
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntriguing enigma of nitrobenzofuroxan's ‘Sphinx’: Boulton–Katritzky rearrangement or unusual evidence of the N-1/N-3-oxide rearrangement?†
Gabriele Micheletti *a,
Leonardo Iannuzzoa,
Matteo Calvaresib,
Silvia Bordonia,
Dario Telesea,
Elena Chugunovacd and
Carla Boga*a
*a,
Leonardo Iannuzzoa,
Matteo Calvaresib,
Silvia Bordonia,
Dario Telesea,
Elena Chugunovacd and
Carla Boga*a
aDepartment of Industrial Chemistry ‘Toso Montanari’, Alma Mater Studiorum – Università di Bologna Viale Del Risorgimento, 4 402136 Bologna, Italy. E-mail: gabriele.micheletti3@unibo.it; carla.boga@unibo.it
bDepartment of Chemistry ‘G. Ciamician’, Alma Mater Studiorum-Università di Bologna, Via F. Selmi 2, Bologna 40126, Italy
cArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, Akad. Arbuzov st. 8, Kazan, Tatarstan 420088, Russia
dLaboratory of Plant Infectious Diseases, FRC Kazan Scientific Center of Russian Academy of Sciences, Lobachevskogo st. 2/31, Kazan, Tatarstan 420111, Russia
First published on 18th September 2020
Abstract
The SEAr/SNAr reaction between 7-chloro-4,6-dinitrobenzofuroxan (ClDNBF) and 2-morpholinyl-, 2-piperidinyl-, or 2-pyrrolidinylthiazole afforded unexpectedly two isomeric products, bearing the benzofuroxanyl moiety bound to the C-5 carbon atom of the thiazole ring. The relative ratio for the two isomers was dependent on temperature and solvent, suggesting the occurrence of an equilibrium between the two novel species. In order to investigate their structure and to design a plausible mechanistic pathway, a series of synthetic and spectroscopic experiments was planned. The isomer's structure was unambigously assigned when the reduction of furoxanyl to the furazanyl ring of the products gave exclusively a single species whose NMR data were coincident with those obtained by reacting the starting 2-aminothiazole derivatives with the 7-chloro-4,6-dinitrobenzofurazan (ClDNBZ). Possible mechanistic pathways might involve N-1-/N-3 oxide tautomerism or Boulton–Katritzky rearrangement and the current study is the first attempt to compare these two reactions. The data collected agree with the first one and DFT calculations permitted also a significant correlation with 13C NMR experimental data and the assignment of the structure of each isomer. Finally, only one Meisenheimer intermediate for each electrophile/nucleophile combination was isolated by coupling the 2-aminothiazole derivatives with 4,6-dinitrobenzofuroxan (DNBF).
Introduction
Benzofuroxan and its derivatives belong to a class of heterocycles of wide and growing interest both in mechanistic and applied fields.1–4 They can be exploited for many applications, ranging from energetic materials5,6 to biologically active compounds, depending on the type of substituents. As bioactive compounds, they can behave as anti-parasitic, anti-microbial, anti-fungal, immunosuppressive and anticancer agents, and have anti-aggregating and vasorelaxant activity owing to their ability to release NO under physiological conditions.7,8Since its discovery,9 the benzofuroxanyl ring has been a considerable challenge to structure attribution.10,11 First, benzofuroxans were proposed to exhibit a dioxime peroxide or o-dinitroso skeleton configuration, until 1912 when the current benzo[1,2-c]1,2,5-oxadiazole N-oxide formulation was suggested,12 which was unequivocally confirmed only 50 years later through NMR and X-ray crystal structure determinations.13,14
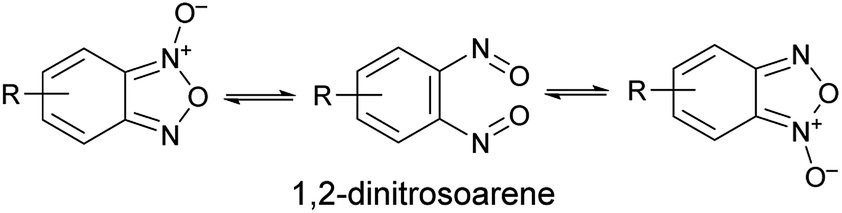
Benzofuroxan derivatives are still an interesting source of peculiar structural features. The first one is the possibility to undergo isomerisation between the N-1-oxide and N-3-oxide form, a transformation that is believed to occur via 1,2-dinitrosoarene as transient intermediate (Scheme 1).
 | ||
| Scheme 1 N-1-Oxide/N-3-oxide tautomerism in benzofuroxan (and its derivatives). | ||
Since the N-1-oxide/N-3-oxide tautomerism is a fast equilibrating system at room temperature, the 1,2-dinitrosoarene constitutes a fugitive species. The existence of this elusive species has been supported by both kinetic studies15,16 and theoretical calculations.17,18 Spectroscopic indications (IR and UV/Vis spectra) of its presence were obtained by photolysis of benzofuroxan in argon matrices at 14 K.19 Further, the reaction products recovered when benzofuroxan was reacted with acetonylmethyl sulfide,20 p-anisyl azide and diphenyldiazomethane21 were explained invoking the intermediacy of 1,2-dinitrosoarene species. A similar dinitroso derivative was also substantiated in the work of Terrier et al., that reported the recovery and characterization of a diadduct from a Diels–Alder cycloaddition as result of the reaction with a o-dinitroso species.22 Recently, a ruthenium complex of the 1,2-dinitrosoarene intermediate derived from the opening of the pentatomic ring of benzofuroxan has been reported.23
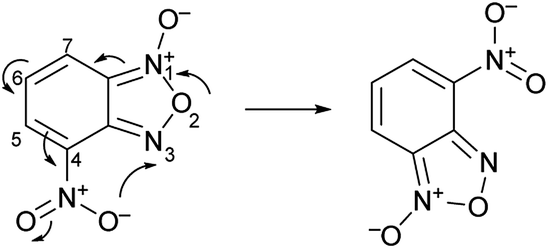
The second characteristic feature of substituted benzofuroxans is their susceptibility to undergo Boulton–Katritsky rearrangement (BKR)24,25 (Scheme 2). The BKR of 4-nitrobenzofuroxan can be considered as a prototype reaction for a class of molecular rearrangements and can compete with the N-1-oxide/N-3-oxide tautomerization; the direction of this rearrangement depends on the substituent in 5 or 7 position.26,27
 | ||
| Scheme 2 BKR rearrangement of 4-nitrobenzofuroxan. | ||
A third peculiar aspect of benzofuroxans resides into their 10π-electron structure which is able to confer electrophilic properties.
