
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomogeneous liquid–liquid microextraction based on liquid nitrogen-induced phase separation followed by GFAAS for sensitive extraction and determination of lead in lead-adulterated opium and refined opium
Toraj Ahmadi-Jouibaria,
Abbas Aghaeib,
Kiomars Sharafic and
Nazir Fattahi *c
*c
aIran West Center for Drug Abuse Studies, Kermanshah University of Medical Sciences, Kermanshah, Iran
bSocial Determinants of Health Research Center, Research Institute for Health Development, Kurdistan University of Medical Sciences, Sanandaj, Iran
cResearch Center for Environmental Determinants of Health (RCEDH), Health Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran. E-mail: nazirfatahi@yahoo.com; Fax: +988338263048; Tel: +989183364311
First published on 10th August 2020
Abstract
Herein, we developed a novel homogeneous liquid–liquid microextraction based on liquid nitrogen-induced phase separation (HLLME-LNPS) for the extraction and determination of lead (Pb) in Pb-adulterated opium and refined opium by GFAAS analysis. In this procedure, first, 400 μl of acetonitrile (extractant) containing 7.0 μl of diethyl dithiophosphoric acid (DDTP) is injected into a sample solution and a homogeneous solution is formed. Subsequently, the homogeneous mixture is cooled using liquid nitrogen for 16 seconds. By this process, due to the difference in the freezing points of the organic and aqueous phases, the homogeneous state is broken and the Pb-DDTP species are extracted into the liquid organic phase collected on top of the frozen aqueous phase. The introduced method exhibited a good linearity with a coefficient of determination (r2) of 0.9988 and an acceptable linear range of 0.6–100 μg l−1. Accordingly, the detection limit was 0.2 μg l−1 (S/N = 3) for Pb ions, and a high enrichment factor was obtained. The proposed method was successfully utilized to determine trace levels of Pb in opium samples. The results of the sample analysis showed that 65% of the opium samples and 85% of the refined opium samples had much higher than expected levels of contaminating Pb, and this contamination poses a serious threat to drug users.
1. Introduction
Drug abuse is one of the most important problems in public health that many developing countries are struggling with.1 Over the past few decades, the production and use of smuggled and illegal drugs has increased worldwide.2 Afghanistan is the world's largest producer of opium and is increasing its production day by day, such that the surface area under poppy cultivation doubled from 2012 to 2019 in this country.3 Iran has a 900 km common border with Afghanistan, which is the main transit route for drug trafficking gangs, making Iran one of the main transit routes in the world.4 This has led to an increase in drug use, especially opium and refined opium, in Iran. In addition, drug profiteers and retailers in Iran use opium and refined opium with contaminants such as burnt oil, soil, chopped liver, dried animal blood, Indian henna, cocoa, tea, artificial leather and heavy metals, especially Pb compounds, to increase the weight and profits.5,6 Pb compounds are one of the most dangerous materials added to opium and refined opium.7Pb is an environmental hazard with potential side effects for human health, affecting the central nervous system, hematopoietic, hepatic and renal systems, causing serious illness as well as death.8 The US Centers for Disease Control and Prevention (CDC) states that the blood Pb level (BLL) in the whole blood of adults and children is 10 and 5 μg dl−1, respectively.9 In Iran, many reports of Pb poisoning caused by drug use have been investigated by researchers, and the use of Pb-contaminated opium and refined opium has become one of the most important sources of Pb poisoning in Iran.10–15 Kermanshah is one of the metropolises in Western Iran where Pb poisoning among drug users has increased sharply in recent years, and the number of people visiting hospitals in the city is increasing day by day. Since most Iranian hospitals do not have the necessary equipment for Pb analysis in biological and drug samples, and there are currently no laboratory kits for detecting and measuring Pb, in most cases the diagnosis and treatment of Pb poisoning is delayed.9 Therefore, an accurate and sensitive measurement of Pb in environmental and biological samples, such as opium and blood, is an important challenge for analytical chemists.
Numerous instrumental techniques, such as flame atomic absorption spectrometry (FAAS),16 graphite furnace atomic absorption spectrometry (GFAAS),17 inductively coupled plasma-optical emission spectrometry (ICP-OES)18 and inductively coupled plasma mass spectrometry (ICP-MS)19 have been used for the sensitive and selective detection of Pb in different matrices. The separation and preconcentration of Pb ions is a critical step for its determination in opium and biological samples. The complexity of these samples, due to the large number of exogenous and mainly endogenous compounds, may hinder the method selectivity. However, in spite of the development of modern analytical instruments, an extraction and preconcentration step for the determination of low concentration of Pb ions is still required. Analytical procedures, such as liquid–liquid extraction (LLE),20 cloud point extraction (CPE),21 solid-phase extraction (SPE),22 solid-phase microextraction (SPME),23 liquid phase microextraction (LPME),24 dispersive liquid–liquid microextraction (DLLME)25–28 and dispersive liquid–liquid microextraction based on the solidification of floating organic droplets (DLLME-SFOD)29,30 have been developed for the extraction and preconcentration of Pb from different samples. Advantages and disadvantages of these techniques have already been discussed.31–34
Homogeneous liquid–liquid microextraction (HLLME) is one of the liquid phase microextraction techniques that has received a lot of attention from researchers in recent years due to its very simple operation, high speed, low cost and the ability to extract and preconcentrate various analytes.35–38 In general, this technique has two steps. In the first step, the sample solution containing the analyte is mixed with a soluble organic extraction solvent in water to form a homogeneous mixture. In this step, the contact surface of the analytes with the extraction solvent is very high and the hydrophobic analytes are extracted into the organic solvent. In the second step, phase separation occurs by changing the conditions or adding a reagent. Adding a reagent usually raises costs and, in some cases, may cause contamination to enter the system, but temperature changes do not have these problems, and HLLME has been extensively developed for phase separation based on temperature reduction and has been used by many researchers.39–42 But the most important problem with this method is the very long time taken to reduce the temperature, which in the above reported cases is about 1 to 12 h. Recently, Dr Farajzadeh et al.43 developed a HLLME method based on temperature reduction for phase separation, in which liquid nitrogen is used to reduce the temperature. One of the most important advantages of this method is the reduction of time required to cool the homogeneous mixture and for phase separation.
