
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis and electrochemical characterization of sodium 1,2-naphthoquinone-4-sulfonate-doped PEDOT/MWCNT composite†
Irina S. Vasil'eva a,
Galina P. Shumakovicha,
Maria E. Khlupovaa,
Roman B. Vasilievb,
Viktor V. Emetsc,
Vera A. Bogdanovskayac,
Olga V. Morozovaa and
Alexander I. Yaropolov*a
a,
Galina P. Shumakovicha,
Maria E. Khlupovaa,
Roman B. Vasilievb,
Viktor V. Emetsc,
Vera A. Bogdanovskayac,
Olga V. Morozovaa and
Alexander I. Yaropolov*a
aBach Institute of Biochemistry, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky Ave. 33, Bld. 2, 119071 Moscow, Russia. E-mail: yaropolov@inbi.ras.ru; alexander-yaropolov52@yandex.ru; Fax: +7 495 954 2732; Tel: +7 495 954 4477
bDepartment of Materials Science, Lomonosov Moscow State University, Leninskie Gory 1/3, 119991 Moscow, Russia
cFrumkin Institute of Physical Chemistry and Electrochemistry of the Russian Academy of Sciences, Leninsky Ave. 31, 119071 Moscow, Russia
First published on 7th September 2020
Abstract
The development of novel materials with improved functional characteristics for supercapacitor electrodes is of current concern and calls for elaboration of innovative approaches. We report on an eco-friendly enzymatic synthesis of a composite based on poly(3,4-ethylenedioxythiophene) (PEDOT) and multi-walled carbon nanotubes (MWCNTs). The redox active compound, sodium 1,2-naphthoquinone-4-sulfonate (NQS), was used as a dopant for the backbone of the polymer. Oxidative polymerization of 3,4-ethylenedioxythiophene (EDOT) was catalyzed by a high redox potential laccase from the fungus Trametes hirsuta. Atmospheric oxygen served as an oxidant. A uniform thin layer of NQS-doped PEDOT formed on the surface of MWCNTs as a result of the enzymatic polymerization. The PEDOT–NQS/MWCNT composite showed a high specific capacitance of ca. 575 F g−1 at a potential scan rate of 5 mV s−1 and an excellent cycling stability within a potential window between −0.5 and 1.0 V, which makes it a promising electrode material for high-performance supercapacitors.
1. Introduction
Supercapacitors (SCs) are promising devices for energy accumulation and storage and can find applications in many fields.1,2 As compared with other energy accumulating devices, SCs have a higher power density, a high discharge rate and an excellent cycling stability.3 SCs are divided into two main groups depending on the energy charge storage mechanism, namely, electrical double layer capacitors and pseudocapacitors.1,4,5 Electrical double layer capacitors use as electrodes various carbon materials with a high specific surface area like activated carbon, carbon nanotubes, carbon fibers, graphene and others,6 and charge is stored on the electrode/electrolyte interface. In contrast, in pseudocapacitors charge storage occurs via the reversible faradaic reaction, and the electrodes are made of transition metal oxides or conducting polymers (CPs).7,8 Numerous studies have been performed recently in order to improve the functional characteristics of electrode materials for SCs.2,4,5,9–12Carbon nanotubes (CNTs) are a promising material for SC electrodes due to a low specific weight and unique electric and mechanical properties.13 The specific capacitance and power density of CNT-based SCs can be increased by combining the capacitance of the nanostructured carbon double layer and the pseudocapacitance of conducting polymers (CPs) due to faradaic processes at the electrode/electrolyte interface.7 Polyaniline (PANI), poly(3,4-ethylenedioxythiophene) (PEDOT) and polypyrrole are most commonly used as CPs in CNT-based composites.7,10,11,14–19 These polymers differ in the theoretical specific capacitance and show a poor stability in charge/discharge cycles.16
CPs and CP-based composites are usually produced by electrochemical or chemical methods,20–22 but they both have some disadvantages. The electrochemical method is not always suitable as monomer polymerization proceeds on electroconducting substrates of limited sizes, and the chemical synthesis requires great amounts of oxidizers, whose reduction products should be disposed of.22 Besides chemically produced composites contain polymers that are not bound to the carbon surface, which worsens the composite cycling stability.23
An alternative to the chemical method is biocatalytic synthesis.24–31 Enzyme-catalyzed polymerization is environmentally friendly and can be performed with a high degree of kinetic reaction control. Moreover, in enzymatic synthesis the amount of the polymer not bound to the surface of nanostructured carbon material is minimal, which has a significant influence on the specific characteristics of both composites themselves and composite-based SCs.23 There are only a few papers in which enzymes are used for oxidative polymerization of EDOT. Nagarajan et al.32 used soybean peroxidase for EDOT polymerization on the polystyrene sulfonate (PSS) soft template. In this work terthiophene served as a redox-mediator/initiator. Horseradish peroxidase was used for polymerization of a water soluble derivative of EDOT without a template.33 The laccase from the fungus Trametes hirsuta was applied to EDOT polymerization on the soft templates (PAMPS, PSS, DNA).34–36
As was mentioned above, CNTs are a promising material for SC electrodes. However, CNTs have a rather low specific capacitance of 5–80 F g−1.37 The combination of the faradaic redox reaction (pseudocapacitive effect of conducting polymers) and the capacitance of carbon material allows increasing the specific capacitance and stability of SC electrodes. Besides the use of redox active compounds as additives to electrolytes38–44 or as dopants for the backbone of CPs45 increases to a great extent the specific characteristics of both composites themselves and composite-based SCs. In our earlier paper39 we reported that the specific capacitance of enzymatically synthesized PANI-TSA/MWCNT composite increased more than 2-fold after adding the redox active compound sodium 1,2-naphthoquinone-4-sulfonate (NQS) into the gel electrolyte H2SO4/PVA. We supported that the use of NQS as dopant can increase the specific capacitance of CP/MWCNT composites. PEDOT as compared with PANI has a lower theoretical specific capacitance (210 F g−1 and 750 F g−1 respectively).16 However, PANI/MWCNT composites described in many papers are unstable at potentials higher than 0.7 V (vs. Ag/AgCl) as PANI transforms into pernigraniline state, which hydrolyzes in water solutions.46 PEDOT-based composites are free of this shortcoming and have a wider potential window.47,48 Therefore in this work we have performed a laccase-mediated synthesis of a composite based on MWCNTs and PEDOT doped with the redox active compound, sodium 1,2-naphthoquinone-4-sulfonate (NQS) and have studied physicochemical properties of the resultant PEDOT–NQS/MWCNT composite.
2. Experimental
2.1 Materials
All commercially available chemicals were of high purity and used without further purification if not stated otherwise: 3,4-ethylenedioxythiophene, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS, Sigma-Aldrich), sodium 1,2-naphthoquinone-4-sulfonate, H2SO4, HNO3 (Chimmed, Russia). All the solutions were prepared using water purified with a MilliQ system (Millipore, USA). Flexible graphite foil (thickness 0.2 mm) was purchased from (Unichimtek, Russia).Multi-walled carbon nanotubes “Taunit M” (NanoTechCentre Ltd, Russia) were used after treatment with hot 70% nitric acid (85 °C, 5 h).
