
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA greatly improved procedure for the synthesis of an antibiotic-drug candidate 2,4-diacetylphloroglucinol over silica sulphuric acid catalyst: multivariate optimisation and environmental assessment protocol comparison by metrics†
Triana Kusumaningsih *,
Wahyu Eko Prasetyo and
Maulidan Firdaus
*,
Wahyu Eko Prasetyo and
Maulidan Firdaus
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Sebelas Maret University, Jl. Ir. Sutami No. 36A, Surakarta, 57126, Indonesia. E-mail: triana_kusumaningsih@staff.uns.ac.id
First published on 27th August 2020
Abstract
Efforts toward the development of a straightforward greener Gram-scale synthesis of the antibiotic compound 2,4-diacetylphloroglucinol (DAPG) have been developed. This beneficial procedure was accomplished through the Friedel–Crafts acylation of phloroglucinol over inexpensive heterogeneous silica sulphuric acid (SSA) catalyst via ultrasound-assisted (US) synthesis under solvent-free condition. The influences of various parameters such as temperature, catalyst loading, and reaction time on the reaction performance were analysed using a multivariate statistical modelling response surface methodology (RSM). A high yield of DAPG (95%) was achieved at 60 °C after 15–20 min reaction with the presence of 10% (w/w) SSA as the catalyst. Column chromatography-free and a Gram scale-up reaction also exhibited the practical applicability of this newly developed protocol. The SSA catalyst was recovered and recycled up to 10 consecutive runs with no appreciable loss of activity. A plausible mechanism for the Friedel–Crafts acylation of phloroglucinol is proposed. Moreover, an environmental assessment has been carried out over this present method and compared with several established literature using the EATOS software and the Andraos algorithm to assess the consumption of the substrates, solvents, catalysts, and the production of coupled products or by-products. In addition, their energy consumptions were also determined. The data collected showed that the present method is the most promising one, characterised by the highest environmental impact profile against all the other reported methods. The physicochemical properties of the synthesised DAPG were assessed and exhibited reasonable oral bioavailability drug property as determined by Lipinski's rules.
Introduction
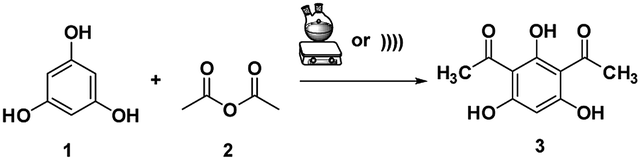
DAPG, an antibiotic produced by Pseudomonas fluorescence 2P24., is one of the most intriguing phenolic secondary metabolite compounds.1–3 Originally, its antimicrobial1,4–7 and fungicide or antifungal2,8,9 agents against various plant pathogens were paid attention to. By virtue of the unique structure of DAPG (Scheme 1), the amphipathic molecules that consist of the acyl group, alkyl group, and hydroxyl group can enable it to interact with various receptors in organisms. Within this context, earlier studies have addressed the antiviral,10 anticancer, antiproliferative, anti-inflammatory, and antimetastatic potential11–13 of the non-nitrogenous polyketide DAPG. This compound was also documented to have beneficial effects on plant growth and it has been shown as a molecular signalling compound.14 Owing to of its extensive applications, the synthetic research of this privileged compound appears to draw the continuing interest of scholars who are expected to be able to afford a conceptual platform for the discovery of novel drugs. Recently, various procedures for producing DAPG using either the biosynthesis or synthetic pathway have been extensively studied.2–4,15,16 The common route to DAPG involves the Friedel–Crafts acylation of either phloroglucinol (PhL)2,16–21 or monoacetylphloroglucinol (MAPG)16 in the presence of Lewis or Brønsted acids, using an acylating agent (acyl halides, acid anhydrides, and/or carboxylic acids).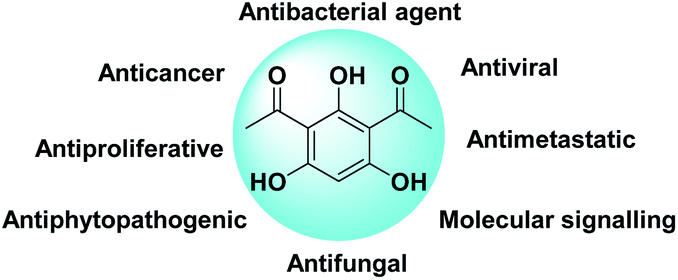 | ||
| Scheme 1 Molecular structure of DAPG and its potential applications. | ||
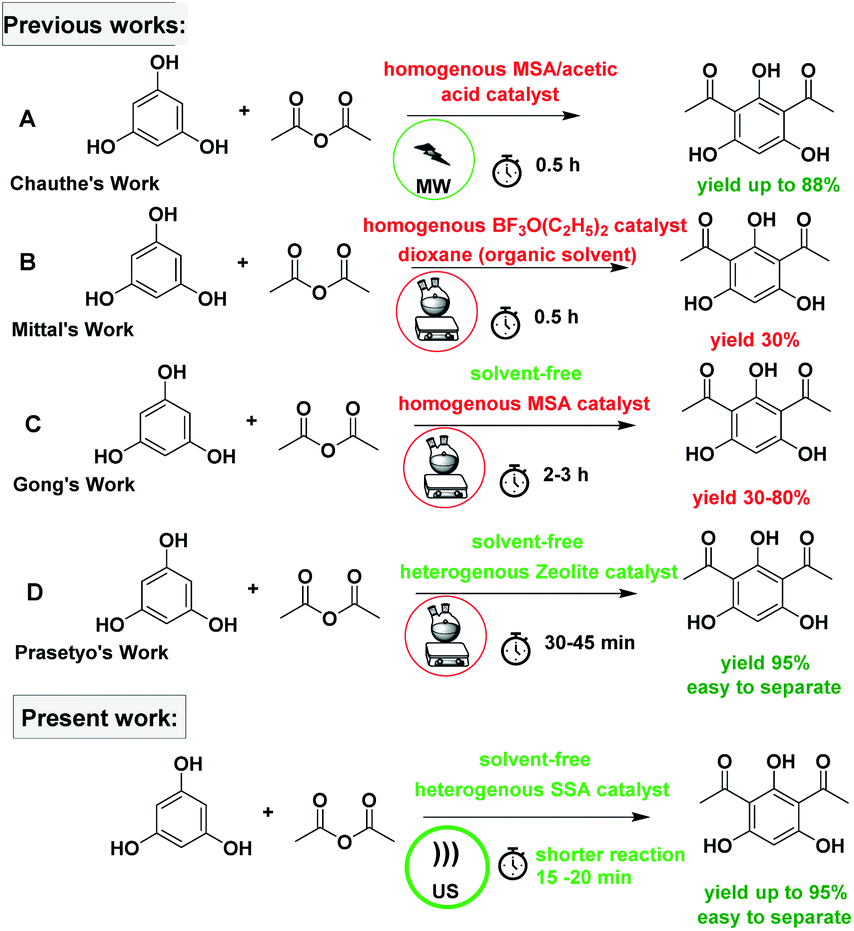
To date, a plethora of procedures have been reported for the synthesis of DAPG and acylated phloroglucinol that utilize AlCl3,17,20 BF3–etherate,5,20,22 and ZnCl2 (ref. 22) as the catalyst. Nevertheless, the foremost drawbacks that were encountered with these last previous routes include variability in the yield, the use of harmful organic solvents, long reaction times, and tedious workup protocols. Hence, as a result of increasing the awareness of environmentally benign chemistry, a solvent-free method has been demonstrated for the synthesis of DAPG using various catalysts such as MSA,2 HCl, TFA, H2SO4, CH3COOH, HBr/alumina, and MSA/CH3COOH.16 Meanwhile, despite the enormous advantages that are offered by a solvent-free reaction, in pursuit of selecting an appropriate catalyst system, we realized that each of these reported procedures have several undesirable aspects, considering the environmental consciousness. Typically, homogeneous catalysts often encounter recovery and reusability problems after the reaction, which leads to high E-factor and results in unacceptably high manufacturing costs. Therefore, it is still desirable to develop various green techniques for the synthesis of DAPG that use highly efficient and reusable catalysts.
Over the past decade, heterogeneous catalysts have drawn increasing attention from several industries due to their number of benefits such as easy recovery and reactivation by a simple treatment, which play an essential green chemistry and sustainability role.18,23–25 In addition, by combining the practice of heterogeneous catalyst and an alternative sustainable synthetic strategy, such as ultrasound-assisted synthesis (US), much attention has been received due to their unique capabilities of providing chemical transformation under milder conditions.26–29 Specifically, along with the utilization of the heterogeneous catalytic system, US irradiation offers several potential effects, such as the promotion of cavitation bubble formation, increase in the active catalyst surface area, and removal of impurities deposited on the catalyst.30,31 Besides, heterogeneous catalysis in the solvent-free condition is considered as a green and sustainable approach since the elimination of solvents and reduces the environmental factor (E-factor). To this endeavour, we developed a novel catalyst system for the synthesis of DAPG, namely, silica sulphuric acid or H2SO4–SiO2 (SSA), which was invented by Zolfigol,32 obtained from silica gel (SG) and supported by sulphuric acid,33 which was used for the first time in promoting the synthesis of disulphide and thionitrites under mild conditions. This catalyst system provided various benefits, including increase in the surface area, improved thermal and mechanical stabilities, as well as simplicity of handling and reusability.32,34 Thus, we would, in the future, be investigating these soon in the study for promoting other chemical reactions.