The purpose of the present study was to develop a HLLME based on liquid nitrogen-induced phase separation (LNPS) for the extraction and preconcentration of Pb ions in the opium and refined opium samples prior to analysis with the GFAAS. In this method, diethyldithio phosphoric acid (DDTP) is used as a complexing agent, which has a high efficiency for complexing Pb ions. In this method, due to the high contact surface between the organic and aqueous phases, the extraction efficiency is very high and the equilibrium is established quickly. Phase separation occurs by reducing the temperature with liquid nitrogen in a few seconds. Another advantage of this method is that it does not use toxic organic solvents, very simple operation, environmentally friendly, a high speed and low cost.
2. Experimental
2.1. Reagents and standards
Methanol (MeOH, for spectroscopy), acetone (AC, SupraSolv), acetonitrile (AN, HPLC grade), NaCl (analytical grade), HNO3 (65%, Suprapur®), and the chelating agent, diethyl dithiophosphoric acid (DDTP) with a density of 1.17 kg l−1 were obtained from Merck company (Darmstadt, Germany). A mixture of Pd(NO3)2 (1000 mg l−1) and Mg(NO3)2 (300 mg l−1) solutions, both from Merck (Darmstadt, Germany), were used as chemical modifiers. The standard solution of Pb was produced by diluting a stock solution of 1000 mg l−1 Pb supplied by Sigma Chemical Co. (St Louis, MO, USA). The working standard solution was prepared by an appropriate dilution of the stock standard solution with ultra-pure water. All the solutions were prepared with ultra-pure water (six times distillated) purchased from Shahid Ghazi Pharmaceutical Co. (Tabriz, Iran).2.2. Instrumentation
Analysis of Pb was done using a Model novAA 400 atomic absorption spectrometer (Analytik Jena AG, Jena, Germany), equipped with deuterium background correction, auto-sampler MPE-60 and a transversely heated graphite tube atomizer. The Pb hollow cathode lamp (Analytik Jena, Jena, Germany) was operated at 5 mA, with a spectral bandwidth of 0.8 nm, and the analytical line at 283.3 nm. Pyrolytic graphite coated graphite tubes with an integrated PIN platform (Analytik Jena part no. 407-A81.026) were used for all measurements. The temperature program and instrumental parameters are given in Table 1. Argon (99.999%) was purchased from Air Products (UK) as a purge and protective gas. A Metrohm pH meter Model 692 (Herisau, Switzerland) combined with a glass-combined electrode was used to determine the sample solution pH. The samples were digested with a Microwave Multiwave 3000 (Anton Paar, Germany). The Hettich Zentrifugen Model EBA20 (Tuttlingen, Germany) was used for centrifugation.| Spectrometer parameter | |
|---|---|
| Wavelength (nm) | 283.3 |
| Spectral bandwidth (nm) | 0.8 |
| Lamp current (mA) | 5.0 |
| Step | Temperature (°C) | Ramp time (s) | Hold time (s) | Argon flow rate (ml min−1) |
|---|---|---|---|---|
| Inject modifier | 80 | 3 | 5 | 1000 |
| Inject sample | ||||
| Drying I | 100 | 1 | 10 | 1000 |
| Drying II | 200 | 2 | 5 | 1000 |
| Pyrolysis | 700 | 10 | 15 | 1000 |
| AZ* | 700 | 0 | 5 | 0 |
| Atomization | 1800 | 0 | 3 | 0 |
| Cleaning | 2200 | 0 | 2 | 1500 |
2.3. Sampling and sample preparation
After obtaining approval to implement the project and obtain a letter of permission as a member of the sample collectors, Kermanshah city was divided into 20 zones (based on consultation with experts from MMT and DIC centers based on different drug-dependent behaviors and also different distributors in these regions). In each region, a sample of opium and a sample of refined opium were collected separately. The approximate weight of each sample was chosen to be exactly 1 g. In total, 20 samples of opium and 20 samples of refined opium to a total of 40 samples from 20 regions of Kermanshah were collected and transferred to the research laboratory.For the preparation and digestion of the opium and refined opium samples, exactly 0.2 g of each sample was carefully weighed and digested using 1.5 ml of HClO4 (70%) and 6 ml of concentrated HNO3 (Suprapur®). After cooling, the resulting solution was filtered using a Whatman No. 42 filter paper and was increased to a volume of 50 ml by distilled water. 5 ml of the resulting solution was used for the proposed extraction method.