The laccase from the fungus Trametes hirsuta (Wulfen) Pilát CF-28 was purified to homogeneity as described previously.49 The specific activity of the enzyme was ca. 131 U mg−1 of protein using ABTS as a chromogenic substrate.50
2.2 Preparation of a PEDOT–NQS/MWCNT composite
The composite was obtained by in situ enzymatic EDOT polymerization. The reaction was performed as follows: 10 mg of acid-treated MWCNTs were dispersed in 20 ml of deionized water and subjected to sonication for 8 h. MWCMTs were separated by centrifugation, added to 10 ml EDOT solution (25 mM) and stirred for 30 min. Afterwards 0.06 g of NQS was added to the dispersion (concentration in the solution 25 mM), which was then stirred for another 30 min. The pH of the solution was brought up to the value of 4.5 with sodium hydroxide, and EDOT polymerization was initiated by adding a laccase stock solution. The specific activity of laccase in the reaction mixture was ca. 1.0 U ml−1. The synthesis was performed in air at room temperature (21–22 °C) and under continuous stirring for 24 h. The PEDOT–NQS/MWCNT composite was separated by centrifugation, repeatedly washed with deionized water and ethanol, dried to constant weight, and then used in further experiments. The content of PEDOT–NQS in the composite was calculated as a difference between the weight of the composite and the weight of MWCNTs used for its synthesis.For the synthesis of PEDOT–NQS, 0.06 g of NQS was added to 10 ml of an EDOT solution (25 mM) and stirred for 30 minutes. The pH of the solution was brought up to 4.5, and polymerization was initiated by adding a laccase stock solution.
2.3 Characterization
SEM and EDX analyses were carried out with a Zeiss Supra 40VP scanning electron microscope (Carl Zeiss AG, Germany).Electrochemical measurements were performed using a BAS CV-50W voltammetric analyzer (Bioanalytical System, USA) and a single-compartment cell. In three-electrode system an Ag/AgCl electrode (BAS) and platinum sheet served as reference and counter electrodes respectively. Flexible graphite foil (0.5 × 2.0 cm) covered with a precise amount of the PEDOT–NQS/MWCNT composite served as a working electrode.
The specific capacitance (Cs) of the composite was calculated using the formula, where I is the current, ΔE is the potential range, v is the potential scan rate, and m is the mass of the electroactive composite. The specific capacitance of the composite was calculated as the average of three independent experiments. Cycling stability of the composite was studied in three-electrode system within a potential window from −0.5 to 1.0 V at a potential scan rate of 100 mV s−1.
To registrate electrochemical impedance spectra (EIS), we used a measuring complex composed of a frequency response analyzer Solartron 1255 B and a potentiostat Solartron 1287 (AMETEK®, USA). The EIS measurements were performed in the three-electrode configuration within a frequency range from 100 kHz to 0.1 Hz with a signal amplitude of 10 mV. The impedance spectra were analyzed, using ZView® software.
Galvanostatic charging/discharging measurements were performed in the two-electrode cell configuration, using a potentiostat/galvanostat Autolab PGSTAT302 (Metrohm AG, Switzerland).
Four-point conductivity measurements were carried out with a Loresta GP MCP-T610 resistivity meter (Mitsubishi, Japan) using MCP-TP06P probe (inter-pin distance 1.5 mm, pin points 0.26 R, spring pressure 70 g per pin). The morphology of MWCNTs and PEDOT–NQS/MWCNT composite was studied using transmission electron microscopy (TEM, JEM-100CX/SFG, Jeol, Japan). FTIR spectra were recorded using KBr pellets on a Frontier FT-IR/FIR spectrometer (PerkinElmer Inc.)
X-ray diffraction (XRD) analysis of finely ground powder samples was performed with CuKα radiation on a Rigaku D/Max 2500V/PC diffractometer (Japan). XRD patterns were recorded in the 2θ range of 10°–70° with a scans step of 0.02° at a scan speed of 0.5° min−1.
3. Results and discussion
In our earlier paper39 we showed that NQS has a high cycling stability in acidic solutions and its behavior is a diffusion-controlled and reversible process. Therefore, in this work we have used NQS as a PEDOT dopant in the enzyme-mediated composite synthesis. Fig. 1 shows the scheme of the enzymatic synthesis of the PEDOT–NQS/MWCNT composite.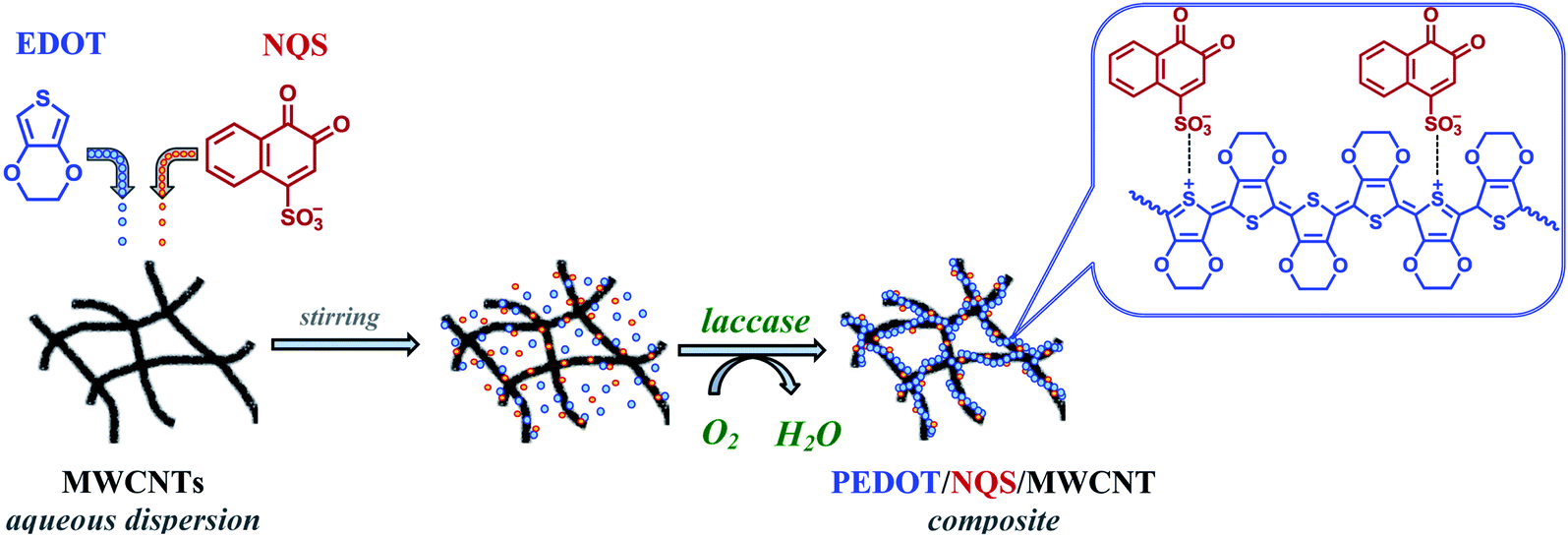 | ||
| Fig. 1 Scheme of the enzymatic synthesis of the PEDOT–NQS/MWCNT composite. | ||
3.1 Characterization of MWCNTs
MWCNTs are a hydrophobic material whose surface are hydrophylized with nitric acid or a mixture of nitric and sulfuric acids, which results in additional defect generation in surficial carbon layers and an increase in the number of carboxyl groups.51–54 At the same time, the metal content of MWCNTs decreases.To modify the surface of nanostructured carbon, we heated MWCNTs in nitric acid. The treatment of MWCNTs with the acid brings about a significant decrease in the contents of amorphous carbon and metals, while the oxygen content increases (see ESI Fig. S1†). Studies on initial and acid-treated MWCNTs by the FTIR method (see ESI Fig. S2†) showed that the acidic treatment led to the formation of carboxyl groups on the MWCNT surface with stretching bands at 1730 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1180 cm−1 (C–O),51–54 while SEM and TEM images revealed no significant difference in their morphology (see ESI Fig. S3†). Thus oxidation of MWCNTs with nitric acid results in hydrophylization of their surface, which enables us to obtain a stable MWCNT water dispersion. Acid-treated functionalized MWCNTs were used for the enzymatical synthesis of the PEDOT–NQS/MWCNT composite.