In this work, attracted by the potential Brønsted acid active sites of SSA and as a part of our on-going endeavours in designing a green synthetic route for a biologically promising compound, we present herein various synthetic procedures to develop a more sustainable route in order to convert phloroglucinol and acetic anhydride into DAPG over SSA as a novel, efficient, and reusable heterogeneous catalyst in an environment-friendly US method. Furthermore, in order to meet the suitable operation conditions for the synthesis of DAPG as well as to explain the effects of the factors on the selected response, the design of experiment (DoE) using response surface methodology (RSM) was performed.35–37 Hence, the three variables, namely, the temperature, catalyst loading, and reaction time, were selected. In addition, the developed procedure for DAPG has been compared with previously established literature (Scheme 2) using the EATOS software and the Andraos' algorithm, along with energy calculation metric, in order to assess any process, information, and versatile outputs related to the “greenness” of the chemical reaction.38–41 Further, in order to provide the potential oral bioavailability of DAPG, a drug-likeness evaluation based on Lipinski's rule was also demonstrated.
 | ||
| Scheme 2 Various selected literature for the synthesis of DAPG.2,16,18,20 | ||
Experimental
General information
All chemicals were reagent grade, which were purchased from Sigma-Aldrich (Singapore) and were used without further purification. Distilled water was obtained from a Sebelas Maret University (UNS) water purification system. Infrared spectra were recorded using a Fourier transform infrared (FT-IR) Prestige-21 Spectrometer, Shimadzu with in the 4000–400 cm−1 region (samples were dispersed into potassium bromide (KBr) pellet at room temperature). X-ray diffraction (XRD) was performed using a Shimadzu Lab-X 6000 diffractometer with Cu Kα radiation (1.5418 Å). Field Emission Scanning Electron Microscopy (FE-SEM) images were obtained using a JEOL JSM-7900F instrument. The 1H and 13C NMR spectra were recorded using an Agilent DD2 NMR Spectrometer using CD3Cl3 as the solvent, operated at 25 °C, 500 MHz for 1H and at 125 MHz for 13C. The collected chemical shifts were reported in parts per million (ppm) relative to that of tetramethylsilane (TMS) as the external reference. Coupling constants (J) are reported in hertz (Hz). Hydrogen coupling patterns were identified as singlet (s), doublet (d), triplet (t), and multiplet (m). Sonication was performed by using an Ultrasonic, Branson B1510R-DTH Ultrasonic Cleaner (frequency 20 kHz and 130 W). The progress of the reactions was observed by thin layer chromatography (TLC), which was performed on gel 60 F254 (Merck) plates and visualised by using UV light (λ = 254 and 365 nm).Preparation and characterisation of the SSA catalyst
The SSA catalyst was prepared from silica gel (SG) (pore size 0.7–0.85 cm3 g−1 pore volume, particle size 230–400 mesh, and surface area ≥ 480 m2 g−1) and concentrated sulphuric acid by following the reported procedure with slight modifications.42,43 SG (12.375 g) was soaked in diethyl ether (50 mL) until a slurry silica gel was formed, followed by the addition of concentrated H2SO4 (6.25 mmol). The obtained mixture was mechanically stirred for 15 min under a nitrogen atmosphere and dried at 60 °C for 18 h to remove the solvent. The obtained residue was heated at 120 °C for 6 h in order to obtain the SSA catalyst as a free-flowing powder. The resultant SSA catalyst was gently stored in a desiccator before further use. Afterwards, the prepared catalyst was further characterized in detail using XRD, FTIR, SEM-EDS, and acid–base titration. The acidity of the SSA catalyst was measured by the acid–base titration method as reported in a previous study.44 The SSA catalyst (100 mg) was stirred in NaOH solution (20 mL of 0.1 M) for 6 h and then titrated with HCl solution (0.1 M) to determine the acidity of the sample. In addition, as a comparison, several protic acids (acetic acid, methanesulfonic acid, and perchloric acid) were also immobilised on silica gel using the same method. The resultant catalysts were then tested on the same typical reaction and conditions as that for the SSA catalyst.General synthesis of DAPG
A mixture of phloroglucinol 1 (1 mmol), acetic anhydride 2 (2 mmol), and SSA was stirred in a round bottom flask at room temperature for 15 min and heated at 80 °C for 1–2 h. For the US method, the mixture was mechanically stirred for 5 min, followed by sonication for 15–20 min at 60 °C under the ambient atmosphere. TLC analysis was used to monitor the progress of the reaction. After the reaction was completed, the obtained mixture was cooled at the ambient temperature, then diluted with EtOAc (15 mL), filtered from the used catalyst, and water (25 mL) was added. The crude solution was extracted with EtOAc (3× 15 mL) and the combined organic layer was dried over anhydrous Na2SO4. The solvent was evaporated under vacuum and the crude product was subjected to recrystallisation using benzene/petroleum ether 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) following the previous literature4 and used without column chromatography treatment (for compound 3, 5a and 5b). Afterwards, the isolated product was subjected to further characterization in detail using 1H NMR and 13C NMR. The collected spectra are shown in the ESI.†
:1 (v/v) following the previous literature4 and used without column chromatography treatment (for compound 3, 5a and 5b). Afterwards, the isolated product was subjected to further characterization in detail using 1H NMR and 13C NMR. The collected spectra are shown in the ESI.†
Reusability and heterogeneity of the SSA catalyst
The recovered SSA catalyst was soaked in ethyl acetate (10 mL) under magnetic stirring for 30 min. It was filtered, followed by washing with ethyl acetate (3× 5 mL), and was soaked again with acetone (3× 5 mL) and stirred for 15 min. After washing, the catalyst was filtered, dried in a rotary evaporator until the solvent was removed, and was then weighed. The amount of catalyst loss was recompensed by adding fresh SSA. Afterwards, the catalyst was reactivated at 120 °C for 3 h. The reaction was repeated following the above procedure.To investigate the possible leaching of the active sites from the catalyst surface of SSA into the reaction medium, a hot filtration method was performed.25,44,45 Under the optimised conditions, the Friedel–Crafts acylation of phloroglucinol was carried out. After 5 min of the reaction, all the catalyst particles were separated by hot filtration and centrifuged. The reaction was continued for 30 min in the absence of the catalyst and was frequently monitored by TLC.
Response surface methodology (RSM)
The reaction optimisation was carried out using multivariate statistical modelling RSM method (operated using Design-Expert Version 11.0.5.0) in order to find out the selected parameters for the synthesis of DAPG using phloroglucinol, acetic anhydride, and SSA as the catalyst. The reaction temperature, catalyst loading, and reaction time were chosen as independent variables based on the previous literature and the results of the preliminary studies. The three independent variables were coded at −1, 0, and +1, which represent low value, centre value, and high value, respectively, as presented in Table 1. According to the model, 20 sets of experiments were carried out to build the desired model.| Run | Experimental factors | Coded variables | Responses | ||||
|---|---|---|---|---|---|---|---|
| T (°C) | Catalyst loading (%w/w) | t (min) | X1 | X2 | X3 | The yield of DAPG (%) | |
| 1 | 25 | 10 | 20 | −1 | 0 | −1 | 10 |
| 2 | 25 | 20 | 60 | −1 | 1 | 1 | 8 |
| 3 | 25 | 5 | 60 | −1 | −1 | 1 | 6 |
| 4 | 80 | 20 | 60 | 1 | 1 | 1 | 68 |
| 5 | 25 | 10 | 30 | −1 | 0 | 0 | 16 |
| 6 | 25 | 20 | 20 | −1 | 1 | −1 | 10 |
| 37 | 80 | 5 | 30 | 1 | −1 | 0 | 52 |
| 8 | 60 | 20 | 30 | 0 | 1 | 0 | 74 |
| 9 | 80 | 20 | 20 | 1 | 1 | −1 | 79 |
| 10 | 60 | 10 | 60 | 0 | 0 | 1 | 80 |
| 11 | 25 | 10 | 30 | −1 | 0 | 0 | 12 |
| 12 | 60 | 10 | 20 | 0 | 0 | −1 | 95 |
| 13 | 25 | 10 | 30 | −1 | 0 | 0 | 14 |
| 14 | 60 | 10 | 60 | 0 | 0 | 1 | 82 |
| 15 | 80 | 5 | 30 | 1 | −1 | 0 | 54 |
| 16 | 80 | 5 | 20 | 1 | −1 | −1 | 46 |
| 17 | 60 | 20 | 20 | 0 | 1 | −1 | 83 |