2.4. HLLME-LNPS procedure
For the HLLME-LNPS, an aliquot of 5 ml of the standard solution of the Pb or real sample was placed in a 10 ml glass centrifuge tube. Four hundred microliters of acetonitrile (extraction solvent), containing 7.0 μl of DDTP (chelating agent) were injected rapidly into a sample solution. For complete homogenization, the mixture was vortexed for 20 seconds and then immersed in liquid nitrogen for 16 seconds to rapidly reduce the temperature of the mixture. Due to the difference between the freezing points of water and acetonitrile, the homogenous state was broken and the aqueous phase was frozen at the bottom of the test tube, while the organic phase (acetonitrile) containing the complexed Pb ions was collected as the liquid above the frozen aqueous phase. The volume of the collected organic phase (acetonitrile) was 50 ± 3 μl. The extraction solvent was then transferred into a conical vial. Finally, 20 μl of this organic phase using an auto-sampler was injected into the GFAAS and was submitted to the temperature program in Table 1.3. Results and discussion
In the proposed procedure, some important parameters which usually affect the extraction performance were optimized. The parameters affecting the HLLME-LNPS procedure, such as the extraction solvent and its volumes, the effect of salt, sample solution pH, chelating agent concentration, vortex and cooling times, and coexisting ions were optimized. The preconcentration factor (PF) was defined as the ratio between the analyte concentration in the organic phase (Corg) and the initial concentration of analytes (C0) within the sample.
 | (1) |
The extraction recovery (ER) was defined as the ratio between the concentration of the analyte in the organic phase (norg) and the initial concentration of the analyte (n0) within the sample.
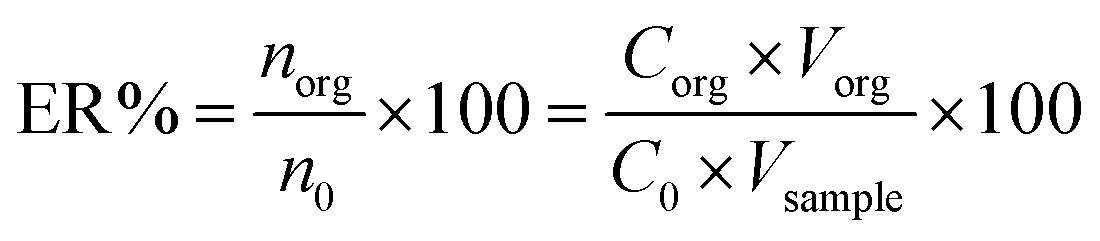 | (2) |
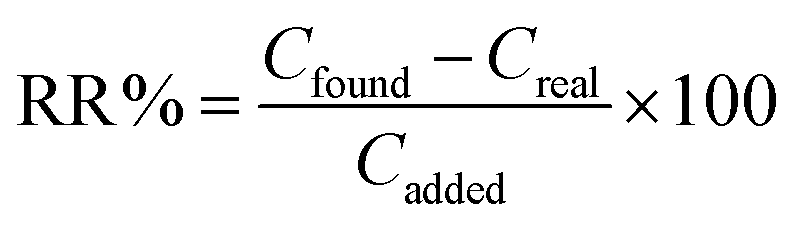 | (3) |
3.1. Effect of the type and volume of extraction solvent
One of the most important factors in the proposed method is the choice of the type of extraction solvent. The extraction solvent must be able to mix completely with the aqueous phase, its freezing point must be much less than the freezing point of the aqueous phase, it must be able to extract the desired compounds and be able to form a two-phase system at low temperatures. These characteristics limit the choice of extraction solvent. However, based on the above characteristics, as well as previous reports,35–39 acetone, acetonitrile, methanol and THF have been used in HLLME. Therefore, a series of tests were conducted and the above 4 solvents were evaluated as extraction solvents. The results showed that in volumes less than 1.0 ml, only acetonitrile could form a two-phase system by reducing the temperature. Although acetone and methanol formed a slightly two-phase system in volumes above 1.0 ml, they did not have a good extraction efficiency. The formation of a two-phase system between acetonitrile and water is likely to be caused by a decrease in temperature because the nitrile groups in acetonitrile have proton-accepting properties and the methyl group has proton-giving properties, and this causes acetonitrile molecules to form dimers with decreasing temperature in the solution and the hydrogen bond between water and acetonitrile is gradually replaced by the dipole–dipole interaction between C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups. Therefore, acetonitrile was chosen for further experiments.
N groups. Therefore, acetonitrile was chosen for further experiments.