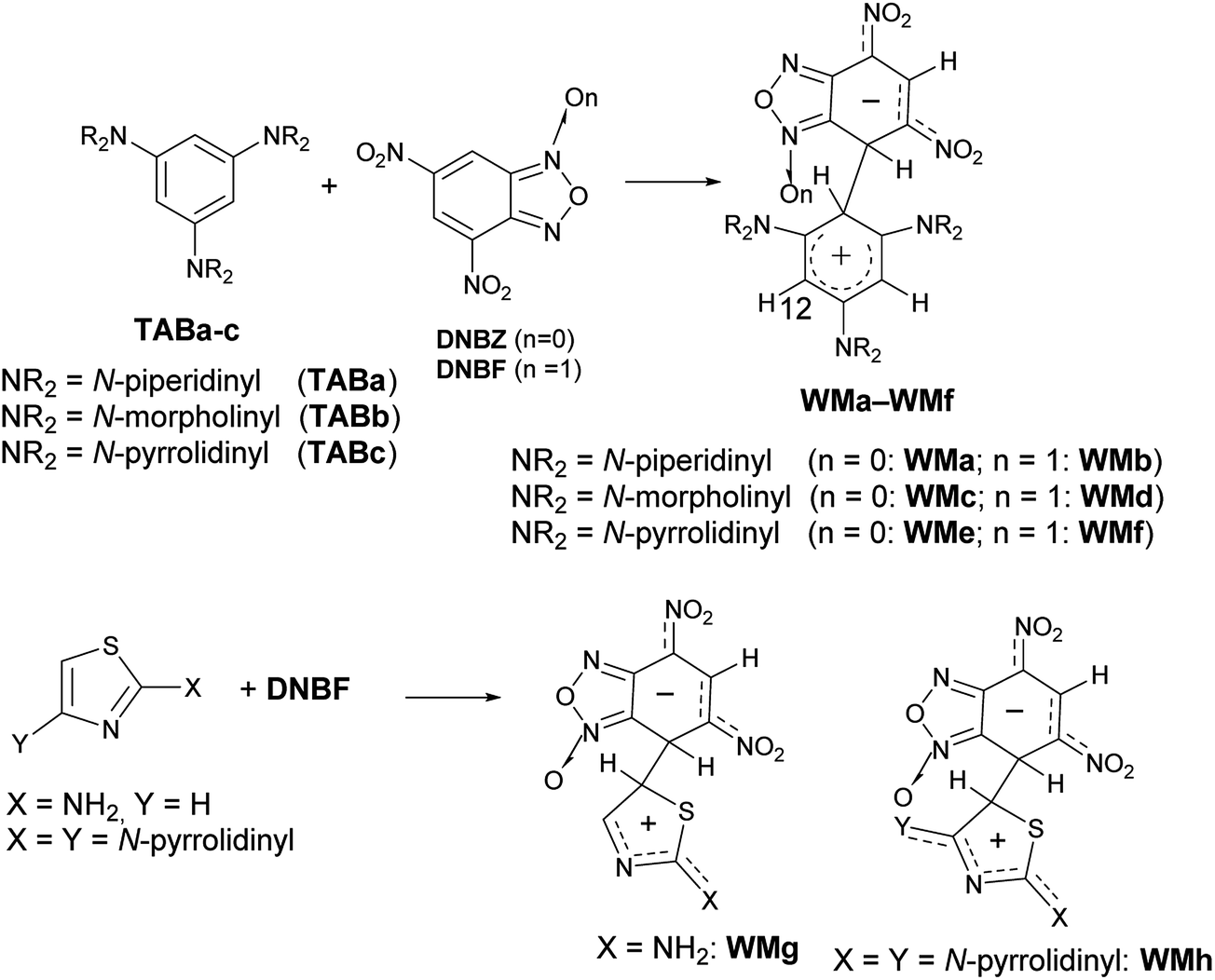
In particular, when two nitro substituents are bound to the carbocyclic ring of the benzofuroxan (or benzofurazan) or their derivatives, these compounds are so much electrophilic to be defined (labeled) as superelectrophiles.28,29 Actually, it is well-known4,30–35 that the reaction of 4,6-dinitrobenzofuroxan (DNBF) produces stable Meisenheimer intermediates, as well as 4,6-dinitrobenzofurazan (DNBZ), with a series of charged or neutral nucleophiles. Recently, the coupling of 1,3,5-triaminobenzene derivatives (TABa-c) or 2-aminothiazoles with a series of electrophiles,35–38 including DNBF or DNBZ, gave evidence of the zwitterionic intermediate – contemporaneously Wheland and Meisenheimer – formed in these SEAr/SNAr aromatic substitution reactions (e.g. WMa-h in Scheme 3). In Scheme 3 we represented the N–O position of WM intermediates on the basis of X-ray diffraction analysis on WMh.
 | ||
| Scheme 3 Examples of WM intermediates detected and characterized using DNBF or DNBZ as electrophiles. | ||
In the thiazole series in particular, the coupling between 2,4-dipyrrolidinylthiazole and DNBF produced the intermediate WMh (Scheme 3, bottom), which was stable enough to provide the first X-ray crystal structure along the class of zwitterionic intermediates, likely due to the high stabilization of the positive charge conferred to the Wheland moiety by both the pyrrolidinyl groups.38 On the other hand, 2,4-dipyrrolidinyl thiazole is such an activated nucleophile that gives a very complicated mixture of products when reacted with electrophiles such as aryldiazonium salts.39
On the contrary, when 2-pyrrolidinylthiazole reacted with aryldiazonium salts, the corresponding azo derivatives bound in position 5 (ref. 39) of the thiazole ring were formed in good yields.
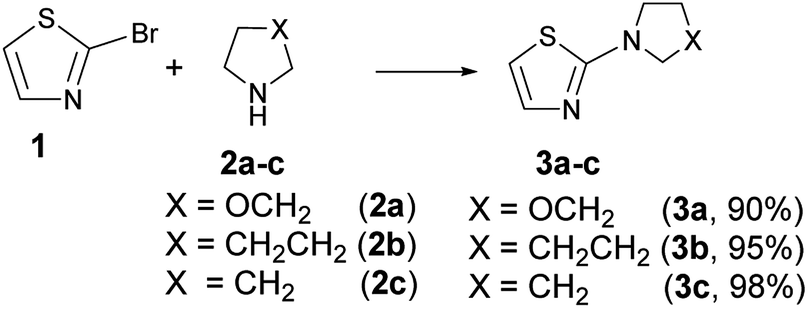
A literature survey revealed that both the 2-pyrrolidinylthiazole and the analogous 2-piperidinyl- and 2-morpholinyl- derivatives (3a–c in Scheme 4) have been poorly studied so far.
 | ||
| Scheme 4 Synthesis of 2-aminothiazole derivatives 3a–c from 2-bromothiazole (1) and amines 2a–c. | ||
Since it is well known that chloronitro- or chlorodinitro-benzofuroxans and -benzofurazans easily give C–C coupling with many nucleophiles30,31,36,40–42 producing a plethora of compounds of interest in applied fields (e.g. pharmaceutical, energetic, optoelectronic), we designed to carry out the reaction of the 2-aminothiazole derivatives with 7-chloro-4,6-dinitrobenzofuroxan (ClDNBF) or 7-chloro-4,6-dinitrobenzofurazan (ClDNBZ) to obtain novel hybrids bearing two moieties, the thiazolyl- and the benzofuroxanyl (or benzofurazanyl-) one.43
Inspired by these considerations, we focused our investigations on the above reactions to obtain novel highly conjugated systems and to exploit new insights on their mechanistic behavior.
Results and discussion
2-Morpholinylthiazole (3a), 2-piperidinylthiazole (3b) and 2-pyrrolidinylthiazole (3c) have been synthesized at room temperature and under solvent-free conditions by reacting 2-bromothiazole (1) with morpholine (2a), piperidine (2b), or pyrrolidine (2c), respectively (Scheme 4).Compounds 3a–c have been synthesized within 24, 4 and 2 h reaction time affording 90, 95 and 98% yield respectively; the different reaction time required to reach the above indicated yields is likely due to the different nucleophilicity of the starting amines whose values, according to the nucleophilicity scale developed by Mayr,44–47 are, in acetonitrile at 20 °C, NMayr = 15.65, 17.35, and 18.64 for morpholine, piperidine, and pyrrolidine, respectively.48
It is known that 2-aminothiazoles can behave as nucleophiles at three distinct positions, namely, exocyclic nitrogen atom, endocyclic nitrogen atom, and C-5. However, the selected substrates 3a–c bear a secondary amino group in position 2 that prevents the tautomeric equilibrium occurring when a –NH2 or –NHR group is present in that position. Thus, only the products 5a–c derived from the nucleophilic attack at C-5 were expected to be formed (Scheme 5), as previously found for aryldiazonium salts.39
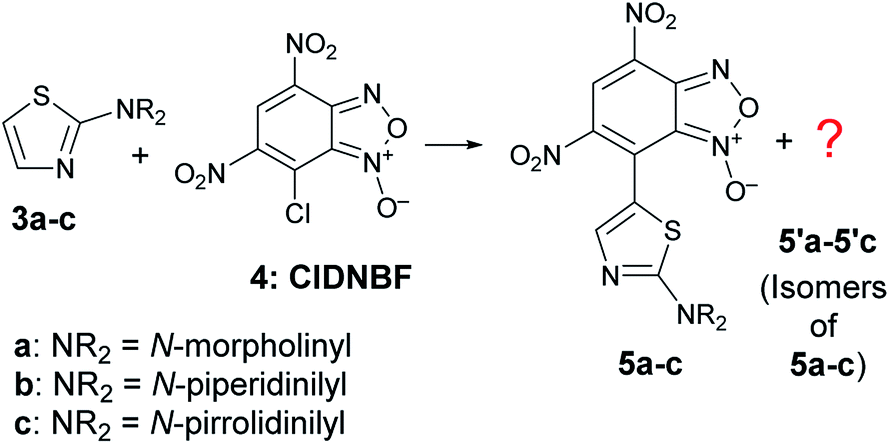 | ||
| Scheme 5 C–C coupling reaction between 3a–c and 4 with formation of the expected products 5a–c together with unexpected isomers. | ||
As soon as the reaction between 3a–c and 7-chloro-4,6-dinitrobenzofuroxan (4, or ClDNBF), run at room temperature in acetonitrile with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, started, the color of the mixture turned from pale yellow to deep blue.
:1 molar ratio, started, the color of the mixture turned from pale yellow to deep blue.