| 18 | 80 | 10 | 20 | 1 | 0 | −1 | 84 |
| 19 | 60 | 20 | 30 | 0 | 1 | 0 | 80 |
| 20 | 60 | 5 | 20 | 0 | −1 | −1 | 68 |
Environmental assessment
For the environmental assessment of the present method, an overview of several relevant methods of synthesis of DAPG are summarised in Scheme 1. The environmental assessment was performed by employing the freeware package Environmental Assessment Tool for Organic Syntheses (EATOS) software developed by Eissen and Metzger40 and the algorithm by Andraos,46 which considers the consumption of substrates, solvents, catalysts, coupled products, and by-products. However, in this assessment, we did not take into account the workup and purification phases due to the lack of literature details. An EATOS is a downloadable Java-archive file, which permits the evaluation of all of the chemicals used in the synthesis of DAPG and its work-up procedures. The data required for the EATOS analysis were attained from the Material Safety Data Sheets (MSDSs) of Sigma-Aldrich combined with the ChemSpider Database to validate their consistency. In particular, the required data deals with risk, human toxicity, chronic toxicity, eco-toxicology, accumulation, and prices. The required data associated with their stoichiometric or amounts of material were obtained from the recent literature.28,31,38,39 In addition, in an attempt to determine the energy consumption of the current reaction, the previous energy calculation metric method was followed.41In silico evaluation of drug-likeness
Drug-likeness study was established based on Lipinski's rule using online Molinspiration software (https://www.molinspiration.com/docu/miscreen/druglikeness.html), combined with SwissADME software (http://www.swissadme.ch/).Results and discussion
Primarily, we began our study by preparing the Brønsted acid SSA catalyst following the reaction pathway (Scheme 3).43 It was further characterised by several techniques including FT-IR, XRD, SEM, EDS analysis, and acid–base titration. The measured XRD patterns for SG and SSA (ESI, Fig. S2†) consist of similar patterns, with a broad peak in the range of 22.6°, which corroborated the non-crystalline nature of SG.47 The decrease in this peak's intensity compared to that of SSA implies that sulphonation was successfully achieved.48 | ||
| Scheme 3 The preparation of SSA involving the addition of sulphuric acid to SG. | ||
Afterwards, FT-IR analysis was employed in order to confirm the successive functionalisation of SG to form SSA. Upon sulphonation treatment, the FTIR spectrum of SSA was identical to that of SG with the presence of a few additional S–O vibrational frequencies, as exhibited in Fig. 1. The band at about 2600 cm−1 was linked to the O–H vibration of the sulphonate group. Other peaks at 1252, 1080, 1024, and 709 cm−1 were attributed to the asymmetric O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO, symmetric OSO, SO stretch, and S–O stretch, respectively.43,49 As a note, after the sulphonation of silica gel by sulphuric acid, some hydroxyl groups of silica gel (the isolated hydroxyl group) may still remain, which was proved from the FTIR analysis, which showed the presence of the silanol group band within the range of 3447–3500 cm−1.43,50,51
SO, symmetric OSO, SO stretch, and S–O stretch, respectively.43,49 As a note, after the sulphonation of silica gel by sulphuric acid, some hydroxyl groups of silica gel (the isolated hydroxyl group) may still remain, which was proved from the FTIR analysis, which showed the presence of the silanol group band within the range of 3447–3500 cm−1.43,50,51
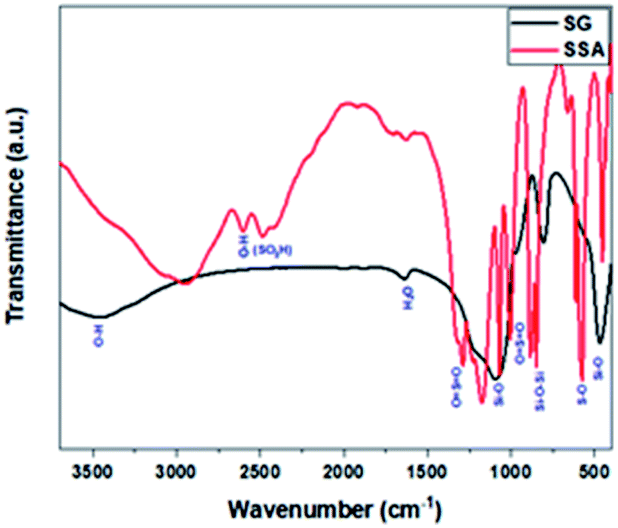 | ||
| Fig. 1 FTIR spectra of SG and SSA. | ||
The morphology of SG and SSA was studied via SEM, as shown in ESI, Fig. S3A and C.† The sulphonation of SG also reveals a similar morphology to that of neat SG, which is reinforced by the XRD and FTIR results. Thus, it might be inferred that the sulphonation of SG does not affect it in terms of the size and the shape. According to the EDS analysis (ESI, Fig. S3B and D†), the existence of the SO3H group on neat SG was also observed, which was indicated by the presence of a strong signal in the region for O and S. In general, these results show that the introduction of the SO3H group does not alter the silica gel framework.43 Also, to investigate the acidity of the SSA catalyst, the acid–base titration method was employed. The acidity was found to be 3.8 mmol H+ per g SiO2, which suggests a strong acidic nature.49
Catalyst screening
In this study, in an endeavour to meet the green chemistry goals,23 we decided to use green and halogen-free acetic anhydride (2) instead of acyl chloride as an acylating agent to synthesize DAPG. Initially, room temperature reaction with and without the presence of the catalyst was carried out under solvent-free condition. Unfortunately, there was no desired product reaction observed for both of these typical reactions. The traditional homogenous catalysts including both the Brønsted and Lewis acids such as HCl, H2SO4, CH3COOH, MSA, and MSA/CH3COOH showed varying catalytic activities under the oil-bath method. According to Table 2, the reactions carried out using HCl, H2SO4, and CH3COOH were found to give low catalytic activities. Even after these reactions were carried out under reflux with vigorous stirring for 12 h, they still gave incomplete reactions (only less than 20% yields of 3). In addition, several unexpected products (phenylacetate) were also observed (as confirmed by TLC and NMR analysis). On the contrary, when MSA and MSA/CH3COOH were introduced as the catalyst, moderate yields of 40–70 and 80% were observed, respectively. The same typical reaction was then screened carefully under the US method, which proceeded smoothly. In contrast, there was no observed product for the use of HCl, H2SO4, and CH3COOH as the catalyst, whereas the presence of MSA and MSA/CH3COOH could improve the yield to 82 and 88%, respectively.
|
|||||
|---|---|---|---|---|---|
| Run | Catalyst | Method | |||
| Conventional method (oil-bath) | US | ||||
| t (min) | Yieldb (%) | t (min) | Yieldb (%) | ||
| a Reaction conditions: 1 mmol of phloroglucinol (1), 2 mmol of acetic anhydride (2) and the above-mentioned catalyst were operated under conventional procedure at 80 °C and US method at 60 °C.b Isolated yield.c No reaction. | |||||
| 1 | 3 mmol of MSA | 30–45 | 40–80 | 20 | 60–86 |
| 2 | 3 mmol of CH3COOH | 45 | c | 20 | c |
| 3 | 3 mmol of TFA | 45 | 10 | 15–20 | 92–94 |
| 4 | 3 mmol of H2SO4 | 45 | 10 | 20 | 25 |
| 5 | 20 wt% silica gel | 45 | <10 | 20 | 12–17 |
| 6 | 3 mmol of HCl | 45 | c | 20 | c |
| 7 | 20 wt% of SSA | 45 | 85 | 15–20 | 87–90 |
| 8 | 10 wt% of SSA | 45 | 88 | 15–20 | 92–95 |
| 9 | 10 wt% of SSA/1 mmol of CH3COOH | 30–45 | 93 | 15–20 | 94–96 |
| 10 | No catalyst | 120 | c | 120 | c |
Afterwards, 20% (w/w) SSA was tested as the catalyst in a similar typical reaction via the conventional method (oil-bath) and US method, which resulted in DAPG yield of 85% and 90%, respectively. As an important aspect, we found that the reaction proceeded faster to give a higher yield of DAPG under the US method compared to the previously employed homogenous catalysts used under oil-bath method. Afterwards, an increase in the catalyst loading to 30% (w/w) resulted in a lower yield of DAPG, accompanied by the formation of triacetylphloroglucinol as a by-product. Fortunately, as the catalyst loading decreases to 10% (w/w), the yield of the product increased to be 95% and no by-product of phenylacetate was observed. Interestingly, it was noticed that the presence of only liquid sulphuric acid or silica gel could lead to rather low yields, followed by the formation of side products. Nevertheless, the high activity of the SSA catalyst may be attributed to its high acidity value caused by the intense dispersion of the SO3H group on the surface of silica.43 In addition, we envisaged that the synergistic effects between the active site of the SSA catalyst and silica gel also could enhance the catalytic activity. It is worth mentioning that silica gel itself has been used as a catalyst in various types of organic reactions through the silanol group as an active site.52,53 Thereby, the presence of two different active sites could possibly improve the collision frequency between the substrates and the catalytic sites of the SSA catalyst.
