
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystal structure, spectroscopic measurement, optical properties, thermal studies and biological activities of a new hybrid material containing iodide anions of bismuth(III)†
Saida Ben Alia,
Amal Feki b,
Valeria Ferrettic,
Moncef Nasrib and
Mohamed Belhouchet*a
b,
Valeria Ferrettic,
Moncef Nasrib and
Mohamed Belhouchet*a
aLaboratory Physico-Chemistry of the Solid State, University of Sfax, Department of Chemistry, Faculty of Sciences of Sfax, B. P. 1171, Sfax, 3000, Tunisia. E-mail: belhouchet2002@yahoo.fr
bLaboratory of Enzyme Engineering and Microbiology, University of Sfax, National School of Engineering of Sfax (ENIS), B.P. 1173-3038 Sfax, Tunisia
cDepartment of Chemical and Pharmaceutical Sciences, Centre for Structural Diffractometry, University of Ferrara, Via L. Borsari 46, I-44121 Ferrara, Italy
First published on 24th September 2020
Abstract
As part of our interest in halogenobismuthate(III) organic–inorganic hybrid materials, a novel compound named bis(4,4′-diammoniumdiphenylsulfone) hexadecaiodotetrabismuthate(III) tetrahydrate with the chemical formula (C12H14N2O2S)2[Bi4I16]·4H2O, abbreviated as (H2DDS)[Bi4I16], has been prepared by a slow evaporation method at room temperature. This compound was characterized by single crystal X-ray diffraction (SCXRD), spectroscopic measurements, thermal study and antimicrobial activity. The examination of the molecular arrangement shows that the crystal packing can be described as made of layers of organic [C12H14N2O2S]2+ entities and H2O molecules, between which tetranuclear [Bi4I16]4− units, isolated from each other, are inserted. The cohesion among the different molecules is assured by N–H⋯I, N–H⋯O and O–H⋯I hydrogen bonding interactions, forming a three-dimensional network. Room temperature IR, Raman spectroscopy of the title compound were recorded and analyzed. The optical properties were also investigated by both UV-vis and photoluminescence spectroscopy. Moreover, the synthesized compound was also screened for in vitro antimicrobial (Gram-positive and Gram-negative) and antioxidant activities (scavenging effect on DPPH free radicals, reducing power and total antioxidant capacity).
1. Introduction
Over the last few years, the chemistry of hybrid compounds has drawn great interest thanks to the possibility of combining the different properties of both inorganic and organic components into one material. Indeed, hybrid compounds are very promising not only for their wide range of technologically advantageous properties and structural flexibility, but also for their interesting applications in optics and acoustics, in particular nonlinear polar and nonlinear optical materials,1–9 as well as for their antimicrobial and antioxidant activities.10 Among them, the halometallates have been extensively investigated owing to their very interesting physical behavior and propensity to form a variety of crystalline structures by self-assembling from suitable solutions at ambient temperature. Hybrids based on trivalent metals halides are known to adopt about 30 variants of the anionic structure types in which organic cations, e.g. amines, of different symmetry, volume and size can be incorporated to give a large diversity of structures.11–20 In particular, Bi(III) ion exhibits a wide range of coordination modes and associated geometries; bismuth halide octahedra can be connected in different ways by face, edge or corner sharing, forming accordingly naturally isolated (0-D) inorganic polyhedra,21–23 extended infinite chains (1-D)24–27 or two dimensional (2-D) networks.28 The choice of organic molecules carrying appropriate functional groups can assure a tight connection with the anionic sublattice via hydrogen bonds, besides electrostatic and van der Waals interactions.29,30In order to combine both the properties of iodobismuthate and the benefits of organic molecules, we chose as counterion the dapsone {4,4′-diammoniumdiphenylsulfone = (DDS)} molecule of the sulfone family, in view of its antibacterial and antibiotic properties. The dapsone (DDS) is used in the treatment of mycobacterium and malaria infection and pharmaceutical dosage formulation.31 In this paper we present a newly synthesized organic–inorganic compound (H2DDS)2[Bi4I16]. It is worth noting that until now, a few works on halobismuthates compounds based on sulfone family have been synthesized and structurally characterized such as: 4,4-diammoniumdiphenylsulfone iodobusmuthate.32 The new compound has been fully characterized through single-crystal X-ray diffraction analysis, and investigating its thermal behavior (via TG-DSC) as well as its vibrational (Infrared and Raman), and optical (photoluminescence and absorption) properties. In addition, the title compound has been tested for its antimicrobial inhibiting potential and antioxidant activity.
2. Experimental
2.1. Synthesis
The title compound was prepared by the reaction of bismuth(III) nitrate pentahydrate (BiNO3·5H2O, 0.485 g, 1 mmol) and 4,4′-diaminodiphenylsulfone (C12H12N2O2S, 0.248 g, 1 mmol) (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in an equal volume of water and ethanol (20 ml) mixed with 2 ml of HI (42%). The resulting aqueous solution was stirred magnetically for 20 min and allowed to stand at room temperature. After few weeks, red prismatic crystals were obtained by slow evaporation with a high yield of 85.5%. Later, the crystals were separated by filtration and dried in air. A single crystal was selected with a polarizing microscope for X-ray diffraction analysis.
:1) in an equal volume of water and ethanol (20 ml) mixed with 2 ml of HI (42%). The resulting aqueous solution was stirred magnetically for 20 min and allowed to stand at room temperature. After few weeks, red prismatic crystals were obtained by slow evaporation with a high yield of 85.5%. Later, the crystals were separated by filtration and dried in air. A single crystal was selected with a polarizing microscope for X-ray diffraction analysis.
2.2. X-ray data collection
A suitable crystal of (H2DDS)[Bi4I16] with dimensions of 0.30, 0.29, 0.09 mm was selected for the X-ray structure analysis. Single-crystal diffraction data were collected at 100 K on a Bruker Smart Apex CCD area detector diffractometer using MoKα radiation (λ = 0.71073 Å). Intensities were corrected for Lorentz, polarization and absorption effects. The structure was solved by direct methods with the SHELXS-97 program33 and refined on F2 by full-matrix least-squares using SHELXL97 (ref. 33) with all non-H atoms anisotropic. The hydrogen atoms of the C–H, N–H (NH3 groups) were included on calculated positions, riding on their carrier atoms, using the appropriate instructions of the SHELXL-97 program. All other hydrogen atoms were located in the difference-Fourier map and refined isotropically. All calculations were performed using the WINGX crystallographic suite of programs.34 The crystallographic parameters and some details of the structural refinement are given in Table 1. The drawings were made by using ORTEPIII,35 DIAMOND36 and Mercury37 programs.| Empirical formula | (C12H14N2O2S)2[Bi4I16]·4H2O |
| Formula weight | 3439.01 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.71073 |
| Crystal system, space group | Monoclinic, P21/n |
| Unit cell dimensions | |
| a (Å) | 12.0453 (14) |
| b (Å) | 20.437 (2) |
| c (Å) | 13.1306 (16) |
| β (°) | 96.880 (2) |
| V (Å3) | V = 3209.1 (7) |
| Z | 2 |
| Density (calculated) (g cm−3) | 3.559 |
| Reflections collected | 48812 |
| Independent reflections | 8017 |
| Reflections observed with I > 2σ(I) | 7328 |
| Number of refined parameters | 274 |
| Goodness-of-fit on F2 | 1.10 |
| Final R indices [I > 2σ(I)] | R1 = 0.051and wR2 = 0.128 |
| CCDC no. | 1909744 |
Crystallographic data for the structural analysis have been deposited at the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge, CB2 1EZ, UK, and are available free of charge from the Director on request quoting the deposition number 1909744.†
2.3. Physical measurements
The infrared absorption spectrum was collected at room temperature on a JASCOFT-IR-420 spectrometer using a crystal sample in the range 400–4000 cm−1.The Raman spectrum was registered between 4000 and 50 cm−1 at room temperature employing a Horiba Jobin-Yvon Lab-RAM HR 800 Dual Spectrophotometer. The excitation line was measured at 630 nm.
The optical absorption spectrum of the films was recorded at room temperature using a UV-vis absorption spectrophotometer (Hitachi, U-3300). Solid photoluminescence spectrum was recorded at room temperature on a Perkin-Elmer LS55 fluorescence spectrometer processed with a 350 nm radiation as the excitation source.
Simultaneous Thermogravimetry – Differential scanning calorimetry (TG-DSC) were carried out using “NETZSCH TASC 409” operating from 20 °C up to 600 °C temperature range with a 5 °C min−1 heating rate. The sample, in form of powder (∼11 mg), was put in an alumina crucible, and the measurements were performed in nitrogen atmosphere.
2.4. In vitro biological activities
DPPH radical scavenging activity. The DPPH free radical-scavenging activity of (H2DDS)2[Bi4I16] compound was determined as described by Bersuder et al. (1998).38 Firstly, sample (500 μl) at different concentrations (0.2 to 1 mg ml−1) was mixed with 375 μl of ethanol (99.5%) and 125 μl of DPPH (0.02%) in ethanol (99.5%). After that, the mixtures were kept at room temperature in the dark for 1 h. The absorbance was measured at 517 nm using a UV-visible spectrophotometer (T70, UV/VIS spectrometer, PG Instruments Ltd., Beijing, China). DPPH radical-scavenging activity was calculated using the following equation:
where, Ac is the absorbance of the control tube; As is the absorbance of (H2DDS)2[Bi4I16] with the DPPH solution. BHA was used as positive control. The experiment was carried out in triplicate.