In order to obtain the optimum volume of acetonitrile, several experiments were performed using different volumes of acetonitrile, which ranged from 100 to 800 μl. The results in Fig. 1A show that in volumes less than 400 μl, a two-phase system is not formed by reducing the temperature, and the operation method is practically inefficient. In volumes of 400 μl and above, a two-phase system is formed by reducing the temperature, but in volumes of 400 μl, the highest extraction efficiency is obtained. At volumes above 400 μl, the extraction efficiency gradually reduces, possibly due to a dilution effect. As a result, the volume of 400 μl was selected as the optimal volume.
 | ||
| Fig. 1 The effects of the (A) extraction solvent volume (acetonitrile), (B) sample solution pH, (C) concentration of DDTP, (D) salt addition, (E) vortex time and (F) cooling time on the extraction recovery or absorbance of Pb. | ||
3.2. Effect of the sample solution pH
The pH of the sample solution plays an important role when extracting Pb ion using the DDTP complex ligand, because the formation of metal–ligand species and their extraction are directly dependent on pH.17 Since the DDTP ligand used in this study had an acidic nature, to investigate the effect of pH it was completely converted into ammonium DDTP using ammonia, and then the pH effect was investigated in the range 1 to 7. The results in Fig. 1B shows that by increasing the pH from 1 to 4, the analytical signal is almost constant and shows the highest value. As the pH increases, the analytical signal decreases. Since the DDTP solution is acidic, therefore adding acid is not necessary to adjust the pH, which may also be a source of contamination.3.3. Effect of the DDTP concentration
DDTP is a well-known chelating agent for Pb extraction with the formation of a stable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with the Pb2+ ion.8 A sufficient amount of DDTP is needed to complex the Pb ion and increase its extraction efficiency. The concentration of DDTP should be high enough for maximum complexation and extraction, but should not be excessive because it enters the extraction phase and may have a quenching effect when analyzed with ETAAS. To investigate the effect of DDTP concentration, experiments with different amounts of DDTP were performed in the range of 0.01 to 0.15% (v/v). The results in Fig. 1C show that with increasing DDTP amounts up to 0.07% (v/v), the analytical signal increases, and with increasing the DDTP amounts further, the analytical signal remains almost constant and in some cases slightly decreases. Therefore, a 0.07% (v/v) concentration was selected as optimal concentration.
:1 complex with the Pb2+ ion.8 A sufficient amount of DDTP is needed to complex the Pb ion and increase its extraction efficiency. The concentration of DDTP should be high enough for maximum complexation and extraction, but should not be excessive because it enters the extraction phase and may have a quenching effect when analyzed with ETAAS. To investigate the effect of DDTP concentration, experiments with different amounts of DDTP were performed in the range of 0.01 to 0.15% (v/v). The results in Fig. 1C show that with increasing DDTP amounts up to 0.07% (v/v), the analytical signal increases, and with increasing the DDTP amounts further, the analytical signal remains almost constant and in some cases slightly decreases. Therefore, a 0.07% (v/v) concentration was selected as optimal concentration.
3.4. Effect of the salt addition
For investigating the effect of salt addition on the performance of HLLME-LNPS, some experiments were performed by adding different amounts of NaCl (0–10%). Other experimental conditions were kept constant. As can be seen from Fig. 1D, with increasing salt from 0 to 4%, the extraction efficiency remains almost constant, because two opposite effects occur in the extraction efficiency, which neutralize each other's effect. On the one hand, with increasing salt, the polarity of the aqueous phase increases and reduces the solubility of the analytes in the aqueous phase, thus increasing the extraction efficiency (salting-out). On the other hand, increasing the salt reduces the solubility of acetonitrile and increases the volume of the obtained organic phase and a dilution effect occurs. At concentrations greater than 4%, the dilution effect prevails on salting-out and the extraction efficiency decreases. Also, at concentrations above 10%, the freezing point of the aqueous phase approaches the freezing point of the acetonitrile phase, and the phase separation is difficult. As a result, all the extraction experiments were carried out without salt addition.3.5. Effect of the vortexing and cooling time
Although acetonitrile is soluble in water and allows DDTP to be completely dispersed in water, it still accelerates the vortexing of the complexing and extracting process of Pb ions. For this purpose, a series of tests were performed in which the resulting mixture was vortexed in the range of 0–60 seconds after the injection of acetonitrile containing DDTP into the aqueous phase. The results in Fig. 1E show that, with increasing vortex time from 0 to 20 seconds, the extraction efficiency improves and no change in the extraction efficiency is achieved in more than 20 seconds. Therefore, 20 seconds was chosen as the optimal vortex time.Cooling time and temperature reduction are very important for phase separation in HLLME, because they affect the total time of preparation and extraction. In other HLLME methods, it takes hours to cool and reduce the temperature to separate the phase, but in the method presented in this study, liquid nitrogen was used to cool and reduce the temperature, which reduces this time to seconds. In order to optimize the cooling time, tests with intervals of 4 to 32 seconds were performed. According to the results in Fig. 1F, phase separation does not occur well in less than 12 seconds and the extraction efficiency is not good, but in intervals between 12 and 20 seconds, the best phase separation and maximum extraction efficiency is obtained. In more than 20 seconds, both organic and aqueous phases are frozen. As a result, 16 seconds was chosen as the optimal cooling time.