O) and 1180 cm−1 (C–O),51–54 while SEM and TEM images revealed no significant difference in their morphology (see ESI Fig. S3†). Thus oxidation of MWCNTs with nitric acid results in hydrophylization of their surface, which enables us to obtain a stable MWCNT water dispersion. Acid-treated functionalized MWCNTs were used for the enzymatical synthesis of the PEDOT–NQS/MWCNT composite.
3.2 Characterization of the enzymatically synthesized PEDOT–NQS/MWCNT composite
Fig. 2 shows TEM images of the PEDOT–NQS/MWCNT composite. As a result of enzymatic EDOT polymerization in the presence of NQS, a rather uniform thin, rough coating layer of the conducting polymer was generated on the surface of MWCNTs. Besides, the composite contained no polymer not bound to the surface of MWCNTs. Compared to the diameter of acid-treated MWCNTs, which is ∼10–15 nm, the diameter of PEDOT–NQS/MWCNT composite increased to 25–30 nm. The PEDOT–NQS content in the composite was ca. 67 wt%.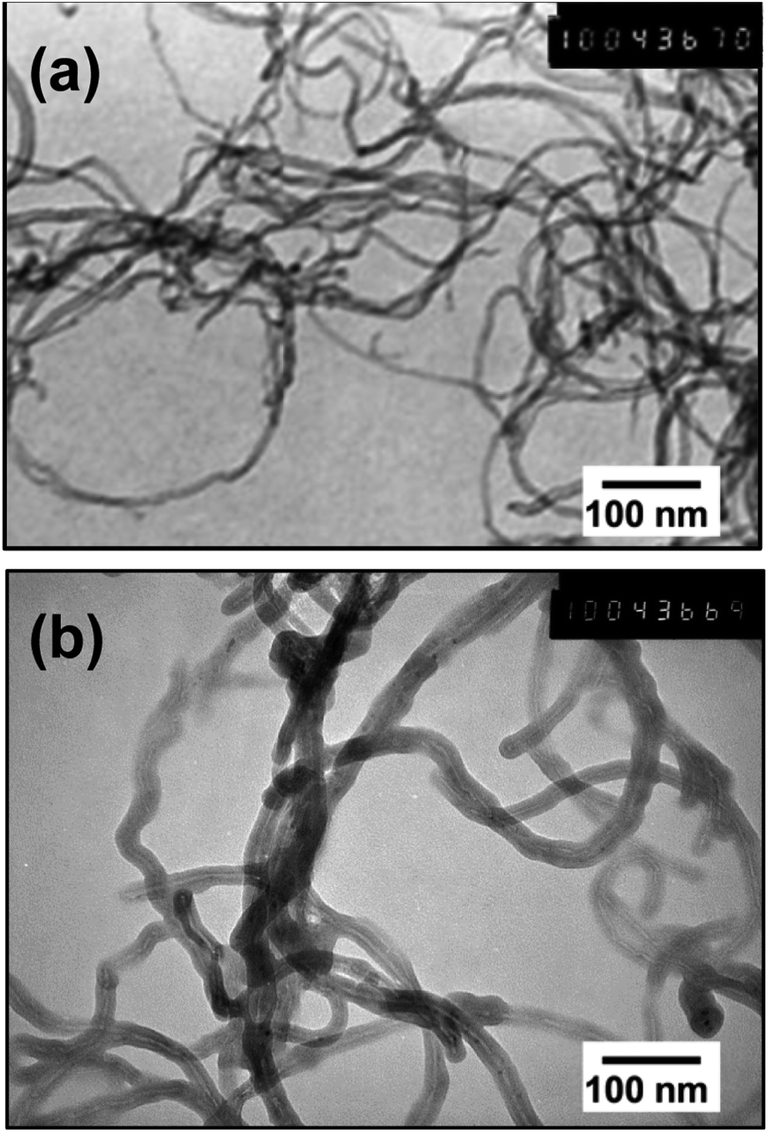 | ||
| Fig. 2 TEM images of the MWCNTs (a) and PEDOT–NQS/MWCNT composite (b). | ||
The functionalized MWCNTs and enzymatically synthesized PEDOT–NQS and PEDOT–NQS/MWCNT composite were studied by X-ray diffraction (Fig. 3). MWCNTs show a sharp and intense peak at 2θ = 26.0°, corresponding to a graphite-like structure, and other peaks at 42.7°and 52.5°, corresponding to catalytic cobalt particles encapsulated within MWCNTs.55–57 Crystal peaks centered at 25.9° and 38.5° in the PEDOT–NQS XRD pattern result from the NQS-doping of PEDOT, which seems to increase the molecular interaction between the stacked π-conjugated polymers molecules.58 The XRD pattern of PEDOT–NQS/MWCNT exhibits characteristic peaks for both MWCNTs and PEDOT–NQS. The results suggest that no additional crystalline order is introduced into the enzymatically synthesized composite.
