital energy levels of σ- and π-type MOs of the NHE–Ag complexes (E = C, Si, and Ge) at the BP86/TZVPP level. | |
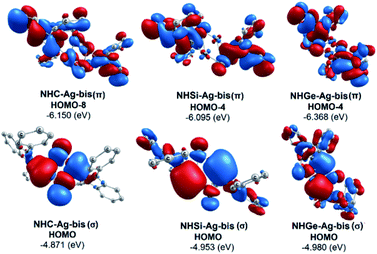 |
| | Fig. 6 Molecular orbitals and orbital energy levels of σ- and π-type MOs of the NHE–Ag-bis complexes (E = C, Si, and Ge) at the BP86/TZVPP level. | |
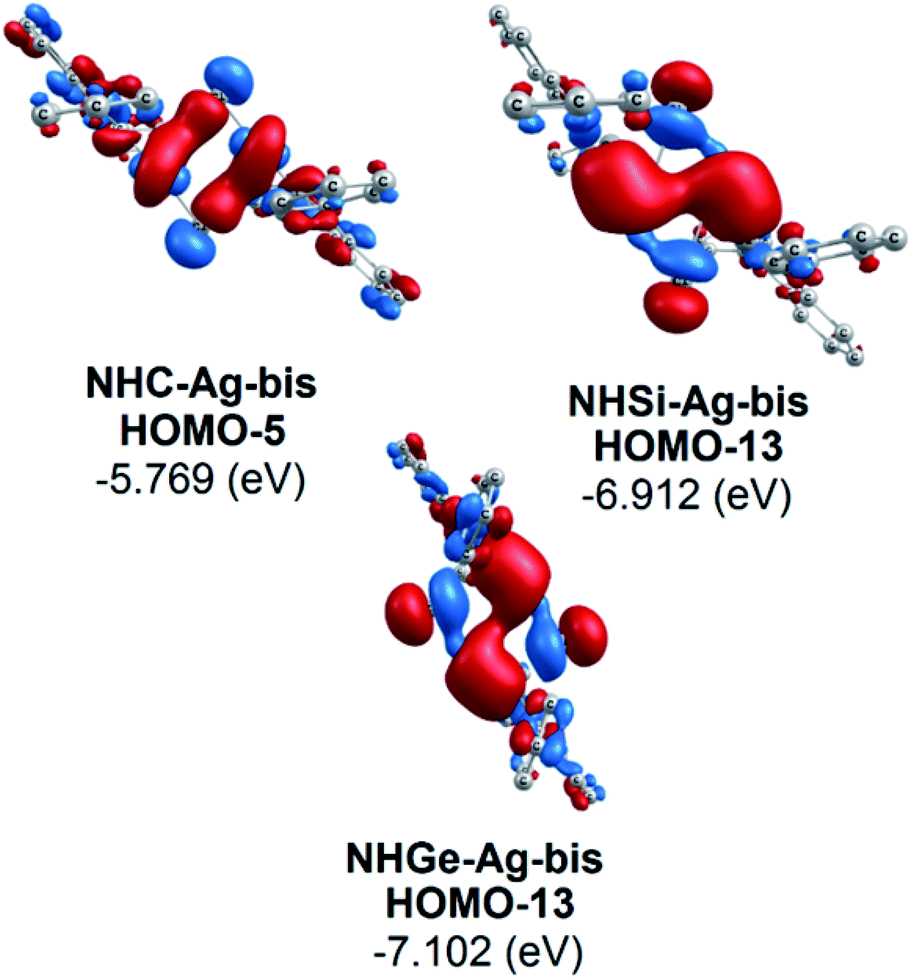
Moreover, Fig. 6 provides information on the interactions of the separated NHE donor–acceptor fragments and AgCl adducts in each side of the NHE–Ag-bis bis-counterparts. The former still excludes the interactions between the two NHE–AgCl bulk-fragments by dative bonds in NHE–Ag-bis. Therefore, HOMO-5 and HOMO-13, illustrating the donor–acceptor bonds in the NHE–Ag-bis complexes, are plotted in Fig. 7 to investigate their intramolecular interactions. The HOMO-5 distribution of NHC–Ag-bis suggests that the NHC←Ag–Cl–Cl–Ag→NHC π back-donation types are insignificant. Also, there is weak σ and π bonding between the carbon central atoms and silver atoms in the metal fragments (AgCl). The profound interaction relates to (AgCl)–(AgCl). Furthermore, the distributions of HOMO-13 for NHSi–Ag-bis and NHGe–Ag-bis suggest that the NHSi←Ag–Cl–Cl–Ag→NHSi and NHGe←Ag–Cl–Cl–Ag→NHGe σ, π, and π back-donation types are substantial. This observation can be explained by the end-on bonded mode characterized for carbene complexes, leading to the major stabilizing strength of the fragments (Ag–Cl–Cl–Ag), and thus is unconducive for mirror-stabilization, i.e. π-back-donation contributions.