Reducing power assay. The ability of (H2DDS)2[Bi4I16] to reduce iron(III) was determined according to Yildirim et al. (2001).39 Briefly, 1 ml of each crystal sample at different concentrations (0.2 to 1 mg ml−1) was mixed with 1.25 ml of phosphate buffer (pH 6.6; 0.2 M) and 2.5 ml of potassium ferricyanide (1% (w/v)) solution. After incubation at 50 °C for 20 min, 2.5 ml of trichloroacetic acid (TCA, 10% (w/v)) was added and the reactions mixtures were centrifuged at 10
000×g for 10 min. From the upper layer, 2.5 ml was mixed with distilled water (2.5 ml) and ferric chloride (0.5 ml of 0.1% (w/v)) solution. The absorbance of the reaction mixtures was calculated at 700 nm. BHA was used as positive control. The experiment was carried out in triplicate.
Determination of total antioxidant capacity. Total antioxidant activity of crystal sample at different concentrations (0.2–2 mg ml−1) was determined according to the method of Prieto et al. (1999).40 Briefly, 0.3 ml of samples was mixed with 1.0 ml reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate and 4 mM ammonium molybdate). After that, the reaction mixture was incubated at 90 °C for 90 min. After cooling at room temperature, the absorbance of each solution was measured at 695 nm. BHA was used as a positive control. The antioxidant activity was expressed as ascorbic acid equivalents.
Microorganisms. Antibacterial activities of the crystal sample were tested against Gram+ and Gram− bacterial strains. Bacteria used were Bacillus cereus (ATCC 11778), Staphylococcus aureus (ATCC 25923), Salmonella enterica (ATCC 43972), Micrococcus luteus (ATCC 4698), Escherichia coli (ATCC 25922), Listeria monocytogenes (ATCC 43251), Enterobacter aeruginosa (ATCC 27853) and Pseudomonas aeruginosa (ATCC 49189).
Agar-well diffusion method. The antibacterial potential was performed by agar-well diffusion method as described by Vanden Berghe & Vlieinck (1991).41 Culture suspensions (200 μl) adjusted to 106 colony-forming units (CFU ml−1) of the microorganisms of bacterial cells were spread on Luria–Bertani (LB) agar. After that, 50 μl of (H2DDS)2[Bi4I16] were loaded into wells (6 mm in diameter) punched in the agar layer. Then, the Petri dishes were kept for 1 h at 4 °C, and incubated for 24 h at 37 °C. Antimicrobial activity was evaluated by determining the zone of growth inhibition (diameter expressed in millimeters) around the wells. All tests were carried out in duplicate.
3. Results and discussion
3.1. Structure description
The ORTEPIII representation of the asymmetric unit, composed by one (C12H14N2O2S)2+ cation, half of a [Bi4I16]4− anion and two water molecules, is shown in Fig. 1. Bond lengths and angles of anionic and cationic moyeties are listed in Tables 2 and 3, respectively. The presence of the inversion center leads to the formation of [Bi4I16]4− anions, constituted by four edge-shared [Bi I6]3− octahedra, as shown in Fig. 2. The overall connectivity of the centrosymmetric tetranuclear anion can be described as a pair of edge-sharing bi-octahedra, which mutually share three cis edges. The analysis of the geometrical features (Table 2) shows that the Bi–I distances fall into two well-defined groups, the shortest distances, ranging from 2.8835 (8) to 2.9335 (8) Å, being found for terminal bonds, while the longest ones, ranging from 2.9950 (8) to 3.4025 (8) Å, involve the bridging iodines. These findings are perfectly in line with what observed in other structures of iodobismuthates(III).42–58 Besides, the I–Bi–I bond angles vary from 82.19 (2)° to 97.63 (2)° for cis and 162.19 (2)° to 174.94 (2)° for trans arrangements (see Table 2).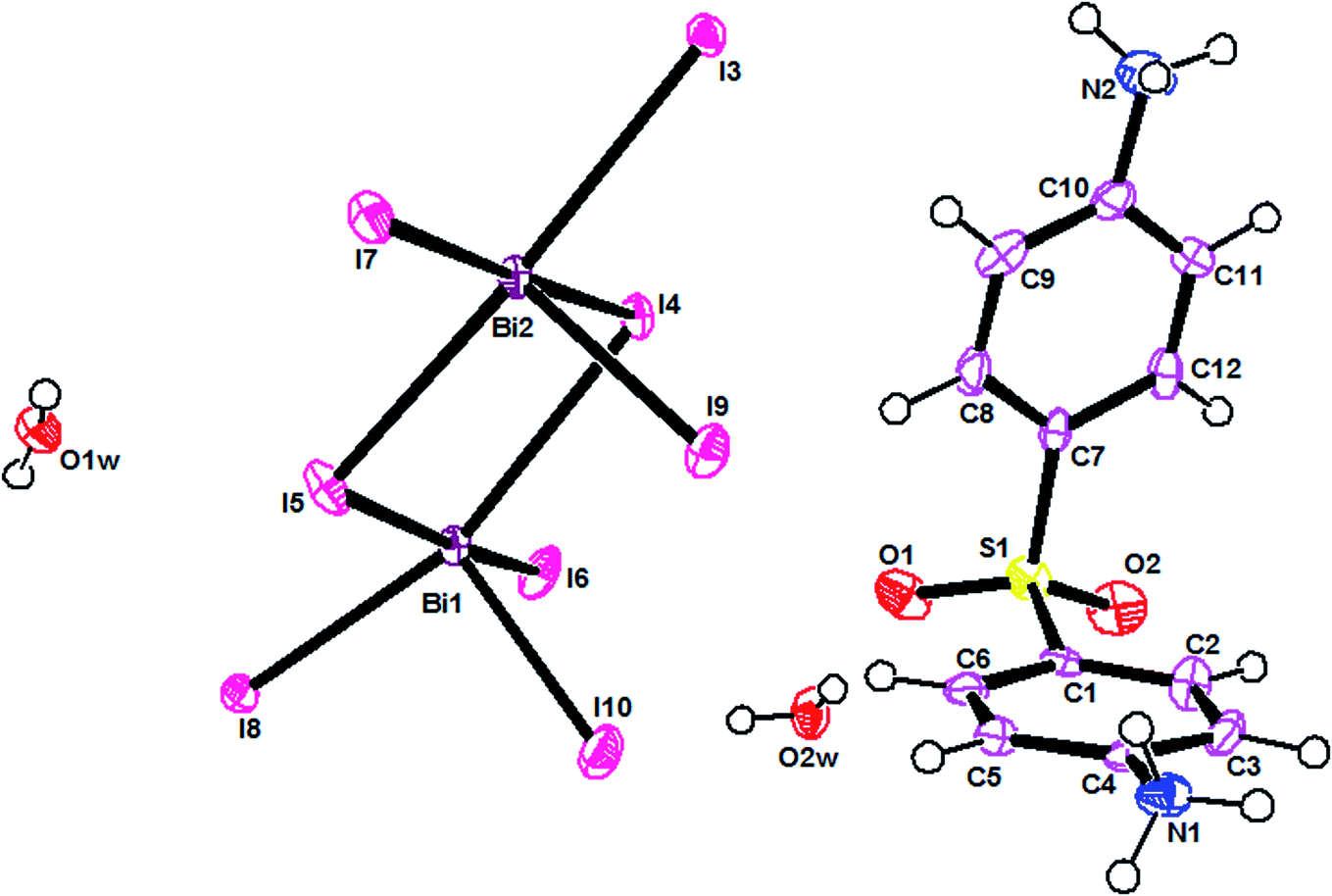 | ||