The reaction was monitored by TLC, showing the spot relative to novel blue species at RF higher than those related to the starting reactants. After 24 h (for the combination 3a/4), 12 h (for 3b/4), or 4 h (for 3c/4), the crude product obtained was subjected to column chromatography on silica gel (FC) leading to isolation of the novel species.
The ESI-MS spectrum of the blue solid isolated by FC was in agreement with that of a product derived from the aromatic substitution between 3a–c and 4, but the 1H- and 13C-NMR spectrum showed a number of signals higher with respect to those expected for species 5a–c. In particular, the 1H NMR spectrum in CD3CN of the solid derived from the reaction between 3c and 4 exhibited four singlets in the aromatic region (Fig. S30, in ESI†). The signal multiplicity and the relative integration suggested the presence of two isomers in 62/38 relative molar ratio bearing the benzofuroxanyl moiety bound to the C-5 of the thiazole moiety, labeled as 5c and 5′c in Scheme 5. After many attempts, the separation of two blue spots was achieved on preparative TLC plate by eluting with a mixture of diethyl ether: light petroleum (7:3 v/v). Then, each spot was scraped and worked-up (see Experimental), and the organic residues were dissolved in CD3CN and analyzed by NMR spectroscopy. Surprisingly, each spectrum showed presence of two compounds, as previously observed after FC, thus suggesting the occurrence of a re-equilibration phenomenon.
This prompted us to plan further experiments to shed some light on this behavior and, for the sake of clarity, below we report in sub-headings the results obtained.
Is the isomeric ratio affected by solvent nature or temperature?
First, we investigated whether the relative ratio between the two isomers 5 and 5′ could be influenced by the reaction conditions such as the solvent or the temperature. For this purpose, we planned to investigate the behavior of the isomeric mixture 5c + 5′c derived from the combination of 3c and 4, since in case c the reaction occurs in a considerably shorter time.A first series of experiments has been run by recording the 1H NMR spectrum of the isomeric mixture, after its isolation by FC, in different solvents at 25 °C. We attributed the two set of signals to the isomers labelled as A and A′ without direct relation to the structure 5c or 5′c of Scheme 5. The results are reported in Table 1.
| Entry | Solvent polarityb | Solvent | Isomerc A (%) | Isomer A′c (%) |
|---|---|---|---|---|
| a After isolation by FC.b Data from ref. 49.c Calculated from the 1H NMR spectrum recorded at 25 °C. | ||||
| 1 | Benzene 0.111 | Benzene-d6 | 53 | 47 |
| 2 | THF 0.207 | Tetrahydrofuran-d8 | 50 | 50 |
| 3 | CHCl3 0.259 | Chloroform-d1 | 55 | 45 |
| 4 | Acetone 0.355 | Acetone-d6 | 57 | 43 |
| 5 | CH3CN 0.460 | Acetonitrile-d3 | 60 | 40 |
| 6 | DMSO 0.444 | DMSO-d6 | 67 | 33 |
| 7 | CH3OH 0.762 | Methanol-d4 | 59 | 41 |
From the relative integration of the signals of the two species, and by supposing the same trend of related chemical shift along the solvents, it can be evinced that their relative ratio is always in favor of the same isomer (except in THF-d8, entry 2 of Table 1). The most relevant difference was observed in DMSO-d6 (entry 6 of Table 1). On going from THF to acetonitrile (entries 2–5 of Table 1) it emerge that the relative ratio is slightly dependent from the solvent even if, from the data obtained, a clear correlation with the solvent polarity cannot be advanced.
In a second series of experiments we analyzed the 1H NMR spectrum of the mixture A + A′, isolated by FC, at variable temperature and in different solvents; the results are reported in Table 2.
| Entry | Solvent | Temperature (°C) | Isomer Ab (%) | IsomerA′b (%) |
|---|---|---|---|---|
| a After isolation by FC.b Calculated from the 1H NMR spectrum. | ||||
| 1 | DMSO-d6 | 25 | 67 | 33 |
| 2 | 35 | 65 | 35 | |
| 3 | 50 | 64 | 36 | |
| 4 | 70 | 60 | 40 | |
| 5 | 80 | 57 | 43 | |
| 6 | CD3CN | 25 | 62 | 38 |
| 7 | 35 | 61 | 39 | |
| 8 | 45 | 59 | 41 | |
| 9 | 55 | 58 | 42 | |
| 10 | 65 | 58 | 42 | |
| 11 | CD3COCD3 | 25 | 57 | 43 |
| 12 | 0 | 60 | 40 | |
| 13 | −20 | 60 | 40 | |
| 14 | −50 | 60 | 40 | |
| 15 | −90 | 60 | 40 | |
In DMSO-d6 the spectrum of the mixture was recorded from 25 °C to 80 °C (Table 2, entries 1–5): on raising the temperature, a gradual increase (until 15%) of the minor isomer was observed. On the contrary, no significant variation of the isomeric ratio was observed both in acetonitrile (from 25 °C to 40 °C, Table 2, entries 6–10) and in acetone (from +25 °C to −90 °C, Table 2, entries 11–15).
In a third series of experiments, the reaction was carried out by mixing the reagents directly in the NMR spectroscopy tube and monitoring it with time. When reagents 3c and 4 (in 2:1 molar ratio) were mixed at 25 °C in CDCl3, CD3CN, or DMSO-d6, the 1H NMR spectra recorded in the first two solvents showed that the A/A′ ratio was the same to what reported in entries 3 and 5 of Table 1, respectively. Surprisingly, no product was formed when the reagents were mixed in DMSO-d6.
In Table 3 are reported the results obtained by 1H NMR spectroscopy, from the reaction between 3c and 4 carried out at low temperature directly in the NMR tube inserted in a cryo-probe. The reaction was monitored with time and at different temperatures.
| Solvent | Temp (°C) | Conversion | A/A′c |
|---|---|---|---|
| a Reaction carried out directly in the NMR spectroscopy tube by mixing, at the lowest temperature indicated in the Table 3, 3c and 4 in 2:1 molar ratio.b Monitoring of the reaction conversion and isomeric ratio of the products through 1H NMR spectroscopy at variable temperature.c The % molar ratio was calculated from the NMR spectrum of the crude reaction mixture.d The conversion and relative ratio values remained unchanged within 10 min after varying the temperature. |
|||
| CDCl3 | −48 | 32 | 43/57 |
| −37 | 40 | 36/64 | |
| −26 | 50 | 34/66 | |
| −15 | 58 | 31/69 | |
| −4 | 71 | 41/59 | |
| +30 | 90 | 55/45 | |
| +40 | 93 | 56/44 | |
| CD3CN | −43 | 36 | 75/25 |
| −32 | 42 | 80/20 | |
| −21 | 45 | 85/15 | |
| −9 | 66 | 78/22 | |
| +6 | 74 | 73/27 | |
| +13 | 84 | 72/28 | |
| +24 | 91 | 62/38 | |
| +24 after 1 day | 99 | 62/38 | |
| CD3COCD3 | −93d | 7 | 95/5 |
| −82d | 7 | 93/7 | |
| −71d | 8 | 92/8 | |
| −59d | 8 | 95/5 | |
| −48d | 11 | 96/4 | |
| −37d | 22 | 96/4 | |
| −26d | 35 | 96/4 | |
| −15 | 51 | 93/7 | |
| −15 after 10 min. | 61 | 87/13 | |
| −4 | 71 | 72/28 | |
| −4 after 10 min. | 78 | 63/37 | |
| +8d | 86 | 59/41 | |
| +25d | 94 | 58/42 | |
The reaction was carried out in acetone-d6 at −93 °C, in CDCl3 at −48 °C, and in CD3CN at −43 °C. In all cases, the conversion was low and the 1H NMR spectrum showed prevalence of signals for one isomer. On raising the temperature, the conversion gradually increased. At +25 °C the conversion was complete and the relative isomeric ratio was in all cases about 6/4, in agreement with results obtained from reaction carried out at room temperature (Table 1). It has to be noted that, in CDCl3, once the reaction was complete, the major isomer was the opposite with respect to that formed at low temperature.
In acetone-d6 the conversion and the isomeric ratio (∼95/5) remained almost unchanged from −93 °C to −60 °C, then they gradually changed. At 25 °C, the reaction goes to completion and the isomeric ratio gradually was 58/42.
The behavior in acetone may suggest that isomer A could be kinetic product (A is almost the exclusive species at lower temperatures), but limited conversion at lower temperatures and trend of the isomer ratio and conversion on raising the temperature (see data of Table 3) do not permit to infer if the formation of A and A′ is ruled by kinetics or thermodynamic control (or by both).