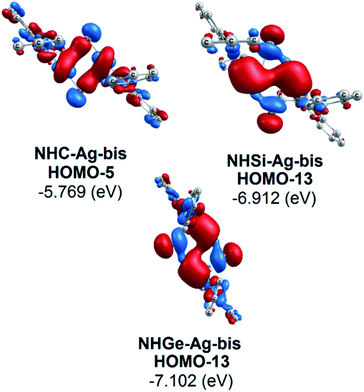 |
| | Fig. 7 Bonding interactions in the NHE–Ag-bis (E = C, Si, and Ge) complexes presented via molecular orbitals and orbital energy levels. | |
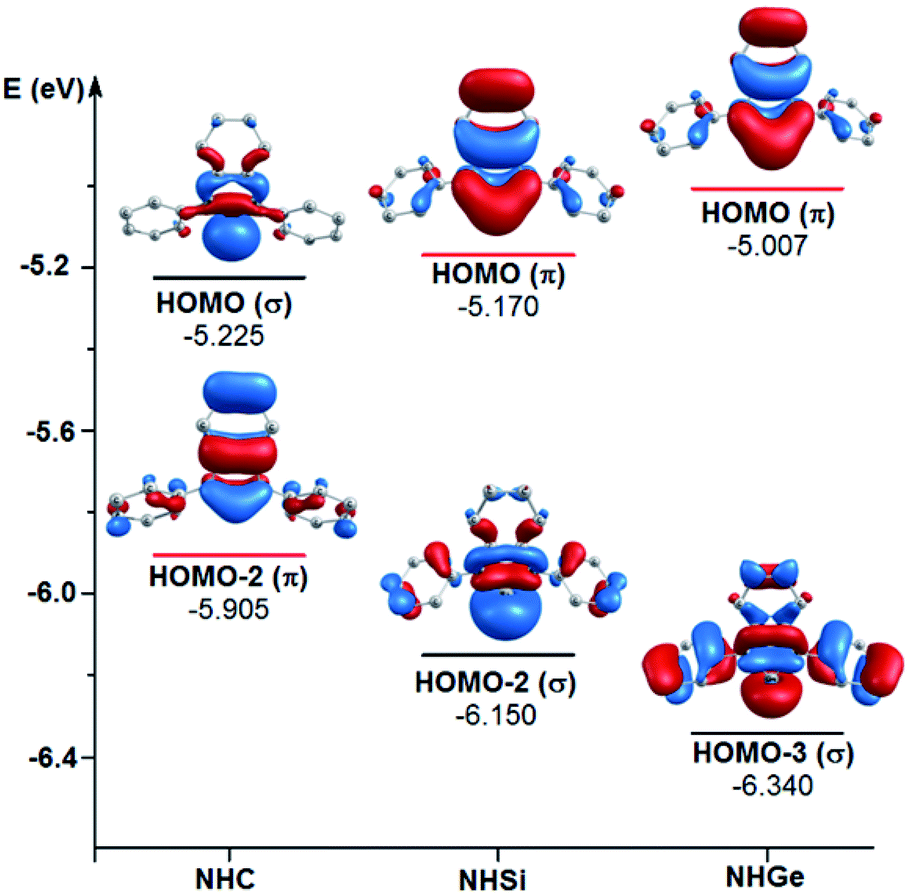
To explain the end-on and side-on structures of the carbenes, silylenes, and germylenes, the highest occupying molecular orbitals according to the σ- and π-orbital of the free NHE (E = C, Si, and Ge) ligands were plotted, as shown in Fig. 8. Fig. 8 displays the shape of HOMO, HOMO-2, and HOMO-3 for the free ligands considering their symmetrical states. Most of the HOMOs of the NHE ligands emerge with π symmetry except for NHC, while all the HOMOs representing σ bonds are symmetrical, including HOMO (NHC), HOMO-2 (NHSi), and HOMO-3 (NHGe). Fig. 8 also shows that the energy level of the π orbitals increases with an increase in the mass of the ligand, while the corresponding figures for the σ orbitals show the reverse tendency. This can be explained by the fact that σ-orbital energy decreases due to the shrinking phenomenon of the σ-orbital resulting from the relativistic effect for the heavier homologues.60,61 These energy level tendencies of the energetically highest-lying σ- and π-orbitals of the NHE ligand verify the preference of the heavier NHSi and NHGe ligands for side-on coordination to the metal, in which the σ-donation takes place through the π orbital of the ligand.60
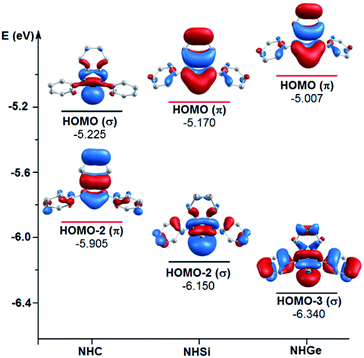 |
| | Fig. 8 Energy levels of the energetically highest lying σ and π orbitals of the free light NHE (E = C, Si, and Ge) ligands. | |
Table 2 summarizes the quantum chemical parameters related to the molecular electronic structures of the studied NHE–Ag, NHE–Ag-bis (E = C, Si, and Ge) complexes. In principle, the EHOMO value represents the electron-accepting capability and ELUMO provides information on the capability of electron donation. Theoretically, either a high HOMO energy or low LUMO energy will be conducive to the binding ability of the complexes to protein since polypeptide molecules have been proposed and well-proven to exhibit electrical conductivity.64,65 The modern explanation for the electron transfer includes the super-exchange theory (or electron tunneling) and the electron hopping model.66 The EHOMO values of the silver–tetrylene complexes follow the decreasing order of NHC–Ag > NHSi–Ag > NHGe–Ag and the EHOMO values of the bis-silver–tetrylene complexes follow a similar trend, i.e. NHC–Ag-bis > NHSi–Ag-bis > NHGe–Ag-bis. These observations are also consistent with the tendencies observed from ELUMO, but with a narrower differential. In detail, the HOMO energy varies from 4.879 eV to 5.617 eV and the LUMO energy is in the range of 2.188–3.178 eV. Overall, the energy gap (ΔEGAP) values of the NHE–Ag-bis complexes are slightly lower than that of their mono counterpart silylenes (2.767 eV to 2.798 eV) and germylene (2.359 eV to 2.439 eV). Conversely, the carbene complexes show the opposite pattern, in which the corresponding value for NHC–Ag (2.653 eV) is smaller than that of NHC–Ag-bis (2.710 eV). In particular, NHGe–Ag-bis exhibits the smallest energy gap (2.359 eV), while NHSi–Ag possesses the largest energy gap (2.798 eV). Table 2 also shows that the electronegativity (χ) value exhibited the decreasing order of NHC–Ag > NHSi–Ag > NHGe–Ag and NHC–Ag-bis > NHSi–Ag-bis > NHGe–Ag-bis and the largest value belongs to NHGe–Ag (4.398). Thus, the noticeable quantum chemical parameters, including energy gap (ΔEGAP), ionization potential (I), electron affinity (A), and electronegativity (χ), imply that all the studied complexes can be considered as versatile ligands and can to create polar interactions with highly polarized amino acids in the protein structure.
Table 2 Quantum chemical parameters of the tetrylene NHE–Ag and NHE–Ag-bis (E = C, Si, and Ge) complexes obtained from the NBO data calculated at the BP86/def2-TZVPP level, including the energy gap (ΔEGAP), ionization potential (I), electron affinity (A) and electronegativity (χ). Energy in eV
| Complex |
ΔE (eV) = (ELUMO − EHOMO) |
I = −EHOMO |
A = −ELUMO |
χ = (I + A)/2 |
| NHC–Ag |
2.653 |
5.222 |
2.569 |
3.896 |
| NHSi–Ag |
2.798 |
5.610 |
2.972 |
4.291 |
| NHGe–Ag |
2.439 |
5.617 |
3.178 |
4.398 |
| NHC–Ag-bis |
2.710 |
4.879 |
2.169 |
3.524 |
| NHSi–Ag-bis |
2.767 |
4.955 |
2.188 |
3.572 |
| NHGe–Ag-bis |
2.359 |
4.966 |
2.607 |
3.787 |
In summary, although the DFT results suggest that the tetrylene complexes containing Si and Ge exhibit desirable quantum properties for practical applications, their potential applicability in physiological systems is limited due to the possible biological toxicity of their elements. Therefore, NHC–Ag and NHC–Ag-bis were selected for further study using molecular docking simulation in an attempt to investigate the deactivation of the SARS-CoV-2 PDB6LU7 enzymes and protection of the host ACE2 receptors. Also, the inhibitory activities of the studied silver complexes will be compared with that of ribavirin and remdesivir, which were considered as references.