In an attempt to improve the yield, the addition of a small amount of acetic acid (∼1 mmol) could increase the yield of DAPG, albeit it was an insignificant improvement. It has been reported that Friedel–Crafts acylation may be enhanced with the presence of hydrogen bonding (H-bond) from a certain solvent and/or catalyst. Acetic acid could provide H-bond donor solvent molecules for Friedel–Crafts acylation.54 Afterwards, when the reaction was extended (overnight condition), it prompted the formation of several by-products (controlled by TLC), which were possibly obtained by decomposition of the product or its intermediates due to rough irradiation. In terms of the reaction time, this fruitful result revealed a significant improvement in the Friedel–Crafts acylation of phloroglucinol to DAPG. The possible interpretation for the positive association of this method is that the ultrasonic waves can boost the number of active cavitation bubbles and the size of the individual bubbles, leading to the improvement in the reaction efficiency with increased reaction rates and reduced reaction time.31,55
Encouraged by this result, we considered several solvents for further investigation (Table 3). Briefly, it is well known that the solvent used is an important factor influencing the chemical reactivity both in the reaction mechanism and the reaction rate.56 However, the result showed that the use of solvents caused the formation of several products including by-products. Accordingly, the implementation of a solvent-free condition is indeed greatly preferable for the synthesis of DAPG promoted by the SSA catalyst. This is evidenced in that the reaction rate observed in the solvent-free condition leads to more favourable reaction kinetics instead of the reactions demonstrated in the presence of any solvent. The main reason why the solvent-free condition could promote this reaction effectively is strongly attributed to the fact that one of the reactants is in the liquid phase (acetic anhydride) at the reaction temperature. Thereby, it must be miscible with phloroglucinol, where the molecular movement and the contact between the reactants with higher concentrations is favoured and the yield of DAPG is improved.29,57,58 Hence, Green Chemistry principle 5, which states that the “use of solvents should be made unnecessary wherever possible and innocuous when used”, is successfully implemented.23
| Run | Solvent | T (°C) | t (min) | Yieldb (%) | Solvent rankingd |
|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol of phloroglucinol (1), 2 mmol of acetic anhydride (2) and 20% of SSA catalyst were used under the US method at 60 °C.b Isolated yield.c No reaction.d Ranking of solvents with respective to CHEM21 solvent guide of “classical” solvents.59e Not reported. | |||||
| 1 | Solvent-free | 60–80 | 20–30 | 92 | e |
| 2 | Chloroform | Reflux | 20–60 | 65 | Highly hazardous |
| 3 | DCM | Reflux | 20–60 | 68 | Hazardous |
| 4 | Toluene | Reflux | 20–120 | c | Problematic |
| 5 | Benzene | Reflux | 20–120 | c | Highly hazardous |
| 6 | Acetonitrile | Reflux | 20–120 | 55 | Problematic |
| 7 | Nitrobenzene | Reflux | 20–60 | 68 | e |
| 8 | Water | Reflux | 20–120 | c | Recommended |
A plausible mechanism for the synthesis of DAPG using SSA
We herein also propose a possible mechanism reaction (Scheme 4) for the ultrasound-promoted one-pot synthesis of DAPG from phloroglucinol and acetic anhydride under solvent-free condition with the presence of SSA as the catalyst. We assume that the reasonable first step of this reaction is that the SSA catalyst activated acetic anhydride through protonation over the Brønsted acid active sites of the catalyst to form an acylium ion (I). Afterwards, the resultant acylium ion takes part in the Friedel–Crafts acylation, which leads to the competition for phenylacetate (PA) through O-acetylation or hydroxhyacetophenone (HAP) C-acetylation (II). Moreover, PA could undergo Fries rearrangement reaction through the O-acylation process, which is processed faster than the C-acylation one, followed by the formation of MAPG and subsequently followed by the formation of DAPG (III). Notably, this Friedel–Crafts acylation reaction is commonly processed faster than the ordinary aromatic compound or phenol due to the presence of strongly activating agents from the three phenol groups of phloroglucinol.60,61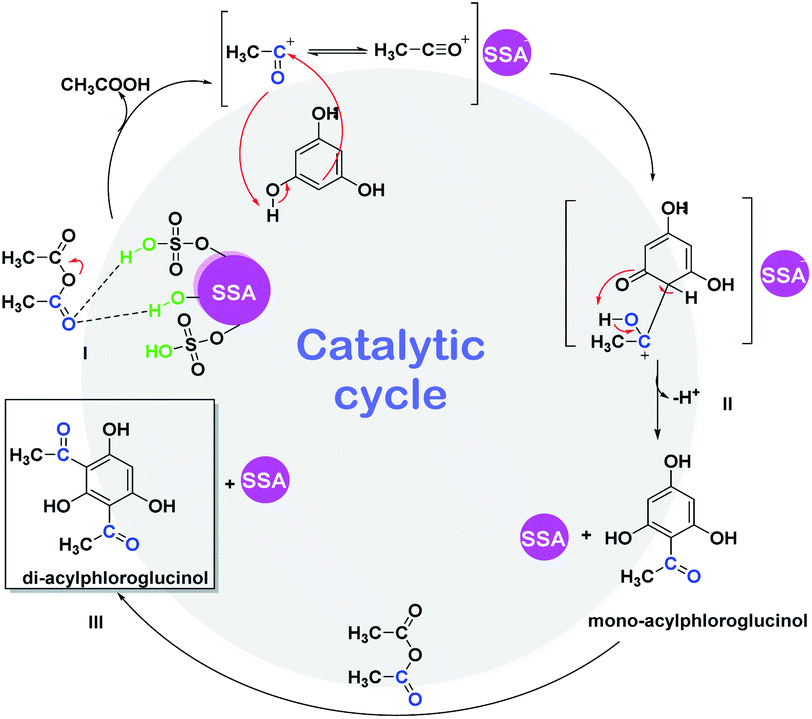 | ||
| Scheme 4 Proposed mechanism for the ultrasound-assisted Friedel–Crafts acylation between phloroglucinol (1) and acetic anhydride (2) promoted by SSA catalyst. | ||
Optimization of the reaction using RSM
In this work, an efficient statistical technique, namely, design of experiment (DoE) approach, has been employed, which is available for establishing the model and optimizing the reaction condition. This optimization approach has been broadly used in the fields of chemical engineering and analytical chemistry. However, it has not been popularly used in organic synthesis. Response surface methodology (RSM) is a part of DoE, which can provide an efficient means of the effects and interaction between multiple system input parameters either separately or in combination through an accurate second-order polynomial equation. In this work, the optimum reaction conditions were studied by varying several significant parameters in this reaction, including the effect of temperature, catalyst loading, and reaction time, according to the prior results and the literature report.18 Eqn (1) shows the second-order quadratic model relationship between the DAPG yield (Y) and the preferred variables.| Y = 84.03 + 29.70A + 6.75B − 1.63C + 3.83AB − 2.79AC − 2.88BC − 39.93A2 − 12.30B2 + 1.58C2 | (1) |
As shown in eqn (1), A (temperature) has a major effect on the DAPG yield (Y). Similarly, the catalyst loading (B), and the interaction between the temperature and catalyst loading (AB) also showed a positive impact on the DAPG yield. In different circumstances, the effects on the model parameters revealed that the reaction temperature (C), the interaction between the temperature and the reaction time (AC), and the interaction between catalyst loading and the reaction temperature (BC) showed a significant negative effect on the DAPG yield. According to Table 4, the p-value can be used as an indicator for checking the significance of each coefficient, where p-values less than 0.05 imply that the model terms are significant. The linear term of A, B, and the quadratic term of A (A2) and B (B2) are significant. Fig. 2 shows good linear correlation between the actual and predicted yield of DAPG. Accordingly, the developed model can possibly predict the yield of the experimental reaction. Considering all the above descriptions, it is strongly evidenced that the developed second-order quadratic model (as presented in eqn (1)) could fruitfully predict the performance of Friedel–Crafts acylation of phloroglucinol by altering the selected operating conditions. To validate the predictability of the model, duplicate runs were performed under the above-optimized conditions. In both the cases, the obtained yields were achieved to be 93% and 91%, which are slightly lower than the predicted ones. Nevertheless, these results still closely match with the predicted result of 95%.
| Source | Sum of squares | df | Mean square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| Model | 19711.72 | 9 | 2190.19 | 85.14 | <0.0001 | Significant |
| A-temperature | 8454.79 | 1 | 8454.79 | 328.68 | <0.0001 | |
| B-catalyst loading | 429.10 | 1 | 429.10 | 16.68 | 0.0022 | |
| C-reaction time | 26.85 | 1 | 26.85 | 1.04 | 0.3310 | |
| AB | 85.70 | 1 | 85.70 | 3.33 | 0.0979 | |
| AC | 52.66 | 1 | 52.66 | 2.05 | 0.1830 | |
| BC | 51.51 | 1 | 51.51 | 2.00 | 0.1874 | |
| A2 | 6787.07 | 1 | 6787.07 | 263.85 | <0.0001 | |
| B2 | 586.87 | 1 | 586.87 | 22.81 | 0.0007 | |
| C2 | 9.96 | 1 | 9.96 | 0.3872 | 0.5477 | |
| Residual | 257.23 | 10 | 25.72 | |||
| Lack of fit | 227.23 | 5 | 45.45 | 7.57 | 0.0222 | Significant |
| Pure error | 30.00 | 5 | 6.00 | |||
| Cor total | 19968.95 | 19 |
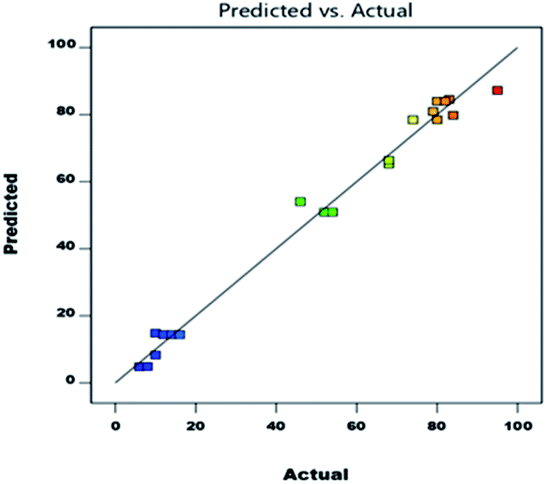 | ||
| Fig. 2 Linear correlation between the actual and predicted yield of DAPG. | ||
The relationship visualisation between the temperature and catalyst loading as the designated variables, and yield as the response is observed in the surface response graphs, which are plotted in 3D surface response, as presented in Fig. 3. Fig. 3A displays the response surface of DAPG yield calculated as a function of the reaction temperature and catalyst loading at the minimum reaction time (x1 = −1). The yield of DAPG increased rapidly to an optimum value with the increase in the reaction temperature and catalyst loading. It was observed that the reaction temperature plays a more dominant role in this typical reaction than the catalyst loading. However, the yield of DAPG slightly dropped after the reaction was operated at 80 °C with the catalyst loading at 20% (w/w). Besides, increasing the reaction temperature to more than 1 h also considerably accelerates the formation of the by-products such as tri-acetylphloroglucinol and several undetectable products, which can reduce the yield and purity of DAPG. The formation of undetectable compounds can be explained on the basis of the collateral effects of the US wave on the thermally unstable organic compounds, which are able to decompose underneath longer reaction times and higher temperature.55 Furthermore, it is evident that the typical reaction of Friedel–Crafts acylation of phenolic compounds only requires a small amount of catalyst loading, which is also reported in several literatures.18,62 In accordance with the green chemistry principles, any reaction processed with a small amount of catalyst loading is desirable to a reaction that requires an extra stoichiometric amount of the catalyst.