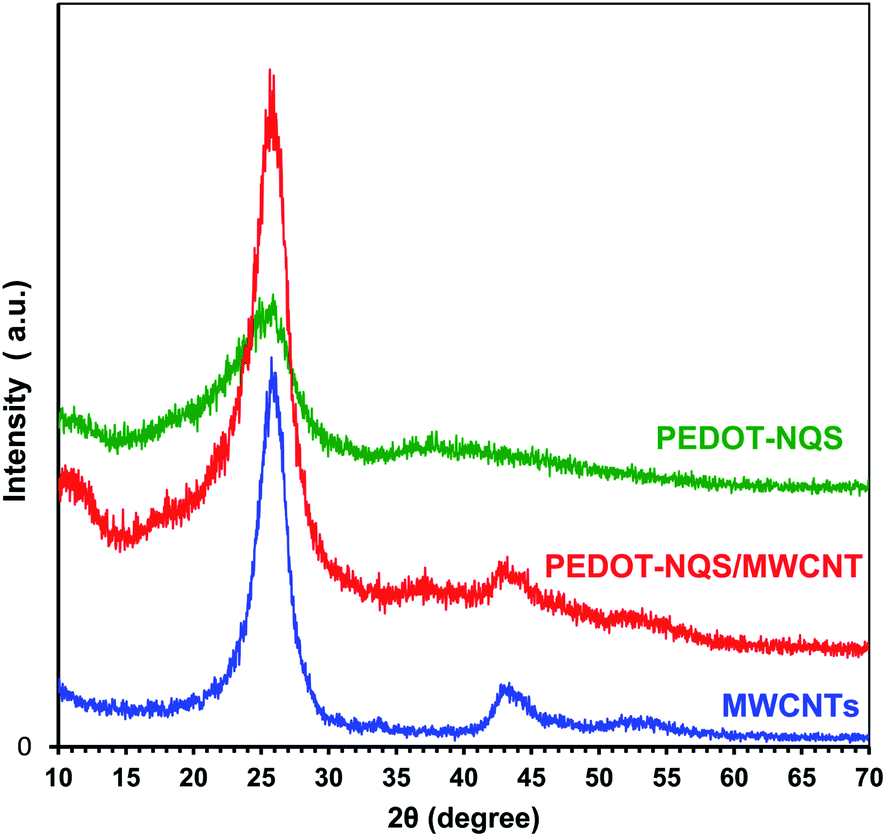 | ||
| Fig. 3 XRD patterns of MWCNTs, PEDOT–NQS and PEDOT–NQS/MWCNT. | ||
The FTIR spectrum of enzymatically synthesized PEDOT–NQS/MWCNT is shown in Fig. 4. The bands at 1520 and 1340–1403 cm−1 are originated from the stretching of CC and C–C in the thiophene ring of PEDOT. Vibrations at 841, 924, 935 and 985 cm−1 are attributed to the C–S bond in the thiophene ring.32,59 The bands at 1053, 1092 and 1145 cm−1 are assigned to the stretching modes of the ethylenedioxy group.59 The bands at 500–800, 1200–1340 and 1600–1700 cm−1 are due to redox-active dopant NQS (Fig. 4).
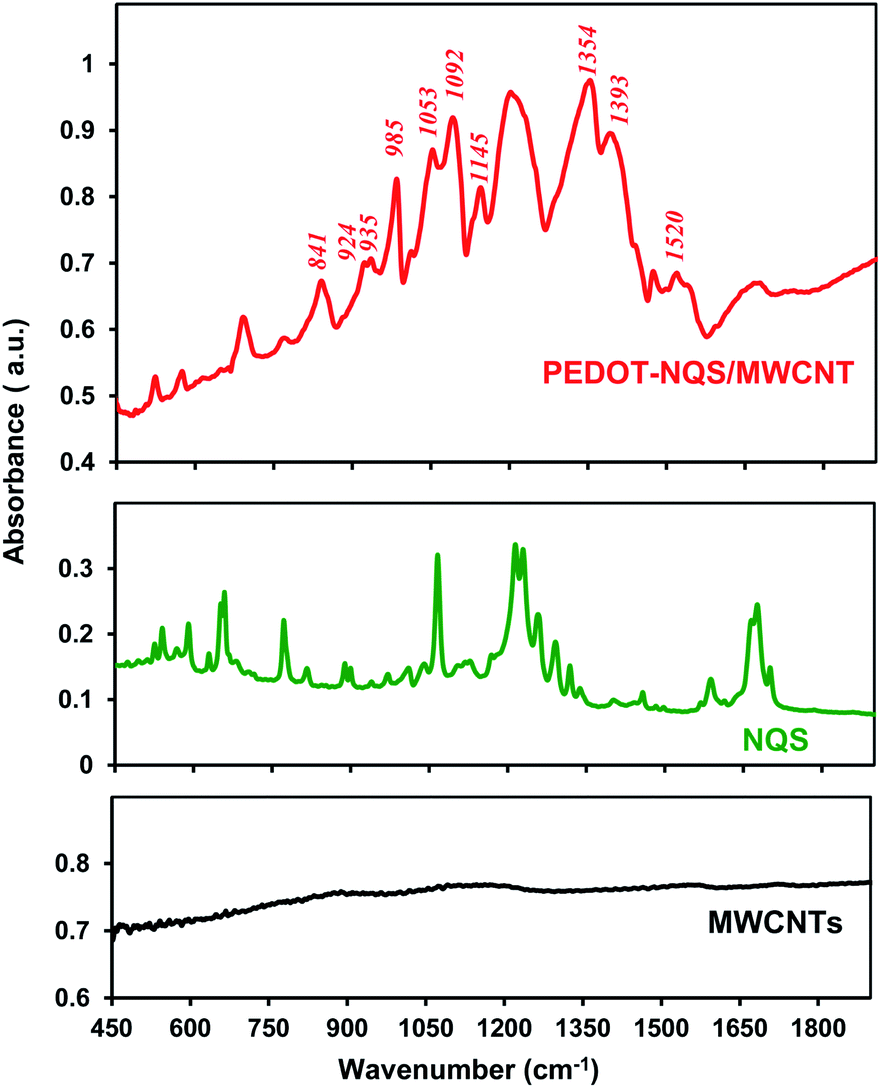 | ||
| Fig. 4 FTIR spectra of acid-treated MWCNTs, redox active dopant NQS and PEDOT–NQS/MWCNT composite. | ||
The conductivity of the enzymatically synthesized PEDOT–NQS/MWCNT composite measured by the four-point probe method was 16.8 ± 0.9 S cm−1, which is significantly higher than that of functionalized MWCNTs (6.9 ± 0.4 S cm−1) and PEDOT–NQS polymer (9.1 ± 0.7 S cm−1).
Thus laccase-catalyzed in situ polymerization of EDOT in the presence of the redox active dopant results in the production of the conducting PEDOT–NQS/MWCNT composite, in which the polymer is bound to the surface of nanotubes.