Moreover, it is worth note that, in the 1H NMR spectra recorded when the conversion was not complete, the signals belonging to the unreacted starting aminothiazole result coincident to the salified one. This observation indicates that the two species (i.e. protonated and not-protonated 2-aminothiazole) are in equilibrium, as we already reported in analogous protonation reactions.39,50
Finally, we want to point out the peculiarity of the behavior showed by the herein reported coupling between 4 and 2-aminothiazole derivatives 3a–c; actually, in our previous studies on the C–C coupling between ClDNBF and diamino-, triamino- or trihydroxybenzene derivatives, solely one product was always isolated.35,42
1H NMR experiments of ClDNBF from +25 to −93 °C in acetone-d6 were also carried out to test the opportunity to detect the presence of two species or to trap the suggested dinitroso intermediate, but no variation was detected.
What about the structure of the two isomers?
The unexpected finding for the reaction between ClDNBF and aminothiazoles 3a–c related to the formation of two isomers (5a–c and 5′a–c) give rise to questions on their structure and mechanism of formation.From the mechanistic point of view, it has to be considered that, in principle, the products from the reaction between 3a–c and ClDNBF might undergo structural rearrangement through two main pathways: (i) N-1-oxide/N-3-oxide tautomerism (via (i) in Scheme 6) or Boulton-Katritzky rearrangement (BKR, via (ii), red arrows in Scheme 6). The first equilibrium is a ring open-closure via the 1,2-dinitroso intermediate which, in the current case, might produce species B1 and B2 (Scheme 6), with a formal shift of oxygen atom from N-1 to N-3.
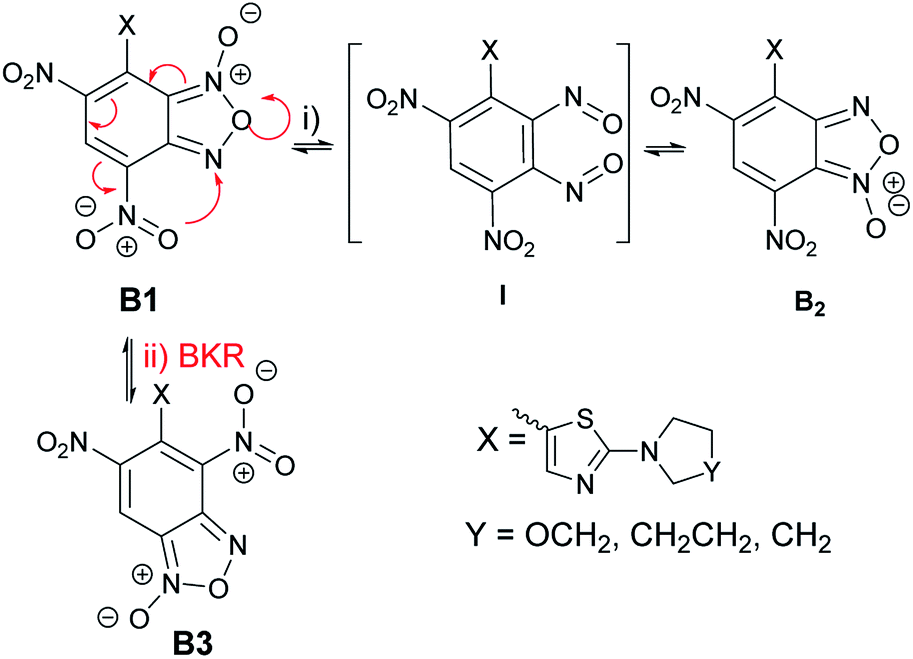 | ||
| Scheme 6 Possible rearrangements of the product derived from the reaction between ClDNBF and 3a–c. via (i): N-1/N-3-oxide equilibrium forming two structural isomers B1 and B2; via (ii): BKR from B1 to B3. | ||
The second reaction pathway, i.e. BKR, occurs when the benzofuroxan moiety bears a nitro group bound to the carbon atom belonging to the carbocyclic ring and adjacent to the fusion carbon, as occurs in current case. In Scheme 6 we indicated the occurrence of BKR on the reaction product B1, since it has been reported that 4 does not undergo BKR rearrangement.51
Although both the mechanisms might simultaneously occur, the presence in the 1H NMR spectrum of the crude reaction mixture of signals ascribable to two novel species prevents this hypothesis. Actually, in case of occurrence of both the rearragements, the signals of three products, namely B1, B2, and B3 must be expected in the spectrum. Therefore, we planned further experiments to discriminate between the two pathways. Focusing attention to the possible N-1/N-3 oxide interconversion, we realized that the reduction of B1 and B2 from furoxan derivatives to furazans would produce the same product. Thus, the isomeric mixture 5c/5′c (also indicated as A and A′ in Tables 1–3, without correspondence of the structure of A or A′ with 5c in Scheme 5) derived from the reaction between 3c and 4 was subjected to reduction with triphenylphosphine in xylene. Since only one compound has been recovered, the occurrence of the 1-oxide/3-oxide tautomerism sketched in Scheme 6 is therefore strongly supported.
However, it is not possible to exclude, ‘a priori’, the occurrence of the preferential reduction of one of the two species involved in the BKR rearrangement; in this case, due to the equilibrium, only one isomer might be recovered after reduction. To shed light on this aspect, it has been necessary to ascertain the structure of the product derived from the reduction of the mixture A + A′. For this purpose, we planned to prepare an authentic sample of the product that might derive from the reduction of B1 and B2, namely 8c, in order to compare the spectroscopic data with those of the product obtained by reduction of the mixture 5c + 5′c. As our first approach, we designed to obtain 8c by nitration of 7c, in turn obtained from 4-chloro-7-nitrobenzofurazan (6, ClNBZ) and 3c (via a in Scheme 7) but the SNAr with ClNBZ did not occur, likely due to the poor electrophilicity of 6 with respect to 4.
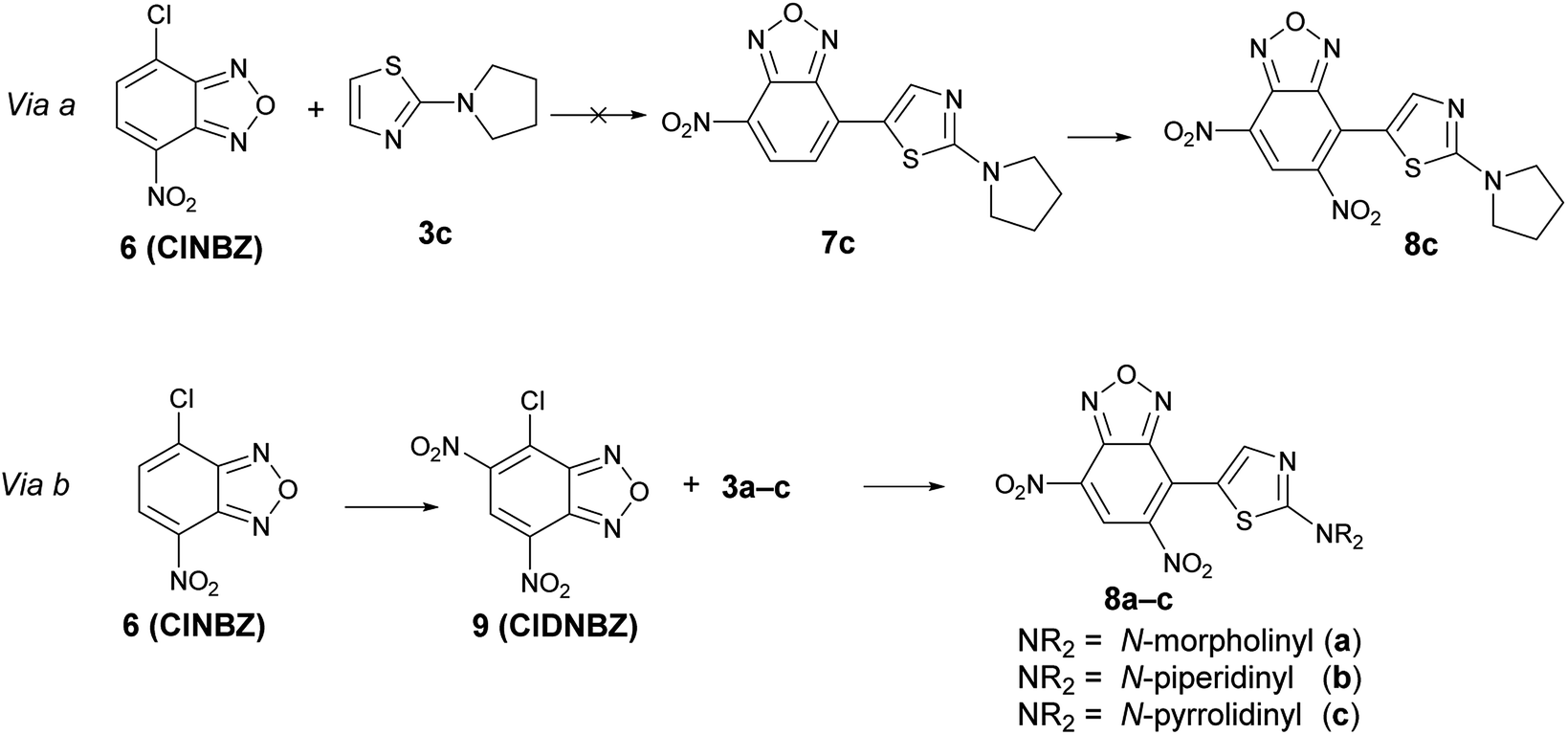 | ||
| Scheme 7 Synthetic routes designed to obtain 8c. | ||
Therefore we used via b as alternative procedure (Scheme 7). 4-Chloro-5,7-dinitrobenzofurazan (9) was prepared by nitration of ClNBZ and reacted with 3c to give 8c, whose chemico-physical data were in agreement with the unique compound obtained by reduction of the two isomers 5c and 5′c. Moreover, by adding a little amount of 8c to the product obtained by reduction of the mixture A + A′, increasing of the related 1H NMR signals was significantly observed. Through an analogous procedure we synthesized also 8a and 8b. Compounds 8a–c are novel and might be of interest in applied field, since they bear contemporarily an electron-donor and an electron-withdrawing moiety.