Docking simulation on ACE2 and PDB6LU7 proteins
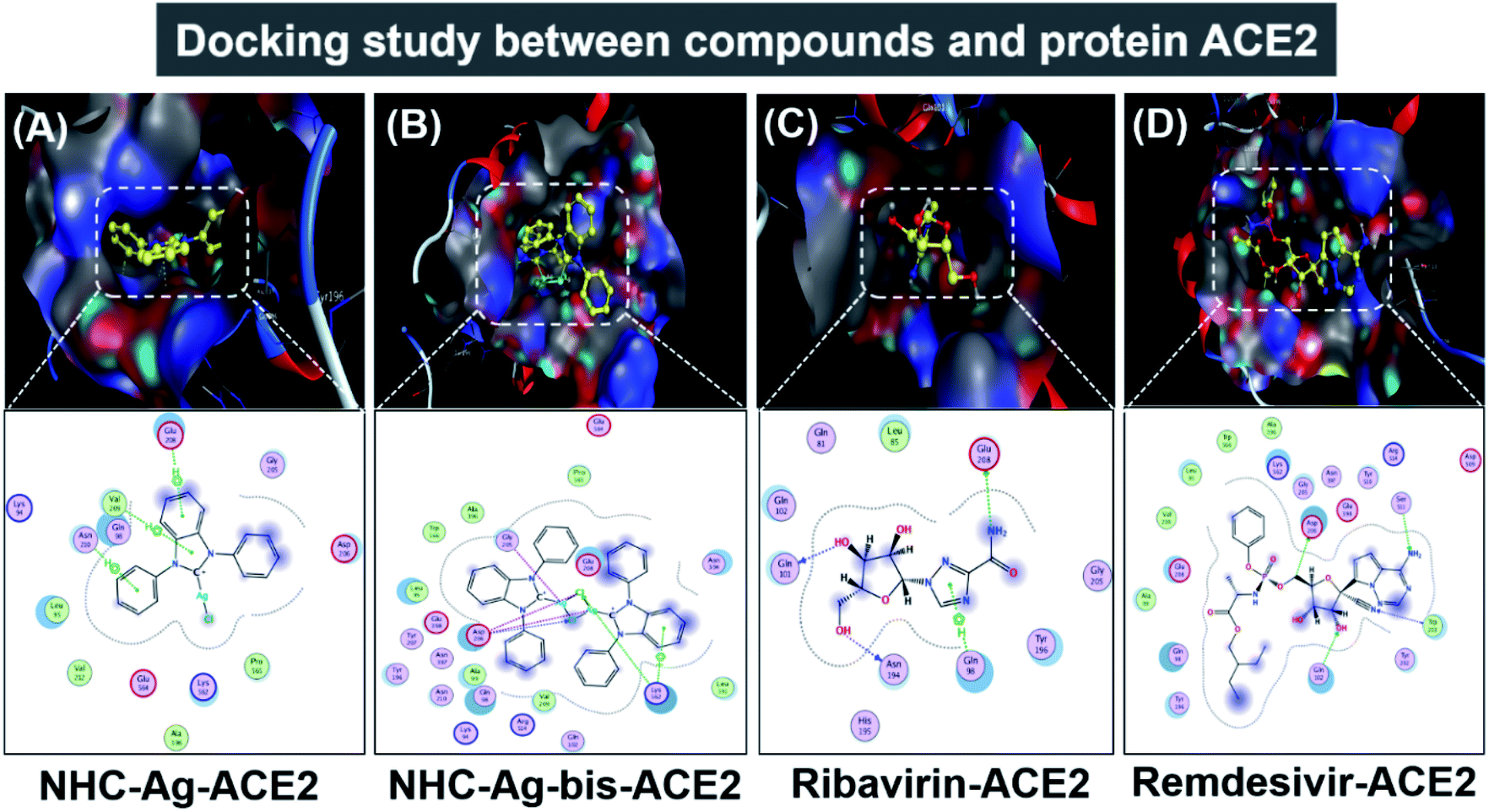
Molecular docking simulation was used to investigate the interactions of the silver–carbene NHC–Ag and bis-silver–carbene NHC–Ag-bis with two proteins, including ACE2 in the human body and PDB6LU7 of SARS-CoV-2. In detail, we determined whether or not the silver–carbene NHC–Ag and bis-silver–carbene NHC–Ag-bis interact with the ACE2 and PDBLU7 proteins and inhibit their actions, and weaken them compared to their original state. The docking score energy (DS) and root-mean-square deviation (RMSD) values as well as various interaction configurations via hydrogen bonds, cation–π, π–π bonds, and ionic interactions, site–site binding, and van der Waals interactions between the carbene complexes, two control drugs and ACE2 and PDB6LU7 proteins are presented in Fig. 9, 10, Tables 3 and 4.