| Fig. 1 ORTEPIII view and atom numbering scheme of the asymmetric unit. | ||
| [Bi4I16]4− anion | |||
|---|---|---|---|
| a Symmetry codes: (i) −x, −y + 1, −z + 1. | |||
| Distances (Å) | |||
| Bi1–I8 | 2.9118 (8) | Bi1–I3i | 3.3750 (8) |
| Bi1–I10 | 2.9168 (9) | Bi2–I3 | 3.1793 (8) |
| Bi1–I6 | 2.9335 (8) | Bi2–I5 | 2.9950 (8) |
| Bi2–I7 | 2.9281 (8) | Bi1–I4 | 3.3062 (8) |
| Bi2–I9 | 2.8835 (8) | Bi2–I4 | 3.2869 (8) |
| Bi1–I5 | 3.2710 (8) | Bi2–I4i | 3.4025 (8) |
|
|||
| Angles (°) | |||
| I8–Bi1–I10 | 92.32 (3) | I4–Bi1–I3i | 83.079 (19) |
| I8–Bi1–I6 | 97.38 (3) | I9–Bi2–I7 | 94.04 (2) |
| I10–Bi1–I6 | 91.09 (2) | I9–Bi2–I5 | 93.90 (3) |
| I8–Bi1–I5 | 82.19 (2) | I7–Bi2–I5 | 89.30 (2) |
| I10–Bi1–I5 | 95.14 (3) | I9–Bi2–I3 | 90.76 (2) |
| I6–Bi1–I5 | 173.76 (2) | I7–Bi2–I3 | 92.34 (2) |
| I8–Bi1–I4 | 162.19 (2) | I5–Bi2–I3 | 174.94 (2) |
| I10–Bi1–I4 | 101.59 (2) | I9–Bi2–I4 | 91.84 (2) |
| I6–Bi1–I4 | 93.47 (2) | I7–Bi2–I4 | 174.12 (2) |
| I5–Bi1–I4 | 85.49 (2) | I5–Bi2–I4 | 90.46 (2) |
| I8–Bi1–I3i | 83.99 (2) | I3–Bi2–I4 | 87.42 (2) |
| I10–Bi1–I3i | 173.62 (3) | I9–Bi2–I4i | 174.37 (2) |
| I6–Bi1–I3i | 84.24 (2) | I7–Bi2–I4i | 89.28 (2) |
| I5–Bi1–I3i | 89.52 (2) | I5–Bi2–I4i | 90.68 (2) |
| Bi2–I4–Bi2i | 95.155 (19) | I3–Bi2–I4i | 84.56 (2) |
| Bi1–I4–Bi2i | 94.696 (19) | I4–Bi2–I4i | 84.845 (19) |
| Bi2–I5–Bi1 | 94.85 (2) | Bi2–I3–Bi1i | 97.63 (2) |
| Bi2–I4–Bi1 | 88.927 (19) | ||
| Organic cation | |||
|---|---|---|---|
| Distances (Å) | |||
| S1–O1 | 1.449 (9) | C3–C4 | 1.382 (14) |
| S1–O2 | 1.451 (9) | C4–C5 | 1.376 (14) |
| S1–C7 | 1.744 (11) | C5–C6 | 1.368 (14) |
| S1–C1 | 1.767 (10) | C7–C8 | 1.406 (14) |
| N1–C4 | 1.469 (13) | C7–C12 | 1.408 (14) |
| C1–C6 | 1.382 (14) | C8–C9 | 1.386 (17) |
| C1–C2 | 1.410 (14) | C9–C10 | 1.376 (16) |
| C2–C3 | 1.386 (15) | C10–C11 | 1.381 (16) |
| N2–C10 | 1.462 (15) | C11–C12 | 1.385 (16) |
|
|||
| Angles (°) | |||
| O1–S1–O2 | 119.5 (5) | C3–C4–N1 | 118.6 (9) |
| O1–S1–C7 | 110.2 (5) | C6–C5–C4 | 119.4 (10) |
| O2–S1–C7 | 108.4 (5) | C5–C6–C1 | 118.8 (10) |
| O1–S1–C1 | 108.1 (5) | C8–C7–C12 | 120.0 (10) |
| O2–S1–C1 | 109.0 (5) | C8–C7–S1 | 119.9 (8) |
| C7–S1–C1 | 99.9 (5) | C12–C7–S1 | 119.8 (8) |
| C6–C1–C2 | 122.2 (9) | C9–C8–C7 | 120.2 (10) |
| C6–C1–S1 | 119.5 (8) | C10–C9–C8 | 118.8 (10) |
| C2–C1–S1 | 117.7 (8) | C9–C10–C11 | 122.2 (11) |
| C3–C2–C1 | 118.0 (9) | C11–C10–N2 | 119.9 (10) |
| C4–C3–C2 | 118.6 (10) | C10–C11–C12 | 119.9 (10) |
| C5–C4–C3 | 122.9 (9) | C9–C10–N2 | 117.8 (10) |
| C5–C4–N1 | 118.4 (9) | C11–C12–C7 | 118.9 (10) |
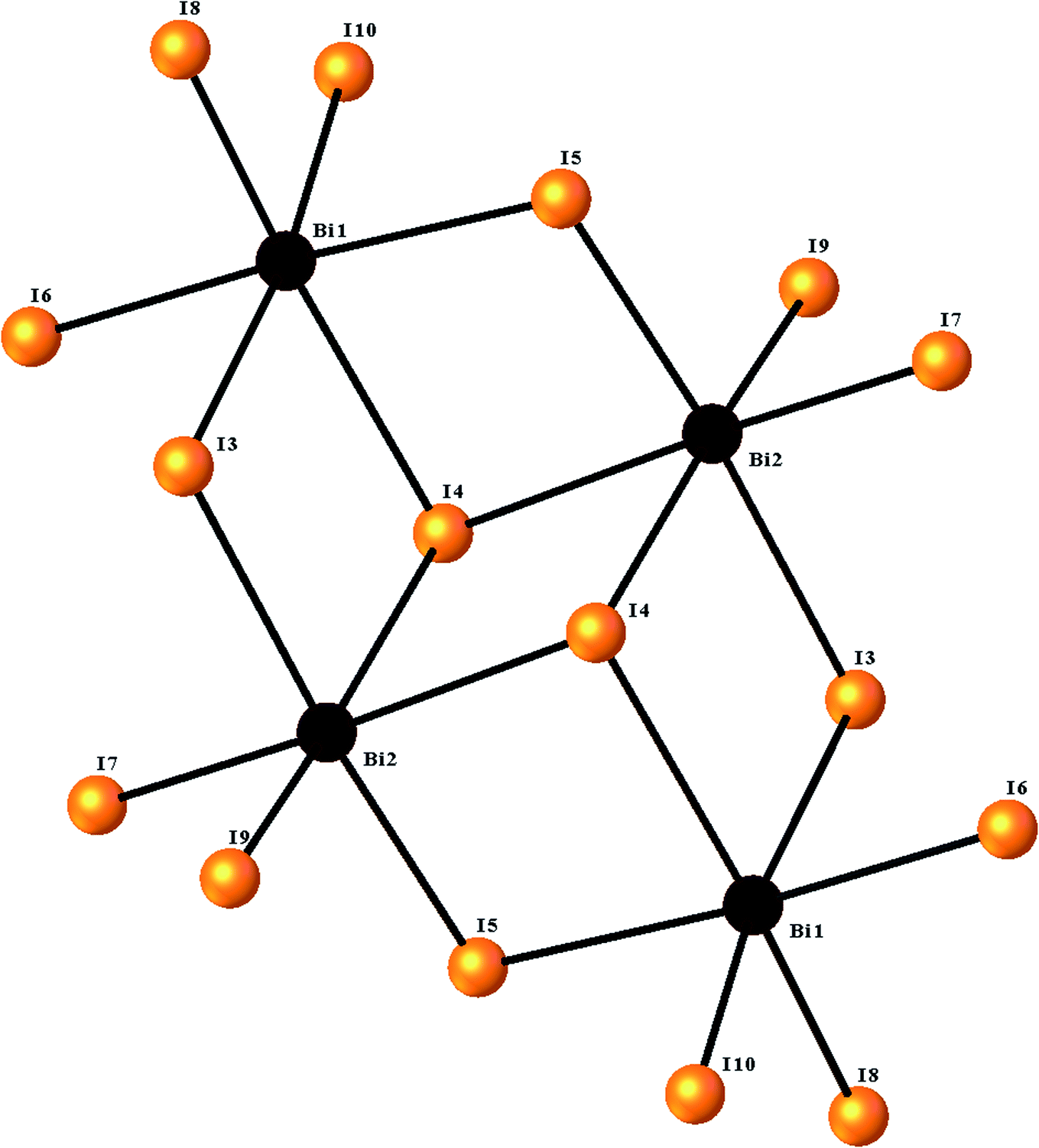 | ||
| Fig. 2 The structure of the [Bi4I16]4− tetramer, showing the octahedral environment around the two bismuth centers. | ||
The main geometrical characteristics of the organic cations are listed in Table 3. In the dapsone cation, the two phenyl rings are almost perpendicular to each other, the C1–S1–C7 angle measuring 99.9(5)°. Both the aminic groups are protonated, leading to a lenghtening of the C–N bond distance (Table 3). All the parameters agree well with what found in the literature.47,50,59,60
Overall, the packing diagram is made of alternating organic and inorganic layers expanding along the a direction (Fig. 3). Inorganic layers can be described as tetranuclear anions [Bi4I16]4− units isolated from each other, located at approximately y = 1/2. Organic layers consists of two organic chains which are hydrogen bonded together through water molecules. These layered organizations are located at around y = 0 and y = 1. In the different layers the cationic groups are linked with the anionic groups through N–H⋯I [N⋯I distances vary between 3.580 (9) Å and 3.662 (9) Å and N–H⋯I angles vary between 112° and 141°].
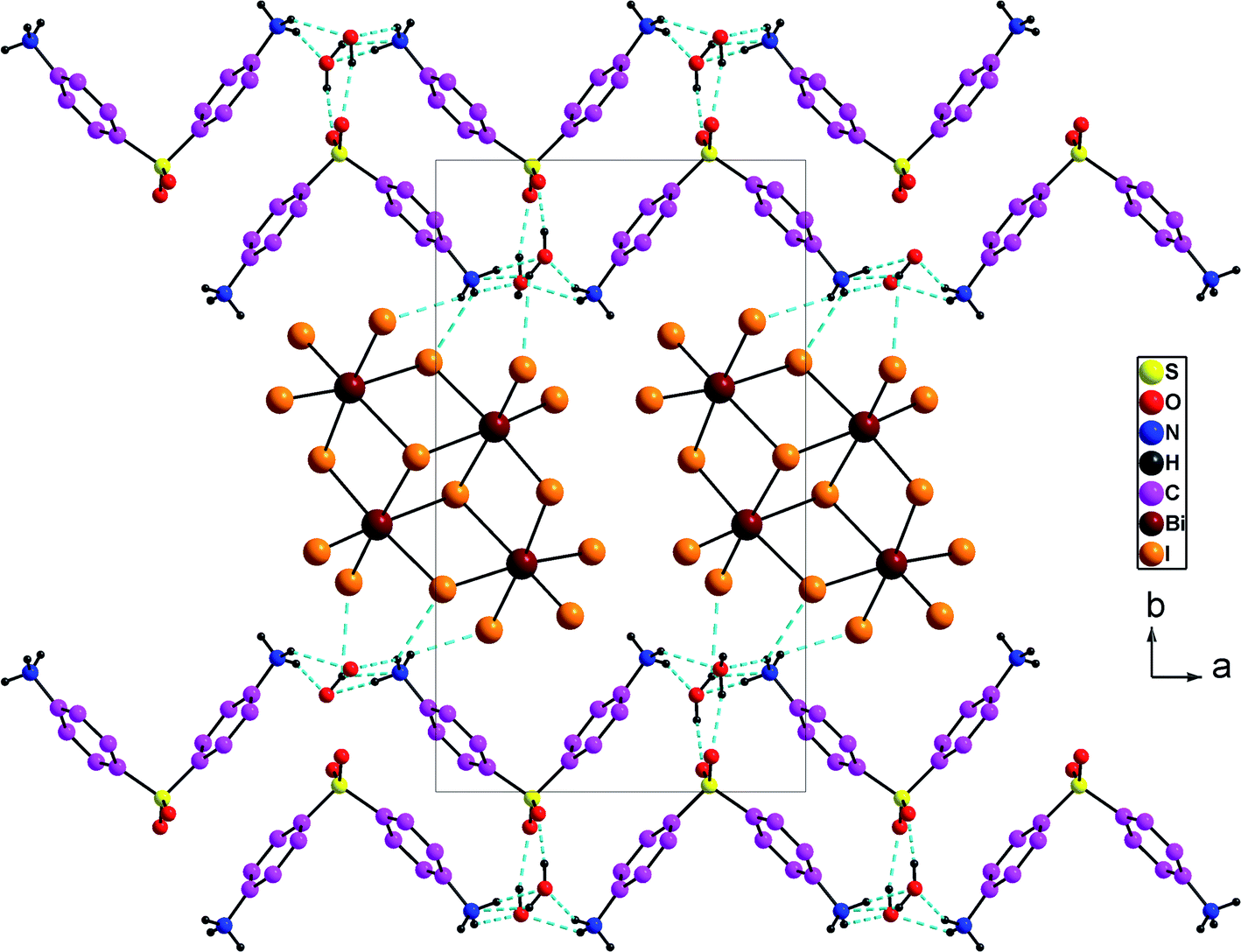 | ||
| Fig. 3 Projection along the c axis of the cell content of (H2DDS)[Bi4I16]. The dotted lines indicate hydrogen bonds. | ||
The oxygen provided from the crystallization water molecules linked with the organic cations through N–H⋯OW [N⋯OW = 2.766 (12) to 2.842 (13) Å; H⋯OW = 1.87–2.19 Å and N–H⋯OW = 132–167°] on the one hand, and through OW–H⋯O [OW⋯O = 2. 718 (11) to 2.845 (12) Å; H⋯O = 1.95 (7) to 2.04 (4) Å, OW–H⋯O = 156 (15) to 166 (16) °], on the other hand. In addition, the two water molecules are associated with the anionic entities via OW–H⋯I [OW⋯I = 3.630 (8) to 3.720 (8) Å; H⋯I = 2.97 (11) to 3.05 (10) Å and OW–H⋯I = 135 (14) to 142 (14)°] (Fig. 3). So, both of the oxygen atoms provided by the crystallization water molecules act as a bridge between the anionic and the cationic groups. They can play as both hydrogen-bond donors and acceptors forming a three-dimensional framework.
As well as, the organic species interact by means of week and strong hydrogen bonds and Vander Waals interactions which participate in the stabilization of the structure.61 The donor⋯acceptor distances, reported in Table 4, are typical of medium-strong charge-assisted hydrogen bonds.
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| a Symmetry codes: (ii) −x + 1, −y + 1, −z; (iii) x + 1/2, −y + 3/2, z − 1/2; (iv) −x + 1, −y + 1, −z + 1; (v) −x + 1/2, y + 1/2, −z + 1/2; (vi) −x, −y + 1, −z; (vii) −x + 1/2, y − 1/2, −z + 3/2; (viii) x, y, z + 1. | ||||
| N1–H1A⋯O2Wii | 0.91 | 2.19 | 2.842 (13) | 129 |
| N1–H1A⋯I3iii | 0.91 | 2.94 | 3.598 (9) | 131 |
| N1–H1B⋯O1Wiv | 0.91 | 1.87 | 2.766 (12) | 167 |
| N1–H1C⋯I6v | 0.91 | 2.91 | 3.662 (9) | 141 |
| N1–H1C⋯I7iii | 0.91 | 3.14 | 3.580 (9) | 112 |
| N2–H2B⋯O2Wvi | 0.91 | 2.15 | 2.842 (14) | 132 |
| O1W–H11⋯I7vii | 0.81 (2) | 3.05 (10) | 3.720 (8) | 142 (14) |
| O1W–H12⋯O2viii | 0.82 (2) | 1.95 (7) | 2.718 (11) | 156 (15) |
| O2W–H21⋯I6 | 0.82 (2) | 2.97 (11) | 3.630 (8) | 138 (14) |
| O2W–H22⋯O1 | 0.82 (2) | 2.04 (4) | 2.845 (12) | 166 (16) |
3.2. Vibrational studies
To gain more information on the crystal structure of the title compound, we have performed a vibrational study using infrared spectroscopy and Raman scattering. The IR and Raman spectra recorded at room temperature are shown in Fig. 4 and 5, respectively. The Raman spectrum of the (H2DDS)[Bi4I16] was observed in the low frequency range (50–200 cm−1). This is most probably due the broad and strong luminescence of the title compound, which makes the Raman signal hardly detectable in the higher 400–4000 cm−1 range. The assignments of the bands are reported in Table 5.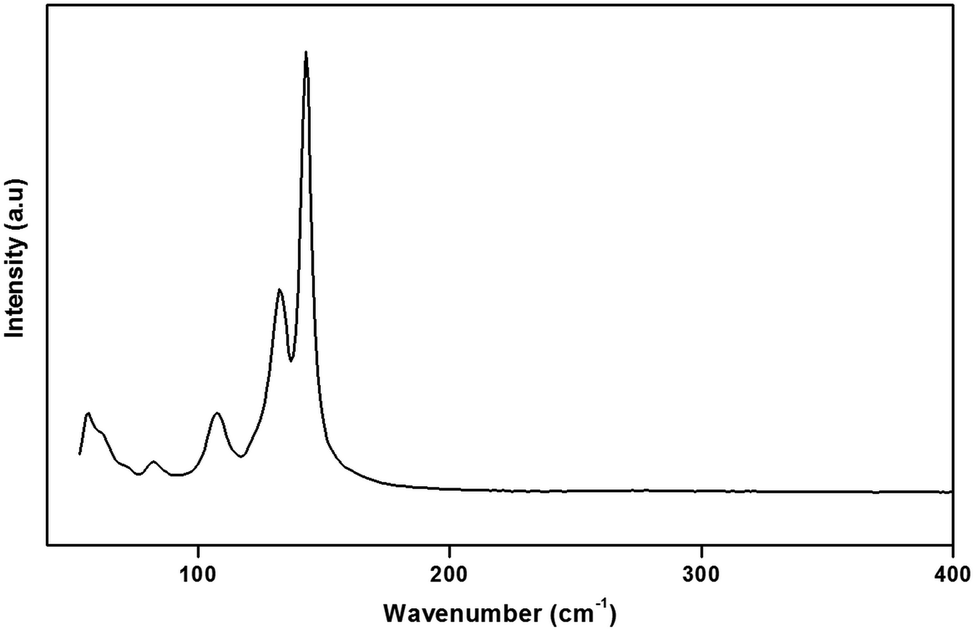 | ||
| Fig. 4 Raman spectra of (H2DDS)[Bi4I16] recorded in the low-frequency range. | ||
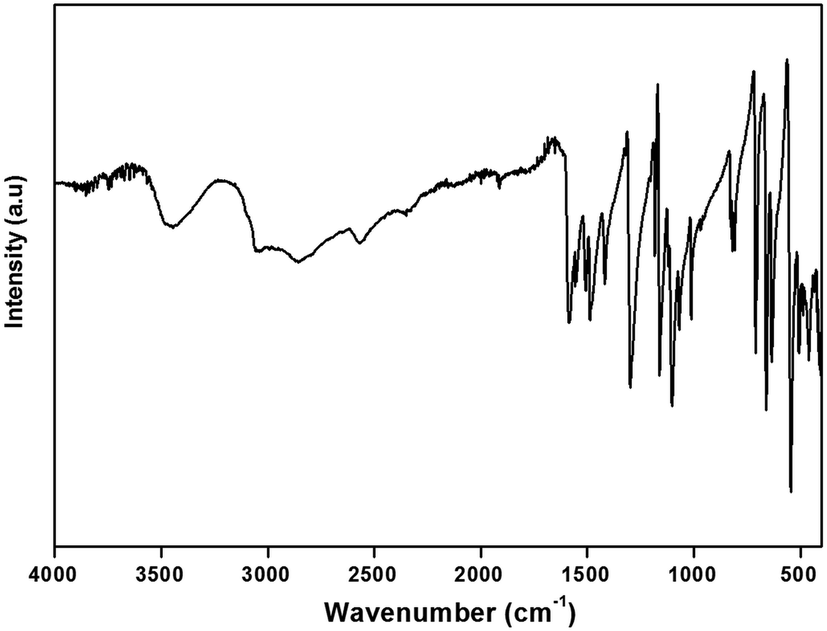 | ||
| Fig. 5 The room temperature IR of (H2DDS)[Bi4I16] compound. | ||
| IR | Raman | Assignment |
|---|---|---|
| a vs: very strong, s: strong, m: medium, vw: very weak, w: weak. | ||
| 3457 L | ν(O–H) | |
| 3048 L | νas(NH3) + νs(NH3) | |
| 2857 L | νas(C–H) | |
| 2569 w | νs(C–H) | |
| 2352 vw | ν(O–H⋯I) | |
| 1918 w | δ(H2O) | |
| 1586 s | δas(NH3) | |
| 1554 s | δs(NH3) | |
| 1508 s | ν(C–S) | |
| 1487 s | ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) C) |
|
| 1418 s | δ(C–C–H) | |
| 1301 vs | ρ(NH3) | |
| 1185 m | γ(C–H) | |
| 1162 vs | ν(SO) |
|
| 1106 vs | ν(C–C) | |
| 1067 s | ν(C–N) | |
| 1012 s | ν(CC) |
|
| 971 m | ν(C–S) | |
| 813 m | δ(SO) |
|
| 657 vs to 710 vs | ρ(H2O) | |
| 636 s | ω(H2O) | |
| 548 vs | δ(C–C–N) | |
| 506 m | δ(C–C–C) | |
| 490–462 | δ(C–S–C) | |
| 142 vs | νas(Bi–I) | |
| 132 s | νs(Bi–I) | |
| 107 m | δ(I–Bi–I) | |
| 82 w | Lattice modes | |
The assignment of the external modes of the inorganic anions, observed in the Raman spectrum, is based on the comparison with previous works reported on homologous compounds.62,63 The Bi–I external asymmetric stretching vibration is seen to give rise to the strongest Raman line at 142 cm−1. The observed band at 132 cm−1 is assigned to the Bi–I external symmetric stretching vibration. Raman line at 107 cm−1 most likely correspond to the δ(I–Bi–I). Frequencies for the lattice modes occur near 82 cm−1.64
The IR spectrum of this compound shows the characteristic signals of the 4,4′(DDS) cation and confirms its presence in the crystal. The assignment of internal modes of the organic cations is based on the comparison of the wavenumbers of others compound associated to the literature data.65–71 The spectrum shows at high wavenumbers very large bands ranging from 3457 cm−1 to 3048 cm−1 describes the stretching vibrations of νas(NH3) νs(NH3) and overlapping with the ν(O–H) modes of the water molecules. Concerning the broad bands observed in 2857–2569 cm−1 spectral region, they correspond to ν(C–H) stretching modes (νas(C–H) and νs(C–H)). The absorption centered at 2352 cm−1 is assignable to ν(O–H⋯I). The weak band observed at 1918 cm−1 may be assigned to the bending vibrations of the water molecules δ(H2O). The strong bands observed at 1586 and 1554 cm−1 are assigned to the asymmetric and symmetric bending vibrations of the NH3 group, respectively. Furthermore, the strong band located at 1508 cm−1 is attributed to the (C–S) stretching mode. The absorption band at 1487 cm−1 corresponds to the stretching vibration of the CC bond. The strong vibration at 1418 cm−1 is due to the deformation of (C–C–H). The very strong band observed at 1301 cm−1 is attributed to the NH3 rocking. The medium-intensity band detected at 1185 cm−1 can be assigned to γ(C–H). Besides, the bands for ν(SO), ν(C–C), ν(C–N), ν(CC) and ν(C–S) are situated at 1162, 1106, 1067, 1012 and 971 cm−1, respectively. The medium band observed at 813 cm−1 correspond to the δ(SO). The bands observed at 710 and 657 cm−1 are attributed to ρ(H2O), while the band at 636 cm−1 is attributed to ω(H2O). Finally, the low frequencies located between 636 and 462 cm−1 are attributed to the following deformations δ(C–C–N), δ(C–C–C) and δ(C–S–C).
3.3. Optical study
The UV-vis absorption and the photoluminescence spectra of the (H2DDS)[Bi4I16] compound, measured at room temperature, are illustrated in Fig. 6. It can be seen that the title compound exhibits two distinct absorption bands around 313 nm and 362 nm. In general, the absorption spectrum of halogenobismuthates(III) based compounds exhibits several bands around 300–400 nm.21,72–74 The first band at 313 nm (3.96 eV) corresponding to the higher energy band should can be assigned to the (LMTC) from the np orbital of I to 6p orbital of Bi(III) since the (LMCT) transitions are from iodine to Bi(III). While, the second absorption band at 362 nm (3.42 eV) can be attributed to the electronic transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). On the other hand, this material exhibits two luminescence bands which could be even observed with the naked eye at room temperature. The broad one with a maximum at 556 nm (2.23 eV) and the weak one with a maximum at 588 nm (2.10 eV), located in the red region, indicate that this compound is a potential candidate for red photoluminescence materials.75 These peaks are attributed to band to band and excitonic emissions within the iodobismuthate inorganic part, respectively. The luminescence originates from electronic transitions within the inorganic part [Bi4I16]4−. As commonly known in organic inorganic compounds, in the bismuth(III) iodide based hybrids, the lowest exciton state arises from excitations between the valence band, which consists of a mixture of Bi(6s) and I(5p) states, and the conduction band, which derives primarily from Bi(6p) states,73,76 and is confined zero-dimensionally in the tetrameric [Bi4I16]4−.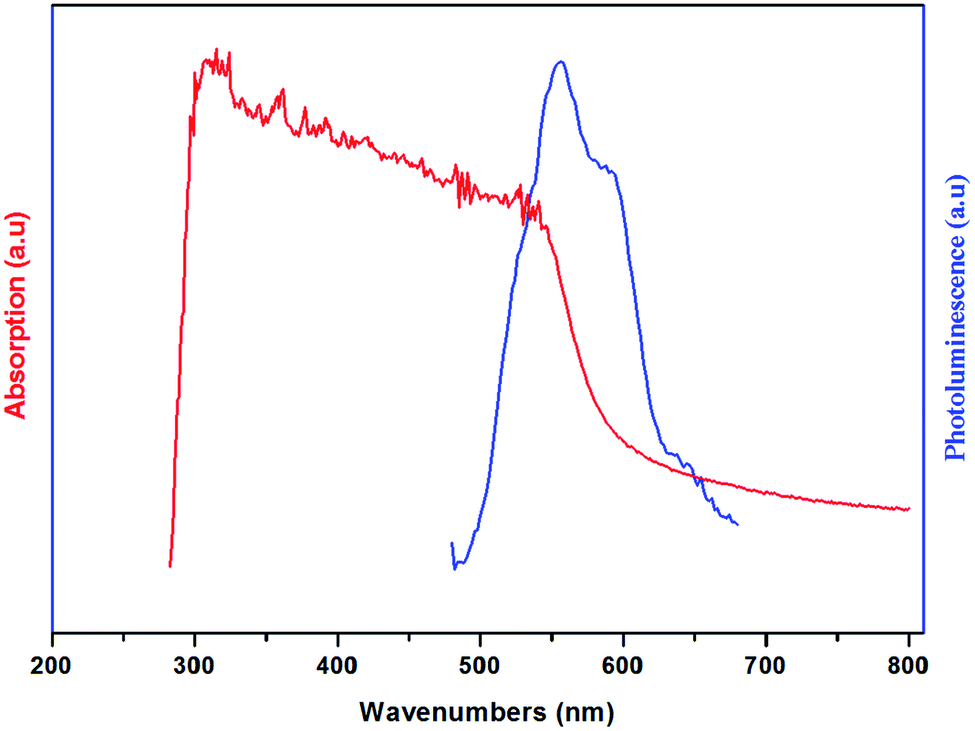 | ||
| Fig. 6 Room temperature absorption (Abs) ((red line) and photoluminescence (PL) (blue line) spectra of (H2DDS)[Bi4I16]. | ||
3.4. Thermal analysis
In order to study the thermal stability and the intervening changes in the (H2DDS)[Bi4I16] crystals, simultaneous TG-DSC analysis were realized with a heating rate of 5 °C min−1 between 20 and 600 °C. The thermal analysis results are reported in Fig. 7. Using precedent works reported in the literature on similar compounds.32,77–80 The DSC thermogram shows a dehydration peak at 125 °C, starting at 70 °C and ending at 150 °C, (observed weight loss, 2%; calculated weight loss, 2.09%), attributed to the elimination of the water molecules. After dehydratation, the DSC curve shows two endothermic peaks, the first peak at 233 °C corresponds to the organic cation degradation, the second one at 253 °C which more intense is attributed to the release of diiodine molecule (I2). This part of decomposition is accompanied by the second weight loss observed in the TG thermogram from the temperature 180 to 285 °C (observed weight loss, 21.3% in good agreement with the calculated value, 21.9%). The remaining peak between 285 °C and 450 °C accompagned by the important weight loss corresponds to the degradation of the remaining inorganic part giving rise to a very small black residue.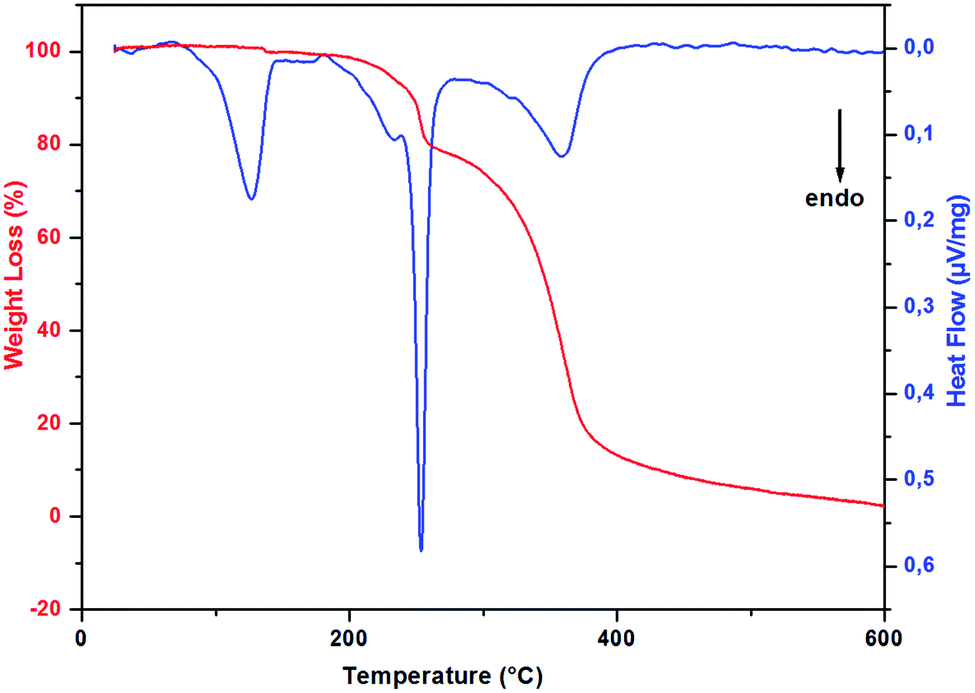 | ||
| Fig. 7 DSC-TGA curves of (H2DDS)[Bi4I16]. | ||
3.5. Evaluation of in-vitro biological activity
DPPH is a stable nitrogen-centered free radical that has been widely used as a quick, reliable compound for estimating the scavenging potential of antioxidants.83 The scavenging activity of (H2DDS)[Bi4I16]compound on DPPH radical was monitored in a dose dependent manner (Fig. 8). At final concentration (1 mg ml−1), the DPPH radical scavenging activity of (H2DDS)[Bi4I16]reached about 60%. Interestingly, (H2DDS)[Bi4I16] could be able to stop the radical chain reaction, retarding as a result the onset of lipid oxidation and providing a potential beneficial health resource. These results were in agreement with previous metallic complexes studies, where the ligand has an antioxidant potential and it is expected that the metal moiety will increase its activity.84
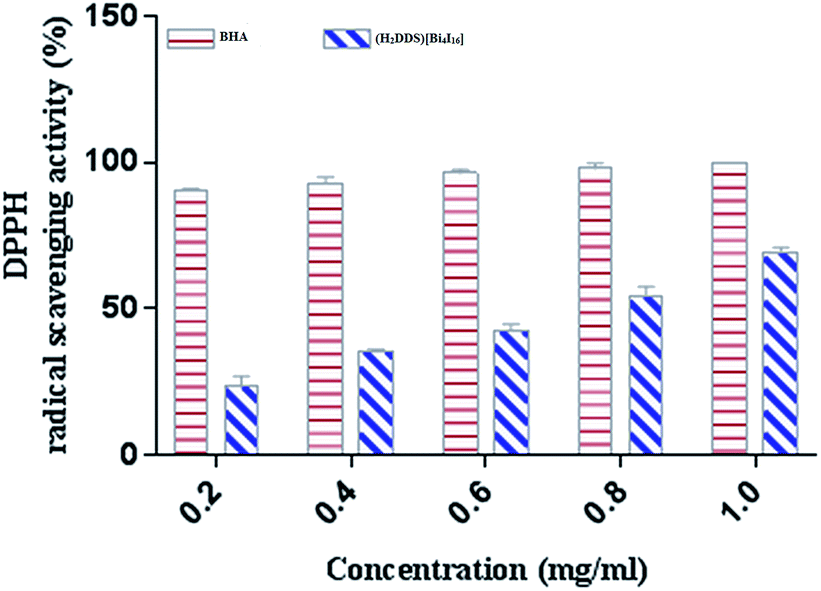 | ||
| Fig. 8 DPPH radical scavenging activity of (H2DDS)[Bi4I16] at different concentrations. | ||
The reducing power assay is based on the reduction of Fe3+/ferricyanide complex to the ferrous form, which serves as a significant indicator of a potent antioxidant activity. As shown in (Fig. 9), (H2DDS)[Bi4I16] reducing activity correlated well with increasing concentrations. The highest value reached OD700 = 3 at a dose of 2 mg ml−1, which is similar to BHA (Fig. 9). In fact, the (H2DDS)[Bi4I16] followed the same scavenging process as DPPH-radical assay, working as reductones by donating a hydrogen atom and breaking the free radical chain.85 The presence of reducers as antioxidants induces the reduction of the Fe3+/ferricyanide complex to the ferrous form. Hence, the Fe2+ complex can be detected by measuring the formation of Perl's Prussian blue at 700 nm. In this trend, some results indicated that there is a direct correlation between antioxidant activities and reducing power assay.86
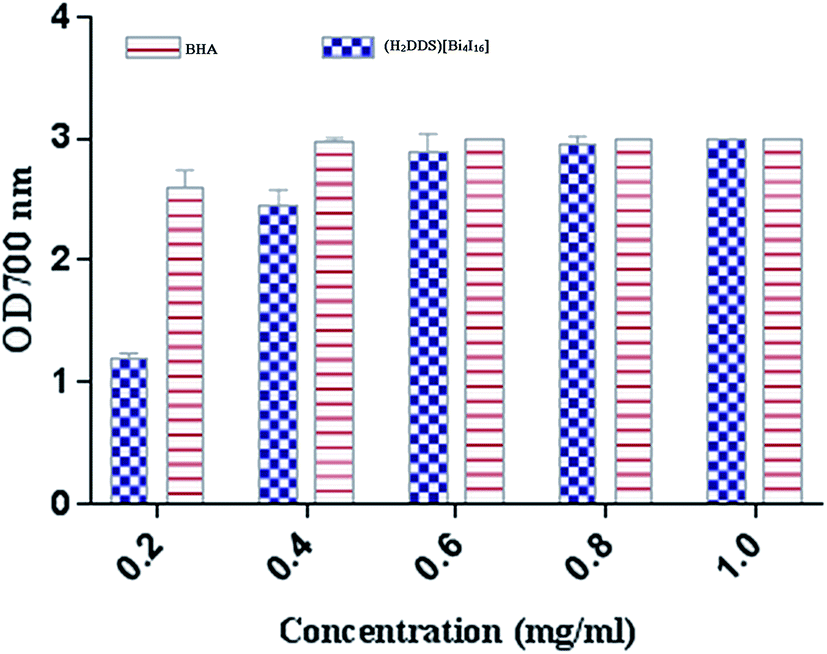 | ||
| Fig. 9 Reducing power effect of (H2DDS)[Bi4I16] at different concentrations. | ||
Additionally, the total antioxidant capacity, which induced the reduction of Mo(VI) to Mo(V) by the sample and the formation of a green phosphate/Mo(V) complex at acid pH, is measured at 695 nm. The total antioxidant capacities of (H2DDS)[Bi4I16] and BHA, used as reference, were performed at different concentrations (0.2–2 mg ml−1) and are illustrated in (Fig. 10). Similarly, the (H2DDS)[Bi4I16] presented increasing antioxidant activity with increasing concentration, and the maximum antioxidative effectiveness (160 μg ascorbic acid equivalents) was reached at 2 mg ml−1. BHA, as positive control, was found to be more efficient (220 μg ascorbic acid equivalents) at the same concentration. Furthermore, even at low concentrations, (H2DDS)[Bi4I16] may serve as a potential antioxidant, with 50 μg ascorbic acid equivalents at 0.4 mg ml−1.
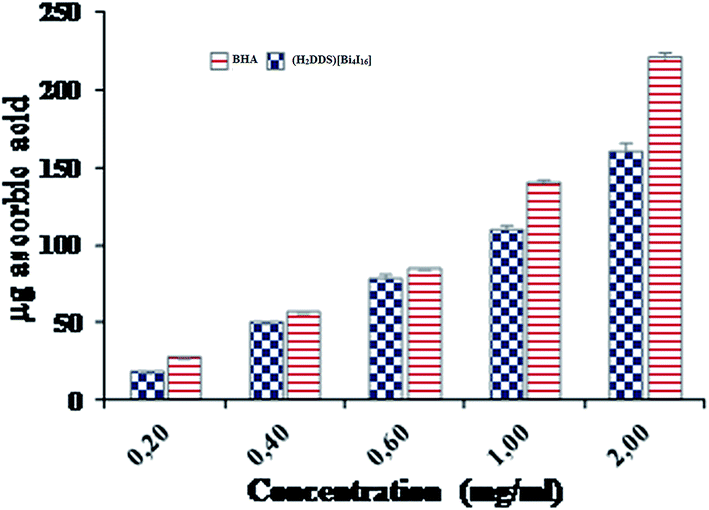 | ||
| Fig. 10 Total antioxidant capacity of (H2DDS)[Bi4I16] at different concentrations. | ||
In conclusion, these results obtained by preliminary screening of antioxidant activity suggested that (H2DDS)[Bi4I16] might serve as interesting compounds for the development of new antioxidant agent, through to the cooperative effects of different antioxidant mechanisms.
| Tested microorganisms | Inhibition zone diameter (mm) |
|---|---|
| Escherichia coli | 11 ± 1.41 |
| Listeria monocytogenes | 12 ± 1.41 |
| Salmonella enterica | 15.5 ± 0.71 |
| Staphylococcus aureus | 11.5 ± 0.71 |
| Micrococcusluteus | 15 ± 1.41 |
| Bacillus cereus | 13.5 ± 0.71 |
| Enterobacteraeruginosa | 8.5 ± 0.71 |
| Pseudomonas aeruginosa | 8 ± 00 |
4. Conclusion
In summary, the structural feature of the new iodobismuthate(III) compound has been presented, which contains a diprotonated bis (4,4′-diammoniumdiphenylsulfone) cation, an inorganic [Bi4I16]4− tetramer and cocrystallized water molecules linked by four types of hydrogen bonds; N–H⋯OW, OW–H⋯O, N–H⋯I and OW–H⋯I, which form a self-assembled three-dimensional layered structure. The vibrational properties of the prepared compound were studied by Raman scattering and infrared spectroscopy. The thermal behavior (TGA and DSC) confirms the stability of this product. The optical properties were investigated by optical absorption and photoluminescence measurements, that show an optical absorption band at 3.42 eV, attributed to the absorption band gap and a band at 3.96 eV, attributed to an excitonic absorption. Moreover, we found that two yellow-green photoluminescence peaks at room temperature are attributed with the band to band and excitonic emissions within the iodobismuthate inorganic part. As for the biological activities of the synthesized compound, the preliminary screening of its antioxidant activity suggested that (H2DDS)[Bi4I16] might serve as interesting compounds for the development of new antioxidant agent, through the cooperative effects of different antioxidant mechanisms. Finally, the antibacterial activity of the crystal compound may pave the way for the exploitation of new antibacterial product.Conflicts of interest
There are no conflicts to declare.References
- A. Piecha-Bisiorek, K. Mencel, V. Kinzhybalo, A. Szota, R. Jakubas, W. Medycki and W. Zawrocki, CrystEngComm, 2018, 20, 2112 RSC
.
- N. A. Yelovik, A. V. Mironov, M. A. Bykov, A. N. Kuznetsov, A. V. Grigorieva, Z. Wei, E. V. Dikarev and A. V. Shevelkov, Journal of Inorganic Chemistry, 2016, 55, 4132 CrossRef CAS
- W. Trigui, A. Oueslati, I. Chaabane, G. Corbel and F. Hlel, Appl. Phys. A: Mater. Sci. Process., 2015, 119, 673 CrossRef CAS
- M. Chanski, A. Białonska, R. Jakubas and A. Piecha-Bisiorek, Polyhedron., 2014, 71, 69 CrossRef CAS
- H. Dammak, S. Triki, A. Mlayah, Y. Abid and H. Feki, J. Lumin., 2015, 166, 180 CrossRef CAS
- C. Hrizi, A. Trigui, Y. Abid, N. Chniba-Boudjada, P. Bordet and S. Chaabouni, J. Solid State Chem., 2011, 184, 3336 CrossRef CAS
- M. Wojtas and R. Jakubas, J. Phys.: Condens. Matter, 2004, 16, 7521 CrossRef CAS
- J. Zhai, R. L. Sang and L. Xu, J. Mol. Struct., 2011, 1006, 553 CrossRef CAS
- S. Chaabouni, A. Hadrich, F. Romain and A. Ben Salah, Solid State Sci., 2003, 5, 1041 CrossRef CAS
- T. J. K. L. G. Marzilli, G. L. Eichhorn and L. Gunther, in Nucleic Acid- Metal Ion Interaction, ed. T.G. Spiro, John Wiley and Sons, New York, 1980 Search PubMed
- Z. Ouerghi, T. Roisnel, R. Fezai and R. Kefi, J. Mol. Struct., 2018, 1173, 439 CrossRef CAS
- S. A. Adonin, M. E. Rakhmanova, D. G. Samsonenko, M. N. Sokolov and V. P. Fedin, Inorg. Chim. Acta, 2016, 450, 232 CrossRef CAS
- I. D. Gorokh, S. A. Adonin, P. A. Abramov, A. S. Novikov, M. N. Sokolov and V. P. Fedin, Inorg. Chem. Commun., 2018, 98, 169 CrossRef CAS
- M. Wojtas, R. Jakubas, Z. Ciunik and W. Medycki, J. Solid State Chem., 2004, 177, 1575 CrossRef CAS
- Z. Aloui, V. Ferretti, S. Abid, M. Rzaigui, F. Lefebvre and C. Ben Nasr, J. Mol. Struct., 2015, 1087, 26 CrossRef CAS
- A. Adonin, I. D. Gorokh, D. G. Samsonenko, I. V. Yushina, M. N. Sokolov and V. P. Fedin, Russ. J. Coord. Chem., 2016, 42, 695 CrossRef
- S. A. Adonin, I. D. Gorokh, D. G. Samsonenko, A. S. Novikov, I. V. Korolkov, P. E. Plyusnina, M. N. Sokolov and V. P. Fedin, Polyhedron, 2019, 1595, 318 CrossRef
- A. P. Bisiorek, A. Ga-gor, R. Jakubas, A. Ci-zman, R. Janickia and W. Medycki, Inorg. Chem. Front., 2017, 4, 1281 RSC
- S. Pandey, A. Nair, A. P. Andrews and A. Venugopal, Eur. J. Inorg. Chem., 2017, 798 CrossRef CAS
- G. Liu, J. Liu, X. Zheng, Y. Liu, D. Yuan, X. Zhang, Z. Gao and X. Tao, CrystEngComm, 2015, 17, 2569 RSC
- M. Essid, Z. Aloui, V. Ferretti, S. Abid, F. Lefebvre, M. Rzaigui and C. Ben Nasr, Inorg. Chim. Acta, 2017, 457, 122 CrossRef CAS
- H. Dammak, H. Feki, H. Boughzela and Y. Abid, Spectrochim. Acta, Part A, 2015, 137, 1235 CrossRef CAS
- S.-Y. Zhang, J. Li, Y. Zeng, H.-R. Wen and Zi-Yi. Du, J. Mol. Struct., 2016, 1125, 227 CrossRef CAS
- A. Adonin, I. D. Gorokh, A. S. Novikov, D. G. Samsonenko, I. V. Korolkov, M. N. Sokolov and V. P. Fedin, Polyhedron, 2018, 139, 282 CrossRef
- B. Wenhua, N. Leblanc, N. Mercier, P. Auban-Senzier and C. Pasquier, J. Chem. Mater., 2009, 21, 4099 CrossRef
- S. A. Adonin, I. D. Gorokh, D. G. Samsonenko, I. V. Korolkov, M. N. Sokolov and V. P. Fedin, Russ. J. Inorg. Chem., 2016, 61, 958 CrossRef CAS
- S. A. Adonin, M. N. Sokolov and V. P. Fedin, Russ. J. Inorg. Chem., 2017, 62, 1789 CrossRef CAS
- A. Piecha-Bisiorek, A. Gągor, R. Jakubas, A. Ci_zman, R. Janickia and W. Medycki, Inorg. Chem. Front., 2017, 4, 1281 RSC
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163 CrossRef CAS
- L. Sobczyk, R. Jakubas and J. Zaleski, Pol. J. Chem., 1997, 71, 265 CAS
- M. Styslo-Zalasik and W. Li, J. Pharm. Biomed. Anal., 2005, 37(3), 529–534 CrossRef CAS
- M. Mansour, T. Ben Issa, N. Issaoui, A. Harchani, E. G. Puebla and B. Ayed, J. Mol. Struct., 2019, 1197, 478 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS
- L. J. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS
- M. N. Burnett and C. K. Johnson, ORTEP-III, Report ORNL-6895, Oak Ridge National Laboratory, Tennessee, USA, 1996 Search PubMed
- K. Brandenburg, Diamond. Visual Crystal Structure Information System, Version 3.2i, Crystal Impact GbR, Bonn, Germany, 2012 Search PubMed
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS
- P. Bersuder, M. Hole and G. Smith, J. Am. Oil Chem. Soc., 1998, 75, 181 CrossRef CAS
- A. Yildirim, A. Mavi and A. A. Kara, J. Agric. Food Chem., 2001, 49, 4083 CrossRef CAS
- P. Prieto, M. Pineda and M. Aguilar, Anal. Biochem., 1999, 269, 337 CrossRef CAS
- D. A Vanden Berghe and A. J. Vlietinck, Methods Plant Biochem., 1991, 6, 47 Search PubMed
- V. V. Sharutin, I. V. Egorova, N. N. Klepikov and O. K. Sharutina, Journal of Inorganic Chemistry, 2010, 55, 1174 Search PubMed
- M. A. Tershansy, A. M. Goforth, J. R. Gardinier, M. D. Smith, L. Peterson Jr and H.-C. zur Loye, Solid State Sci., 2007, 9, 410 CrossRef CAS
- V. V. Sharutin, I. V. Egorova, M. V. Levchuk, B. V. Bukvetskii and D. Yu. Popov, J. Coord. Chem., 2002, 285, 613 CrossRef
- H. Jeghnou, A. Ouasri, A. Rhandour, M. C. Dhamelincourt, P. Dhamelincourt, A. Mazzah and P. Roussel, J. Raman Spectrosc., 2005, 36, 1023 CrossRef CAS
- A. Ouasri, H. Jeghnou, A. Rhandour and P. Roussel, J. Solid State Chem., 2013, 200, 22 CrossRef CAS
- D. B. Mitzi, D. R. Meseiros and P. R. L. Malenfant, Journal of Inorganic Chemistry, 2002, 41, 2134 CrossRef CAS
- A. Khalid, A. I. Farhan and I. Abdulaziz AI-Wassf, J. Chem. Crystallogr., 1995, 25, 12 Search PubMed
- V. V. Sharutin, I. V. Egorova, E. A. Boyarkina and O. K. Sharutina, Russ. J. Gen. Chem., 2008, 78, 1320 CrossRef CAS
- A. M. Goforth, L. Peterson, M. D. Smith and H. C. zur Loye, J. Solid State Chem., 2005, 178, 3529 CrossRef CAS
- Y. Chen, Z. Yang, C. X. Guo, C. Y. Ni, Z. G. Ren, H. X. Li and J. P. Lang, Eur. J. Inorg. Chem., 2010, 5326 CrossRef CAS
- C. J. Carmalt, L. J. Farrugia and N. C. Norman, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 1591 CAS
- W. H. Bi and N. Mercier, Chem. Commun., 2008, 5743 RSC
- A. M. Goforth, J. R. Gardinier, M. D. Smith, L. Peterson and H. C. zur Loye, Inorg. Chem. Commun., 2005, 8, 684 CrossRef CAS
- R. Kubiak and K. Ejsmont, J. Mol. Struct., 1999, 474, 275 CrossRef
- V. V. Sharutin, I. V. Egorova, O. K. Sharutina, O. A. Dorofeeva, T. K. Ivanenko, A. V. Gerasimenko and M. A. Pushilin, Russ. J. Coord. Chem., 2004, 30, 874 CrossRef CAS
- V. V. Sharutin, I. V. Egorova, N. N. Klepikov, E. A. Boyarkina and O. K. Sharutina, Russ. J. Inorg. Chem., 2009, 54, 1768 CrossRef
- S. A. Adinin, M. N. Sokolov and V. P. Fedin, Coord. Chem. Rev., 2016, 312, 1 CrossRef
- A. Kessentini, T. Dammak and M. Belhouchet, J. Mol. Struct., 2017, 1149, 818 CrossRef CAS
- A. Rafael, C. All-ao, A. Jackson, L. C. Resende, S. S. Jr, L. Sorace, M. Andruhad and G. F. M. Vaz, CrystEngComm, 2013, 15, 8422 RSC
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC
- T. Sun, F. Liang, X. Zhang, H. Tu, Z. Lin, G. Zhang and Y. Wu, Polyhedron., 2017, 127, 478 CrossRef CAS
- A. Samet, A. Ben Ahmed, A. Mlayah, H. Boughzala, E. K. Hlil and Y. Abid, J. Mol. Struct., 2010, 977, 72 CrossRef CAS
- H. Ltaief, A. Mahroug, P. Paoli, P. Rossi and M. Belhouchet, J. Mol. Struct., 2020, 1220, 128760 CrossRef CAS
- S. Gatfaoui, N. Issaoui, A. Mezni, F. Bardak, T. Roisnel, A. Atac and H. Marouani, J. Mol. Struct., 2017, 1150, 242 CrossRef CAS
- M. S. Lassoued, W. Ben Soltan, M. S. M. Abdelbaky, S. Ammar, A. Gadri, A. Ben Salah and S. Garcia-Granda, J. Mater. Sci.: Mater. Electron., 2017, 28, 12698 CrossRef CAS
- P. Krishnan, K. Gayathri, G. Bhagavannarayana, S. Gunasekaran and G. Anbalagan, Spectrochim. Acta, Part A, 2013, 102, 379 CrossRef CAS
- J. Tarasiewicz, R. Jakubas, G. Bator, J. Zaleski, J. Baran and W. Medycki, J. Mol. Struct., 2009, 932, 6 CrossRef CAS
- P. Krishnan, K. Gayathri, G. Bhagavannarayana, S. Gunasekaran and G. Anbalagan, Spectrochim. Acta, Part A, 2013, 102, 379 CrossRef CAS
- N. Elleuch, W. Amamou, A. Ben Ahmed, Y. Abid and H. Feki, J. Spectrochim. Acta, Part A, 2014, 128, 781 CrossRef CAS
- A. Kessentini, A. Ben Ahmed, T. Dammak and M. Belhouchet, Spectrochim. Acta, Part A, 2018, 191, 241 CrossRef CAS
- X. H. Zhu, N. Mercier, P. Frere, P. Blanchard, J. Roncali, M. Allain, C. Pasquier and A. Riou, Journal of Inorganic Chemistry, 2003, 42, 5330 CrossRef CAS
- H. Dammak, A. Yengui, S. Triki, Y. Abid and H. Feki, J. Lumin., 2015, 161, 214 CrossRef CAS
- T. Kawai and S. Shimanuki, Phys. Status Solidi B, 1993, 177, K43 CrossRef CAS
- N. Elfaleh and S. Kamoun, J. Organomet. Chem., 2016, 819, 95 CrossRef CAS
- C. Hrizi, A. Samet, Y. Abid, S. Chaabouni, M. Fliyou and A. Koumina, J. Mol. Struct., 2011, 992, 96 CrossRef CAS
- I. Abdehalim Ahmed, R. Blachnik and H. Reuter, Z. Anorg. Allg. Chem., 2001, 627, 2057 CrossRef
- Z. Ouerghi, H. Gornitzka, E. Temel, I. Dridi and R. Kefi, J. Mol. Struct., 2019, 1181, 338 CrossRef CAS
- R. Jakubas, A. Piecha, A. Pietraszko and G. Bator, Phys. Rev. B, 2005, 72, 104107 CrossRef
- R. Hajji, A. Oueslati, N. Errien and F. Hlel, Polyhedron, 2014, 795, 97 CrossRef
- A. Feki, H. Ben Saad, I. Bkhairia, N. Ktari, M. Naifar, O. Boudawara, M. Droguet, C. Magné, M. Nasri and I. Ben Amara, J. Envir. Toxicology, 2018, 1 DOI:10.1002/tox.22682
- E. Akila, M. Usharani and R. Rajavel, Int. J. Pharm. Pharm. Sci., 2013, 5, 573 Search PubMed
- J. Wang, S. Hu, S. Nie, Q. Yu and M. Xie, Oxid. Med. Cell. Longevity, 2016, 5692852 Search PubMed
- A. M. Ben Salah, N. Sayari, H. Naïli and A. J. Norquistc, RSC Adv., 2016, 64, 59055 RSC
- N. Hfidhi, I. Bkhairia, D. Atoui, J. Boonmak, M. Nasri, R. B. Salem, S. Youngme and H. Naïli, Appl. Organomet. Chem., 2019, 4793 CrossRef
- Z. Miao, L. Xu, H. Song, H. Zhao and L. Chou, Catal. Sci. Technol., 2013, 1942 RSC
- A. L. Koch, Clin. Microbiol. Rev., 2003, 16, 673 CrossRef
Footnote |
| † CCDC 1909744. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05646d |
| This journal is © The Royal Society of Chemistry 2020 |