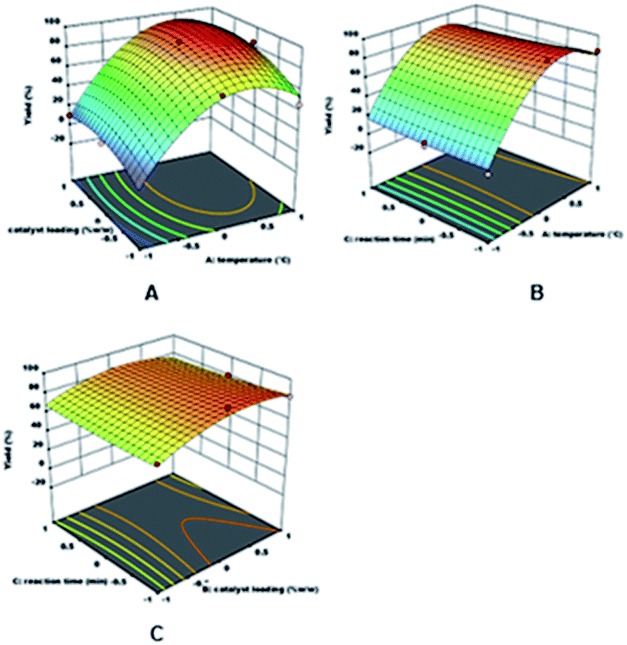 | ||
| Fig. 3 Dependence of DAPG yield (3D surface interaction plots) on (A) the reaction temperature and catalyst loading calculated for the minimum temperature (x1 = −1); (B) the reaction temperature and reaction time calculated for the intermediate catalyst loading (x2 = 0); (C) catalyst loading and reaction time calculated for the intermediate reaction temperature (x3 = −1). | ||
Fig. 3B reports the response surface of the DAPG yield calculated as a function of temperature and reaction time at the catalyst loading (x2 = 0), whereas Fig. 3C shows the dependence of DAPG yield on the catalyst loading and reaction time calculated for the intermediate reaction temperature (x3 = −1). Both these graphs highlight that no noteworthy effects were observed on the DAPG yield by varying the reaction time within the predetermined range. Finally, several runs were carried out in order to confirm the validity of the optimum condition, which was predicted by the statistical model. Therefore, the real experiment was performed in the presence of 10% SSA and operated at 60 °C for 20 min with three repetitions. The obtained yield was found to be 94%, which is relatively close to the predicted result of 95%. Accordingly, this model was found to be effective for predicting the DAPG yield precisely due to the less percentage of error (less than 6%).18
With the optimized conditions in hand, we further explored the scope of this reaction using several selected acylating agents (Scheme 5). Besides acetic anhydride, several acyl sources were selected for the synthesis of DAPG derivatives bearing different alkyl moieties. Generally, it was observed that this reaction took a slightly longer time to produce the expected products when the acetyl group was replaced with longer alkyl chains. The different reactivity due to electronic and steric reasons might be influenced by the selectivity of this reaction. However, these results are still tolerable within the above-mentioned optimized conditions. Furthermore, we also investigated the selectivity of the SSA catalyst for the synthesis of monoacylphloroglucinol derivatives (an analogue of the commercial antispasmodic agent, Flopropione63,64) by reacting 1 mmol equivalent of the acylating agent. For this reaction, a high yield of monoacylphloroglucinol was observed in less than 10 min without the formation of the by-product. This clearly proved that the SSA catalyst can play an important role as a selective catalyst for this typical reaction.
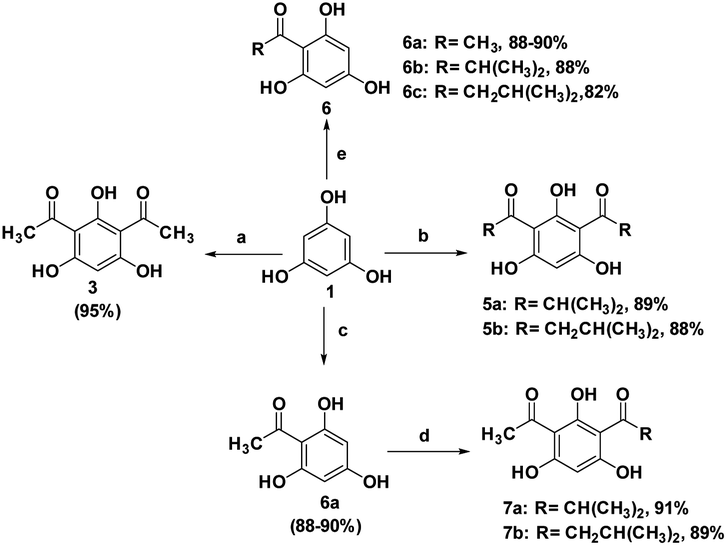 | ||
| Scheme 5 The scope studied under the optimised conditions. Reagents and conditions: (a) Ac2O (2 mmol), SSA (10%), 60 °C, US, 15–20 min, (b) RCOCl (2 mmol), SSA (10%), 60 °C, US, 20–25 min, (c) Ac2O (1.2 mmol), SSA (10%), 60 °C, US, 10–15 min, (d) RCOCl (1.1 mmol), SSA (10%), 60 °C, US, 20–25 min. | ||
In addition, in order to assess the feasibility of this method on a preparative scale, Gram-scale synthesis was then examined under the optimal reaction conditions. For the small scale Friedel–Crafts acylation reaction, 1 mmol equivalent of phloroglucinol (1) and 2 mmol equivalents of acetic anhydride (2) was required with the yield of 95%. Fortunately, when the reaction was scaled-up ten times to the previous stoichiometric ratio, DAPG was also afforded in a reproducible yield of about 93% (Scheme 6).
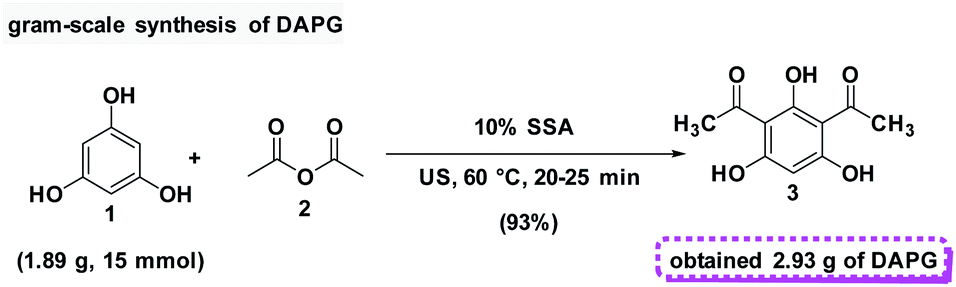 | ||
| Scheme 6 Preparative scale synthesis of DAPG under the above optimised conditions. | ||
Hence, DAPG, which is a privileged building block and versatile synthetic intermediate, could be simply synthesised by the present method. In addition, to our great delight, when the same type of reaction was carried out under an inert atmosphere, there were no significant differences. This indicates that this typical reaction is relatively constant under ambient atmosphere conditions. In the best cases, especially for products 3, 5a, and 5b, they can be purified only using recrystallisation technique or without additional chromatographic treatment.
Reaction schemes of the five synthetic pathways leading to DAPG assessed by the EATOS tool, Andraos algorithm, and energy requirement
The subject of metrics in chemical transformations is increasingly becoming important for measuring the greenness of a reaction. In this study, our main scope encompasses a comparative environmental assessment of the five synthetic pathways leading to DAPG (Scheme 1): (1) synthesis of A: reaction of phloroglucinol with acetic anhydride in the presence of MSA and acetic acid under solvent-free microwave-assisted (MW) (88% yield);16 (2) synthesis of B: phloroglucinol with acetic anhydride in the presence of BF3-etherate as the catalyst and dioxane as the organic solvent under conventional heating (30% yield);20 (3) synthesis of C: the reaction between phloroglucinol and acetic anhydride in the presence of MSA as the catalyst under solvent-free conventional heating (30–80% yield);2 (4) synthesis of D: the reaction of phloroglucinol with acetic anhydride in the presence of natural activated zeolite mordenite (nHZMOR) as the heterogeneous catalyst under solvent-free conventional heating (95% yield);18 and (5) synthesis of E (present work): reaction of phloroglucinol with acetic anhydride in the presence of SSA as the heterogeneous catalyst under solvent-free ultrasound-assisted (95% yield).To obtain the entire representation for the quantification of all the substances involved in the synthetic process, the reaction was evaluated by EATOS.40,65–67 It was feasible to measure four indexes, namely, the mass index (S−1), environmental factor (E-factor), environmental index input (EIin), and environmental index output (EIout). S−1 and EIin of all the investigated strategies are presented in Fig. 4. S−1 refers to the quantity of chemical used, whereas EIin calculates the price of the starting materials used and their detailed risk information (collected from MSDS). The highest S−1 value of 108 resulted for the synthesis of B, which is mainly ascribed to a large amount of solvent (dioxane) and substrates used. This synthetic method was also exemplified by the highest EIin (419.86 PEI per kg). The synthesis of A, C, D, and E, which differ from method B due to the use of solvent-free condition, are characterised by a lower S−1 equal to 3.44, 12.14, 1.76, and 1.61, respectively and lower EIin equal to 13.71, 22.87, 5.60, and 5.25, respectively. Remarkably, the present method (synthesis of E) is characterised by the lowest S−1 and Eiin due to a relatively low number of input materials and low-risk value of the substrates used.