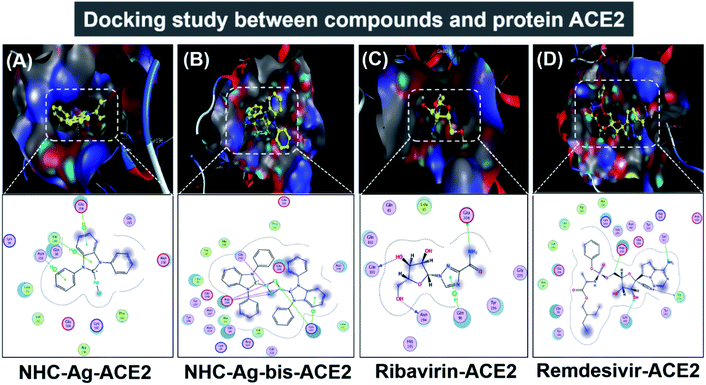 |
| | Fig. 9 Protein ACE2 docked with (A) silver carbene complex (NHC–Ag–ACE2), (B) bis-silver carbene complex (NHC–Ag-bis–ACE2), (C) ribavirin (Ribavirin–ACE2), and (D) remdesivir (Remdesivir–ACE2). | |
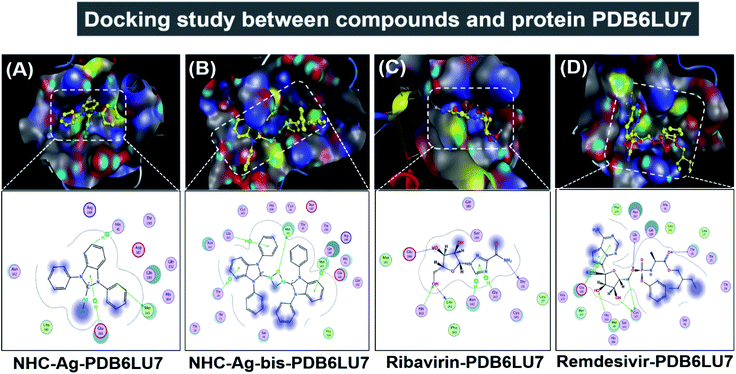 |
| | Fig. 10 Protein PDB6LU7 docked with (A) silver carbene complex (NHC–Ag–PDB6LU7), (B) bis-silver carbene complex (NHC–Ag-bis–PDB6LU7), (C) ribavirin (Ribavirin–PDB6LU7), and (D) remdesivir (Remdesivir–PDB6LU7). | |
Table 3 Docking simulation results with docking score energy (DS), root-mean-square deviation (RMSD), and van der Waals (VDW) interactions of NHC–Ag, NHC–Ag-bis, ribavirin, and remdesivir with the amino acids of the ACE2 and PDB6LU7 proteins
| Compound |
Symbol (compound–protein) |
DS (kcal mol−1) |
RMSD (Å) |
VDW interaction with amino acid |
| Compound – ACE2 |
| NHC–Ag |
NHC–Ag–ACE2 |
−14.1 |
1.13 |
Gln 98; Gly 205; Asp 206; Pro 565; Lys 562; Ala 396; Glu 564; Val 212; Leu 95; Lys 94 |
| NHC–Ag-bis |
NHC–Ag-bis–ACE2 |
−17.5 |
1.54 |
Glu 564; Pro 565; Asn 394; Leu 391; Gln 102; Val 209; Arg 514; Gln 98; Lys 94; Ala 99; Asn 210; Asn 397; Tyr 196; Tyr 207; Glu 398; Leu 95; Trp 566; Ala 396 |
| Ribavirin |
Ribavirin–ACE2 |
−16.5 |
1.54 |
Gln 102; Leu 85; Gln 81; Gly 205; Tyr 196; His 195 |
| Remdesivir |
Remdesivir–ACE2 |
−16.9 |
1.70 |
Tyr 196; Gln 98; Ala 99; Glu 208; Val 209; Leu 95; Trp 566; Ala 296; Lys 562; Gly 205; Asn 397; Glu 398; Arg 514; Tyr 510; Asp 509 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Compound – PDB6LU7 |
| NHC–Ag |
NHC–Ag–PDB6LU7 |
−14.1 |
1.75 |
Thr 190; Asp 187; Gln 189; Leu 141; Asn 142; Arg 188 |
| NHC–Ag-bis |
NHC–Ag-bis–PDB6LU7 |
−16.8 |
1.98 |
Asp 187; Glu 166; Arg 188; Pro 168; Ser 46; Thr 25; Thr 24; Asn 142; Cys 145; His 164; Cys 44; Thr 45; Thr 190; His 41; Gln 192 |
| Ribavirin |
Ribavirin–PDB6LU7 |
−16.6 |
1.52 |
Met 165; Phe 140; Leu 27; Gln 189; Cys 145; Ser 144 |
| Remdesivir |
Remdesivir–PDB6LU7 |
−16.9 |
1.67 |
Ser 144; Met 49; His 164; Met 165; His 172; Phe 140; Asn 142; Gln 189; His 41; Leu 141; Leu 27; Thr 25; Thr 24; Ser 46 |
Table 4 Molecular docking simulation results with critical interactions between the complexes and the two proteins (ACE2 and PDB6LU7), including interaction and distance, site–site binding, energy, cation–π, π–π bonds, ionic interactions, and total hydrogen bonds
| Symbol (compound–protein) |
Ligand |
Protein |
Interaction |
Distance (Å) |
Energy (kcal mol−1) |
Hydrogen bonds |
| NHC–Ag–ACE2 |
6-ring |
C |
Glu 208 |
π–H |
3.67 |
−0.6 |
3 |
| 5-ring |
C |
Val 209 |
π–H |
4.34 |
−1.8 |
| 6-ring |
C |
Asn 210 |
π–H |
3.78 |
−0.9 |
| NHC–Ag-bis–ACE2 |
Cl |
C |
Lys 562 |
H-acceptor |
3.74 |
−1.8 |
6 |
| Cl |
C |
Asp 206 |
H-acceptor |
3.77 |
−2.1 |
| Ag |
O |
Gly 205 |
Metal |
2.88 |
−1.3 |
| Ag |
O |
Asp 206 |
Metal |
2.75 |
−2.2 |
| Cl |
O |
Asp 206 |
Ionic |
3.38 |
−2.4 |
| 6-ring |
N |
Lys 562 |
π–cation |
3.82 |
−1.3 |
| Ribavirin–ACE2 |
O |
O |
Asn 194 |
H-donor |
2.89 |
−2.3 |
4 |
| O |
O |
Gln 101 |
H-donor |
2.93 |
−1.5 |
| N |
O |
Glu 208 |
H-donor |
3.35 |
−1.9 |
| 5-ring |
C |
Gln 98 |
π–H |
3.59 |
−0.6 |
| Remdesivir–ACE2 |
O |
N |
Asn 394 |
H-acceptor |
2.86 |
−2.4 |
6 |
| O |
N |
Arg 514 |
H-acceptor |
2.78 |
−1.7 |
| O |
N |
Arg 514 |
H-acceptor |
3.46 |
−1.6 |
| N |
N |
Asn 394 |
H-acceptor |
3.26 |
−0.9 |
| 5-ring |
5-ring |
His 401 |
π–π |
3.95 |
−0.1 |
| 6-ring |
5-ring |
His 401 |
π–π |
3.51 |
−0.3 |
| NHC–Ag–PDB6LU7 |
C |
S |
Met 165 |
H-donor |
3.84 |
−0.8 |
3 |
| C |
5-ring |
His 41 |
π–H |
3.91 |
−1.3 |
| 5-ring |
N |
Glu 166 |
π–H |
4.22 |
−2.5 |
| NHC–Ag-bis–PDB6LU7 |
Cl |
S |
Met 49 |
H-donor |
3.34 |
0.1 |
4 |
| 6-ring |
N |
Thr 26 |
π–H |
4.68 |
−0.7 |
| 6-ring |
N |
Gly 143 |
π–H |
4.51 |