3.3 Electrochemical properties of the PEDOT–NQS/MWCNT composite
Electrochemical performance of the PEDOT–NQS/MWCNT composite was tested in 1 M H2SO4 aqueous electrolyte by cyclic voltammetry (CV) using three-electrode cell configuration at various scan rates (Fig. 5a). The cyclic voltammogram recorded at 5 mV s−1 (Fig. 5b) clearly demonstrates three redox peak couples whose characteristics are given in Table 1, where ΔE is the difference in potentials for the maximum of anodic (Ea) and cathodic (Ec) peaks and Emp is the middle point potential.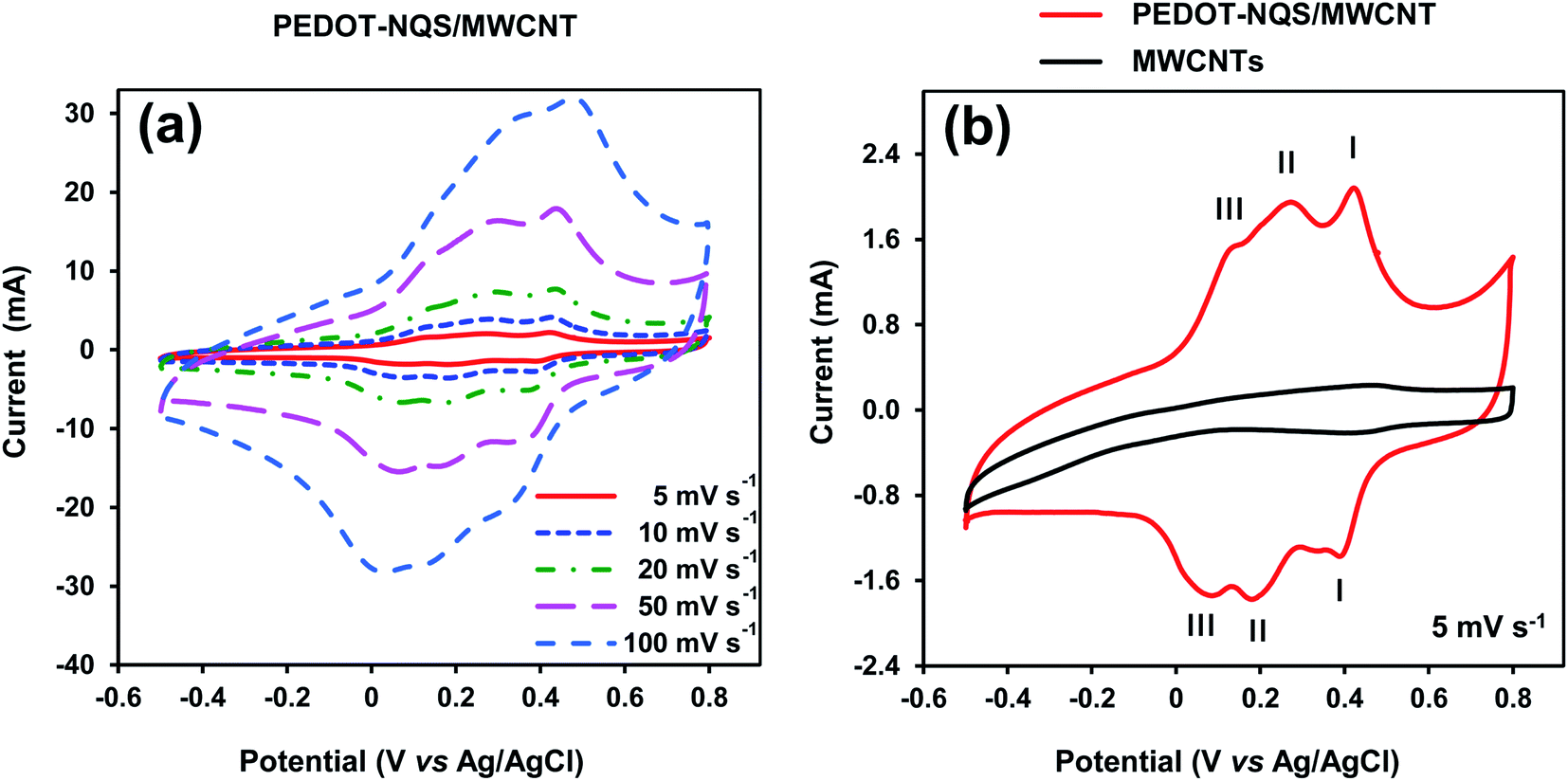 | ||
| Fig. 5 CV curves of the PEDOT–NQS/MWCNT composite at different scan rates (a) and at 5 mV s−1 (b). Roman figures I, II, III show redox peak couples, whose characteristics are given in Table 1. The mass of the electroactive composite was 1.0 mg. | ||
| Redox peak couple | Ea, V | Ec, V | ΔE, V | Emp, V |
|---|---|---|---|---|
| a CV recorded at 5 mV s−1. | ||||
| I | 0.427 | 0.395 | 0.032 | 0.410 |
| II | 0.284 | 0.169 | 0.115 | 0.227 |
| III | 0.148 | 0.076 | 0.072 | 0.112 |
Couple I of redox peaks correspond to the electrochemical conversion of NQS weakly bound to the surface of MWCNTs.39 Couple II of redox peaks seems to correspond to the redox reaction of NQS, which is the counter ion of PEDOT. The linear dependence of the cathode and anode currents of couple II of redox peaks on the scan rate (see ESI Fig. S4†) suggests the adsorption character of the redox reaction. Couple III of redox peaks correspond to the oxidation/reduction of PEDOT.16
The specific capacitance of the PEDOT–NQS/MWCNT composite depended on the scan rate and was 575, 544, 503, 460 and 425 F g−1 at 5, 10, 20, 30 and 50 mV s−1, respectively (see ESI Fig. S5†). It is noteworthy that the values of the specific capacitance of the composite are higher than those reported in the literature, which seems to be due the fact that the redox active compound NQS was used as a PEDOT dopant. For reference, the specific capacitance of the enzymatically synthesized PEDOT/PSS/MWCNT in 1 M H2SO4 (ref. 35) was 246 F g−1 at a potential scan rate of 5 mV s−1. The capacitance of chemically synthesized PEDOT/MWCNT was 61 F g−1 in 0.1 M KCl at a scan rate of 50 mV s−1,60 95 F g−1 in 1 M H2SO4 at a scan rate of 2 mV s−1,16 and 133 F g−1 in 1 M NaNO3 at a scan rate of 5 mV s−1.61
The PEDOT–NQS/MWCNT composite was studied in 1 M H2SO4 at a potential window from −0.5 to 1.0 V (vs. Ag/AgCl) (Fig. 6). First the CV curve was recorded at a potential scan range of 50 mV s−1. Then a potentiostatic pause was performed at 1.0 V for 10 min, and a potential cycle was recorded again within the same potential window. The cyclic voltammogram recorded after the potentiostatic pause almost completely coincides with the initial one, which means that at high potentials the PEDOT–NQS/MWCNT composite is markedly more stable as compared to PANI-based composites.46 Hence, the extension of the potential window to 1.0 V had no effect on the composite stability. A little shift of the peaks after the potentiostatic pause seems to be due to some changes in the polymer layer. However, the specific capacitance of the composite before and after the potentiostatic pause changes by less than 4%.