3.6. Effect of the coexisting ions
The DDTP ligand has a high selectivity and does not react with the alkaline and alkaline earth elements, which are the most common constituent elements in a real sample matrix.30 To evaluate the selectivity of the HLLME-LNPS method, the recovery of a 5 μg l−1 Pb solution in the presence of different amounts of intrusive ions was performed by the proposed method. The tolerable amount is defined as the maximum concentration that could cause a change of less than 5% in signal compared to the signal of Pb without any interference. Table 2 shows tolerance limits of the interfering ions.| Interferent | Potentially interfering ions to analyte ratio | Range of recoveries (%) |
|---|---|---|
| Na+, K+, Li+, Ca2+, Mg2+, Cl−, SO42−, NO3−, SCN− | 20000 |
91.5–105.0 |
| Co(II), Fe(II), Zn(II), Hg(II), Cu(II), Cr(III), Ag(I) | 1000 | 93.1–106.4 |
| Cd(II), Sn(IV) | 400 | 90.0–98.5 |
| As(V), Cr(VI) | 200 | 92.2–101.6 |
| As(III) | 100 | 94.2 |
3.7. Analytical figures of merit
Validation of the HLLME-LNPS procedure was carried out by investigating some analytical figures of merit, such as repeatability (intra-day), reproducibility (inter-day), linear dynamic range (LR), limit of detection (LOD), extraction recovery (ER), preconcentration factor (PF) and enhancement factor (EF). The repeatability (intra-day) and reproducibility (inter-day) of the HLLME-LNPS coupled with GFAAS for 5.0 μg l−1 Pb were determined to be 2.8 and 4.2%, respectively. The correlation coefficient (r2) of the calibration curve was 0.9988. The LOD, defined as CL = 3Sb/m (where CL, Sb, and m are the LOD, SD of the blank and slope of the calibration graph, respectively), was 0.2 μg l−1. An LR of 0.6–100 μg l−1 for Pb was obtained. The ER% and PF were both 85. The enhancement factor was obtained from the slope ratio of the calibration graph after and before extraction, which was about 91 (Table 3).3.8. Determination of Pb in opium
The efficiency of the HLLME-LNPS procedure in the measurement of Pb ions was investigated by analyzing Pb-adulterated opium and refined opium samples. Samples of opium and refined opium were obtained from 20 regions of Kermanshah, where most drug trafficking and retail transaction take place. One sample of opium and one sample of refined opium was collected from each region and a total of 20 samples of opium and 20 samples of refined opium were collected and analyzed. The results are summarized in Table 4. The results show that the amount of Pb in opium and refined opium samples are in the ranges of 18.9–313.5 and 25.5–408.5 μg g−1, respectively. Although some Pb naturally enters opium when planting and harvesting, high concentrations of Pb in 13 of the opium samples and 17 of the refined opium samples indicate a very large threat in the addition of Pb compounds by traffickers and retailers.| Sample type | Sample no. | Added (μg g−1) | Found mean ± SD (μg g−1) (n = 3) | Relative recovery (%) |
|---|---|---|---|---|
| a These data are based on the diluted volumes of aqueous samples, and the dilution effect was considered for their calculation.b Certified value of Pb ions. | ||||
| Opium | 1 | 0 | 137.4 ± 6.5 | — |