−0.6 |
| 6-ring |
C |
Gln 189 |
π–H |
4.25 |
−0.7 |
| Ribavirin–PDB6LU7 |
O |
O |
Leu 141 |
H-donor |
3.14 |
−0.7 |
6 |
| N |
O |
Thr 26 |
H-donor |
3.13 |
−1 |
| O |
N |
His 163 |
H-acceptor |
3.1 |
−1.5 |
| O |
N |
Glu 166 |
H-acceptor |
3.01 |
−1.0 |
| 5-ring |
C |
Asn 142 |
π–H |
3.95 |
−1.0 |
| 5-ring |
N |
Gly 143 |
π–H |
3.28 |
−2.4 |
| Remdesivir –PDB6LU7 |
C |
S |
Cys 145 |
H-donor |
4.09 |
−0.7 |
7 |
| O |
S |
Cys 145 |
H-donor |
3.22 |
−2.1 |
| O |
N |
Thr 26 |
H-acceptor |
3.13 |
−2.8 |
| O |
N |
Gly 143 |
H-acceptor |
3.34 |
−0.7 |
| O |
N |
His 163 |
H-acceptor |
3.46 |
−0.7 |
| 5-ring |
N |
Glu 166 |
π–H |
4.17 |
−3 |
| 6-ring |
N |
Glu 166 |
π–H |
4.1 |
−3.1 |
Firstly, the docking simulations on the ACE2 protein were investigated, and the obtained results are presented in Fig. 9 and Tables 3 and 4. It can be seen that all the root-mean-square deviation values are smaller than 2 Å. The NHC–Ag-bis complex docked with the ACE2 protein exhibits a stronger inhibitory effect in comparison to the that derived from NHC–Ag with the docking score energy values of −17.5 kcal mol−1 and −14.1 kcal mol−1, respectively. This can be explained by the fact that NHC–Ag-bis possesses a larger volume and a higher molecular mass, leading to a significant polarizability and strong binding capacity with amino acids. Furthermore, Tables 3 and 4 show that the NHC–Ag complex forms 10 van der Waals interactions with the protein and 3 hydrogen bonds with its π–H bonds between the phenyl ring and N-heterocyclic of the carbene complex and –C– of different amino acids, including Glu 208 (3.67 Å), Val 209 (4.34 Å), and Asn 210 (3.78 Å). Significantly, NHC–Ag-bis exhibited excellent docking capacity on the ACE2 protein based on its interactions with the host amino acids via 18 van der Waals interactions and 6 chemical interactions. The latter includes hydrogen bonds in the cation–π bonds, π–π bonds, and ionic interactions. The NHC–Ag-bis bis-silver carbene complex with the carbon center atoms, AgCl metal compound, and phenyl rings as ligands mainly interact with the –O– and –C–, and –N– of the amino acids Lys 562 (3.74 Å), Asp 206 (3.77 Å), Gly 205 (2.88 Å), Asp 206 (2.75 Å), Asp 206 (3.38 Å), and Lys 562 (3.82 Å) in ACE2. Besides, ribavirin and remdesivir were successfully docked on the ACE2 protein by forming 6 and 15 van der Waals interactions, respectively. These drugs show strong interactions with the amino acids of the proteins including 4 interactions for Ribavirin–ACE2 and 6 for Remdesivir–ACE2. Indeed, the docking complex Ribavirin–ACE2 has a DS value of −16.5 kcal mol−1, RMSD of 1.45 Å, and site–site bonding interaction including –O–, –N–, and –N-heterocyclic of ribavirin and –O–, and –C– of the amino acids Asn 194 (2.89 Å), Gln 101 (2.93 Å), Glu 208 (3.35 Å), and Gln 98 (3.59 Å). The complex Remdesivir–ACE2 has a DS value of −16.9 kcal mol−1, RMSD of 1.70 Å, and site–site bonding interaction including –O–, –N–, N-aromatic ring, and N-heterocyclic of remdesivir and –N–, and N-aromatic rings of the amino acids Asn 394 (2.86 Å), Arg 514 (2.78 Å), Arg 514 (3.46 Å), Asn 394 (3.26 Å), His 401 (3.95 Å), and His 401 (3.51 Å).
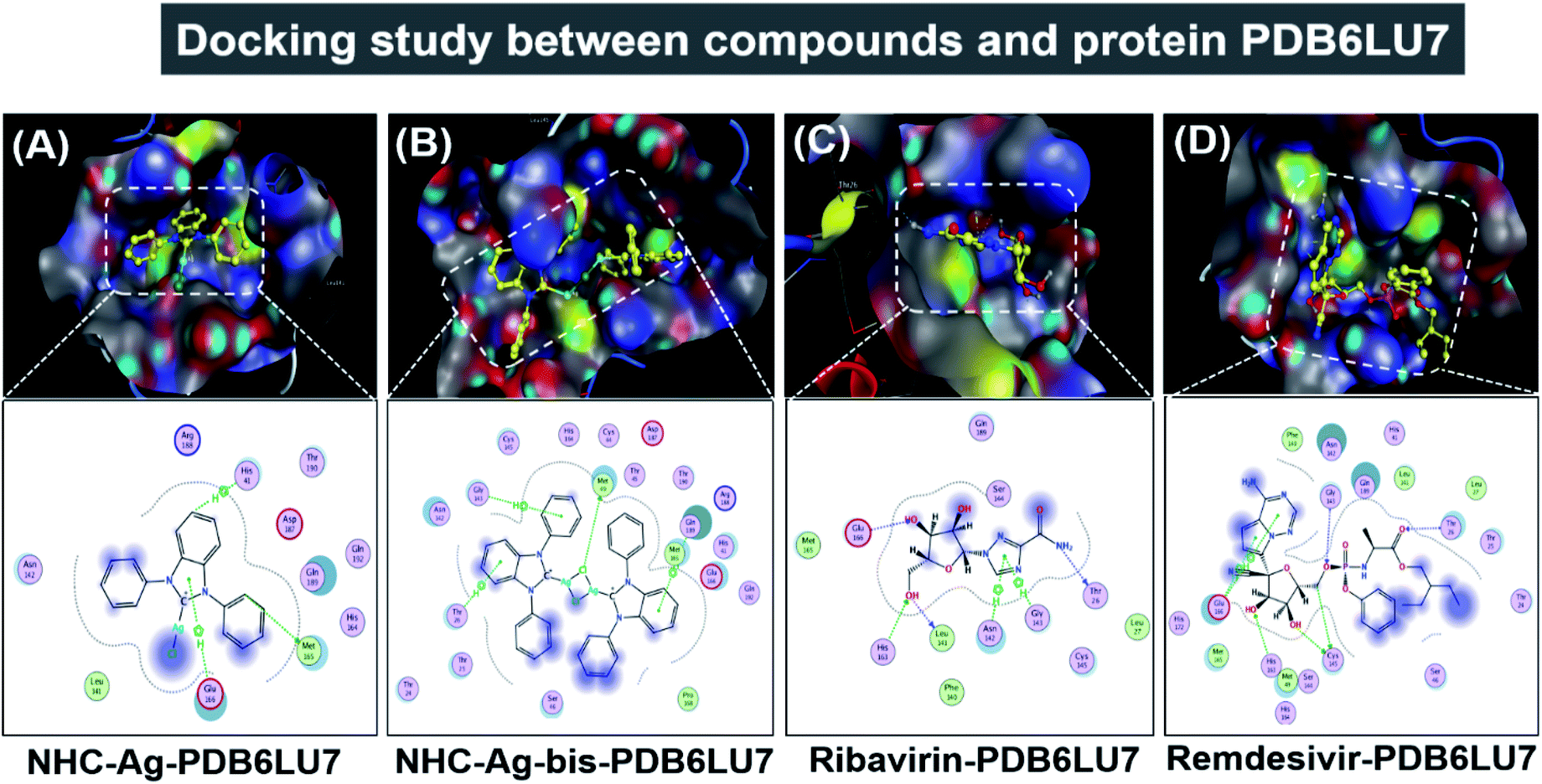