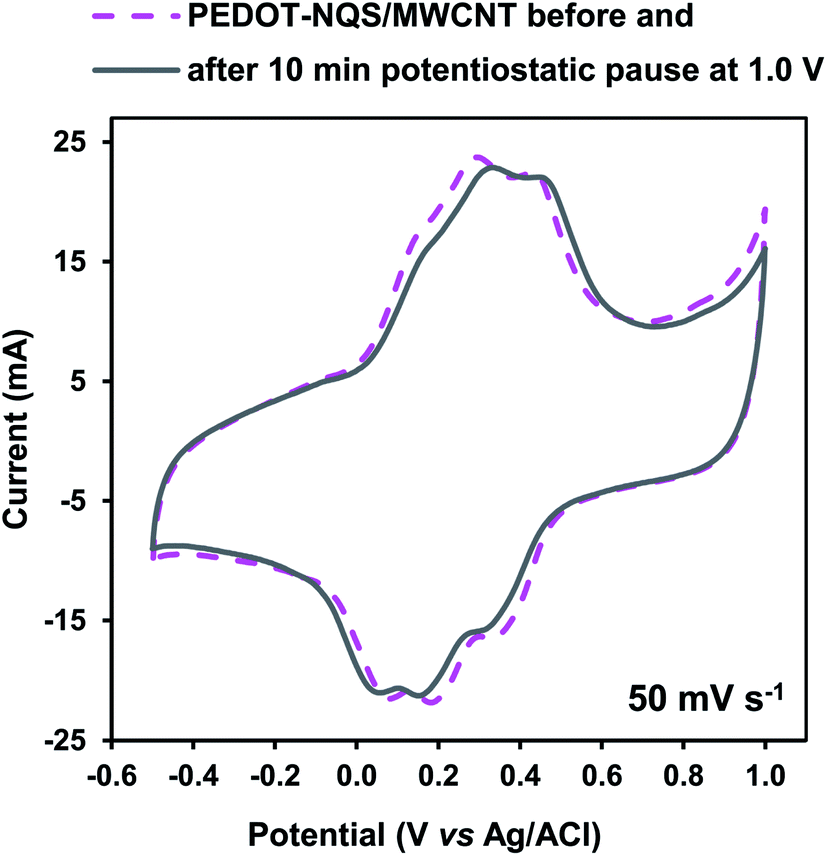 | ||
| Fig. 6 CV curves of the PEDOT–NQS/MWCNT composite at 50 mV s−1 within a potential window from −0.5 to 1.0 V. The mass of the electroactive composite was 1.2 mg. | ||
The PEDOT–NQS/MWCNT composite showed an excellent cycling stability in 1 M H2SO4: after 1000 cycles its specific capacitance decreased by less than 5% (see ESI Fig. S6†), which is a great deal better than in the case of PEDOT/MWCNT composites described in the literature: the specific capacitance of PEDOT-based composites decreased by 8% (0.1 M H2SO4, 1000 cycles),62 by 15% (1 M LiClO4, 1000 cycles),63 by 26.9% (0.5 M H2SO4, 2000 cycles),47 by 33% (1 M HCl, 1000 cycles).60
In order to perform electrochemical tests, the composite was applied onto the surface of the electrode made of graphite foil. Therefore, it is virtually impossible to separate the composite from the electrode after electrochemical testing and analyze it by TEM.
The PEDOT–NQS/MWCNT composite was further examined by electrochemical impedance spectroscopy (EIS), which enabled us to investigate both the ion transport and electron transfer via the electrode/electrolyte interface, and the pseudocapacitance of the composite. The Nyquist plots of EIS spectra of the PEDOT–NQS/MWCNT composite in 1 M H2SO4 at different potential values are shown in Fig. 7a and b. The EIS spectra have a single semicircle in the high frequency region, which may be attributed to the redox reactions of the composite, while the inclined line at the low frequencies seems to be due to the pseudocapacitance of the polymer layer. The electrochemical impedance spectra were analyzed by an equivalent circuit model (Fig. 7c), where Rs is mainly arising from the electrolyte resistance, the intrinsic resistance of the active material and the contact resistance at the electroactive material/current collector interface; Cdl and Rct represent the double-layer capacitance at the electrode/electrolyte interface and the charge transfer resistance on the electrode (the resistance of electrochemical reactions). The Warburg open element W0 models the diffusion of ions in the polymer layer and is characterized by the Warburg constant (W). The constant phase element (CPE) is applied to denote the pseudocapacitance of the polymer layer. CPE is defined by:
| ZCPE = CPET(jω)−p, |
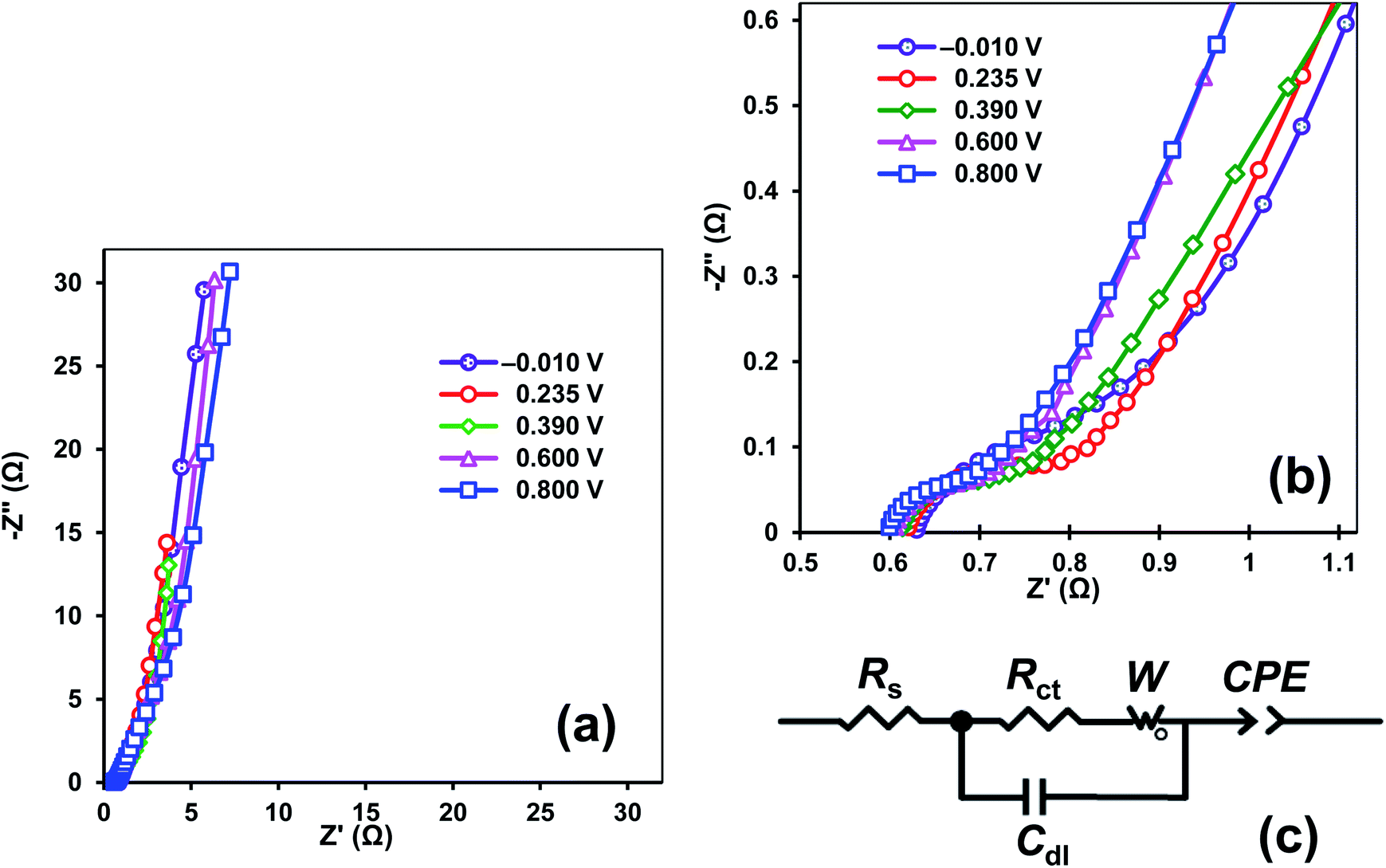 | ||
| Fig. 7 (a) Nyquist plots of PEDOT–NQS/MWCNT composite in 1 M H2SO4 at different potential values; (b) expanded high frequency region of Nyquist plots; (c) electrical equivalent circuit used for fitting the impedance spectra. | ||
| Potential, V | Rs, Ω | Rct, Ω | Cdl, F | W, (Ω s−1/2) | CPET, (Ω s−1) | p |
|---|---|---|---|---|---|---|
| −0.010 | 0.637 | 0.143 | 0.000655 | 2.93 | 0.060 | 0.922 |
| 0.235 | 0.625 | 0.125 | 0.000632 | 2.03 | 0.122 | 0.921 |
| 0.390 | 0.615 | 0.097 | 0.000633 | 2.46 | 0.170 | 0.846 |
| 0.600 | 0.610 | 0.103 | 0.000633 | 2.72 | 0.061 | 0.852 |
| 0.800 | 0.602 | 0.099 | 0.000626 | 3.02 | 0.059 | 0.842 |
The p values given in Table 2 indicate the capacitive nature of the CPE element. The CPET values that characterize the pseudocapacitance of the PEDOT–NQS layer are 200 times as high as Cdl. Hence, the EIS data support the conclusion that the NQS-doped polymer makes the main contribution to the capacitance of the PEDOT–NQS/MWCNT composite. As the electrode potential increases from −0.010 to 0.800 V, the Rs, Rct and Cdl values remain virtually the same, and the W constant changes insignificantly. The CPET value varies the most and reaches the maximum at 0.390 V, which is due to summing the contributions of the redox transformations of PEDOT, PEDOT–NQS and NQS. The CPET value drastically decreases within the potential window 0.600–0.800 V, where the main contribution is made by PEDOT. The change of the CPET value with the potential is consistent with the results of cyclic voltammetry (Fig. 5b). As is seen from Table 2, the PEDOT–NQS/MWCNT composite has low Rs, Rct, and W values, which assures high power characteristics of supercapacitors designed on its basis.