Summarizing, the above findings bring to exclude structure B3 in favor of 1/3-oxide rearrangement, by considering that upon reduction of the furoxan ring of A + A′ to furazan one, only product 8c was formed. One plausible explanation for the not formation of B3 might be ascribed to the steric hindrance of the two nitro substituents in ortho position with respect to the thiazole moiety in the BKR product.
These findings are in agreement with those previously reported by Chistyakov et al. on the reaction between 4 and N-methylpyrrole that gave two 1,3-N-oxide tautomers.52
DFT calculations
The relative 13C NMR signals of A and A′, owing to their different relative ratio were attributed by g-HSQC NMR experiments (Table 4). However, the tautomeric structure of A and A′ (due to the 1/3-oxide rearrangement) in solution cannot be unambigously assigned by NMR experiments solely.
|
|
|||
|---|---|---|---|---|
| Energy (kcal mol−1) | 0.0 | 0.23 | ||
| 13C chem. shifts | Calc. | Exp.a (A, major isomer) | Calc. | Exp.a (A′, minor isomer) |
| a Data from spectra recorded at T = 25 °C in CDCl3. | ||||
| C3a | 106.12 | 105.95 | 144.84 | 144.89 |
| C4 | 127.03 | 124.97 | 126.21 | 125.38 |
| C5 | 125.03 | 127.87 | 128.71 | 128.53 |
| C6 | 138.13 | 137.95 | 137.51 | 138.93 |
| C7 | 127.38 | 128.04 | 129.73 | 130.72 |
| C7a | 151.20 | 151.26 | 114.73 | 113.43 |
| C8 | 115.67 | 113.83 | 110.92 | 109.06 |
| C9 | 157.53 | 156.57 | 156.59 | 153.85 |
| C10 | 171.60 | 172.54 | 173.37 | 172.38 |
| C11 | 50.64 | 50.76 | 50.97 | 50.76 |
| C12 | 27.23 | 25.61 | 27.21 | 25.55 |
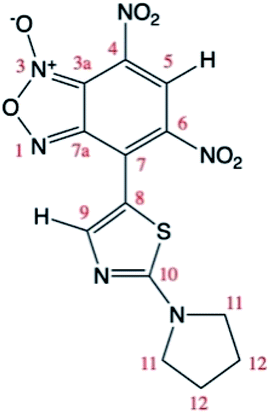
One of the main difficulties in studying these compounds spectrometrically resides mainly in the lack of spin markers (protons). The g-HSQC experiments allowed to assign with confidence only correlations with the H5 and H9 signals while other correlations are usually too weak to be used in structural elucidation. Quantum mechanical calculations of 13C NMR chemical shifts are widely used to make easier signal assignment. Thus, a joint analysis of experimental and calculated NMR 13C chemical shifts was used for the assignment of the 13C chemical shifts of the two tautomers. The 13C theoretical chemical shifts (Table 4) correlate very well with the experimental data, with correlation coefficients of least-squares linear fits (R2) close to unity (0.9995 and 0.9992 respectively for A and A′).
Theoretical calculations allowed also the determination of the energy of the tautomers A and A′. In agreement with the assignment of the 13C chemical shifts of the two tautomers, the most abundant tautomer A (N-3-oxide) is also energetically more stable than A′ (N-1-oxide). The energetic difference between the two tautomers is very small (0.23 kcal mol−1), explaining their presence in a isomeric mixture (Table 1).
The two tautomers show a similar structure (Fig. 1), the main factor that control their relative stability is (i) the presence of an non-standard intramolecular hydrogen bond between C9–H9 and N-1 in tautomer A and N-1-oxide in tautomer A′, (ii) the steric/electrostatic repulsions between the N-oxide moieties with the nitro-group in tautomer A or C9–H9 bond in tautomer A′.
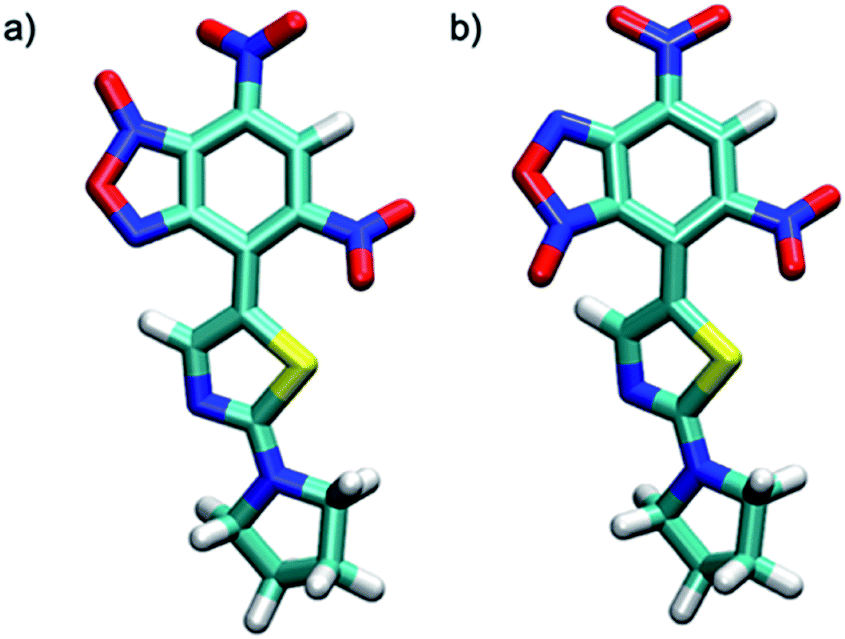 | ||
| Fig. 1 Molecular structure of (a) N-3-oxide, (b) N-1-oxide tautomers. | ||
Further mechanistic insights: reaction of 3a–c with 4,6-dinitrobenzofuroxan
The above finding of two isomeric species derived from the aromatic substitution reaction involving ClDNBF has never reported before.By using nucleophilic species such as diamino- or triamino-benzenes, only one substituted product has been recovered from the reaction with ClDNBF. Further, no evidence of isomeric intermediates such as Meisenheimer (M) or Wheland-Meisenheimer (WM) species was so far reported from C–C coupling reactions of 4,6-dinitrobenzofuroxan (10, DNBF) either with amino- (or hydroxy)-benzenes or 2,4-dipyrrolidinylthiazole (11 in Scheme 8). In the latter case, the isomeric structure of the isolated intermediate (WM in Scheme 8) was ascertained by X-ray diffraction analysis. The particular behavior found by coupling 2-aminothiazoles 3a–c with ClDNBF suggested us to investigate the reaction with DNBF. Equimolar amount of DNBF and 3a (or 3b, 3c) were mixed in acetone-d6 at −93 °C in a NMR spectroscopy tube and the mixture was analyzed by NMR at −93 °C. Immediately after mixing, new born 1H NMR signals appeared in the spectrum, compatible with those of the σ-anionic intermediate Ma (or Mb, Mc) (Scheme 8). In particular, diagnostic was the signal at high field (5–6 ppm) and ascribable to the sp3 hybridized carbon atom bound to H-7, which is involved in the newly formed C–C bond. Moreover, two singlets belonging to H-5 and H-4′ were present in the 5–10 ppm region (see Spectra in ESI†).