| 20 | 156.8 ± 55.7 | 97 | ||
| 2 | 0 | 211.2 ± 15.3 | — | |
| 30 | 243.5 ± 19.4 | 108 | ||
| 3 | 0 | 313.5 ± 22.4 | — | |
| 40 | 355.1 ± 28.6 | 104 | ||
| 4 | 0 | 28.3 ± 1.7 | — | |
| 50 | 77.8 ± 3.1 | 99 | ||
| 5 | 0 | 96.7 ± 5.6 | — | |
| 60 | 155.1 ± 8.7 | 97 | ||
| 6 | 0 | 120.5 ± 8.1 | — | |
| 70 | 185.2 ± 10.9 | 92 | ||
| 7 | 0 | 198.3 ± 13.4 | — | |
| 80 | 281.5 ± 14.8 | 104 | ||
| 8 | 0 | 218.3 ± 11.5 | — | |
| 90 | 305.4 ± 22.7 | 97 | ||
| 9 | 0 | 155.8 ± 13.5 | — | |
| 100 | 251.7 ± 19.6 | 96 | ||
| 10 | 0 | 67.2 ± 7.5 | — | |
| 10 | 76.8 ± 8.1 | 96 | ||
| 11 | 0 | 88.1 ± 6.8 | — | |
| 20 | 109.2 ± 7.3 | 105 | ||
| 12 | 0 | 18.9 ± 1.0 | — | |
| 30 | 46.3 ± 3.8 | 91 | ||
| 13 | 0 | 76.4 ± 5.9 | — | |
| 40 | 113.6 ± 7.3 | 93 | ||
| 14 | 0 | 115.4 ± 9.2 | — | |
| 50 | 165.3 ± 8.1 | 100 | ||
| 15 | 0 | 66.1 ± 5.2 | — | |
| 60 | 123.6 ± 11.7 | 96 | ||
| 16 | 0 | 159.1 ± 12.2 | — | |
| 60 | 223.2 ± 16.7 | 107 | ||
| 17 | 0 | 301.4 ± 15.2 | — | |
| 80 | 387.6 ± 21.6 | 108 | ||
| 18 | 0 | 266.1 ± 19.2 | — | |
| 80 | 350.6 ± 22.7 | 106 | ||
| 19 | 0 | 209.1 ± 13.2 | — | |
| 100 | 305.6 ± 24.7 | 97 | ||
| 20 | 0 | 144.1 ± 9.2 | — | |
| 100 | 239.6 ± 17.3 | 95 | ||
| Refined opium | 1 | 0 | 56.3 ± 4.2 | — |
| 10 | 66.6 ± 5.7 | 103 | ||
| 2 | 0 | 354.5 ± 25.7 | — | |
| 10 | 363.6 ± 29.2 | 91 | ||
| 3 | 0 | 223.5 ± 14.4 | — | |
| 10 | 232.6 ± 18.3 | 91 | ||
| 4 | 0 | 408.5 ± 32.4 | — | |
| 50 | 461.2 ± 38.6 | 105 | ||
| 5 | 0 | 258.5 ± 20.2 | — | |
| 50 | 304.8 ± 18.8 | 93 | ||
| 6 | 0 | 167.5 ± 12.1 | — | |
| 50 | 220.3 ± 18.5 | 106 | ||
| 7 | 0 | 302.5 ± 26.2 | — | |
| 50 | 353.6 ± 31.3 | 102 | ||
| 8 | 0 | 25.5 ± 2.4 | — | |
| 70 | 97.1 ± 7.6 | 102 | ||
| 9 | 0 | 288.5 ± 22.9 | — | |
| 70 | 353.3 ± 27.4 | 93 | ||
| 10 | 0 | 367.0 ± 33.8 | — | |
| 70 | 434.2 ± 35.5 | 96 | ||
| 11 | 0 | 208.4 ± 12.9 | — | |
| 70 | 281.0 ± 20.1 | 104 | ||
| 12 | 0 | 155.7 ± 11.4 | — | |
| 70 | 230.1 ± 16.2 | 106 | ||
| 13 | 0 | 325.5 ± 22.4 | — | |
| 80 | 409.3 ± 35.6 | 105 | ||
| 14 | 0 | 163.5 ± 9.8 | — | |
| 80 | 239.6 ± 20.7 | 95 | ||
| 15 | 0 | 297.9 ± 25.4 | — | |
| 80 | 382.3 ± 29.3 | 106 | ||
| 16 | 0 | 144.5 ± 6.8 | — | |
| 80 | 219.2 ± 13.5 | 93 | ||
| 17 | 0 | 98.5 ± 8.4 | — | |
| 100 | 201.1 ± 15.6 | 103 | ||
| 18 | 0 | 377.4 ± 32.3 | — | |
| 100 | 483.6 ± 27.5 | 106 | ||
| 19 | 0 | 264.8 ± 21.7 | — | |
| 100 | 372.6 ± 19.8 | 108 | ||
| 20 | 0 | 175.5 ± 10.2 | — | |
| 100 | 276.4 ± 25.3 | 101 | ||
| NIST 1571, Orchard Leaves | 45 ± 3.1b | 42.8 ± 3.6 | 95 | |
Also, to investigate the accuracy and matrix effect, the added-found method was employed. All the opium and refined opium samples were spiked with the Pb at different concentration levels and the HLLME-LNPS was applied to these (n = 3). The results showed (Table 4) that the relative recoveries of Pb were in the range of 91–108%. In addition, the accuracy of the HLLME-LNPS method was also tested by measuring the concentration of the Pb in a standard reference material (NIST 1571, Orchard Leaves) with a certified Pb content of 45 ± 3.1 μg g−1, and the results are given in Table 4. The results showed that the obtained values are in satisfactory agreement with the certified values.