The PEDOT–NQS/MWCNT composite was further studied by the galvanostatic method (Fig. S8†). The composite capacitance calculated by the formula described in65 was 487 and 534 F g−1 at 1.51 and 0.56 A g−1, respectively. For comparison, Snook et al. reported in the review48 on composites containing PEDOT/PSS and carbon nanotubes with specific capacitance varying from 85 to 150 F g−1.
4. Conclusions
We have performed an enzymatic oxidative EDOT polymerization on the surface of acid-treated MWCNTs using the redox active compound NQS as dopant and showed that laccase-mediated EDOT polymerization in atmospheric oxygen enables the production of a stable, electrochemically active PEDOT–NQS/MWCNT composite with a high specific capacitance of 534 F g−1 (at 0.56 A g−1). The enzymatically synthesized PEDOT–NQS/MWCNT composite has a significantly broader potential window as compared to PANI-based composites, which can increase the energy density of supercapacitors on the basis of the PEDOT–NQS/MWCNT composite.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study was partially supported by the Russian Foundation for Basic Research, project no. 18-29-23042.References
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498, DOI:10.1016/S0013-4686(00)00354-6.
- S. Poonam, K. Sharma, A. Arora and S. K. Tripathi, J. Energy Storage, 2019, 21, 801–825, DOI:10.1016/j.est.2019.01.010.
- M. Winter and R. Brodd, Chem. Rev., 2004, 104, 4245–4269, DOI:10.1021/cr020730k.
- S. M. Chen, R. Ramachandran, V. Mani and R. Saraswathi, Int. J. Electrochem. Sci., 2014, 9, 4072–4085 Search PubMed , http://www.electrochemsci.org/papers/vol9/90804072.pdf.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828, 10.1039/C1CS15060J.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531, 10.1039/B813846J.
- Q. F. Meng, K. F. Cai, Y. X. Chen and L. D. Chen, Nano Energy, 2017, 36, 268–285, DOI:10.1016/j.nanoen.2017.04.040.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672, 10.1039/C7TA00863E.
- C. Lei, P. Wilson and C. Lekakou, J. Power Sources, 2011, 196, 7823–7827, DOI:10.1016/j.jpowsour.2011.03.070..
- H. Zhou, H. J. Zhai and G. Han, J. Power Sources, 2016, 323, 125–133, DOI:10.1016/j.jpowsour.2016.05.049.
- H. Zhou and X. Zhi, Synth. Met., 2017, 234, 139–144, DOI:10.1016/j.synthmet.2017.10.011.
- S. N. J. S. Z. Abidin, N. H. N. Azman, S. Kulandaivalu and Y. Sulaiman, J. Nanomater., 2017, 5798614, DOI:10.1155/2017/5798614.
- Z. Yang, J. Tian, Z. Yin, C. Cui, W. Qian and F. Wei, Carbon, 2019, 141, 467–480, DOI:10.1016/j.carbon.2018.10.010.
- E. Frackowiak, V. Khomenko, K. Jurewicz, K. Lota and F. Beguin, J. Power Sources, 2006, 153, 413–418, DOI:10.1016/j.jpowsour.2005.05.030.
- A. Eftekhari, L. Li and Y. Yang, J. Power Sources, 2017, 347, 86–107, DOI:10.1016/j.jpowsour.2017.02.054.
- K. Lota, V. Khomenko and E. Frackowiak, J. Phys. Chem. Solids, 2004, 65, 295–301, DOI:10.1016/j.jpcs.2003.10.051.
- P. Liu, J. Yan, Z. Guang, Y. Huang, X. Li and W. Huang, J. Power Sources, 2019, 424, 108–130, DOI:10.1016/j.jpowsour.2019.03.094.
- S. Dhibar, A. Roy and S. Malik, Eur. Polym. J., 2019, 120, 109203, DOI:10.1016/j.eurpolymj.2019.08.030.
- T. Abdiryim, A. Ubul, R. Jamal, F. Xu and A. Rahman, Synth. Met., 2012, 162, 1604–1608, DOI:10.1016/j.synthmet.2012.07.007.
- A. A. Syed and M. K. Dinesan, Talanta, 1991, 38, 815–837, DOI:10.1016/0039-9140(91)80261-W.
- I. Sapurina and J. Stejskal, Polym. Int., 2008, 57, 1295–1325, DOI:10.1002/pi.2476.
- G. G. Wallace, P. R. Teasdale, G. M. Spinks and L. A. P. Kane-Maguire, Conductive electroactive polymers: intelligent polymer systems, CRC Press, Boca Raton, 2002 Search PubMed.
- G. V. Otrokhov, G. P. Shumakovich, M. E. Khlupova, I. S. Vasil'eva, I. B. Kaplan, B. T. Zaitchik, E. A. Zaitseva, O. V. Morozova and A. I. Yaropolov, RSC Adv., 2016, 6, 60372–60375, 10.1039/c6ra12352j.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78, DOI:10.1021/ja982270b.
- P. Walde and Z. Guo, Soft Matter, 2011, 7, 316–331, 10.1039/C0SM00259C.
- R. Cruz-Silva, C. Ruiz-Flores, L. Arizmendi, J. Romero-García, E. Arias-Marin, I. Moggio, F. F. Castillon and M. H. Farias, Polymer, 2006, 47, 1563–1568, DOI:10.1016/j.polymer.2005.12.082.
- F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793, DOI:10.3390/polym4010759.