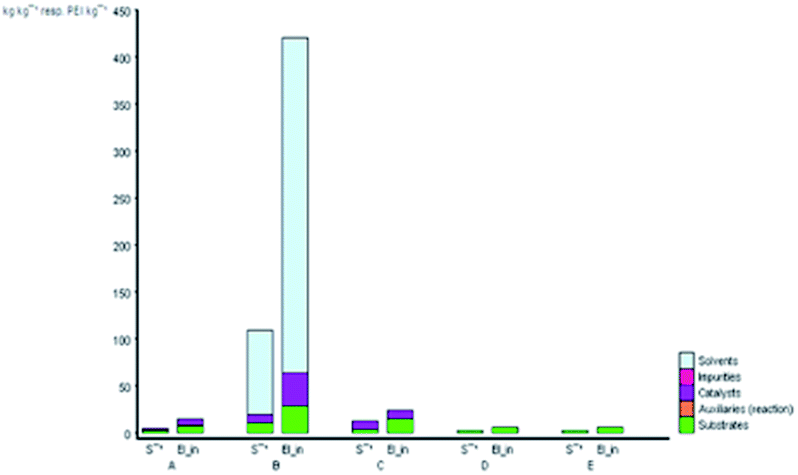 | ||
| Fig. 4 S−1 and EIin values for the synthetic routes of A, B, C, D, and E investigated with the EATOS software. | ||
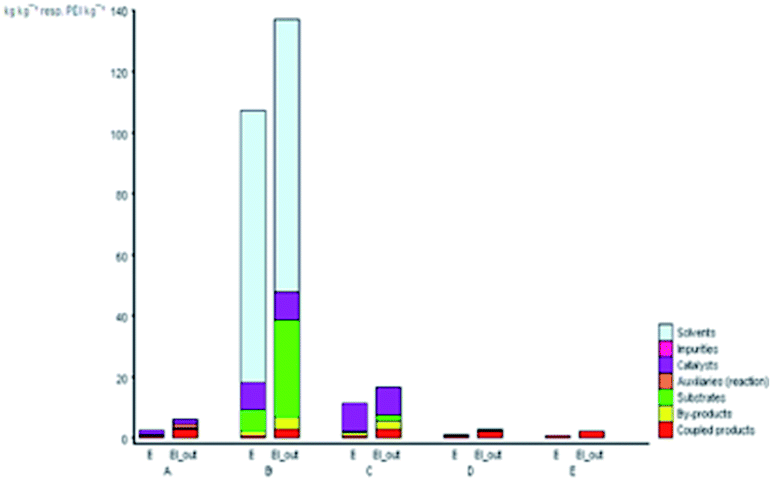
Fig. 5 depicts the E-factor and the EIout of the five studied synthetic pathways. These calculations represent the amount of waste produced and take into account its nature during the chemical process. The synthesis of B is the most mass-intensive compared to the other methods due to the large amount of waste produced (107 kg g−1 product), which is mainly ascribed to the amount of solvent (dioxane) and substrates used. According to green chemistry principles, the remediation of waste is not preferable but the prevention of waste from the source is mostly preferable.23,68 Therefore, solvent reduction or elimination in organic synthesis can considerably minimize the environmental impact of a given chemical process by developing cleaner processes.
 | ||
| Fig. 5 E and EIout values for the synthetic routes of A, B, C, D, and E investigated with the EATOS software. | ||
On the other hand, performing the reaction under solvent-free protocol (synthesis of A, C, D, and E) dramatically reduces the amount of waste. The synthesis of A and C produced 2.43 and 11.14 kg waste per kg product, respectively. Meanwhile, the lesser E-factor value in the synthesis of D and E (0.76 and 0.61 kg waste per kg product) is due to no significant excess of the solvent and/or reagents used and the higher yield isolated compared to the other methods. Besides, it is worthy to note that not only can the solvent and/or reagents be saved according to the present protocol but also the catalyst loading has been minimally employed. The heterogenous catalyst can be collected and reused several times without having a negative effect on the yield of DAPG.
The value of EIout can be considered as the character of the waste produced. The EIout value of synthesis B clearly confirms that the use of dioxane as the solvent and BF3OEt2 as the catalyst presents the lowest environmental impact than those of the other synthesis methods, which are characterised by the highest value of environmental impact (16.6 PEI per kg). The synthesis of A and C, which use MSA as the catalyst, present an EIout equal to 5.72 and 16.6 PEI per kg, respectively. Meanwhile, the EIout obtained for the synthesis of D and E, which employed eco-friendly heterogeneous catalyst systems, are considered to be lower values of environmental impact (2.57 and 2.05, respectively) compared to all of the studied methods.
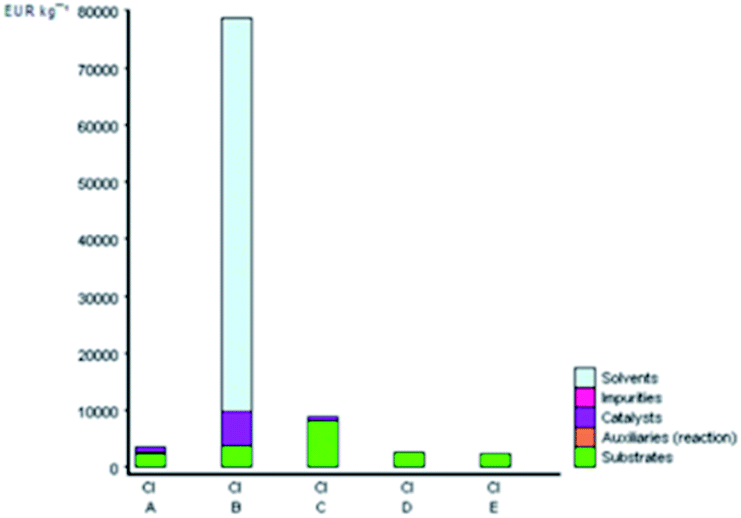
Furthermore, the production costs are reveal the environmental and social costs in a reliable way.40 The EATOS tool can be used to establish the environmental impact of their production through the costs of the substrates involved. Fig. 6 shows the value of the cost index (CI) for the five studied methods, which represents the material costs during the chemical process. The comparison of the CI clearly shows that synthesis B presents the highest CI equal to 78532.22-unit EATOS compared to the other reactions. On the contrary, the synthesis of A, C, D, and E is characterised by lower CI equal to 3395.44, 8755.13, 2395.08, and 2237.31-unit EATOS, respectively. However, the synthesis of C presents higher CI compared to the other solvent-free methods, which is attributed to the use of excess catalyst. Also, in this case, it is important to underline that the synthesis of D and E (present method) show better CI because of the use of a highly efficient and low-cost catalyst and the use of solvent-free condition with no excess reagents compared to the other studied methods.
 | ||
| Fig. 6 CI values for the synthetic routes of A, B, C, D, and E investigated with the EATOS software. | ||
In order to consider the quantity of material footprint, Andraos algorithm39,46,69 was employed to the five syntheses for the production of DAPG (Table 5, Fig. 7). According to this calculation, the synthesis of E (present work) is closer to the ideal conditions compared to the other investigated methods. The synthesis of D is also closer to the ideal conditions but is characterised by lower Material Recovery Parameter (MRP) and Reaction Mass Efficiency (RME) than the synthesis of E. The synthesis of A is characterised by lower value in all the parameters compared to the synthesis of D and E. Nevertheless, this method presents better environmental profile than the synthesis of C and B. These methods are in accordance with green chemistry principles,23 which stress in solvent-free conditions. The synthesis of B recorded the lowest value in all the parameters, which was mainly ascribed to a large amount of solvent (dioxane) and substrates used. As an important aspect, we found that the reactions performed in solvent-free conditions proved to have a better environmental profile in terms of the mass flows, environmental impacts of the substances involved, and waste produced.
| A | B | C | D | E | |
|---|---|---|---|---|---|
| AE | 0.424 | 0.182 | 0.633 | 0.633 | 0.633 |
| Rxn yield | 0.881 | 0.300 | 0.600 | 0.942 | 0.947 |
| 1/SF | 0.391 | 0.111 | 0.617 | 0.617 | 0.617 |
| MRP | 0.904 | 0.207 | 0.053 | 0.867 | 1.000 |
| RME | 0.132 | 0.001 | 0.012 | 0.319 | 0.370 |
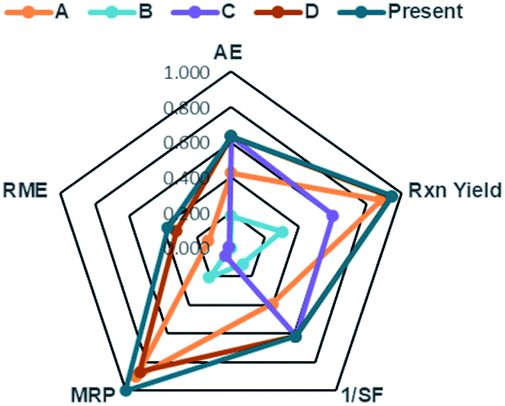 | ||
| Fig. 7 Comparison of the raw material footprint for the synthetic routes A, B, C, D, and E to DAPG. | ||
Despite the enormous benefits offered by EATOS and Andraos algorithm, both these tools do not take into account the energy required in the process. The energy intake during the process can be calculated by a simple energy calculation metric developed by Clark and co-workers.41 Table 6 shows the energy intake requirement obtained for the Friedel–Crafts acylation reaction between phloroglucinol and acetic anhydride for the methods studied. Because of the lack of detailed data regarding the heating power used in the synthesis A, B, and C, we assumed that the conventional (oil-bath) method was employed with a similar heating power as that in synthesis D (our previous work).18
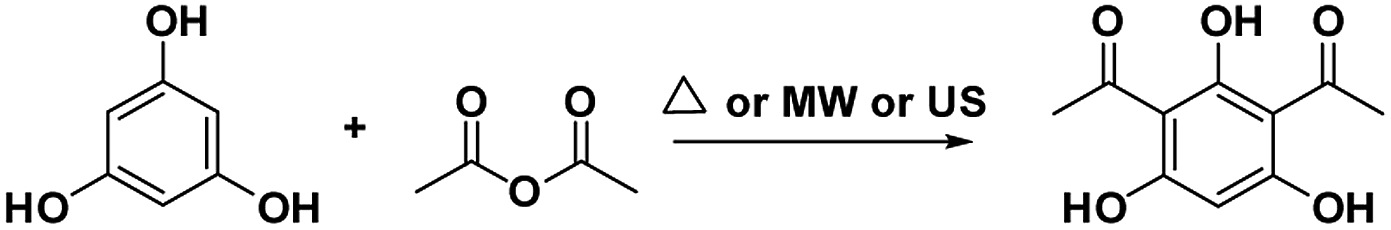
The MW reaction or the synthesis of A has been stated to give 88% yield of DAPG, which could associate to the lowest energy usage of about 13.26 kW mol−1. By taking all of the conventional methods into account, the synthesis of B, C, and D significantly corresponded to higher energy usage of 2400, 12000, and 757.89 kW mol−1, respectively. The low yield in the synthesis of B and the long-time reaction in the synthesis of C cause the energy requirement per mol to be extremely high as compared to the energy usage in the synthesis of C. The oil-bath method is characterised by the requirement of prolonged time and inefficient strategy for transferring heat into the system. In contrast, the present method (synthesis of E), which was assisted by US, corresponded to a lower energy usage of 56.14 kW mol−1 compared to the oil-bath methods. It is interesting to notice that the application of US for the synthesis of DAPG could increase the rate of reaction and required a smallest amount of energy throughout compared to the oil-bath methods. Nevertheless, the present method required slightly higher energy as compared with the MW method (synthesis of A). Accordingly, the MW method could demonstrate a shorter reaction time than the US method. Besides the lowest energy required, the obtained yield of method A is lower than that of our developed method. Finally, reflecting on all the used green parameters, it is obvious that the present method significantly improved the synthetic procedure for DAPG compared to the conventional and MW techniques.