Secondly, the docking simulations for the PDB6LU7 protein are presented in Fig. 10, Tables 3 and 4. The docking score energy (DS) values are −14.1, −16.8, −16.6, and −16.9 kcal mol−1 for NHC–Ag, NHC–Ag-bis, ribavirin and remdesivir, respectively. The RMSD values vary between 1.52 and 1.98 Å. The NHC–Ag complex forms 6 van der Waals interactions and 3 hydrogen-bond interactions with PDB6LU7. The latter includes –C– in the N-heterocyclic ring of the carbene ligand interacting with the –S–, –N–, and N-aromatic rings, while the former embraces 3 hydrogen bond interactions with the PDB6LU7 amino acids, i.e. Met 165 (3.84 Å), His 41 (3.91 Å), and Glu 166 (4.22 Å). Tables 3 and 4 also reveal the slightly stronger inhibitory effect of the bis-silver–carbene adduct NHC–Ag-bis when it is docked with the PDB6LU7 protein. The obtained results include 15 van der Waals interactions and 4 hydrogen-bond interactions. In the latter, the hydrogen bond interaction contains strong interactions between AgCl, phenyl rings, N-heterocyclic rings of NHC–Ag-bis and H-donor and π–H bonds via –S–, –N–, and –C– of the amino acids Met 49 (3.34 Å), Thr 26 (4.68 Å), Gly 143 (4.51 Å), and Gln 189 (4.25 Å). This reveals the significant contribution of the N-heterocyclic and phenyl rings in the versatile bis-carbene ligands and AgCl metal compound with the presence of Ag+ and Cl− ions. Besides, the inhibition towards the PDB6LU7 protein by ribavirin and remdesivir resulted in the DS values of −16.6 and −16.9 kcal mol−1, respectively. This observation shows the relatively strong inhibitory effect of the two carbene complexes in comparison to the referenced drugs. This can be explained by the flexible properties of the compounds with –OH groups, and N-aromatic rings. Furthermore, the –NH, –O−, O-heterocyclic, –NH2, and N-heterocyclic groups strongly interact with the amino acids of the SARS-CoV-2 main protease PDB6LU7. The Ribavirin–PDB6LU7 complex forms 6 van der Waals interactions and 6 interactions of hydrogen bonds, including H-donors, H-acceptors, and π–H bonds of –O–, –N–, and five nitrogen aromatic interactions with –O–, –N–, and –C– of the amino acids Leu 141 (3.14 Å), Thr 26 (3.13 Å), His 163 (3.10 Å), Glu 166 (3.01 Å), Asn 142 (3.95 Å), and Gly 143 (3.28 Å). The Remdesivir–PDB6LU7 complex exhibits 14 van der Waals interactions and 7 hydrogen bonds including H-donors, H-acceptors, and π–H bonds of –O–, –C–, five aromatic nitrogen, and six aromatic carbon interactions with –S– and –N– of the amino acids Cys 145 (4.09 Å), Cys 145 (3.22 Å), Thr 26 (3.13 Å), Gly 143 (3.34 Å), His 163 (3.46 Å), Glu 166 (4.17 Å), and Glu 166 (4.10 Å).
Table 5 summarizes the docking parameters including DSaverage (kcal mol−1), polarizability (Å3) and total hydrogen bonds when each complex/drug is docked with the ACE2 and PDB6LU7 proteins. These parameters were calculated using the QSARIS system with the Gasteiger–Marsili method.67 The ACE2 and PDB6LU7 inhibition ability of NHC–Ag, NHC–Ag-bis, ribavirin, and remdesivir is expressed by the average DS values of −14.1, −17.2, −16.6, and −16.9 kcal mol−1, respectively. These indicate that all the compounds can be considered to exhibit strong inhibitory effects on ACE2 and PDB6LU7. The justification is based on their relative significance in comparison to the DS values representing the inhibitory capability of other duo-systems previously reported in the literature.42,43,68,69 In addition, the polarizability values of NHC–Ag-bis and remdesivir are significantly higher (i.e. 71.3 and 57.1 Å3) than the corresponding values for NHC–Ag and ribavirin (35.7 and 21.1 Å3). The reason for this may be related to the fact that they possess the largest molecular mass and volume, which lead to high binding capacity with amino acids. Although NHC–Ag and ribavirin possess a smaller molecular mass and polarizability values, they also exhibit relatively strong site–site binding due to their hydrogen bond interactions, i.e. 6 and 10 hydrogen bonds, respectively. In summary, the results imply a feasible correlation between the structures of the silver–carbene and bis-silver–carbene complexes, and anti-SARS-CoV-2 activities considering their docking capability towards both the virus main protease PDB6LU7 and the host complementary receptor ACE2.