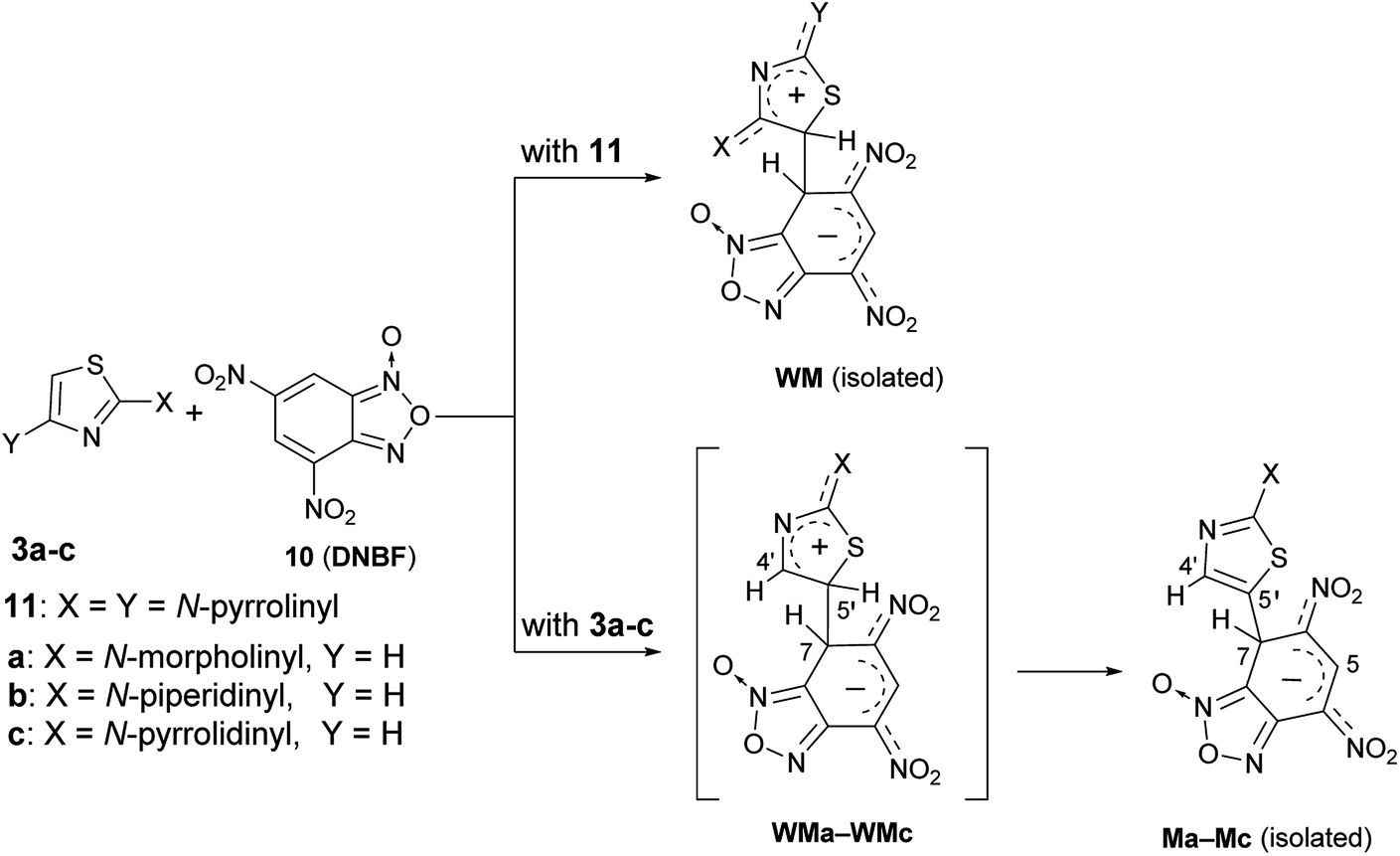 | ||
| Scheme 8 Formation of stable intermediates of reactions between DNBF and 2,4-dipyrrolidinylthiazole (11) or 2-aminothiazoles 3a–c. | ||
On gradually raising the temperature, the intensity of signals of Ma–Mc increased, whereas those of reactants gradually disappear (at 25 °C and 0 °C completely for cases b and c, respectively; whereas, in case of a traces of reagents were present at 25 °C).
Once the conversion has been complete, a red solid appeared at the bottom of the tube extracted from the NMR probe. The solid was collected, then dissolved in DMSO-d6 for 1H NMR, 13C NMR and g-HSQC experiments that confirmed the Meisenheimer structure. The solid was stable enough to be analyzed also through ESI mass spectrometry (negative ion mode).
In principle, in the reactions between 3a–c and 10 the species WMa-WMc (Scheme 8) are the first covalent intermediates but only M species were spectroscopically detected.
Conversely, the presence of two amino groups on the thiazole moiety (as in compound 11), stabilizes so strongly the corresponding zwitterionic WM intermediate to allow its isolation and the first example of X-ray structural determination in such elusive species38 (WM in Scheme 8). This indicates that the lack of the second amino group give rise to zwitterionic fugitive species WMa-WMc undetectable on the NMR time scale. Further, as a matter of facts, only one M species has been detected and isolated for each electrophile/nucleophile combination by coupling 3a–c with 10. In Scheme 8 (as well as in Scheme 3) we have represented the structure of WMa-WMc and Ma-Mc intermediates as N-1 oxides by analogy with the structure of WM, and also in agreement with literature data from which it emerges that the N-oxide in ClDNBF and DNBF is situated on the nitrogen farthest from the nitro group in peri position (and adjacent to the junction carbon). Moreover, in current case, as well as in those represented in Scheme 3, in which DNBF is used (and chlorine is absent), the experimental finding of only one intermediate might be explained taking into consideration that the formation of the two isomers (N-1-oxide and N-3-oxide) through the passage to dinitrosobenzene is disadvantaged or impeded as the tautomeric process involves rearomatization of the carbocyclic ring which is unlikely when a negative charge still exists on the latter as in M or WM and the departure of the hydride ion is generally difficult. On the contrary, in the case in which the starting electrophile is ClDNBF, as in current case, likely the isomerization occurs immediately after the departure of the chloride, which would make stable, even if for a very short life time, the dinitrosobenzene, thus making possible the formation of the two isomers A and A′.