Comparison experiments of the SSA catalyst
As a further effort, we compared the activity of SSA catalyst with a variety of protic acids (acetic acid, methanesulfonic acid, and perchloric acid) immobilised on silica gel, following the same typical reaction, as presented in Table 7. Moreover, we also undertook the comparative study of their catalytic performance with a reported heterogeneous catalyst such as zeolite.8 In the comparison of the activity of SSA with the other protic acids immobilised on silica gel, the highest yield of DAPG was obtained by using SSA as the catalyst (Table 7). When the activity of SSA was compared with that of the heterogeneous catalyst system employed in the previous study,16,18 it was found that the present method demonstrated higher yield and faster reaction.| Runs | Catalyst | Cat. loading | T (°C) | t (min) | Yield (%) | Ref |
|---|---|---|---|---|---|---|
| a Not reported. | ||||||
| 1 | H2SO4–SiO2 | 20% | 60 | 15–20 | 95 | Present work |
| 2 | HClO4–SiO2 | 20% | 60 | 40 | 64 | Present work |
| 3 | HBr–SiO2 | 20% | 60 | 60 | 54 | Present work |
| 4 | TFA–SiO2 | 20% | 60 | 120 | 70 | Present work |
| 5 | Zeolite Y | 20% | 80 | 120 | 10 | Present work |
| 6 | MSA/alumina | a | 80 | 30 | 0 | 16 |
| 7 | MSA/graphite | a | 80 | 30 | 0 | 16 |
| 8 | Activated natural zeolite mordenite | 20% | 80 | 30–45 | 95 | 18 |
Recycling experiment of the SSA catalyst
To increase the environmental benefits of the current developed method, we investigated the reusability of the SSA catalyst in the model reaction. The ease of recovery and recycling of the SSA catalyst can be easily demonstrated. After the synthesis of DAPG was completed, the SSA catalyst was recovered by extracting the mixture with acetone and ethyl acetate (1:2) and was subjected to subsequent cycles of the same typical reaction. It was found to retain the catalytic activity for up to ten consecutive runs, thus showing remarkably constant catalytic activity (Fig. 8).
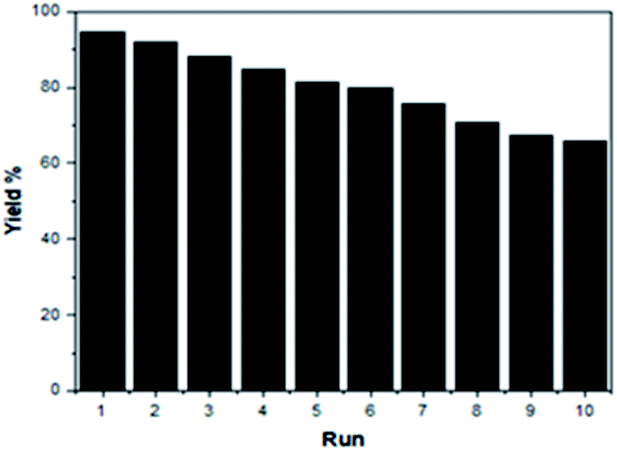 | ||
| Fig. 8 Recycling of the SSA catalyst on the DAPG yield under the optimum conditions. | ||
The recycled catalyst was characterised by XRD, FTIR, and SEM analyses. The XRD pattern of the recycled catalyst was found to be the same as that of the fresh one, indicating that there was no significant structural change in the catalyst after the tenth run (ESI, Fig. S4†). According to the FTIR spectrum, there was no appreciable change in the recycled SSA catalyst. All the characteristic bands in the fresh SSA catalyst were retained in the spectrum, corroborating that there was no loss in the structure of the recovered catalyst (ESI, Fig. S5†). In addition, SEM analysis confirmed that the surface morphology of the recycled catalyst did not significantly change after the tenth run (ESI, Fig. S6†).
In an attempt to check the catalyst leaching during reaction time, hot filtration test was performed.25,44,45 After the SSA catalyst was filtrated under hot condition, followed by the continuation of the reaction for 30 min, no significant improvement in the yield of DAPG was observed. Thereby, it obviously confirmed that no effective leaching of the active species from the SSA catalyst took place into the reaction medium. Furthermore, this observation also strengthens the heterogeneity of the SSA catalyst.
Drug-likeness screening
Drug-likeness is a qualitative method employed in drug design, which is designated to optimize pharmacodynamics, pharmacokinetics, and pharmaceutical properties, such as the solubility, bioavailability, chemical stability, and distribution profile in the human body.70 The most common rule used to evaluate the drug-likeness aspect is Lipinski's rule of five.71 According to the results (Table 8), all the synthesised compounds, either diacylphloroglucinols or monoacylphloroglucinol, are in accordance with the adequate criteria of Lipinski's rule of five, i.e., molecular weight (MW), hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), and logP; therefore, they can be considered as oral drug candidates.
| Compound | Lipinski's rule of five | nVd | |||
|---|---|---|---|---|---|
| MWa | HBDb | HBAc | LogP |
||
| <500 | <5 | <10 | <5 | ||
| a MW: molecular weight.b HBD: hydrogen bond donor.c HBA: hydrogen bond acceptor.d nV: number of violations. | |||||
| 3 | 210.19 | 3 | 5 | 0.83 | 0 |
| 5a | 266.29 | 3 | 5 | 2.25 | 0 |
| 5b | 294.35 | 3 | 5 | 2.80 | 0 |
| 6a | 168.15 | 3 | 4 | 0.70 | 0 |
| 6b | 196.20 | 3 | 4 | 1.40 | 0 |
| 6c | 210.23 | 3 | 4 | 1.72 | 0 |
| 7a | 238.24 | 3 | 5 | 1.57 | 0 |
| 7c | 252.27 | 3 | 5 | 1.81 | 0 |
Conclusions
In summary, we have developed a sustainable and highly efficient synthetic method of the highly valuable organic compound DAPG from phloroglucinol over the eco-friendly heterogeneous SSA catalyst under milder conditions. The present method shows significant advantages, both in terms of sustainability and efficiencies. It uses no solvent and is a metal-free catalyst, and uses US waves as an alternative green source of energy. It also has rapid reaction times, an excellent yield, a simple workup, and an inexpensive and recyclable catalyst. RSM analysis and the confirming experiment of the Friedel–Crafts acylation reaction run indicated that the highest DAPG yield (95%) was achieved at 60 °C after 20 min employing the SSA catalyst at a concentration of 10% (w/w). According to the environmental assessment profile, the sustainable reaction conditions, viz., solvent-free condition and ultrasound assistance for the synthesis of DAPG under the optimum conditions were evaluated to be a convenient tool for meeting the green chemistry principles23 of waste reduction and energy requirement reduction. However, as compared with the MW method (synthesis of A), the newly developed method required slightly higher energy. Nonetheless, the practicality and sustainability significantly improved the synthetic procedure for DAPG, as established using the EATOS software, Andraos algorithm, and the energy requirement calculations. These results have evaluated the most substantial contribution and improvements of this method against the others reported in the literature concerning the synthesis of DAPG. Furthermore, all DAPG derivatives satisfy the Lipinski's rule of five; thus, they would not exhibit problems in the oral bioavailability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Ministry of Research, Technology and Higher Education, the Republic of Indonesia for providing financial support through Penelitian Dasar Unggulan Perguruan Tinggi (PDUPT) 2019 grant (contract agreement number: 719/UN27.21/PN/2019). The authors also wish to gratefully acknowledge to the Institute of Research and Community of Services Sebelas Maret University for partially finance through Hibah Penelitian Unggulan (PU-UNS) 2020 (contract agreement number: 452/UN27.21/PN/2020), Hibah Pascasarjana (PPS) 2019 grant (contract agreement number: 516/UN27.21/PP/2019) and Hibah Pascasarjana (PPS) 2020 grant (contract agreement number: 452/UN27.21/PN/2020). We are also grateful to Septin D Anggraini S. Si and Ita Apriana, S. Si for being assistant during the preparation of catalyst.Notes and references
- M. M. Zhao, N. Lyu, D. Wang, X. G. Wu, Y. Z. Zhao, L. Q. Zhang and H. Y. Zhou, Sci. Rep., 2020, 10, 4296 CrossRef PubMed.
- L. Gong, H. Tan, F. Chen, T. Li, J. Zhu, Q. Jian, D. Yuan, L. Xu, W. Hu, Y. Jiang and X. Duan, Sci. Rep., 2016, 6, 32266 CrossRef CAS PubMed.
- K. Gutiérrez-García, A. Neira-González, R. M. Pérez-Gutiérrez, G. Granados-Ramírez, R. Zarraga, K. Wrobel, F. Barona-Gómez and L. B. Flores-Cotera, J. Nat. Prod., 2017, 80, 1955–1963 CrossRef PubMed.
- P. A. Marchand, D. M. Weller and R. F. Bonsall, J. Agric. Food Chem., 2000, 48, 1882–1887 CrossRef CAS PubMed.
- J. Zakrzewski, M. Karpińska and Z. Maliński, Arch. Pharm., 2007, 340, 103–106 CrossRef CAS PubMed.
- B. Syed, M. N. Nagendra Prasad, K. Mohan Kumar and S. Satish, Sci. Total Environ., 2018, 637–638, 274–281 CrossRef CAS PubMed.