Table 5 Molecular docking parameters including the average docking score energy values (DSaverage, kcal mol−1), polarizability (Å3), and total hydrogen bonds of NHC–Ag, NHC–Ag-bis, ribavirin and remdesivir docked with the ACE2 and PDB6LU7 proteins
| Compound |
DSaverage |
Polarizability |
Hydrogen bond |
| NHC–Ag |
−14.1 |
35.7 |
6 |
| NHC–Ag-bis |
−17.2 |
71.3 |
10 |
| Ribavirin |
−16.6 |
21.1 |
10 |
| Remdesivir |
−16.9 |
57.1 |
13 |
Conclusions
The calculated equilibrium structures of the NHC–Ag and NHC–Ag-bis complexes reveal that the NHC tetrylene ligands bond in a head-on mode to the AgCl and (AgCl)2 bis-silver chloride fragments, while the NHE (E = Si and Ge) ligands bond in a side-on configuration to AgCl and (AgCl)2. The calculated BDEs suggest that the NHE–AgCl bond strength decreases in the order of NHC–Ag > NHSi–Ag > NHGe–Ag. Meanwhile, the corresponding calculation implies that the bis-silver chloride–NHE bond strength decreases in the order of NHC–Si-bis > NHGe–Ag-bis > NHC–Ag-bis. The NBO analysis indicates that [NHEPh→AgCl] and [(NHEPh)2→(AgCl)2] donations of the complexes are mainly derived from the σ- and π-contributions of the ligands.
The docking simulation results suggest that the studied carbene complexes (NHC–Ag and NHC–Ag-bis) are active for inhibiting the host receptor ACE2 and PDB6LU7 protein of SARS-CoV-2. However, NHC–Ag-bis has stronger inhibitory effects on both the ACE2 and PDB6LU7 proteins. The NHC–Ag-bis bis-silver–carbene complex, ribavirin, and remdesivir exhibit similar efficacy in inhibiting either ACE2 or PDB6LU7. Their corresponding docking score energies are −17.5 to −16.5 kcal mol−1 and −16.9 to −16.6 kcal mol−1, respectively. The root-mean-square deviation values are always less than 2 Å in all the calculated systems. The structures of the potential drugs fit well with the site–site binding of the host receptor ACE2 and the viral protease PDB6LU7 based on the hydrogen bond interactions. Interestingly, the results also indicate that the inhibitory ability of potential drugs on the ACE2 and PDB6LU7 proteins seems to be correlated among the average of docking score energy, site–site active interactions of the potential complexes/drugs-proteins, and polarizability of the potential carbene and bis-carbene complexes and drugs. Thus, the obtained results in this study suggest that the NHC–Ag carbene complex and NHC–Ag-bis bis-carbene adduct are promising complexes, which can serve as a reference source of data for further research on the development of new agents to inhibit the host receptor ACE2 and the main protease PDB6LU7 of SARS-CoV-2.
The results in this study are highly conducive for the preparation of therapeutic drugs for SARS-CoV-2 considering the theoretical demonstration of the stability of the potential drugs based on the DFT results and their inhibitory effectiveness by molecular docking simulation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Nguyen Thi Ai Nhung thanks Prof. Dr Gernot Frenking for allowing the continuous use of her own resources within Frenking's group. The programs used in the studies were run via the Erwin/Annemarie clusters operated by Reuti (Thomas Reuter) at the Philipps-Universität Marburg, Germany. This research is funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.06-2017.303.
Notes and references
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef
 .
. - A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913–921 CrossRef CAS .
- L. Benhamou, E. Chardon, G. Lavigne, S. p. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed .
- M.-T. Lee and C.-H. Hu, Organometallics, 2004, 23, 976–983 CrossRef CAS .
- V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714–5724 CrossRef CAS PubMed .
- W. Hermann, T. Weskamp and V. P. Bohm, Adv. Organomet. Chem., 2001, 48, 1–71 CrossRef .
- J. P. Fackler, Inorg. Chem., 2002, 41, 6959–6972 CrossRef CAS PubMed .
- A. J. Arduengo III, H. R. Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405–3409 CrossRef .
- A. Liu, X. Zhang, W. Chen and H. Qiu, Inorg. Chem. Commun., 2008, 11, 1128–1131 CrossRef CAS .
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed .
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS .
- A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 CrossRef CAS .
- S. Roland, C. Jolivalt, T. Cresteil, L. Eloy, P. Bouhours, A. Hequet, V. Mansuy, C. Vanucci and J. M. Paris, Chem.–Eur. J., 2011, 17, 1442–1446 CrossRef CAS .
- W. Streciwilk, J. Cassidy, F. Hackenberg, H. Mueller-Bunz, F. Paradisi and M. Tacke, J. Organomet. Chem., 2014, 749, 88–99 CrossRef CAS .
- C. N. Banti and S. K. Hadjikakou, Metallomics, 2013, 5, 569–596 RSC .
- L. Eloy, A. S. Jarrousse, M. L. Teyssot, A. Gautier, L. Morel, C. Jolivalt, T. Cresteil and S. Roland, ChemMedChem, 2012, 7, 805–814 CrossRef CAS PubMed .
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042–15053 CrossRef CAS PubMed .
- M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer and R. Mahiou, Dalton Trans., 2009, 6894–6902 RSC .
- A. Melaiye, R. S. Simons, A. Milsted, F. Pingitore, C. Wesdemiotis, C. A. Tessier and W. J. Youngs, J. Med. Chem., 2004, 47, 973–977 CrossRef CAS PubMed .
- R. A. Haque, S. Budagumpi, H. Z. Zulikha, N. Hasanudin, M. B. K. Ahamed and A. M. A. Majid, Inorg. Chem. Commun., 2014, 44, 128–133 CrossRef CAS .
- S. Budagumpi, R. A. Haque, S. Endud, G. U. Rehman and A. W. Salman, Eur. J. Inorg. Chem., 2013, 2013, 4367–4388 CrossRef CAS .
- C. L. Cannon, L. A. Hogue, R. K. Vajravelu, G. H. Capps, A. Ibricevic, K. M. Hindi, A. Kascatan-Nebioglu, M. J. Walter, S. L. Brody and W. J. Youngs, Antimicrob. Agents Chemother., 2009, 53, 3285–3293 CrossRef CAS PubMed .
- W. J. Youngs, A. R. Knapp, P. O. Wagers and C. A. Tessier, Dalton Trans., 2012, 41, 327–336 RSC .
- D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53–57 RSC .