3.9. Comparison of the presented method with other methods
The proposed method is compared with other analytical methods for the extraction, preconcentration and measurement of Pb ions in different samples and the results are summarized in Table 5. One of the important advantages of this method compared to other methods is the very short extraction time, which is less than one minute in total. The extraction solvent used in this method is cheaper, less toxic and less dangerous to the environment than other solvents. It does not require complex equipment and facilities, and its operation is very simple. The efficiency of the extraction, detection limit and accuracy of the presented method can be compared with other methods and, in some cases, it is even better. All these results indicate that HLLME-LNPS is a very fast, reproducible and simple method that can be used for the extraction and preconcentration of Pb ions from opium samples.| Methods | LODa (μg l−1) | LRb (μg l−1) | RSDc% | Extractant volume (μl) | Extraction time (min) | Samples | Reference |
|---|---|---|---|---|---|---|---|
| a LOD, limit of detection.b LR, linear range.c RSD, relative standard deviation. | |||||||
| MADLLME-SFO-GFAAS | 0.1 | 0.3–50 | 3.2 | 530 | <10 | Lipsticks and hair dyes | 8 |
| DLLME-SFO-GFAAS | 0.05 | 0.1–50 | 4.7 | 1 ml + 40 | <10 | Rice, wheat, barley, peas, beans, corn and lentil | 17 |
| DLLME-FAAS | 0.5 | 1–70 | 2.0 | 2.5 ml + 52 | <3 | Water | 25 |
| SPE-DLLME-GFAAS | 0.001 | 0.003–0.06 | 5.2 | 1.5 ml + 18 | <15 | Water | 26 |
| DLLME-SDES-GFAAS | 0.01 | 0.02–200 | 2.3 | 50 | <15 | Soil and vegetables | 29 |
| DLLME-SFO-GFAAS | 0.04 | 0.1–100 | 3.5 | 1 ml + 40 | <10 | Trout fish | 30 |
| HLLME-LNPS-GFAAS | 0.2 | 0.6–100 | 2.8 | 400 | <1 | Opium and refined opium | This work |
4. Conclusions
This study is the first attempt to investigate the performance of HLLME-LNPS for the extraction and preconcentration of Pb in Pb-adulterated opium and refined opium. In this method, unlike other microextraction methods, centrifugation or a reagent additive is not used to separate the phase, and phase separation occurs only with decreasing temperature. Liquid nitrogen is also used to reduce the temperature, which occurs in a very short time, and the extraction and preparation time of the sample is reduced to less than one minute. The presented method is very simple, low cost, fast, environmentally friendly and applicable in any laboratory. The proposed method has the potential to be used as one of the alternatives to conventional methods for the extraction of organic and inorganic compounds from different matrices and can be coupled with other instruments, such as GC, HPLC, FAAS, etc. Due to the high selectivity of this method for the extraction, preconcentration and determination of Pb ions, it was successfully applied for Pb determination in Pb-adulterated opium and refined opium. The results of the sample analysis showed that 65% of the opium samples and 85% of the refined opium samples had a much higher than expected level of contaminated Pb, and this contamination poses a serious threat to drug users. The high concentration of Pb in drugs is mainly due to the addition of Pb compounds to drugs by traffickers and sellers of the drugs to increase weight and profits.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Research Council of Kermanshah University of Medical Sciences (Grant Number: 97765) for financial support.References
- E. R. Zaaijer, J. Bruijel, P. Blanken, V. Hendriks, M. W. J. Koeter, M. J. Kreek, J. Booij, A. E. Goudriaan, J. M. Van Ree and W. Van den Brink, Drug Alcohol Depend., 2014, 145, 101–105 CrossRef PubMed.
- A. L. Van Nuijs, S. Castiglioni, I. Tarcomnicu, C. Postigo, M. L. De Alda, H. Neels, E. Zuccato, D. Barcelo and A. Covaci, Sci. Total Environ., 2011, 409, 3564–3577 CrossRef CAS PubMed.
- R. Aghababaei, I. Javadi, A. Nili-Ahmadabadi, S. Parsafar and D. Ahmadimoghaddam, Daru, J. Pharm. Sci., 2018, 26, 77–83 CrossRef CAS PubMed.
- K. Oskarsson, The role of Iran in Afghanistan's reconstruction & development, Civil-Military Fusion Center, 2013 Search PubMed.
- Opium Impurities Declared by a Drug Dealer. Tabnak, 2016, accessed 10 November 2019, http://www.tabnak.ir/fa/news/612436/.
- Lethal Impurities of Opioids, JameJam online, 2014, accessed 10 November 2019, http://jamejamonline.ir/online/1765934059982350484/.
- G. Flora, D. Gupta and A. Tiwari, Interdiscip. Toxicol., 2012, 5, 47–58 CAS.
- K. Sharafi, N. Fattahi, M. Pirsaheb, H. Yarmohamadi and M. Fazlzadeh Davil, Int. J. Cosmet. Sci., 2015, 37, 489–495 CrossRef CAS PubMed.
- N. Zamani, H. Hassanian-Moghaddam, H. Bahrami-Motlagh, S. Ahmadi and S. Phillips, J. Trace Elem. Med. Biol., 2019, 55, 26–32 CrossRef CAS PubMed.
- R. Afshari and A. Emadzadeh, Drug Chem. Toxicol., 2010, 33, 48–49 CrossRef CAS PubMed.
- H. Khatibi-Moghadam, M. Khadem-Rezaiyan and R. Afshari, Hum. Exp. Toxicol., 2016, 35, 861–865 CrossRef CAS PubMed.
- M. M. Hayatbakhsh, Z. Oghabian, E. Conlon, S. Nakhaee, A. R. Amirabadizadeh, M. J. Zahedi, S. DarvishMoghadam, B. Ahmadi, S. Soroush, J. Aaseth and O. Mehrpour, Subst. Abuse Treat. Prev. Pol., 2017, 12, 43–50 CrossRef PubMed.
- S. Alinejad, J. Aaseth, M. Abdollahi, H. Hassanian-Moghaddam and O. Mehrpour, Basic Clin. Pharmacol. Toxicol., 2018, 122, 56–64 CrossRef CAS PubMed.
- A. Mandegary, S. Rostami, K. Alimoghaddam, A. Ghavamzadeh and M. H. Ghahremani, Asian Pac. J. Cancer Prev., 2011, 12, 1279–1282 Search PubMed.