Experimental
General
The 1H and 13C NMR spectra were recorded at 300, 400, or 600 MHz (1H NMR) and 75.46, 100.56, or 150.80 MHz (13C NMR), respectively. J values are given in Hz. Signal multiplicities were established by DEPT-135 experiments. Chemical shifts were referenced to the solvent [δ = 7.27 and 77.0 ppm for CDCl3), (δ = 1.96 and 118.26 ppm for CD3CN), (δ = 2.05 and 30.2 ppm for CD3COCD3), (δ = 2.50 and 39.5 ppm for DMSO-d6) for 1H and 13C NMR, respectively]. ESI-MS spectra were recorded using a Waters 2Q 4000 instrument. Chromatographic purifications (FC) were carried out on silica gel columns at medium pressure. TLC was carried out on aluminum coated silica gel with 254 nm fluorescence indicator (Fluka, DC-Alufolien-Kieselgel).ClNBZ was commercially available, whereas 2-pyrrolidinylthiazole (3c),39 7-Chloro-4,6-dinitrobenzofurazan (ClDNBZ, 9),35 7-Chloro-4,6-dinitrobenzofuroxan (ClDNBF, 4),43 and 4,6-dinitrobenzofuroxan (DNBF, 10),34 were synthesized and purified as previously described. All novel solid compounds decomposed when heated in melting point apparatus. Although the reactions carried out in NMR spectroscopy tube showed conversion almost quantitative, the yields in isolated products were lower, likely due to purification methods, but we did not focus our efforts on yield optimization. Computational and variable temperature NMR experimental details are reported in ESI.†
Synthesis of compounds 3a–c
Reaction between 3a–c and ClDNBF
:ethyl acetate 9:1. The collected fractions were concentrated in vacuo and analyzed through 1H NMR and 13C NMR, and mass spectrometry. The NMR analysis indicated presence of two species. Many attempts to obtain crystals suitable for X-ray diffraction analysis failed. The mixture was subjected to preparative TLC and the two separated spot were scraped but, once each was dissolved in CD3CN to be analyzed, the 1H NMR revealed presence of a mixture of two compounds in relative ratio equal to that found prior to separation. Thus, below we report the physico-chemical data of the mixture of isomers and, due to the presence of two isomers, we considered sufficient to record ESI-MS data. In particular, when, from the 1H NMR spectrum, it was evident that the two species were present in different amount, we indicated as Maj or Min the signals of the isomer present in major or minor amount, respectively. On the basis of the analysis derived from experimental data/DFT calculations, below we report the names of the isomers attributing structure A′ to 5c and A to 5′c, and assuming analogous behaviour also for cases a and b.:33) δ (ppm): 8.73 (s, 1H, Maj), 8.47 (s, 1H, Min), 8.32 (s, 1H, Min), 7.71 (s, 1H, Maj), 3.85–3.75 (m, 8H, Maj + Min), 3.67–3.59 (m, 8H, Maj + Min); 1H NMR (399.9 MHz, CDCl3, 25 °C, isomeric Maj/Min ratio = 50:50) δ (ppm): 8.69 (s, 1H), 8.65 (s, 1H), 8.24 (s, 1H), 7.79 (s, 1H), 3.89–3.84 (m, 8H), 3.78–3.74 (m, 4H), 3.71–3.67 (m, 4H); 13C NMR: (150.8 MHz, CDCl3, 25 °C) δ (ppm, selected data): 176.0, 175.9, 154.9 (CH), 152.1 (CH), 151.1, 144.6, 139.5, 132.4, 127.9 (CH), 125.0, 124.6, 124.2 (CH), 113.5, 113.3, 108.6, 105.8, 66.0 (OCH2), 65.9 (OCH2), 48.9 (NCH2), 48.8 (NCH2); ESI-MS (m/z): 393 (M − H)−; HRMS (ESI+) m/z: [M + H]+ calcd for C13H11N6O7S+: 395.0404; found: 393.0412.:44) δ (ppm): 8.79 (s, 1H, Maj), 8.71 (s, 1H, Min), 8.29 (s, 1H, Maj), 7.94 (s, 1H, Min), 3.80–3.73 (m, 4H, Min), 3.73–3.68 (m, 4H, Maj), 1.83–1.73 (m, 12H, Maj + Min); 13C NMR: (150.8 MHz, CDCl3, 25 °C) δ (ppm): 176.1, 175.8, 157.1 (CH), 155.1 (CH), 151.3, 145.0, 138.6, 138.0, 130.4, 128.7 (CH), 127.9, 125.4, 125.0, 124.9 (CH), 113.8, 113.3, 109.4, 105.9, 50.5 (NCH2, 2 signals overlapped), 25.4 (NCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 23.8 (NCH2CH2H2), 23.7 (NCH2CH2H2); HRMS (ESI+) m/z: [M + H]+ calcd for C14H13N6O6S+: 393.0612; found: 393.0632.:41) δ (ppm): 8.81 (s, 1H, Maj), 8.71 (s, 1H, Min), 8.28 (s, 1H, Maj), 7.96 (s, 1H, Min), 4.00–3.35 (m, 8H, Maj + Min), 2.27–2.03 (m, 8H, Maj + Min); 1H NMR: (300 MHz, CD3CN, 25 °C, isomeric Maj/Min ratio = 56:44) δ (ppm): 8.73 (s, 1H, Maj), 8.66 (s, 1H, Min), 8.35 (s, 1H, Min), 7.90 (s, 1H, Maj), 3.72–3.33 (m, 8H, Maj + Min), 2.14–1.94 (m, 8H, Maj + Min); 13C NMR: (150.8 MHz, CDCl3, 25 °C) δ (ppm): 172.8, 172.7, 156.8 (CH), 154.9 (CH), 151.3, 145.0, 138.6, 137.9, 130.3, 128.6 (CH), 127.9, 125.6, 125.0, 124.9 (CH), 113.9, 113.3, 109.2, 106.0, 50.6 (NCH2, 2 broad signals overlapped), 29.7 (NCH2H2), 25.6 (NCH2H2); HRMS (ESI+) m/z: [M + H]+ calcd for C13H11N6O6S+: 379.0455; found: 379.0466.
H2), 23.8 (NCH2CH2H2), 23.7 (NCH2CH2H2); HRMS (ESI+) m/z: [M + H]+ calcd for C14H13N6O6S+: 393.0612; found: 393.0632.:41) δ (ppm): 8.81 (s, 1H, Maj), 8.71 (s, 1H, Min), 8.28 (s, 1H, Maj), 7.96 (s, 1H, Min), 4.00–3.35 (m, 8H, Maj + Min), 2.27–2.03 (m, 8H, Maj + Min); 1H NMR: (300 MHz, CD3CN, 25 °C, isomeric Maj/Min ratio = 56:44) δ (ppm): 8.73 (s, 1H, Maj), 8.66 (s, 1H, Min), 8.35 (s, 1H, Min), 7.90 (s, 1H, Maj), 3.72–3.33 (m, 8H, Maj + Min), 2.14–1.94 (m, 8H, Maj + Min); 13C NMR: (150.8 MHz, CDCl3, 25 °C) δ (ppm): 172.8, 172.7, 156.8 (CH), 154.9 (CH), 151.3, 145.0, 138.6, 137.9, 130.3, 128.6 (CH), 127.9, 125.6, 125.0, 124.9 (CH), 113.9, 113.3, 109.2, 106.0, 50.6 (NCH2, 2 broad signals overlapped), 29.7 (NCH2H2), 25.6 (NCH2H2); HRMS (ESI+) m/z: [M + H]+ calcd for C13H11N6O6S+: 379.0455; found: 379.0466.Synthesis of 7-(2-dialkylaminothiazol)-4,6-dinitrobenzo[c][1,2,5]oxadiazoles
Conclusions
The SEAr/SNAr reaction by combination of Cl-DNBF and 2-aminothiazole derivatives 3a–c gave two isomeric products, bearing the benzofuroxanyl moiety bound to the C-5 carbon atom of the thiazole ring. Experiments carried out by changing the reaction conditions showed that the two isomers were formed in relative ratio depending on the reaction temperature and the solvent nature. The behavior observed suggests the occurrence of an equilibrium between the two novel species. Further experiments have been performed in order to shed some light on the reaction mechanism and to elucidate the structure of the two isomers. The reduction of the furoxanyl ring to furazanyl one of the isomeric mixture (5c + 5′c) gave the same product derived from the reaction between 3c and 7-chloro-4,6-dinitrobenzofurazan (Cl-DNBZ). This permitted to solve the dilemma of a unambiguous assignment of the isomeric forms structure supporting their origination from N-1/N-3-oxide isomerization. DFT calculations confirm the correlation between the NMR signals and the isomeric structures. Current study is, to the best of our knowledge, the first attempt to compare N-1/N-3-oxide and BKR isomerizations. Finally, a unique Meisenheimer intermediate Ma, Mb, or Mc, were isolated by coupling the 2-aminothiazole derivative 3a, 3b, or 3c with 4,6-dinitrobenzofuroxan (DNBF).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful, for the financial support, to Alma Mater Studiorum – Università di Bologna (RFO funds). E. Chugunova is grateful for the financial support to the Ministry of Science and Higher Education of the Russian Federation (grant No. 075-15-2019-1881). The authors thank Dr Daniele Padovan and Mr Luca Zuppiroli for mass spectra.Notes and references
- A. J. Boulton and P. B. Ghosh, Advances in Heterocyclic Chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, New York, 1969, vol. 10, pp. 1–41 Search PubMed.
- A. Gasco and A. J. Boulton, Furoxans and Benzofuroxans in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, New York, 1981, vol. 29, pp.251–340 Search PubMed.
- G. N. Nikonov and S. Bobrov, 1,2,5-Oxadiazoles, in Comprehensive Heterocyclic Chemistry III, Five-membered Rings: Triazoles, Oxadiazoles, Thiadiazoles and Their Fused Carbocyclic Derivatives, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, 2008, vol. 5, pp. 315–395 Search PubMed.
- F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley VCH, Weinheim, 2013 Search PubMed.
- J. W. Fronabarger, M. D. Williams, W. B. Sanborn, D. A. Parrish and M. Bichay, Propellants, Explos., Pyrotech., 2011, 36, 459–470 CrossRef CAS.
- J. Šarlauskas, Ž. Anusevičius and A. Misiūnas, Cent. Eur. J. Energ. Mater., 2012, 9, 365–386 Search PubMed.
- H. Cerecetto and M. González, Top. Heterocycl. Chem., 2007, 10, 265–308 CAS.
- E. A. Chugunova and A. R. Burilov, Curr. Top. Med. Chem., 2017, 17, 986–1005 CrossRef CAS.