- T. A. Al-Allaf, M. T. Ayoub and L. J. Rashan, J. Inorg. Biochem., 1990, 38, 47–56 CrossRef CAS .
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed .
- W. A. Herrmann, K. Öfele, M. Elison, F. E. Kühn and P. W. Roesky, J. Organomet. Chem., 1994, 480, c7–c9 CrossRef CAS .
- K. Lv and D. Cui, Organometallics, 2008, 27, 5438–5440 CrossRef CAS .
- İ. Özdemir, E. Ö. Özcan, S. Günal and N. Gürbüz, Molecules, 2010, 15, 2499–2508 CrossRef PubMed .
- M. L. Agostini, E. L. Andres, A. C. Sims, R. L. Graham, T. P. Sheahan, X. Lu, E. C. Smith, J. B. Case, J. Y. Feng and R. Jordan, mBio, 2018, 9, e00221–00218 CrossRef CAS PubMed .
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li and C.-L. Huang, Nature, 2020, 1–4 Search PubMed .
- A. Zumla, J. F. Chan, E. I. Azhar, D. S. Hui and K.-Y. Yuen, Nat. Rev. Drug Discovery, 2016, 15, 327 CrossRef CAS .
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res., 2020, 30, 269–271 CrossRef CAS PubMed .
- J. S. Khalili, H. Zhu, N. S. A. Mak, Y. Yan and Y. Zhu, J. Med. Virol., 2020, 92, 740–746 CrossRef CAS PubMed .
- J. Fischer, C. R. Ganellin and D. P. Rotella, Analogue-based Drug Discovery III, John Wiley & Sons, 2012 Search PubMed .
- W. H. Organization, World Health Organization model list of essential medicines: 21st list 2019, World Health Organization, 2019 Search PubMed .
- M. K. Lo, R. Jordan, A. Arvey, J. Sudhamsu, P. Shrivastava-Ranjan, A. L. Hotard, M. Flint, L. K. McMullan, D. Siegel and M. O. Clarke, Sci. Rep., 2017, 7, 43395 CrossRef PubMed .
- T. P. Sheahan, A. C. Sims, R. L. Graham, V. D. Menachery, L. E. Gralinski, J. B. Case, S. R. Leist, K. Pyrc, J. Y. Feng and I. Trantcheva, Sci. Transl. Med., 2017, 9, eaal3653 CrossRef PubMed .
- C. Tikellis and M. Thomas, Int. J. Pept., 2012, 2012, 1–8 CrossRef PubMed .
- A. Roberts, D. Deming, C. D. Paddock, A. Cheng, B. Yount, L. Vogel, B. D. Herman, T. Sheahan, M. Heise and G. L. Genrich, PLoS Pathog., 2007, 3, e5 CrossRef PubMed .
- Homo Sapiens (Human), https://www.uniprot.org/uniprot/Q9BYF1 Search PubMed.
- R. R. Narkhede, R. S. Cheke, J. P. Ambhore and S. D. Shinde, Eurasian Journal of Medicine and Oncology, 2020, 4, 185–195 Search PubMed .
- T.-D. Tran, T.-C.-V. Nguyen, N.-S. Nguyen, D.-M. Nguyen, T.-T.-H. Nguyen, M.-T. Le and K.-M. Thai, Appl. Sci., 2016, 6, 198 CrossRef .
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji and X. Li, Gaussian 09, Gaussian Inc, Wallingford CT, 2009 Search PubMed .
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef CAS .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed .
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef .
- B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS .
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC .
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS .
- I. Lukovits, E. Kalman and F. Zucchi, Corrosion, 2001, 57, 3–8 CrossRef CAS .
- O. Tarasova, V. Poroikov and A. Veselovsky, Molecules, 2018, 23, 1233 CrossRef PubMed .
- T. M. C. Babu, S. S. Rajesh, B. V. Bhaskar, S. Devi, A. Rammohan, T. Sivaraman and W. Rajendra, RSC Adv., 2017, 7, 18277–18292 RSC .
- T.-D. Ngo, T.-D. Tran, M.-T. Le and K.-M. Thai, Mol. Diversity, 2016, 20, 945–961 CrossRef CAS PubMed .
- K.-M. Thai, D.-P. Le, T.-D. Tran and M.-T. Le, J. Theor. Biol., 2015, 385, 31–39 CrossRef CAS PubMed .
- The crystal structure of COVID-19 main protease in complex with an inhibitor N3, https://www.wwpdb.org/pdb?id=pdb_00006lu7, initial deposition on: 26 January 2020 Search PubMed.
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS .
- D. Nemcsok, K. Wichmann and G. Frenking, Organometallics, 2004, 23, 3640–3646 CrossRef CAS .
- T. A. N. Nguyen and G. Frenking, Chem.–Eur. J., 2012, 18, 12733–12748 CrossRef CAS PubMed .
- T. A. N. Nguyen, D. S. Tran, T. P. L. Huynh, T. H. Le, T. Q. Duong, T. T. Nguyen, T. C. Vo, V. T. Pham and T. H. Dang, Z. Anorg. Allg. Chem., 2017, 643, 826–838 CrossRef CAS .
- T. A. N. Nguyen, T. P. L. Huynh and V. T. Pham, J. Vietnam. Environ., 2014, 6, 142–148 Search PubMed .
- T. A. N. Nguyen, T. P. L. Huynh, T. Q. Duong, D. S. Tran and T. H. Dang, Z. Phys. Chem., 2017, 231, 1467–1487 CAS .
- B. Rosenberg, Nature, 1962, 193, 364–365 CrossRef CAS PubMed .
- V. Kharkyanen, E. Petrov and I. Ukrainskii, J. Theor. Biol., 1978, 73, 29–50 CrossRef CAS PubMed .
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC .
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS .
- T. H. M. Vu, D. D. Vu, D. Van Truong, P. T. V. Nguyen and D. T. Tran, MedPharmRes, 2017, 1, 26–36 Search PubMed .
- R. Yu, L. Chen, R. Lan, R. Shen and P. Li, Int. J. Antimicrob. Agents, 2020, 106012 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05159d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tran Thi Ai Mya,
Duong Tuan Quangb,
Bui Thi Phuong Thuy*c,
Vo Duy Nhand,
Phan Tu Quye,
Pham Van Tatf,
Duy Quang Dao
*a,
Tran Thi Ai Mya,
Duong Tuan Quangb,
Bui Thi Phuong Thuy*c,
Vo Duy Nhand,
Phan Tu Quye,
Pham Van Tatf,
Duy Quang Dao