- S. Shojaeepour, M. Fazeli, Z. Oghabian, L. Pourgholi and A. Mandegary, Food Chem. Toxicol., 2018, 120, 571–577 CrossRef CAS PubMed.
- B. T. Zaman, A. F. Erulaş, D. S. Chormey and S. Bakirdere, Food Chem., 2020, 303, 125396 CrossRef CAS PubMed , in press.
- M. Ataee, T. Ahmadi-Jouibari and N. Fattahi, Int. J. Environ. Anal. Chem., 2016, 96, 271–283 CrossRef CAS.
- I. Rehan, M. A. Gondal and K. Rehan, Talanta, 2018, 182, 443–449 CrossRef CAS PubMed.
- J. Terán-Baamonde, S. Bouchet, E. Tessier and D. Amouroux, J. Chromatogr. A, 2018, 1547, 77–85 CrossRef PubMed.
- A. L. D. Comitre and B. F. Reis, Talanta, 2005, 65, 846–852 CrossRef CAS PubMed.
- Y. Wang, J. Han, Y. Liu, L. Wang, L. Ni and X. Tang, Food Chem., 2016, 190, 1130–1136 CrossRef CAS PubMed.
- A. Mehdinia, M. Ramezani and A. Jabbari, Food Chem., 2017, 237, 1112–1117 CrossRef CAS PubMed.
- S. Liu, H. Yang, S. Yang, Y. Huang, Z. Xiang and G. Ouyang, Anal. Chim. Acta, 2020, 1111, 147–154 CrossRef CAS PubMed.
- T. Borahan, T. Unutkan, N. B. Turan, F. Turak and S. Bakırdere, Food Chem., 2019, 299, 125065 CrossRef CAS PubMed , in press.
- M. T. Naseri, P. Hemmatkhah, M. R. M. Hosseini and Y. Assadi, Anal. Chim. Acta, 2008, 610, 135–141 CrossRef CAS PubMed.
- M. Shamsipur, N. Fattahi, M. Sadeghi and M. Pirsaheb, J. Iran. Chem. Soc., 2014, 11, 249–256 CrossRef CAS.
- S. Erarpat, G. Özzeybek, D. S. Chormey and S. Bakırdere, Chemosphere, 2017, 189, 180–185 CrossRef CAS PubMed.
- D. Martínez, G. Grindlay, L. Gras and J. Mora, J. Food Compost. Anal., 2018, 67, 178–183 CrossRef.
- M. H. Habibollahi, K. Karimyan, H. Arfaeinia, N. Mirzaei, Y. Safari, R. Akramipour, H. Sharafi and N. Fattahi, J. Sci. Food Agric., 2018, 99, 656–665 CrossRef PubMed.
- M. Pirsaheb and N. Fattahi, Anal. Methods, 2015, 7, 6266–6273 RSC.
- M. Sadeghi, Z. Nematifar, M. Irandoust, N. Fattahi, P. Hamzei, A. Barati, M. Ramezani and M. Shamsipur, RSC Adv., 2015, 5, 100511–100521 RSC.
- S. Taheri, F. Jalali, N. Fattahi, R. Jalili and G. Bahrami, J. Sep. Sci., 2015, 38, 3545–3551 CrossRef CAS PubMed.
- M. Rezaee, F. Khalilian, H. A. Mashayekhi and N. Fattahi, Anal. Methods, 2014, 6, 3456–3461 RSC.
- M. Pirsaheb and N. Fattahi, RSC Adv., 2018, 8, 11412–11418 RSC.
- M. Gupta and A. Dsouza, J. Food Compost. Anal., 2020, 87, 103396 CrossRef CAS , in press.
- X. Di, X. Wang, Y. Liu, X. Guo and X. Di, J. Chromatogr. B: Biomed. Sci. Appl., 2019, 1118–1119, 109–115 CrossRef CAS PubMed.
- M. Torbati, M. A. Farajzadeh, M. Torbati, A. A. Alizadeh Nabil, A. Mohebbi and M. R. Afshar Mogaddam, Talanta, 2018, 176, 565–572 CrossRef CAS PubMed.
- M. Karimi, S. Dadfarnia, A. M. Haji Shabani, F. Tamaddon and D. Azadi, Talanta, 2015, 144, 648–654 CrossRef CAS PubMed.
- S. M. Goulart, M. E. L. R. de Queiroz, A. A. Neves and J. H. de Queiroz, Talanta, 2008, 75, 1320–1323 CrossRef CAS PubMed.
- F. Wang, S. Li, H. Feng, Y. Yang, B. Xiao and D. Chen, Food Chem., 2019, 275, 530–538 CrossRef CAS PubMed.
- H. Zhang, S. Li, X. Liu, F. Yuan, Y. Liang and Z. Shi, J. Liq. Chromatogr. Relat. Technol., 2015, 38, 584–590 CrossRef CAS.
- F. Ahmadi, Y. Shahbazi and N. Karami, Food Anal. Methods, 2015, 8, 1883–1891 CrossRef.
- T. Okhravi, S. M. Sorouraddin, M. A. Farajzadeh and A. Mohebbi, Anal. Bioanal. Chem., 2020, 412, 1675–1684 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |