DOI:
10.1039/D0RA05604A
(Paper)
RSC Adv., 2020,
10, 35820-35830
Investigation of some benzoquinazoline and quinazoline derivatives as novel inhibitors of HCV-NS3/4A protease: biological, molecular docking and QSAR studies†
Received
26th June 2020
, Accepted 14th September 2020
First published on 30th September 2020
Abstract
Morbidity and mortality due to hepatitis C virus (HCV) is a globe health concern. Hence, there is a persistent demand to design and optimize current HCV therapy and develop novel agents. HCV NS3/A4 protease plays an essential role in HCV life cycle and replication. Thus, HCV NS3/A4 protease inhibitors are one of the best therapeutic targets for the identification of novel candidate drugs. Recent studies have shown some benzoquinazolines as potent antiviral agents and promising HAV-3C protease inhibitors. In the present study, a series of benzo[g]quinazolines (1–13) and their quinazoline analogues (14–17) were evaluated for their HCV-NS3/4A inhibitory activities using in vitro assay. Our results revealed that the target compounds inhibited the activity of the NS3/4A enzyme, (IC50 = 6.41 ± 0.12 to 78.80 ± 1.70 μM) in comparison to telaprevir (IC50 = 1.72 ± 0.03 μM) as a reference drug. Compounds 1, 2, 3, 9, 10 and 13 showed the highest activity (IC50 = 11.02 ± 0.25, 6.41 ± 0.12, 9.35 ± 0.19, 9.08 ± 0.20, 16.03 ± 0.34 and 7.21 ± 0.15 μM, respectively). Molecular docking was performed to study the binding modes of the docked-chosen benzo[g]quinazolines, hydrogen bonding, and amino acid residues at the catalytic triad of the NS3/4A enzyme of HCV. The QSAR was determined to explore the relationships between the molecular structures of the targets and their biological activities by developing prediction models among the known HCV NS3/A4 inhibitors and then to predict the inhibitory activity of the target molecules synthesized.
Introduction
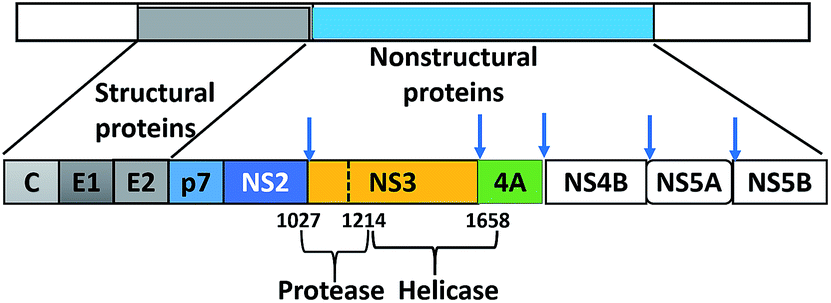
Hepatitis C virus (HCV), a positive-stranded RNA (enveloped) belonging to the Flaviviridae family, was discovered in 1989 and identified as the causative agent for “non-A, non-B hepatitis”.1,2 HCV causes chronic liver disease (hepatitis C) and is the major cause of liver cirrhosis and hepatocellular carcinoma.3 Annually, almost 3 million people are infected by HCV, and 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–500000 die from liver disease related to HCV.3 Unfortunately, the development of an HCV vaccine is hindered by viral genetic heterogeneity due to immune elusion and the lack of a relevant animal model.4 HCV-RNA is translated by host ribosomes into a polyprotein that is subsequently processed into 10 proteins (four structural and six non-structural) by host and viral proteases. The structural proteins (core, E1, E2, and p7) display an effective role in assembly of new virus particles and non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) participate in viral genome replication (Fig. 1).5
000–500000 die from liver disease related to HCV.3 Unfortunately, the development of an HCV vaccine is hindered by viral genetic heterogeneity due to immune elusion and the lack of a relevant animal model.4 HCV-RNA is translated by host ribosomes into a polyprotein that is subsequently processed into 10 proteins (four structural and six non-structural) by host and viral proteases. The structural proteins (core, E1, E2, and p7) display an effective role in assembly of new virus particles and non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) participate in viral genome replication (Fig. 1).5
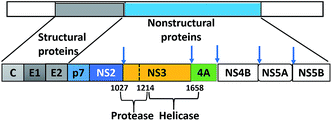 |
| | Fig. 1 Schematic of the HCV polyprotein with cleavage sites of the two proteases, NS2 and NS3. | |
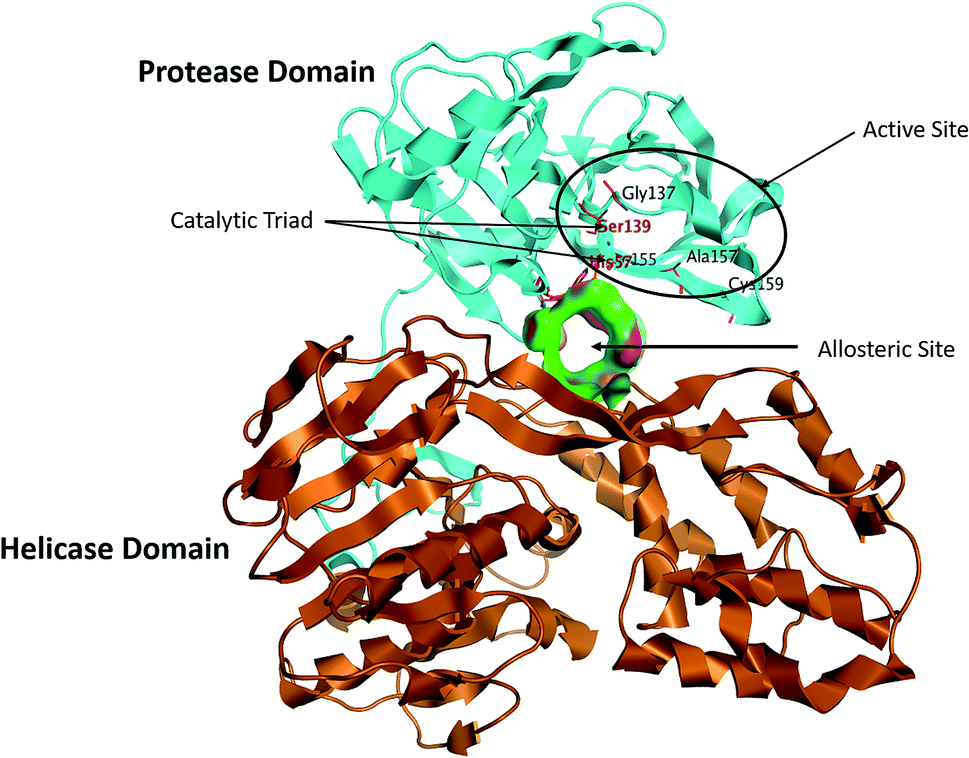
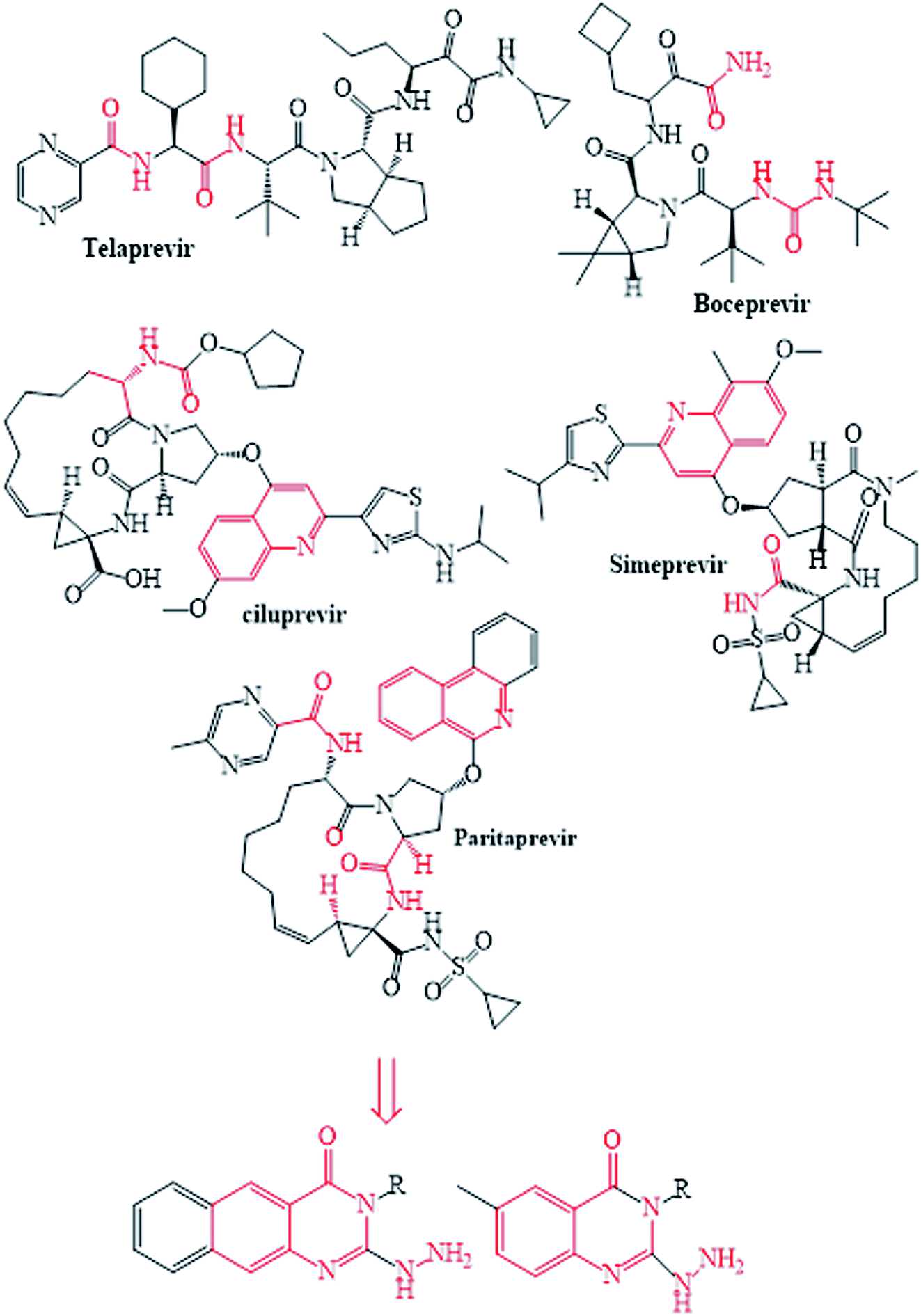
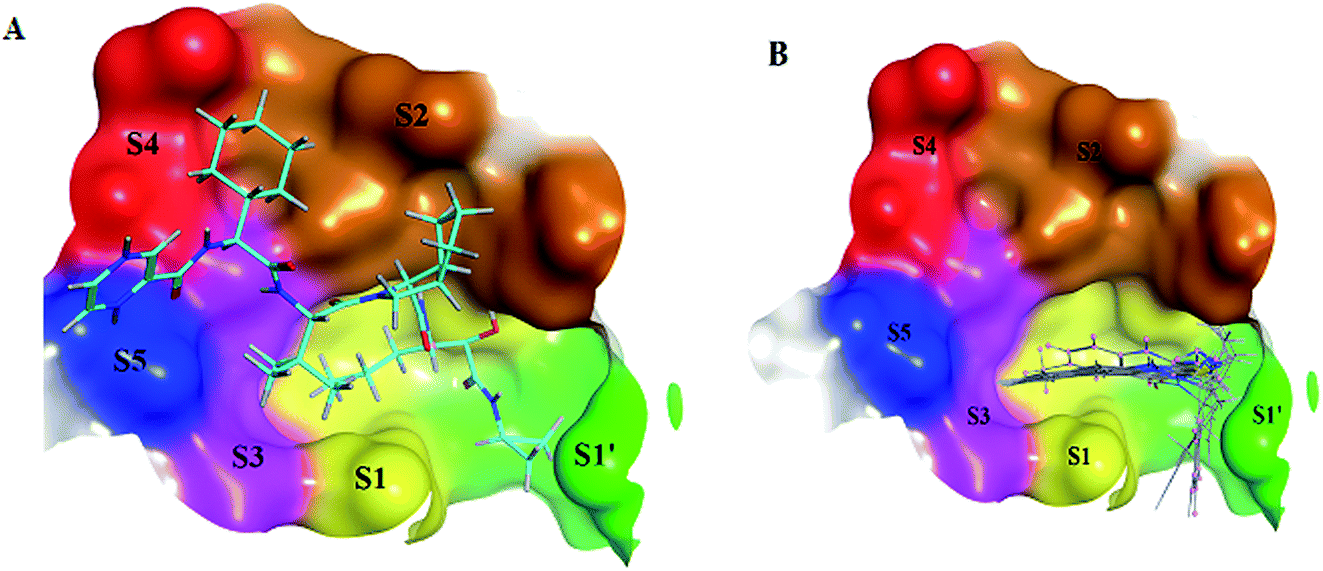
The NS3/4A is a multifunctional protein comprising two domains: a helicase and a protease (Fig. 2).6,7 The proteolytic cleavage process of the viral polyprotein into NS4A, NS4B, NS5A, and NS5B is catalysed by NS3/4A protease. Therefore, it is important for viral replication and production of infectious viral particles.4 Thus, inhibiting NS3/4A protease serves a dual purpose by inhibiting viral maturation and is considered to be a valid drug target for anti-HCV treatment. Different NS3/4A protease inhibitors (PIs) have been explored and based on their modes of action and structures, they can be classified into covalent (reversible) and non-covalent inhibitors. Boceprevir and telaprevir are linear and binds covalently with the catalytic serine (Ser139) of NS3/4A protease. Whereas, simeprevir, ciluprevir, and paritaprevir are macrocyclic noncovalent binding PIs (Fig. 3).4,8,9 Also, allosteric inhibitors for NS3/4A protease have been identified and their location is very close to the catalytic triad (His, Asp, and Ser), but their modes of action and structures are different compared to the active site binding inhibitors.10 Although protease inhibitors have been very efficient, long term effects may be restricted due to a prompt appearance of drug resistance.11,12 In majority of cases, resistance substitutions develop in close proximity to the NS3/4A catalytic triad, slightly outside the natural substrate.13 Such changes in the protease domain reduce the affinity for drugs, but permit the recognition and cleavage of substrates, usually with a lower catalytic rate.14 Fig. 3 shows that the structures of NS3/4A PIs are similar, but their biological activities are different. Many potent NS3/4A PIs have been identified by various research laboratories worldwide. However, most of these PIs are poorly tolerated and accompanied with constant viral resistance. Furthermore, the X-ray structure of NS3/NS4A protease shows that the S1 and S3 subsites (Fig. 4A) are in close proximity and characterized as hydrophobic pockets, which are lined with the hydrophobic residues of Val132, Leu135, Phe154, Ala156 and Ala157. To explore further mechanism of inhibition, docking studies were also presented to disclose the interactions binding between HCV protease and ligand, which could supply the molecular basis for further lead optimization and SAR analysis.7
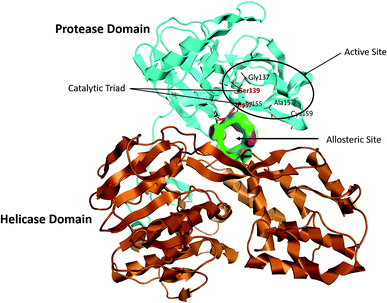 |
| | Fig. 2 Structure of the NS3/4A serine protease, with the NS3 protease domain coloured in cyan, helicase domain coloured in brown and allosteric pocket coloured in green, the active site residues, Ser139, His57 and Asp81, sit on the protein–protein interaction surface and are shown as stick figures in black. | |
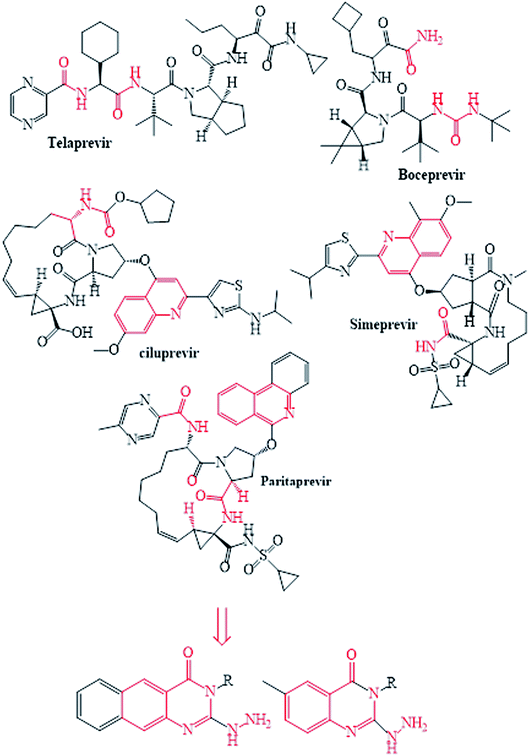 |
| | Fig. 3 The reported and designed HCV-NS3/4A inhibitors. | |
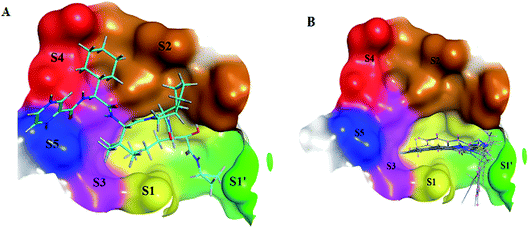 |
| | Fig. 4 (A) 3D structural representation of NS3/4A complexed with telaprevir and binding site divided at S1′ (green; Gln1041, Thr1042, Phe1043 and Gly1137), S1 (yellow; Leu1135, Lys1136, Ser1139 and Phe1154), S2 (brown; His1057, Arg1155, Ala1156 and Asp1168), S3 (Pink; Ile1132 and Ala1157), S4 (red; Arg1123 and Val1158) and S5 (blue; Ser1159) subsites. (B) 3D structural representation of NS3/4A complexed with superposing selected synthesized compounds (1–10), with binding site divided at S1′, S1, S2, S3, S4 and S5. | |
Benzoquinazoline, a drug of immense research interest, has found a conspicuous place in medicinal chemistry owing to its diverse pharmacological properties.15–20 Many benzoquinazolines were reported to show anti-HSV, -CVB4, viral activity and demonstrated promising 3C protease inhibition activity against HAV with IC50 values of 3.30, 5.92, and 22.38 μM, respectively.15,18 The results encouraged us to extend our research on benzoquinazolines chemistry to investigate their activity against HCV. The present study aims at the investigation of the inhibitory effects for seventeen benzoquinazolines and quinazolines against NS3/4A protease. The SAR rationalization of the investigated molecules was explained on the basis of a molecular docking study to analyse the binding modes between the docked-selected compounds and amino acid residues in the active site of the NS3/4A protease enzyme. In addition, QSAR was performed to explore relationships between the molecular structures of the targets and their biological activities. It is designed also to develop prediction models with robust accuracy among the known inhibitors, and then to predict the biological activities for unknown or novel probable compounds.
Results and discussion
HCV-NS3/4A protease inhibition
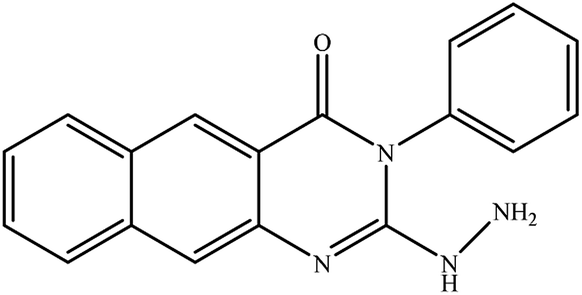
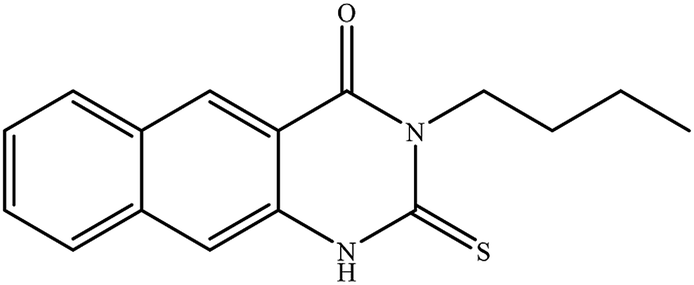
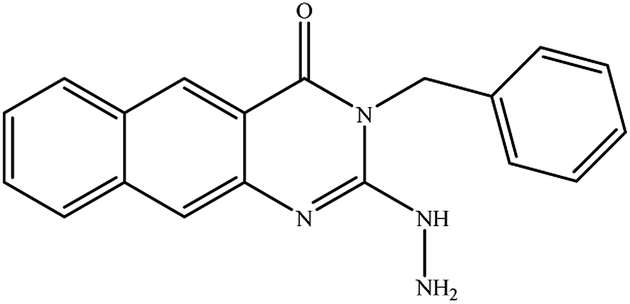
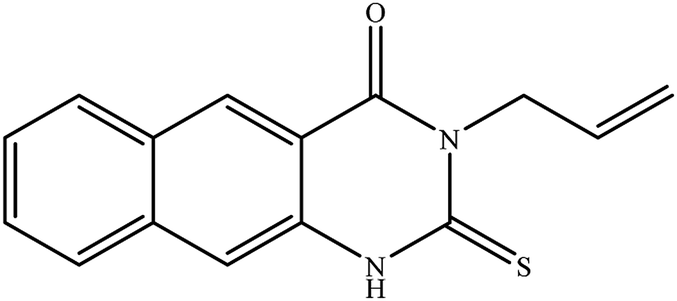
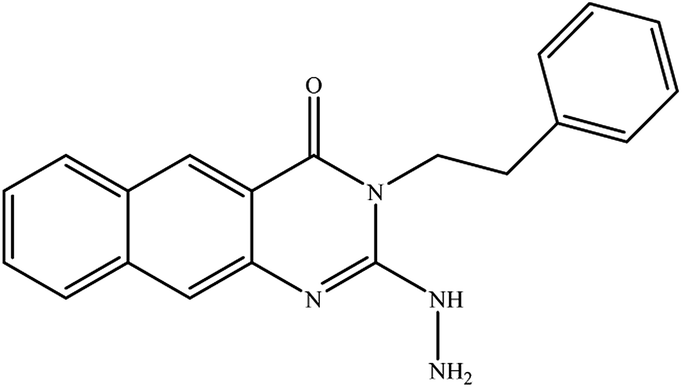
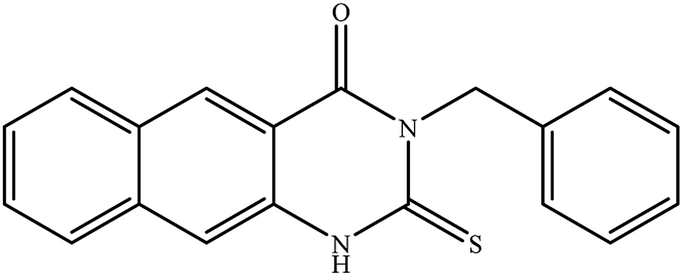
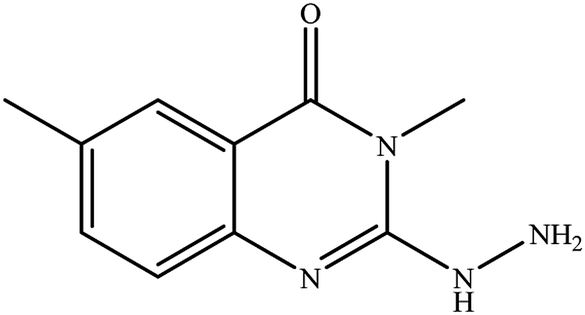
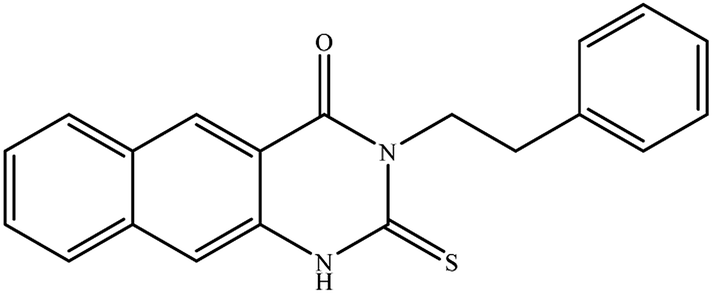
In our literature,17,19,21 the convenient alkyl isothiocyanates were treated with 3-aminonaphthaoic acid to afford the corresponding benzo[g]quinazolines 1–6, that in turn transformed into 7–13 by hydrazinolysis. Similarly, 14–17 were synthesized by hydrazinolysis of their thioxoquinazolines (Scheme 1 and Table 1).
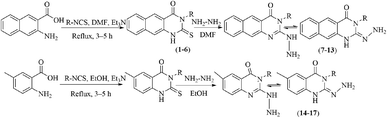 |
| | Scheme 1 Synthetic routes for the target benzoquinazolines and quinazolines 1–17. | |
Table 1 Synthesized benzoquinazolines 1–17
| Cp |
Structure |
Cp |
Structure |
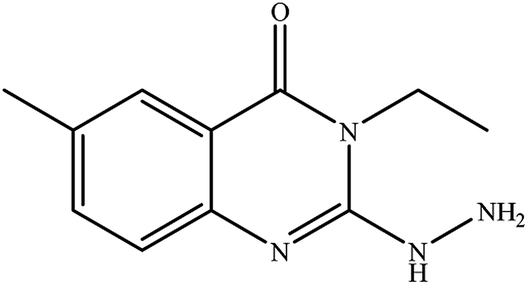
| 1 |
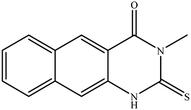 |
10 |
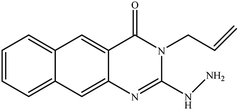 |
| 2 |
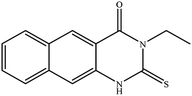 |
11 |
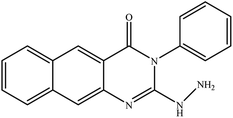 |
| 3 |
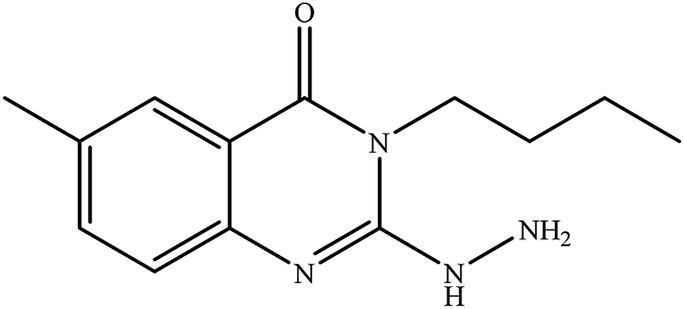
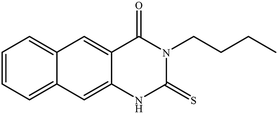 |
12 |
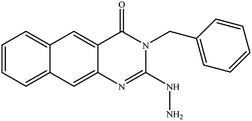 |
| 4 |
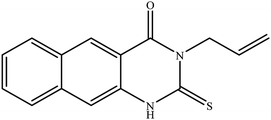 |
13 |
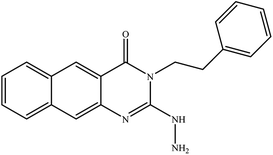 |
| 5 |
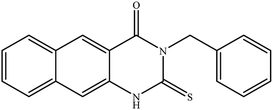 |
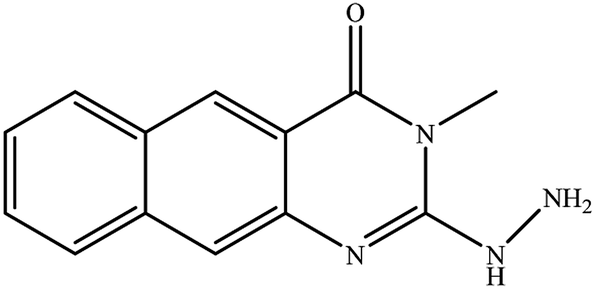
14 |
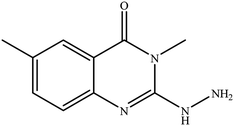 |
| 6 |
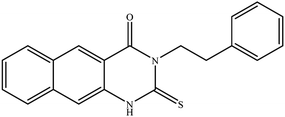 |
15 |
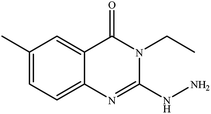 |
| 7 |
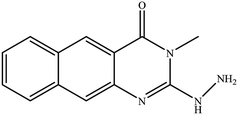 |
16 |
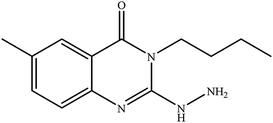 |
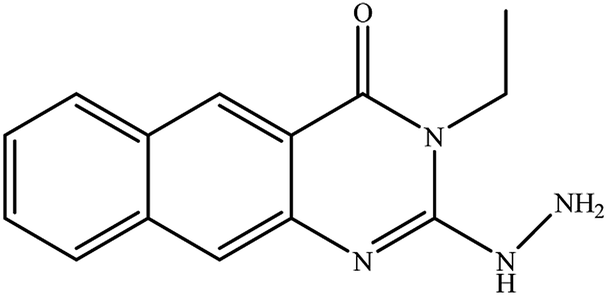
| 8 |
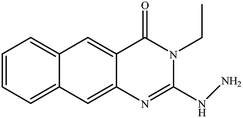 |
17 |
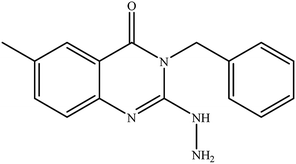 |
| 9 |
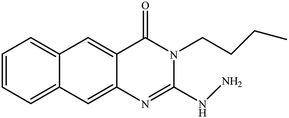 |
The targets 1–17 were investigated in an enzymatic inhibitory activity against the purified HCV-NS3/4A protease by an in vitro assay system and their potency, expressed as IC50 values in comparison with telaprevir as a reference drug (Table 2). The active compounds showed inhibitory effects against HCV-NS3/4A protease giving IC50 values in the range of 6.41 ± 0.12 to 36.81 ± 0.79 μM with respect to telaprevir as a potent HCV-NS3/4A inhibitor (IC50 = 1.72 ± 0.03 μM). To examine the inhibitory effects of the new molecules, the activity of the parent benzo[g]quinazolines 1–6 was firstly determined in terms of IC50 values (Table 2).
Table 2 Inhibitory activity of the targets 1–17 towards HCV-NS3/4A protease
| Cp |
HCV-NS3 protease, IC50 μM |
Cp |
HCV-NS3 protease, IC50 μM |
| 1 |
11.02 ± 0.25 |
10 |
16.03 ± 0.34 |
| 2 |
6.41 ± 0.12 |
11 |
33.98 ± 073 |
| 3 |
9.35 ± 0.19 |
12 |
222.80 ± 4.87 |
| 4 |
20.36 ± 0.42 |
13 |
7.21 ± 0.15 |
| 5 |
36.81 ± 0.79 |
14 |
25.52 ± 0.55 |
| 6 |
31.95 ± 0.69 |
15 |
177.94 ± 3.90 |
| 7 |
22.31 ± 0.46 |
16 |
23.40 ± 0.54 |
| 8 |
78.80 ± 1.70 |
17 |
29.64 ± 0.64 |
| 9 |
9.08 ± 0.20 |
Telaprevir |
1.72 ± 0.03 |
All parent compounds exhibited good activity, particularly compounds 1, 2, and 3 showed potent inhibitory activity in comparison to telaprevir. Our previous antiviral activity results,15,18 revealed that incorporation of hydrazine moiety into benzo[g]quinazoline core resulted in a promising compound of high activity against HSV, CVB4, and HAV (3C proteinase). In this study, such modification enhanced positively the inhibition of HCV-NS3/4A, as it was demonstrated by compounds 9, 10 and 13, as well. This indicates the insertion of the hydrazine group increased the interactions with the active site of the enzyme. Thus, compounds 7, 9, 10, 11 and 13 showed good inhibitory activity against HCV-NS3/4A, however compounds 8 and 12 showed lower activity than their parent compounds 2 and 5. Furthermore, the replacement of the benzo[g]quinazoline core, by the quinazoline scaffold, has led to a series of structurally related products (14–17) that were evaluated for their HCV-NS3/4A inhibitory activities (Table 2). It was noticed that compound 14 exhibited almost the same behaviour as its related compound 7. However, 17 showed sharply higher activity (IC50 = 29.64 ± 0.64 μM) than its analogue 12 (IC50 = 2222.80 ± 4.87 μM), and vice versa was observed for compound 15 that exhibited a sharply lower activity (IC50 = 177.94 ± 3.90 μM) than 8 (IC50 = 78.80 ± 1.70 μM). Modifications at positions 2 and 3 of the benzo[g]quinazoline structure led to the following conclusions: the replacement of methyl group in 1 (IC50 = 11.02 ± 0.25 μM) with the ethyl and butyl groups in 2 and 3 significantly increased HCV-NS3/4A inhibitory activity (IC50 = 6.41 ± 0.12 & 9.35 ± 0.19 μM). This could be attributed to the increase of the hydrophobic character. Whereas, the insertion of an aromatic group (benzyl/phenethyl) in compounds 5 and 6 led to sharp decreasing of the inhibition potency in comparison with corresponding targets of aliphatic chain (1, 2, and 3). Moreover, exploring the effect of the nitrogen in thiocarbamide group could be used to engage the enzyme in further adequate interactions with possibly positive effects on its activity.
Docking study
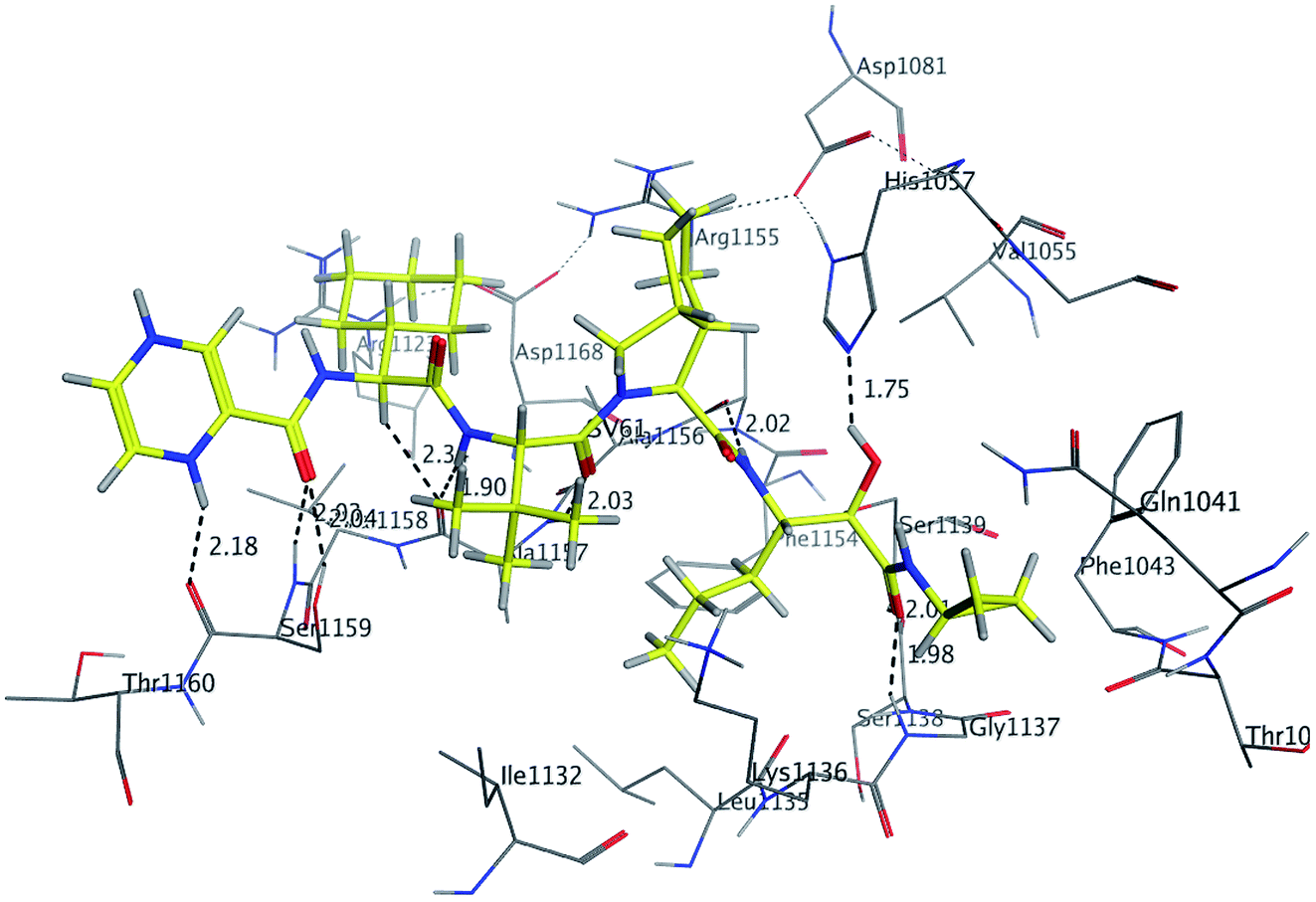
In molecular docking study, the structure of the HCV NS3/4A serine protease substrate with 10 residues is given in Fig. 4A with NS4A/NS4B as the representative peptide. In this study, a molecular modelling model was applied using the molecular operating environment (MOE), to clarify the activity of the targets (1–17) and determine their probably bioactive conformations. We found important subsites for the HCV NS3/4A inhibition by three-dimensional telaprevir-complexed structure (PDB: 3SV6, resolution 1.4 Å),22 which was obtained from the PDB database, and the validation of the molecular docking protocol done by molecular redocking, with a position structure of RMSD lower than 2 Å (Fig. 4A and 5). Generally, stronger interactions with the catalytic triad would provide a good inhibition, therefore the NS3 of 3SV6 was docked with all synthesized compounds, and the results were compared. The interaction of telaprevir with active site of the HCV NS3/4A enzyme showed four hydrogen bonds with His1057, Gly1137, Ser1139, Arg1155, Ala1157, and Ser1159 (Table 3, Fig. 5), and demonstrated the ability to bind with two catalytic triad residues (His1057 and Ser1139) and Gly1137 in the oxyanion hole (Fig. 4A and 5).
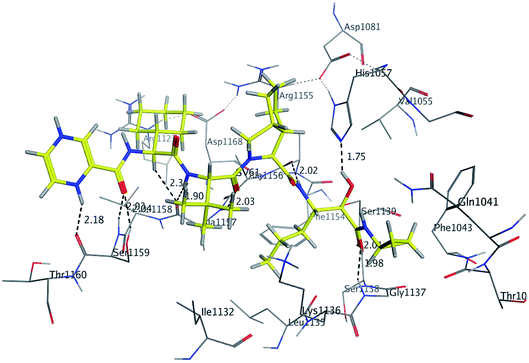 |
| | Fig. 5 3D Interaction of reference compound (telaprevir) with active site of HCV NS3/4A enzyme. Telaprevir shows four hydrogen bonds with Ser1159, Ala1157, Arg1155, His1057, Ser1159, Gly1137 and Ser1139 of the target protein active site residue. | |
Table 3 Analysis of the interaction between HCV NS3/4A protease enzyme and synthesized compounds or telaprevir
| Cp |
Ligand |
Receptor |
Interaction |
Distance (Å) |
Binding affinity (kcal mol−1) |
Docking score (S) |
Binding energy kcal mol−1 |
| 1 |
O 14 |
N⋯Gly 1137 |
H-acceptor |
2.31 |
−5.22 |
−8.0175 |
−26.74 |
| S 19 |
NE2⋯Gln1041 |
H-acceptor |
2.65 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
3.43 |
| 6-Ring |
NZ⋯Lys1136 |
pi-cation |
2.83 |
| 2 |
S22 |
NE2⋯Gln1041 |
H-acceptor |
2.73 |
−5.71 |
−8.701 |
−28.26 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
2.13 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
3.40 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
2.81 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
2.80 |
| 3 |
S 28 |
NE2⋯Gln1041 |
H-acceptor |
2.57 |
−6.23 |
−8.798 |
−29.997 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
2.17 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
3.38 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
3.82 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
2.88 |
| 4 |
S23 |
NE2⋯Gln1041 |
H-acceptor |
2.55 |
−5.911 |
−8.422 |
−28.876 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
2.22 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
2.84 |
| 6-Ring |
NZ⋯Lys1136 |
pi-cation |
3.40 |
| 5 |
S29 |
NE2⋯Gln1041 |
H-acceptor |
2.71 |
−5.943 |
−9.0854 |
−32.601 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
2.25 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
2.84 |
| 6-Ring |
NZ⋯Lys1136 |
pi-cation |
3.43 |
| 6 |
S32 |
NE2⋯Gln1041 |
H-acceptor |
2.07 |
−6.024 |
−8.71 |
−31.179 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
3.04 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.73 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
4.0 |
| 6-Ring |
HE3⋯Lys1136 |
pi-H |
2.81 |
| 7 |
O22 |
HN⋯Gly1137 |
H-acceptor |
2.3 |
−5.3285 |
−7.758 |
−27.88 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.73 |
| 6-Ring |
HE3⋯Lys1136 |
pi-H |
2.81 |
| 8 |
O22 |
HN⋯Gly1137 |
H-acceptor |
2.32 |
−5.7982 |
−9.21 |
−30.39 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.39 |
| 6-Ring |
HE3 Lys 1136 |
pi-H |
2.83 |
| 9 |
O22 |
HN⋯Gly1137 |
H-acceptor |
2.06 |
−6.014 |
−9.657 |
−30.975 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.38 |
| 6-Ring |
HE3⋯Lys1136 |
pi-H |
2.77 |
| 10 |
O22 |
HN⋯Gly1137 |
H-acceptor |
2.17 |
−5.7693 |
−9.209 |
−30.931 |
| 6-Ring |
HZ2⋯Lys 1136 |
pi-cation |
3.44 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
3.77 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
3.82 |
| 11 |
N 24 |
O⋯Leu1135 |
H-donor |
2.15 |
−5.0924 |
−8.1919 |
−24.4278 |
| N 23 |
N⋯Gly 1137 |
H-acceptor |
2.68 |
| 6-Ring |
5-Ring His1057 |
pi-pi |
4.06 |
| 6-Ring |
5-Ring His1057 |
pi-pi |
3.38 |
| 12 |
O22 |
HN⋯Gly1137 |
H-acceptor |
2.58 |
−5.327 |
−10.359 |
−25.736 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
4.64 |
| 13 |
O22 |
N⋯Gly1137 |
H-acceptor |
2.01 |
−6.0885 |
−9.8605 |
−33.0523 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.40 |
| 6-Ring |
CE⋯Lys1136 |
pi-H |
3.82 |
| 14 |
HN10 |
NE2⋯His1057 |
H-donor |
2.13 |
−4.905 |
−7.474 |
−21.144 |
| O15 |
HN⋯Gly1137 |
H-acceptor |
2.09 |
| 15 |
HN10 |
NE2⋯His1057 |
H-donor |
2.09 |
−5.237 |
−7.265 |
−24.615 |
| O15 |
HN⋯Gly1137 |
H-acceptor |
2.16 |
| 16 |
HN30 |
OE1⋯Gln1041 |
H-donor |
1.93 |
−5.8285 |
−8.969 |
−30.375 |
| O14 |
HN⋯Gly1137 |
H-acceptor |
2.08 |
| 6-Ring |
CD⋯Lys1136 |
pi-H |
3.79 |
| 6-Ring |
HZ2⋯Lys1136 |
pi-cation |
3.56 |
| 17 |
O 15 |
HN⋯Gly1137 |
H-acceptor |
1.99 |
−5.506 |
−9.385 |
−30.062 |
| HN10 |
NE2⋯His1057 |
H-donor |
2.10 |
| Telaprevir |
HAF10 |
O⋯Ser1159 |
H-donor |
2.18 |
−7.3906 |
−9.408 |
−40.347 |
| HAY16 |
O⋯Ala1157 |
H-donor |
2.34 |
| HAC37 |
O⋯Ala1157 |
H-donor |
1.9 |
| HAE79 |
O⋯Arg1155 |
H-donor |
2.02 |
| OBR103 |
NE2⋯His1057 |
H-donor |
1.75 |
| OBW9 |
HG⋯Ser1159 |
H-acceptor |
2.04 |
| OBW9 |
HN⋯Ser1159 |
H-acceptor |
2.03 |
| OBT55 |
HN⋯Ala1157 |
H-acceptor |
2.03 |
| OBS105 |
N⋯Gly1137 |
H-acceptor |
1.98 |
| OBS105 |
HN⋯Ser1139 |
H-acceptor |
2.01 |
Initially, the molecular docking of benzo[g]quinazolines 1–6 in hydrophobic pocket of the HCV NS3/4A enzyme was studied. Where, the strongest binding affinity (about −6 kcal mol−1), lower binding energy (about −30 kcal mol−1) and good docking score (about −9) were observed. Furthermore, it was found that all six benzo[g]quinazolines formed hydrogen bonds with Gly1137 in the S1′ subsites with distance less than 3 Å (in the oxyanion hole), and with Gln1041 in the S1′ subsites. In addition two π-bonds with Lys1136 in the S1 subsites. Moreover, these compounds made one or more hydrophobic binding contacts in the hydrophobic pocket with His1057 in S2 subsites (catalytic triad residue); Val1055; Phe1043; Ile1132; Lys1136; and Ala1157. The orientation of that compounds 1–6 were occupied in S1′ and S1 subsites at the active site of the HCV NS3/4A enzyme (Table 3; Fig. 4B and 6).
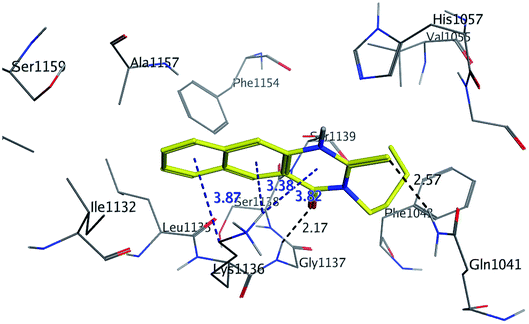 |
| | Fig. 6 3D Interaction of compound 3 with active site of HCV NS3/4A enzyme. Compound 3 shows four hydrogen bonds with Gln1041 and Gly1137 and two π-bonds with Lys1136 of the target protein active site residue. | |
By docking, a good binding affinity was observed with the HCV NS3/4A enzyme and the targets 1–6. In other hand, 1, 2, 3, and 4 showed high potent inhibitory activities, this could be attributed to their good binding affinity due to the aliphatic substituents (methyl, ethyl, butyl and allyl, respectively). Moreover, these alkyl substitutions are electron donors (+I effect), thus increasing the strength of the hydrogen bond (increasing the electron density on the molecule). The presence of the alkyl group in the molecule enhances its movement towards the hydrophobic pocket, thus the molecule comes closer to the catalytic triad and forms hydrophobic binding contacts with His1057 in addition to Val1055; Phe1043; Ile1132; Lys1136; and Ala1157. Because of their aromatic substituents, 5 and 6 didn't form hydrophobic forces with His1057, thus, they were less active than the aliphatic substituted derivatives 1–4 (Table 2).
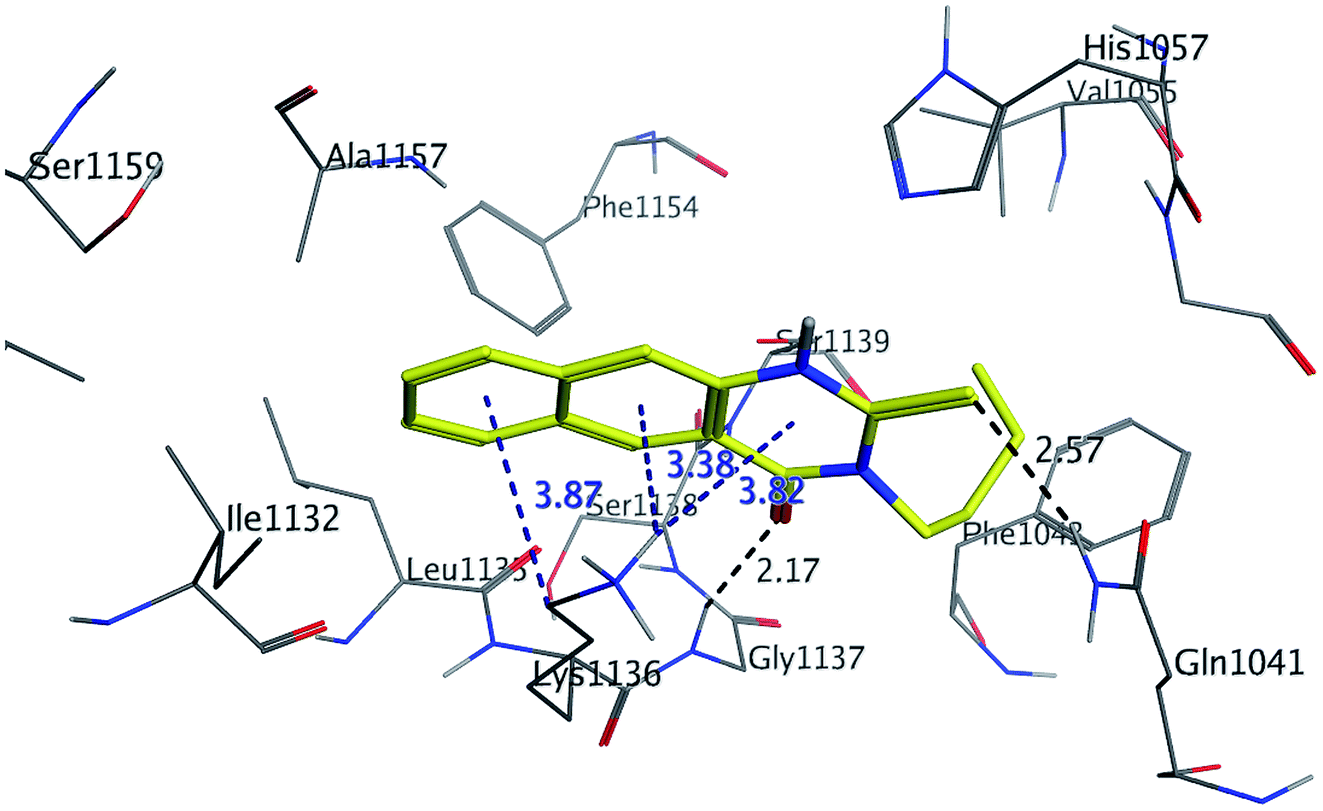
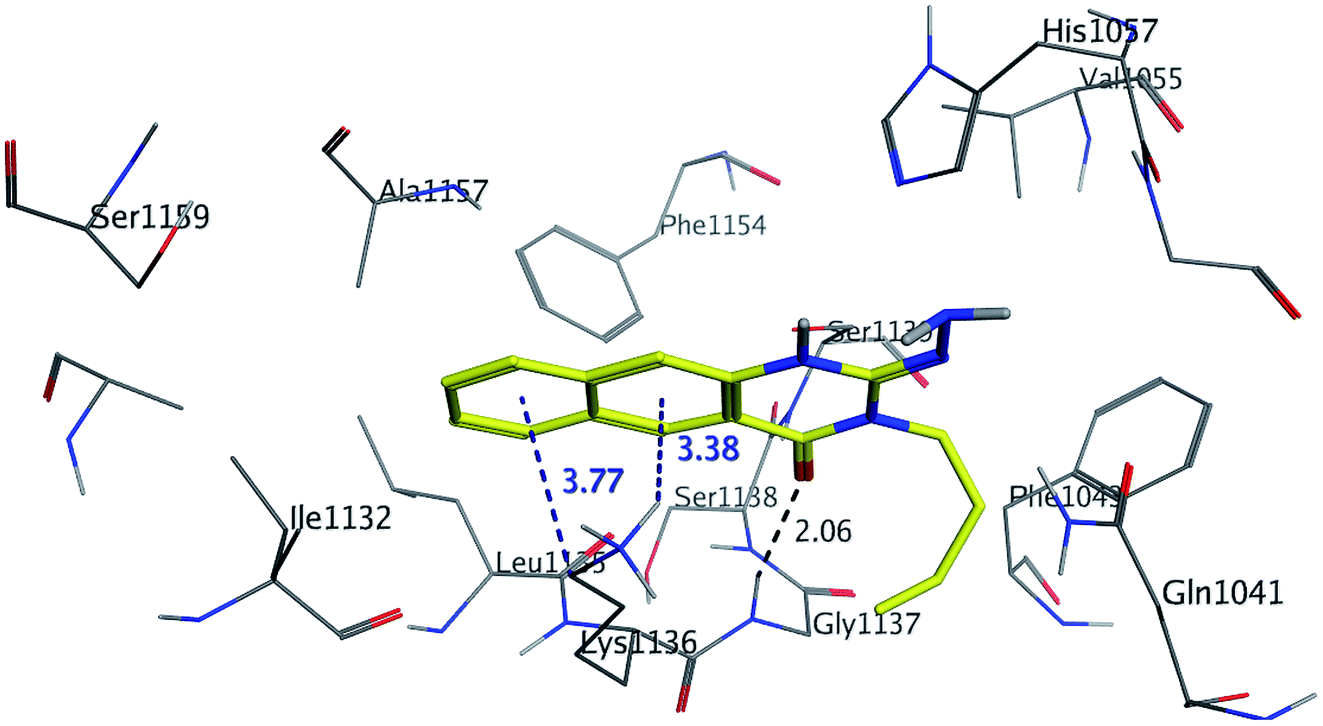
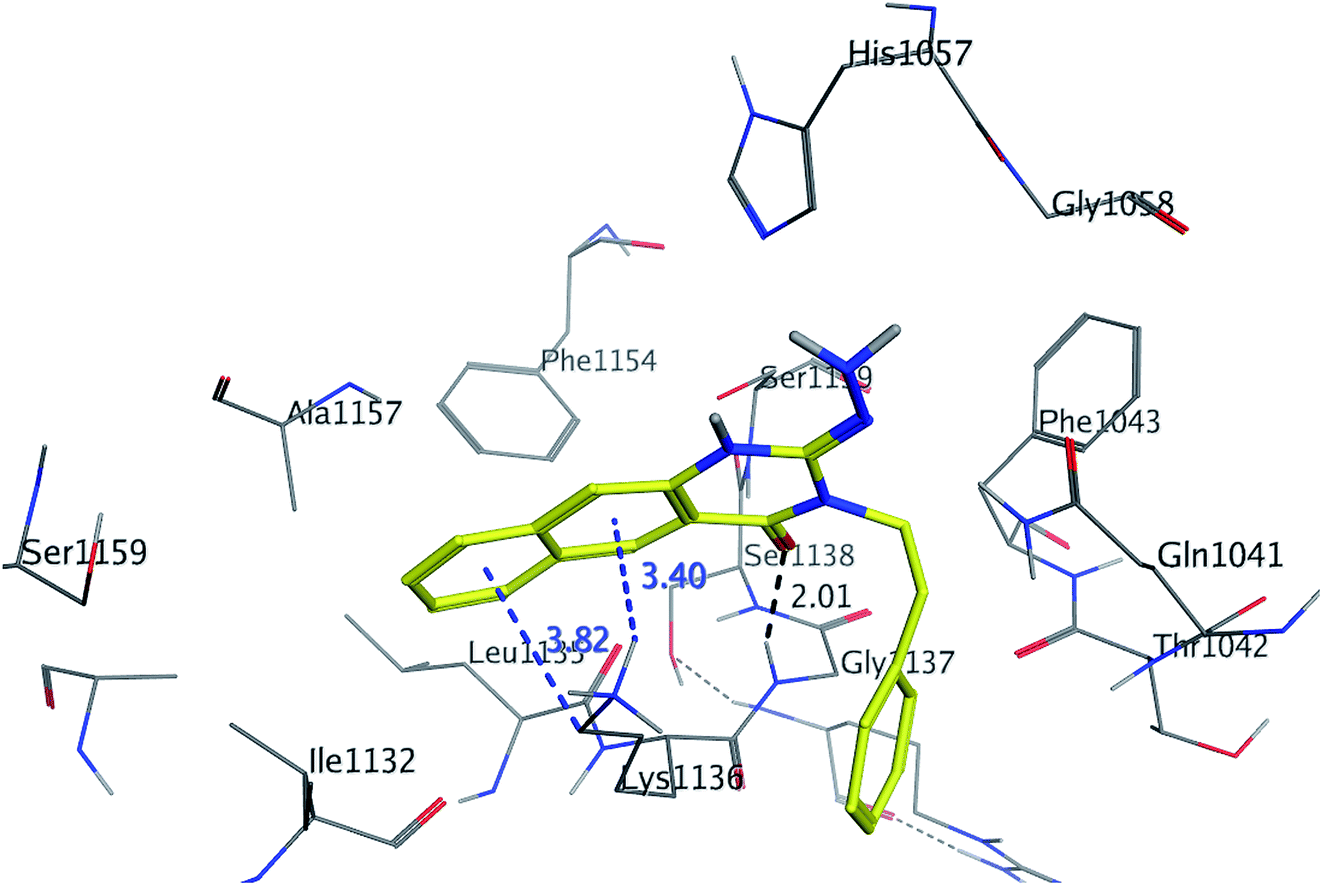
The binding mode of benzo[g]quinazolines 7–10 within the HCV NS3/4A enzyme shows that the quinazoline moiety was oriented closer to the catalytic site and stacked against Gly1137 (distance < 5 Å), hydrogen bond in the oxyanion hole, (Table 3, Fig. 7) and Lys1136 (cation binding site). All these compounds showed good binding affinity, in particular compounds 8–10, which was in good agreement with the biological results. Compound 13 formed hydrogen bond with Gly1137 (distance < 3 Å) and two π-H bonds with Lys1136 (distance < 5 Å), which gave good binding affinity (Table 3, Fig. 8). Therefore, such bonds play a fundamental role in the enzyme activity and are strictly related to the biological inhibition process of the target. In the other hand, benzo[g]quinazolines 11 and 12 made weak hydrogen bonds. The conformation predicted that compounds 14–17 were oriented perpendicular to the plane of quinazoline and was closer to the catalytic triad residual as His1057. Therefore, compounds 16 and 17 showed good inhibitory activity (Table 2) and this result was in accordance with the molecular docking outputs.
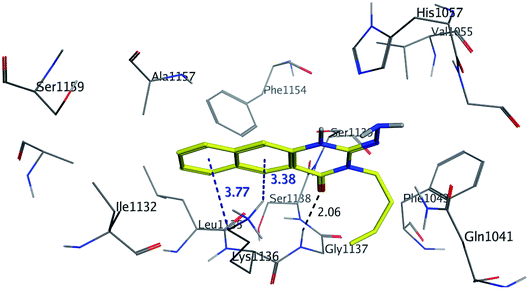 |
| | Fig. 7 3D Interaction of compound 9 with active site of HCV NS3/4A enzyme. Compound 9 shows four hydrogen bonds with Gly1137 and two π-bonds with Lys1136 of the target protein active site residue. | |
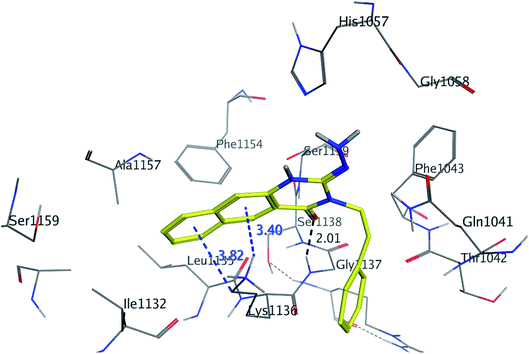 |
| | Fig. 8 3D interaction of compound 13 with active site of HCV NS3/4A enzyme. Compound 13 shows four hydrogen bonds with Gly1137 and two π-bonds with Lys1136 of the target protein active site residue. | |
As shown in Table 3, compounds 3, 8, 9, 13 and 16 have lower binding energy than telaprevir and formed two hydrogen bonds or more with active site of HCV NS3/4A protease enzyme. While other targets appeared to have good binding affinity with active site of HCV NS3/4A, but lower than 3, 8, 9, 13 and 16.
As illustrated in the Fig. 5–8, formation the hydrogen bonds with Gly1137 and Gln1041 in the S1′ subsites; two π-bonds with Lys1136 in the S1 subsites by the target benzo[g]quinazolines. Moreover, hydrophobic binding contacts in the hydrophobic pocket with His1057 in S2 subsites; Val1055; Phe1043; Ile1132; Lys1136; and Ala1157. This orientation in S1′ and S1 subsites at the active site of the HCV NS3/4A enzyme indicated that the target benzo[g]quinazolines behave as competitive inhibitors against HCV NS3/A4 protease.
QSAR analysis
By Hansch analysis, a multiple linear regression analysis was performed, using a set of 2D physicochemical descriptors to identify the most important physicochemical features of the targets 1–17 towards HCV NS3/4A serine protease. The following equation is solely based on molar refractory (MR) and was derived (using the contingency analysis tool in MOE) to identify the most important descriptors (eqn (1)):| | |
pIC50 = 0.390 × MR + 1.235 where r2 = 0.83155; q(LOO)2 = 0.87462; RMSE = 0.60071
| (1) |
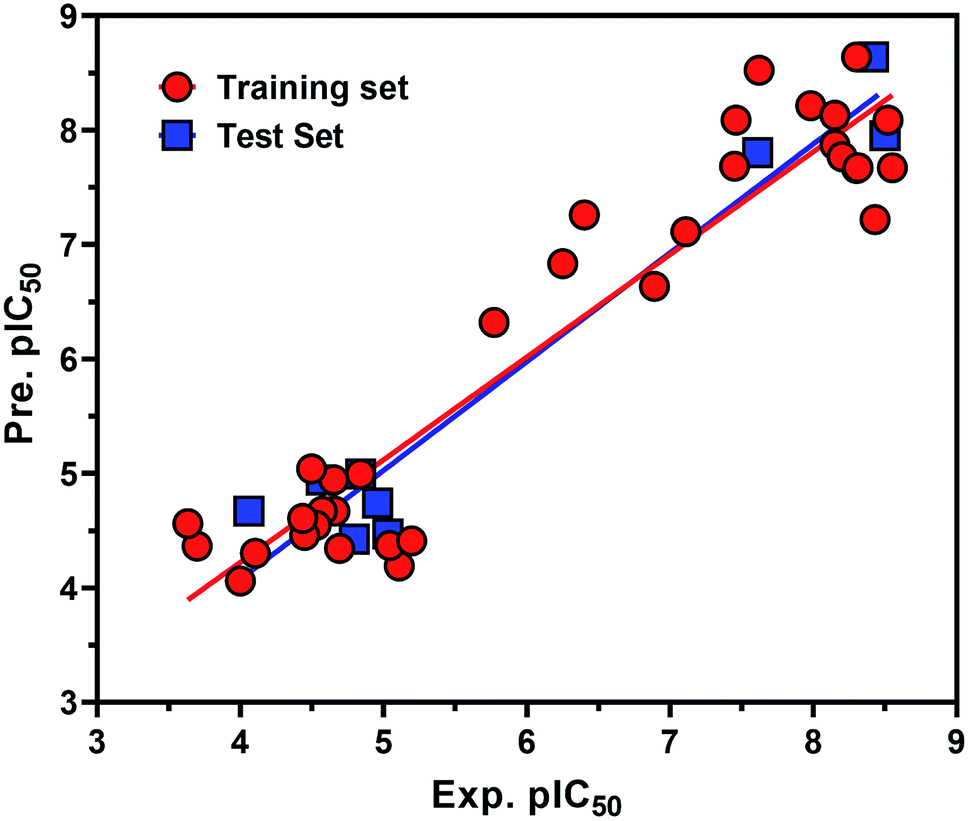
From the eqn (1), it is evident that MR is positively correlated with the inhibitory potency of training set against the HCV NS3/4A serine protease. The graph of the experimental and predicted inhibitory potency (pIC50) values of the training and test set compounds was provided in Fig. 9. No outliers were observed in the training data (square), and the prediction of all compounds were perfect with a value of residual of less than one log unit and q2 and r2 values of 0.87107 and 0.83155, respectively.
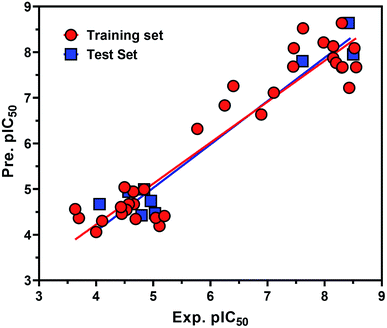 |
| | Fig. 9 Experimental versus predicted (LOO) inhibitory potency pIC50 values of the HCV NS3/4A serine protease inhibitors. The data points in red circle and blue square represent training and test set compounds, respectively. | |
Also the external test set was utilized to additional confirm the final model, and the prediction of all test set compounds (circle) was studied with variance of lower than one log unit among the experimental and predicted inhibitory potency pIC50 for the HCV NS3/4A serine protease (Fig. 9).
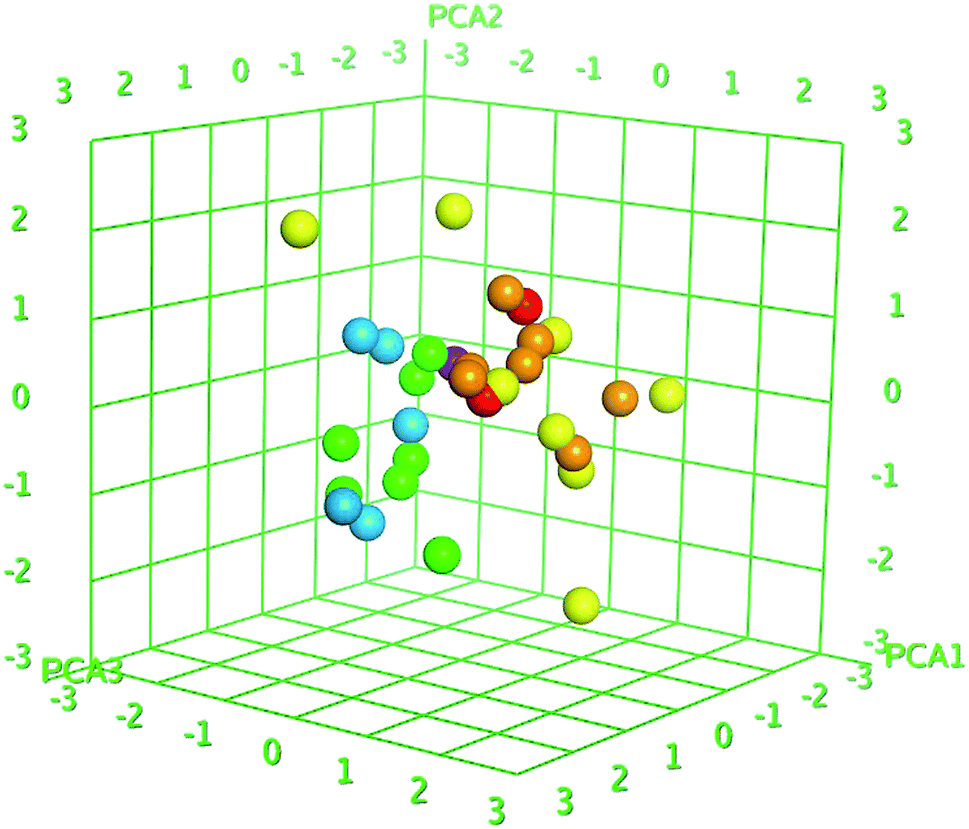
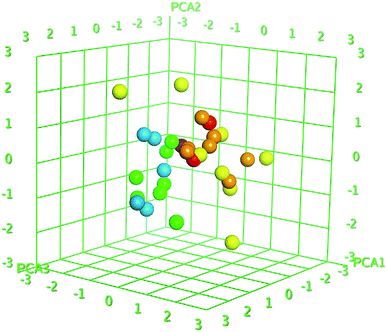
Z-score was predicted to training set including synthesized compounds 1–17 for the HCV NS3/4A serine protease. All the compounds have shown good Z-score value except compound 12. It has shown Z-score value beyond the limit (1.91275704) with 3.5. The compounds 13, 8, 17, and 11 have shown more significant Z-scores with 0.23, 0.14, 0.17 and 0.04, respectively for the HCV NS3/4A serine protease (Table S1†). Using the three eigenvectors, PCA1, PCA2 and PCA3, the principal component analysis was carried out and the 3D graphical plots were generated involved 98% of variance. In the plot, all the values were appeared to lie in the range of −3 to +3 and found with different colored spots and each color spot represents one compound (Fig. 10). The good predictive efficiency of the selected QSAR model was demonstrated far enough.
 |
| | Fig. 10 PCA plots of training set compounds for data obtained for the HCV NS3/4A serine protease. The 3D graphical plot was constructed with three eigenvectors PCA1, PCA2 and PCA3 in the range of +3 to −3 and coordinate value for each compound has represented with colored spots. | |
Conclusions
In conclusion, the findings of this study show that compounds 1, 2, 3, 9 and 13 exhibited the highest inhibitory activity towards HCV-NS3/4A enzyme. Their inhibition activities represented in terms of average IC50 values in the range 6.41 ± 0.12 and 11.02 ± 0.25 μM. In light of the molecular docking analysis, this result suggests that the aliphatic functionality group of the active compounds could impart better binding and make closer to the catalytic triad and form hydrophobic binding contacts with His1057 in addition to Val1055; Phe1043; Ile1132; Lys1136; and Ala1157 of the enzyme. Moreover, QSAR and molecular docking studies confirmed these observations. HCV-NS3/4A was still an essential target for anti-HCV agents design. Considering the current anti-HCV situation, it remains necessary to discover novel and potential NS3/4A inhibitors. Our findings provide a rationale for further exploring useful proposals to design and develop more active and potent HCV-NS3/4A inhibitory agents.
Materials and methods
The synthesized targets 1–17
All compounds were previously synthesized and fully characterized.17,19,21,23 The general procedures for preparation of compounds 7, 8, 14 and 15 were reported,17,19 however, their characterization data are reported herein for the first time as following:
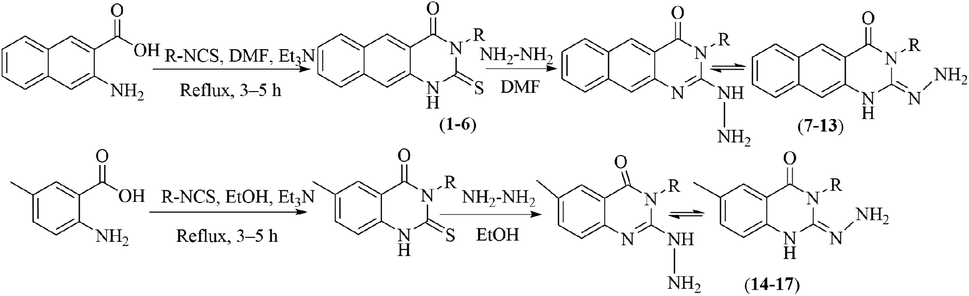
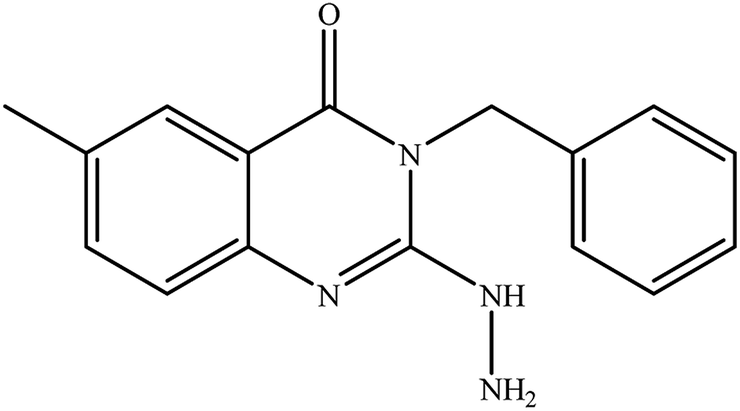
3-Methyl-2-hydrazinylbenzo[g]quinazolin-4(3H)-one (7). Yield 70%; mp 284–286 °C; 1H NMR (700 MHz, DMSO-d6): δ 10.04 (s, 1H, –NH), 8.84 (s, 1H), 8.60 (s, 1H), 8.21 (br d, J = 8.2 Hz, 1H), 8.00 (br d, J = 8.2 Hz, 1H), 7.90 (br t, J = 7.5 Hz, 1H), 7.68 (br t, J = 7.5 Hz, 1H), 6.23 (s, 2H), 3.6 (s, 3H); 13C NMR (175 MHz, DMSO-d6): δ 167.7, 157.8, 144.45, 135.6, 130.5, 130.1, 127.6, 127.3, 113.21, 29.28; ESI-MS (m/z): 239.10 [M−H]− (negative mode) for MW = 240.26.
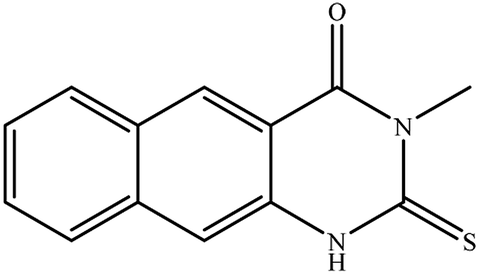
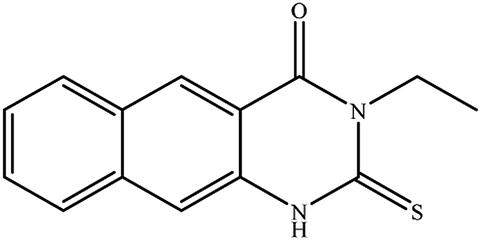
3-Ethyl-2-hydrazinyl-benzo[g]quinazolin-4(3H)-one (8). Yield 71%; mp 245–247 °C; 1H NMR (700 MHz, DMSO-d6): δ 9.60 (s, 1H, –NH), 8.68 (s, 1H), 8.43 (s, 1H), 8.26 (br s, 1H), 8.04 (br s, 1H), 7.79 (br t, J = 8 Hz, 1H), 7.68 (br s, 1H), 6.25 (s, 2H, –NH2), 4.08 (q, J = 7 Hz, 2H), 1.28 (t, J = 7 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 159.3, 148.3, 144.4, 136.8, 135.5, 131.1, 130.2, 130.0, 127.5, 127.2, 116.1, 113.1, 40.9, 13.7; ESI-MS (m/z): 253.2 [M−H]− (negative mode) for MW = 254.29.
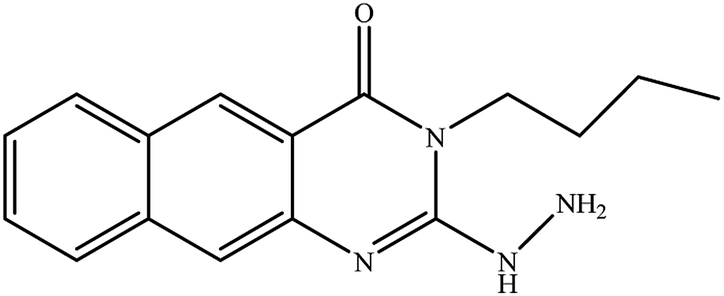
3-Methyl-2-hydrazinyl-6-methyl-3H-quinazolin-4-one (14). Yield: 55%; mp: 269–270 °C; 1H NMR (DMSO-d6): δ 7.74 (br s, 1H), 7.44 (br d, J = 7 Hz, 1H), 7.25 (d, J = 8 Hz, 1H), 3.5 (s, 3H, N–CH3), 2.35 (s, 3H, Ar–CH3); 13C NMR (175 MHz, DMSO-d6): δ 155.1, 146.8, 138.2, 136.7, 131.9, 128.3, 116.9, 115.3, 32.0 (N–CH3), 21.3 (Ar–CH3); ESI-MS (m/z): 203.1 [M−H]- (negative mode) for MW = 204.23.
3-Ethyl-2-hydrazinyl-6-methyl-3H-quinazolin-4-one (15). Yield: 58%; mp: 198–200 °C; 1H NMR (DMSO-d6): δ 7.69 (br s, 1H), 7.41 (br d, J = 7 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4,01 (q, J = 7.0 Hz, 2H), 2.33 (s, 3H, Ar–CH3), 1.14 (t, J = 7.0 Hz, 3H); 13C NMR (175 MHz, DMSO-d6): δ 157.2, 147.3, 136.8, 131.7, 130.5, 129.0, 116.1, 115.7, 41.4 (N–CH2CH3), 21.0 (Ar–CH3), 12.9 (N–CH2CH3); ESI-MS (m/z): 217.10 [M−H]− (negative mode) for MW = 218.26.
NS3/4A protease inhibition protocol
The SensoLyte® 490 HCV protease kit was optimized to detect the activity of hepatitis C virus NS3/4A protease using an EDANS/DABCYL FRET peptide substrate, with its fluorescence monitored at Ex/Em = 340 nm/490 nm upon proteolytic cleavage. NS3-containing cellular membrane fractions were prepared using a previously published protocol.24,25 The 96-well or 384-well microplate can provide better signal-to-noise ratio. The fluorescence microplate reader was capable of detecting emissions at 490 ± nm with excitation at 340 ± 30 nm. HCV NS3/4A protease can be produced using Escherichia coli. AnaSpec provided highly active recombinant HCV NS3/4A protease.
Molecular docking
Molecular operating environment (MOE) software was employed.26 To study the binding modes between the docked-selected compounds and amino acid residues at the active site of the NS3/4A protease, the 3SV622 (telaprevir) crystal structures of HCV NS3/4A serine protease were used. HCV NS3/4A protease was obtained from Protein Data Bank [PDB code 3SV6]. The detailed methodology was reported in accordance to the literature.22,26,27
QSAR analysis
From the literature,28,29 a series of active inhibitors against the HCV NS3/4A serine protease was extracted. The dataset is illustrated in Tables S1 and S2,† and contains 42 compounds which include of synthesized compounds 1–17, ethyl pyrrolidine-5,5-trans-lactams28 and benzofuran-3-carboxamide29 derivatives. For the current QSAR study, the described inhibitory potencies (pIC50) of the inhibitors range from 4 μM to 8.52. A various subset selection method30 was applied to divide the compounds into a training (80%) and test set (20%). Briefly, about 300 2D and 3D descriptors available in the MOE version 2015.0802 software31 were employed for the distance calculation of each database entry. The test set comprised 20% of the data structures (8 compounds) that had greater distance values from each other and the remaining 34 compounds (80%) were taken as the training set.
The QSAR 2D was carried out with the QuaSAR module in the MOE software for training set and test set and their detailed processes were reported in accordance to the literature.31–33
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through the research group No. RG-1435–068.
References
- L. Chan, O. Pereira, T. J. Reddy, S. K. Das and C. Poisson, et al., Bioorg. Med. Chem. Lett., 2004, 14, 797 CrossRef CAS.
- S. Colarusso, B. Gerlach, U. Koch, E. Muraglia and I. Conte, et al., Bioorg. Med. Chem. Lett., 2002, 12, 705 CrossRef CAS.
- L. Chan, S. K. Das, T. J. Reddy, C. Poisson and M. Proulx, et al., Bioorg. Med. Chem. Lett., 2004, 14, 793 CrossRef CAS.
- F. Fontaine, M. Pastor, I. Zamora and F. Sanz, J. Med. Chem., 2005, 48, 2687 CrossRef CAS.
- E. Nizi, U. Koch, J. M. Ontoria, A. Marchetti and F. Narjes, et al., Bioorg. Med. Chem. Lett., 2004, 14, 2151 CrossRef CAS.
- T. J. Reddy, L. Chan, N. Turcotte, M. Proulx and O. Z. Pereira, et al., Bioorg. Med. Chem. Lett., 2003, 13, 3341 CrossRef CAS.
- J. Li, X. Liu, S. Li and Y. Wan, et al., Int. J. Mol. Sci., 2013, 14, 22845 CrossRef.
- V. A. Vaillancourt, M. M. Cudahy, S. A. Staley, R. J. Brideau and S. J. Conrad, et al., Bioorg. Med. Chem. Lett., 2000, 10, 2079 CrossRef CAS.
- M. Hagel, D. Niu, T. Martin, M. P. Sheets, L. Qiao and B. Hugues, et al., Nat. Chem. Biol., 2011, 7, 22 CrossRef CAS.
- S. M. Saalau-Bethell, A. J. Woodhead, G. Chessari and M. G. Carr, et al., Nat. Chem. Biol., 2012, 8, 920 CrossRef CAS.
- N. K. Yilmaz, R. Swanstrom and C. A. Schiffer, Trends Microbiol., 2016 Jul, 24(7), 547–557 Search PubMed.
- P. Halfon and S. Locarnini, J. Hepatol., 2011 Jul, 55(1), 192–206 Search PubMed.
- K. P. Romano, A. Ali, W. E. Royer and C. A. Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20986 CrossRef CAS.
- J.-M. Pawlotsky, Hepatology, 2011, 53, 1742 CrossRef CAS.
- R. Al-Salahi, E. H. Anouar, M. Marzouk and H. A. Abuelizz, Bioorg. Med. Chem. Lett., 2019, 29, 1614 CrossRef CAS.
- R. Al-Salahi, E. M. Moustapha, H. A. Abuelizz, A. I. Alharthi and K. A. Alburikan, et al., Saudi Pharm. J., 2018, 26, 1120 CrossRef.
- R. Al-Salahi, R. Ahmad, E. H. Anouar, N. I. Nor Azman, M. Marzouk and H. A. Abuelizz, Future Med. Chem., 2018, 10, 1889 CrossRef CAS.
- R. Al-Salahi, H. A. Abuelizz, H. Ghabbour, R. Eldib and M. Marzouk, Chem. Cent. J., 2016, 10, 1 CrossRef.
- R. Al-Salahi, R. El Dib and M. Marzouk, Heterocycles, 2015, 91, 1735 CrossRef CAS.
- R. Al-salahi, H. A. Abuelizz, R. Eldib and M. Marzouk, Med. Chem., 2017, 13, 85–92 CrossRef CAS.
- H. A. Abuelizz, M. Marzouk, H. Ghabbour and R. Al-Salahi, Saudi Pharm. J., 2017, 25, 1047 CrossRef.
- K. P. Romano, A. Ali, C. Aydin, D. Soumana, A. Özen and L. M. Deveau, et al., PLoS Pathog., 2012, 8(7), e1002832 CrossRef CAS.
- A. I. Khodair, M. A. Elsafi and S. A. Al-Issa, J. Heterocycl. Chem., 2019, 56, 2358 CrossRef CAS.
- P. Örtqvist, J. Gising, A. E. Ehrenberg, A. Vema, A. Borg, A. Karlén and A. Sandström, Bioorg. Med. Chem., 2010, 18, 6512 CrossRef.
- A. Wadood, M. Riaz, R. Uddin and Z. Ul-Haq, PLoS One, 2014, 9, e89109 CrossRef.
- Molecular Operating Environment (MOE), 2015.10, Chemical Computing Group Inc., 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015 Search PubMed.
- P. J. Labute, Comput. Chem., 2008, 29, 1693 CrossRef CAS.
- M. J. Slater, D. M. Andrews, G. Baker, S. S. Bethell and S. Carey, et al., Bioorg. Med. Chem. Lett., 2002, 12, 3359 CrossRef CAS.
- X. Dai, H. Liu, A. Palani, S. He, R. Nargund, D. Xiao, and N. Zorn, et al., US Pat. 9364482B2, 2016.
- M. Schmuker, A. Givehchi and G. Schneider, Mol. Diversity, 2004, 8, 421 CrossRef CAS.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7, 146 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS.
- A. Elisseeff and M. Pontil, ATO Sci. Ser., Ser. III, 2003, 190, 111 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05604a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mohamed Marzoukb,
Ahmed H. Bakheitac and
Rashad Al-Salahi
a,
Mohamed Marzoukb,
Ahmed H. Bakheitac and
Rashad Al-Salahi