- E. Noelting and O. Kohn, Chemiker-Zeitung, Chemische Apparatur, 1894, 18, 1095 Search PubMed.
- P. Drost, Justus Liebigs Ann. Chem., 1899, 307, 49–69 CrossRef CAS.
- P. Drost and T. Zincke, Justus Liebigs Ann. Chem., 1900, 313, 309–325 CrossRef.
- A. G. Green and F. M. Rowe, J. Chem. Soc., 1912, 101, 2452–2459 RSC.
- R. K. Harris, A. R. Katritzky, S. Oksne, A. S. Bailey and W. G. Pateson, J. Chem. Soc., 1963, 197–203 RSC.
- R. Guntram, A. A. Jarzecki and P. Pulay, J. Comput. Chem., 1997, 18, 489–500 CrossRef.
- F. B. Mallory, S. L. Manatt and C. S. Wood, J. Am. Chem. Soc., 1965, 87, 5433–5438 CrossRef CAS.
- F. B. Mallory and A. Cammarata, J. Am. Chem. Soc., 1966, 88, 61–64 CrossRef CAS.
- W. Friedrichsen, J. Phys. Chem., 1994, 98, 12933–12937 CrossRef CAS.
- J. Stevens, M. Schweizer and G. Rauhut, J. Am. Chem. Soc., 2001, 123, 7326–7333 CrossRef CAS.
- I. R. Dunkin, M. A. Lynch, A. J. Boulton and N. Henderson, Chem. Commun., 1991, 1178–1179 RSC.
- E. Abushanab and N. D. Alteri, J. Org. Chem., 1975, 40, 157–160 CrossRef CAS.
- A. B. Bulacinski, E. F. V. Scriven and H. Suschitzky, Tetrahedron Lett., 1975, 41, 3577–3578 CrossRef.
- (a) E. Buncel and F. Terrier, Org. Biomol. Chem., 2010, 8, 2285–2308 RSC and ref therein; (b) C. Jovené, M. Sebban, J. Marrot and R. Goumont, The Diels-alder reactivity of the furoxan ring of substituted benzofuroxans. Synthesis of substituted imines and evidence of the intermediacy of ortho-dinitrosoarenes in the 1-oxide/3-oxide interconversion, in Targets in Heterocyclic Systems Chemistry and Properties, ed. O. A. Attanasi and D. Spinelli, Società Chimica Italiana, Roma, 2012, vol. 16, pp. 90–112 Search PubMed.
- S.-C. Chan, J. England, K. Wieghardt and C. Y. Wong, Chem. Sci., 2014, 5, 3883–3887 RSC.
- A. J. Boulton, A. R. Katritsky and A. M. Hamid, J. Chem. Soc. C, 1967, 2005–2007 RSC.
- A. R. Katritzky, C. A. Ramsden, J. A. Joule, and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, Amsterdam, 3rd edn, 2010, ch. 2.4, pp. 139–209 Search PubMed.
- G. Rauhut and F. Eckert, Quantum Chemical Studies on Heterocyclic Rearrangements in Benzofuroxans: Reaction Paths, Vibnrational Spectra, and Rate Constants in High Performance Computing in Science and Engineering ’99, ed. E. Krause and W. Jäger, Springer-Verlag, Heidelber, 2000, pp. 183–211 Search PubMed.
- G. Rauhut, Recent Advances in Computing Heteroatom-Rich Five- and Six-Membered Ring Systems in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic Press London, 2001, vol. 81, p. 37 Search PubMed.
- F. Terrier, S. Lakhdar, T. Boubaker and R. Goumont, J. Org. Chem., 2005, 70, 6242–6253 CrossRef CAS.
- S. Lakhdar, R. Goumont, F. Terrier, T. Boubaker, J. M. Dust and E. Buncel, Org. Biomol. Chem., 2007, 5, 1744–1751 RSC.
- F. Terrier, Nucleophilic Aromatic Displacement, ed. H. Feuer, VCH, New York, 1991 Search PubMed.
- R. Read, R. J. Spear and W. P. Norris, Aust. J. Chem., 1984, 37, 985–999 CrossRef CAS.
- F. Terrier, Chem. Rev., 1982, 82, 77–152 CrossRef CAS.
- E. Buncel, J. M. Dust and F. Terrier, Chem. Rev., 1995, 95, 2261–2280 CrossRef CAS.
- C. Boga and L. Forlani, J. Chem. Soc., Perkin Trans. 2, 2001, 1408–1413 RSC.
- (a) G. Micheletti, C. Boga, S. Cino, S. Bordoni and E. Chugunova, RSC Adv., 2018, 8, 41663–41674 RSC; (b) E. Del Vecchio, C. Boga, L. Forlani, S. Tozzi, G. Micheletti and S. Cino, J. Org. Chem., 2015, 80, 2216–2222 CrossRef CAS.
- (a) C. Boga, E. Del Vecchio, L. Forlani, A. Mazzanti and P. E. Todesco, Angew. Chem., Int. Ed., 2005, 44, 3285–3289 CrossRef CAS; (b) C. Boga, G. Micheletti, S. Cino, S. Fazzini, L. Forlani, N. Zanna and D. Spinelli, Org. Biomol. Chem., 2016, 14, 4267–4275 RSC.
- C. Boga, E. Del Vecchio, L. Forlani, R. Goumont, F. Terrier and S. Tozzi, Chem.–Eur. J., 2007, 13, 9600–9607 CrossRef CAS.
- L. Forlani, C. Boga, A. Mazzanti and N. Zanna, Eur. J. Org. Chem., 2012, 6, 1123–1129 CrossRef.
- C. Boga, S. Cino, G. Micheletti, D. Padovan, L. Prati, A. Mazzanti and N. Zanna, Org. Biomol. Chem., 2016, 14, 7061–7068 RSC.
- G. Micheletti and C. Boga, Synthesis, 2017, 49, 3347–3356 CrossRef CAS.
- G. Micheletti, C. Boga, M. Pafundi, S. Pollicino and N. Zanna, Org. Biomol. Chem., 2016, 14, 768 RSC.
- G. Micheletti, S. Bordoni, E. Chugunova and C. Boga, Molecules, 2017, 22, 684 CrossRef.
- E. Chugunova, C. Boga, I. Sazykin, S. Cino, G. Micheletti, A. Mazzanti, M. Sazykina, A. Burilov, L. Khmelevtsova and N. Kostina, Eur. J. Med. Chem., 2015, 93, 349–359 CrossRef CAS , and ref. therein.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938–957 CrossRef.
- H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS.
- H. Mayr, M. Patz, M. F. Gotta and A. R. Ofial, Pure Appl. Chem., 1998, 70, 1993–2000 CAS.
- H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remmenikov and N. Schimmel, J. Am. Chem. Soc., 2001, 123, 9500–9512 CrossRef CAS.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org. Chem., 2009, 6379–6385 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Publishers, 3rd edn, 2003 Search PubMed.
- C. Boga, L. Forlani, S. Tozzi, E. Del Vecchio, A. Mazzanti, M. Monari and N. Zanna, Curr. Org. Chem., 2014, 18, 512–523 CrossRef CAS.
- R. W. Read and W. P. Norris, Aust. J. Chem., 1985, 38, 435–445 CrossRef CAS.
- V. A. Chistyakov, Yu. A. Semenyk, P. G. Morozov, E. V. Prazdnova, V. K. Chmykhalo, E. Yu. Kharchenko, M. E. Kletskii, G. S. Borodkin, A. V. Lisovin, O. N. Burov and S. V. Kurbatov, Russ. Chem. Bull. Int. Ed., 2015, 64, 1369–1377 CrossRef CAS.
- (a) S. K. Verma, B. N. Acharya and M. P. Kaushik, Org. Biomol. Chem., 2011, 95, 1324–1327 RSC; (b) P. K. Dutta, S. Sen, D. Saha and B. Dhar, Eur. J. Org. Chem., 2018, 657–665 CrossRef CAS.
- (a) D. Keil and H. Hartmann, Liebigs Ann., 1995, 6, 979–984 CrossRef; (b) H. Fei-Dong, X. Chang, L. Dong-Dong, S. Dong-Sheng, L. Tian and L. Feng-Shou, J. Org. Chem., 2018, 83, 9144–9155 CrossRef.
- F. Terrier, A. P. Chatrousse, Y. Soudais and M. Hlaibi, J. Org. Chem., 1984, 49, 4176–4181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, mass spectra of new compounds and cartesian coordinates for A and A′. See DOI: 10.1039/d0ra05249c |
| This journal is © The Royal Society of Chemistry 2020 |