- Y. Kamei and A. Isnansetyo, Int. J. Antimicrob. Agents, 2003, 21, 71–74 CrossRef CAS PubMed.
- Y.-S. Kwak, P. A. H. M. Bakker, D. C. M. Glandorf, J. T. Rice, T. C. Paulitz and D. M. Weller, Phytopathology, 2009, 99, 472–479 CrossRef CAS PubMed.
- D. Haas and C. Keel, Annu. Rev. Phytopathol., 2003, 41, 117–153 CrossRef CAS PubMed.
- M. Tada, T. Takakuwa, M. Nagai and T. Yoshii, Agric. Biol. Chem., 1990, 54, 3061–3063 CAS.
- I. P. Singh, J. Sidana, P. Bansal and W. J. Foley, Expert Opin. Ther. Pat., 2009, 19, 847–866 CrossRef CAS PubMed.
- V. K. Veena, K. Kennedy, P. Lakshmi, R. Krishna and N. Sakthivel, Mol. Cell. Biochem., 2016, 414, 47–56 CrossRef CAS PubMed.
- V. K. Veena, R. N. Popavath, K. Kennedy and N. Sakthivel, Apoptosis, 2015, 20, 1281–1295 CrossRef CAS PubMed.
- M. Maurhofer, E. Baehler, R. Notz, V. Martinez and C. Keel, Appl. Environ. Microbiol., 2004, 70, 1990–1998 CrossRef CAS PubMed.
- F. Yang and Y. Cao, Appl. Microbiol. Biotechnol., 2012, 93, 487–495 CrossRef CAS PubMed.
- S. K. Chauthe, S. B. Bharate, G. Periyasamy, A. Khanna, K. K. Bhutani, P. D. Mishra and I. P. Singh, Bioorg. Med. Chem. Lett., 2012, 22, 2251–2256 CrossRef CAS PubMed.
- S. K. Chauthe, S. B. Bharate, S. Sabde, D. Mitra, K. K. Bhutani and I. P. Singh, Bioorg. Med. Chem., 2010, 18, 2029–2036 CrossRef CAS PubMed.
- W. E. Prasetyo, T. Kusumaningsih and M. Firdaus, Synth. Commun., 2019, 49, 3352–3372 CrossRef CAS.
- M. O. Duarte, S. Lunardelli, C. J. Kiekow, A. C. Stein, L. Müller, E. Stolz, S. M. K. Rates and G. Gosmann, Nat. Prod. Commun., 2014, 9, 1934578X1400900 CrossRef.
- N. Mittal, H. H. Tesfu, A. M. Hogan, S. T. Cardona and J. L. Sorensen, J. Antibiot., 2019, 72, 253–259 CrossRef CAS PubMed.
- T. Kusumaningsih, M. Firdaus, A. N. Artanti and W. E. Prasetyo, IOP Conf. Ser. Mater. Sci. Eng., 2019, 578, 012057 CrossRef CAS.
- A. S. R. Anjaneyulu, U. V. Mallavadhani, Y. Venkateswarlu and A. V. R. Prasad, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1987, 26, 823–826 Search PubMed.
- P. T. Anastas and J. C. Warner, Principles of green chemistry, Oxford University Press New York, 1998 Search PubMed.
- M. Firdaus and M. D. Prameswari, Bull. Chem. React. Eng. Catal., 2019, 14, 9 CrossRef CAS.
- K. Rajkumari, D. Das, G. Pathak and L. Rokhum, New J. Chem., 2019, 43, 2134–2140 RSC.
- G. Y. Bai, Y. H. Yang, Z. Ma, J. Z. Xu, M. De Qiu, F. He, X. L. Yan and H. Y. Dou, Res. Chem. Intermed., 2012, 38, 795–806 CrossRef CAS.
- M. Fujita, J. M. Lévêque, N. Komatsu and T. Kimura, Ultrason. Sonochem., 2015, 27, 247–251 CrossRef CAS PubMed.
- M. Satish Kumar, K. Chinna Rajanna, P. Venkanna and M. Venkateswarlu, Tetrahedron Lett., 2014, 55, 1756–1759 CrossRef CAS.
- H. Ahankar, A. Ramazani, K. Ślepokura, T. Lis and S. W. Joo, Green Chem., 2016, 18, 3582–3593 RSC.
- H. Naeimi, V. Nejadshafiee and S. Masoum, RSC Adv., 2015, 5, 15006–15016 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559–1584 RSC.
- M. A. Zolfigol, Tetrahedron, 2001, 57, 9509–9511 CrossRef CAS.
- M. Gawande, R. Hosseinpour and R. Luque, Curr. Org. Synth., 2014, 11, 526–544 CrossRef CAS.
- M. Dabiri, P. Salehi, M. Baghbanzadeh, M. A. Zolfigol and M. Bahramnejad, Synth. Commun., 2007, 37, 1201–1209 CrossRef CAS.
- A. L. García-Cabeza, L. P. Ray, R. Marín-Barrios, M. J. Ortega, F. J. Moreno-Dorado, F. M. Guerra and G. M. Massanet, Org. Process Res. Dev., 2015, 19, 1662–1666 CrossRef.
- U. Mondal and S. Sen, Org. Process Res. Dev., 2017, 21, 23–30 CrossRef CAS.
- J. Fan, C. Yi, X. Lan and B. Yang, Org. Process Res. Dev., 2013, 17, 368–374 CrossRef CAS.
- A. Rybak and M. A. R. Meier, Green Chem., 2008, 10, 1099 RSC.
- S. Toniolo, F. Aricò and P. Tundo, ACS Sustain. Chem. Eng., 2014, 2, 1056–1062 CrossRef CAS.
- M. Eissen and J. O. Metzger, Chem. –Eur J., 2002, 8, 3580–3585 CrossRef CAS.
- M. J. Gronnow, R. J. White, J. H. Clark and D. J. Macquarrie, Org. Process Res. Dev., 2005, 9, 516–518 CrossRef CAS.
- S. K. Karmee, Biofuels, 2017, 7269, 1–7 Search PubMed.
- J. Manna, B. Roy and P. Sharma, J. Power Sources, 2015, 275, 727–733 CrossRef CAS.
- M. S. Tiwari, J. S. Dicks, J. Keogh, V. V. Ranade and H. G. Manyar, Mol. Catal., 2020, 488, 110918 CrossRef CAS.
- R. Goyal, S. Sameer, B. Sarkar, A. Bag, N. Singhal and A. Bordoloi, Chem. –Eur J., 2017, 23, 16555–16565 CrossRef CAS PubMed.
- J. Andraos, Org. Process Res. Dev., 2009, 13, 161–185 CrossRef CAS.
- N. R. E. Radwan, M. Hagar, T. H. Afifi, F. Al-Wadaani and R. M. Okasha, Catalysts, 2018, 8, 36 CrossRef.
- G. N. Shao, R. Sheikh, A. Hilonga, J. E. Lee, Y. H. Park and H. T. Kim, Chem. Eng. J., 2013, 215–216, 600–607 CrossRef CAS.
- K. A. Shah, J. K. Parikh and K. C. Maheria, Bioenergy Res., 2014, 7, 206–216 CrossRef CAS.
- D. V. Quang, T. A. Hatton and M. R. M. Abu-Zahra, Ind. Eng. Chem. Res., 2016, 55, 7842–7852 CrossRef CAS.
- N. C. Marziano, L. Ronchin, C. Tortato, A. Zingales and A. A. Sheikh-Osman, J. Mol. Catal. Chem., 2001, 174, 265–277 CrossRef CAS.
- Y. Jin, J. Li, L. Peng and C. Gao, Chem. Commun., 2015, 51, 15390–15393 RSC.
- O. V. Bityukov, V. A. Vil’, V. M. Merkulova, G. I. Nikishin and A. O. Terent’ev, Pure Appl. Chem., 2018, 90, 7–20 CAS.
- G. Liu and B. Xu, Tetrahedron Lett., 2018, 59, 869–872 CrossRef CAS.
- D. Quiroga, L. D. Becerra and E. Coy-Barrera, ACS Omega, 2019, 4, 13710–13720 CrossRef CAS PubMed.
- B. Guo, E. Middha and B. Liu, ACS Nano, 2019, 13, 2675–2680 CrossRef CAS PubMed.
- W. Li, H. Jin, S. Yang, X. Guo, G. He and R. Zhang, Green Process. Synth., 2019, 8, 474–479 CAS.
- G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 21, 2159–2169 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- J. Criquet, E. M. Rodriguez, S. Allard, S. Wellauer, E. Salhi, C. A. Joll and U. von Gunten, Water Res., 2015, 85, 476–486 CrossRef CAS PubMed.
- H. Jirglová, A. F. Pérez-Cadenas and F. J. Maldonado-Hódar, Langmuir, 2009, 25, 2461–2466 CrossRef PubMed.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077–1104 CrossRef CAS PubMed.
- N. Li, S. Khan, S. Qiu and X.-C. Li, Molecules, 2018, 23, 3232 CrossRef PubMed.
- T. Fukuyama, Y. Takahashi, M. Kuze and E. Arai, Acta Urol. Jpn., 1969, 15, 818–823 CAS.
- D. Ravelli, S. Protti, P. Neri, M. Fagnoni and A. Albini, Green Chem., 2011, 13, 1876 RSC.
- S. Protti, D. Ravelli, M. Fagnoni and A. Albini, Chem. Commun., 2009, 7351 RSC.
- Y. Gaber, U. Törnvall, C. Orellana-Coca, M. Ali Amin and R. Hatti-Kaul, Green Chem., 2010, 12, 1817 RSC.
- R. A. Sheldon, ACS Sustain. Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
- G. Vistoli, A. Pedretti and B. Testa, Drug Discov. Today, 2008, 13, 285–294 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 2012, 64, 4–17 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05424k |
| This journal is © The Royal Society of Chemistry 2020 |