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209, DOI:10.1002/adsc.200800775.
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927, DOI:10.1002/adsc.201300960.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353, DOI:10.1021/cr900165z.
- P. Xu, A. Singh and D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 69–94, DOI:10.1007/12_036.
- S. Nagarajan, J. Kumar, F. F. Bruno, L. A. Samuelson and R. Nagarajan, Macromolecules, 2008, 41, 3049–3052, DOI:10.1021/ma0717845.
- H. Zhou, Y. Zhao, X. Shen and Z. Ni, Mater. Chem. Phys., 2018, 208, 91–96, DOI:10.1016/j.matchemphys.2018.01.022.
- G. Shumakovich, G. Otrokhov, I. Vasil'eva, D. Pankratov, O. Morozova and A. Yaropolov, J. Mol. Catal. B: Enzym., 2012, 81, 66–68, DOI:10.1016/j.molcatb.2012.05.008.
- I. S. Vasil'eva, G. P. Shumakovich, O. V. Morozova, M. E. Khlupova, R. B. Vasiliev, E. A. Zaitseva and A. I. Yaropolov, Chem. Pap., 2018, 72, 1499–1505, DOI:10.1007/s11696-018-0396-8.
- I. S. Vasil'eva, M. E. Khlupova, G. P. Shumakovich, E. A. Zaitseva, O. V. Morozova and A. I. Yaropolov, Moscow Univ. Chem. Bull., 2019, 74, 186–190, DOI:10.3103/S0027131419040096.
- E. Frackowiak and F. Béguin, Carbon, 2002, 40, 1775–1787, DOI:10.1016/S0008-6223(02)00045-3.
- D. Vonlanthen, P. Lazarev, K. A. See, F. Wudl and A. J. Heeger, Adv. Mater., 2014, 26, 5095–5100, DOI:10.1002/adma.201400966.
- G. P. Shumakovich, O. V. Morozova, M. E. Khlupova, I. S. Vasil'eva, E. A. Zaitseva and A. I. Yaropolov, RSC Adv., 2017, 7, 34192–34196, 10.1039/C7RA04801G.
- H. Zhang, J. Li, C. Gu, M. Yao, B. Yang, P. Lu and Y. Ma, J. Power Sources, 2016, 332, 413–419, DOI:10.1016/j.jpowsour.2016.09.137.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, ChemSusChem, 2012, 5, 1181–1185, DOI:10.1002/cssc.201200227.
- S. T. Senthilkumar, R. K. Selvan, N. Ponpandian, J. S. Melo and Y. S. Lee, J. Mater. Chem. A, 2013, 1, 7913–7919, 10.1039/C3TA10998D.
- Y. Tian, M. Liu, R. Che, R. Xue and L. Huang, J. Power Sources, 2015, 324, 334–341, DOI:10.1016/j.jpowsour.2016.05.086.
- L.-Q. Fan, J. Zhong, J.-H. Wu, J.-M. Lin and Y.-F. Huang, J. Mater. Chem. A, 2014, 2, 9011–9014, 10.1039/C4TA01408A.
- K. Shi, X. Pang and I. Zhitomirsky, J. Appl. Polym. Sci., 2015, 132, 42376, DOI:10.1002/app.42376.
- G. Otrokhov, D. Pankratov, G. Shumakovich, M. Khlupova, Y. Zeifman, I. Vasil'eva, O. Morozova and A. Yaropolov, Electrochim. Acta, 2014, 123, 151–157, DOI:10.1016/j.electacta.2013.12.089.
- L. Chen, C. Yuan, H. Dou, B. Gao, S. Chen and X. Zhang, Electrochim. Acta, 2009, 54, 2335–2341, DOI:10.1016/j.electacta.2008.10.071.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12, DOI:10.1016/j.jpowsour.2010.06.084.
- E. S. Gorshina, T. V. Rusinova, V. V. Biryukov, O. V. Morozova, S. V. Shleev and A. I. Yaropolov, Appl. Biochem. Microbiol., 2006, 42, 558–563, DOI:10.1134/S0003683806060056.
- G. Shumakovich, V. Kurova, I. Vasil'eva, D. Pankratov, G. Otrokhov, O. Morozova and A. Yaropolov, J. Mol. Catal. B: Enzym., 2012, 77, 105–110, DOI:10.1016/j.molcatb.2012.01.023.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840, DOI:10.1016/j.carbon.2008.02.012.
- B. Scheibe, E. Borowiak-Palen and R. J. Kalenczuk, Mater. Charact., 2010, 61, 185–191, DOI:10.1016/j.matchar.2009.11.008.
- F. Avilés, J. V. Cauich-Rodríguez, L. Moo-Tah, A. May-Pat and R. Vargas-Coronado, Carbon, 2009, 47, 2970–2975, DOI:10.1016/j.carbon.2009.06.044.
- T. A. Saleh, Appl. Surf. Sci., 2011, 257, 7746–7751, DOI:10.1016/j.apsusc.2011.04.020.
- S. Liu, J. Yue and R. J. Wehmschulte, Nano Lett., 2002, 2, 1439–1442, DOI:10.1021/nl0257869.
- H. Fan, H. Wang, N. Zhao, X. Zhang and J. Xu, J. Mater. Chem., 2012, 22, 2774–2780, 10.1039/C1JM14311E.
- U. Male, B. K. Shin and D. S. Huh, Macromol. Res., 2017, 25, 1121–1128, DOI:10.1007/s13233-017-5163-0.
- Y. Lei, H. Oohata, S. Kuroda, S. Sasaki and T. Yamamoto, Synth. Met., 2005, 149, 211–217, DOI:10.1016/j.synthmet.2005.01.004.
- C. Kvarnstroöm, H. Neugebauer, S. Blomquist, H. J. Ahonen, J. Kankare and A. Ivaska, Electrochim. Acta, 1999, 44, 2739–5270, DOI:10.1016/S0013-4686(98)00405-8.
- A. Dettlaff, P. R. Das, L. Komsiyska, O. Osters, J. Łuczak and M. Wilamowska-Zawłocka, Synth. Met., 2018, 244, 80–91, DOI:10.1016/j.synthmet.2018.07.006.
- D. Antiohos, G. Folkes, P. Sherrell, S. Ashraf, G. G. Wallace, P. Aitchison, A. T. Harris, J. Chen and A. I. Minett, J. Mater. Chem., 2011, 21, 15987–15994, 10.1039/C1JM12986D.
- A. Dettlaff, M. Sawczak, E. Klugmann-Radziemska, D. Czylkowski, R. Miotk and M. Wilamowska-Zawłocka, RSC Adv., 2017, 7, 31940–31949, 10.1039/C7RA04707J.
- X. Bai, X. Hu, S. Zhou, J. Yan, C. Sun, P. Chen and L. Li, Electrochim. Acta, 2013, 87, 394–400, DOI:10.1016/j.electacta.2012.09.079.
- H. Li, J. Wang, Q. Chu, Z. Wang, F. Zhang and S. Wang, J. Power Sources, 2009, 190, 578–586, DOI:10.1016/j.jpowsour.2009.01.052.
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025–4031, DOI:10.1021/nl1019672.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05589a |
| This journal is © The Royal Society of Chemistry 2020 |