
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO2 activation by core–shell Ru13@Pt42 particles in comparison with Pt55 particles: a DFT study†
Jing Lua,
Bo Zhu b and
Shigeyoshi Sakaki*bc
b and
Shigeyoshi Sakaki*bc
aHubei Key Laboratory of Advanced Textile Materials & Application, Hubei International Scientific and Technological Cooperation Base of Intelligent Textile Materials & Application, Wuhan Textile University, Wuhan 430200, China
bElement Strategy Initiative for Catalysts and Batteries, Kyoto University, Goryo-Ohara 1-30, Nishikyo-ku, Kyoto 615-8245, Japan. E-mail: sakaki.shigeyoshi.47e@st.kyoto-u.ac.jp; Fax: +81-75-383-3047; Tel: +81-75-383-3036
cFukui Institute for Fundamental Chemistry (FIFC), Kyoto University, Takano-Nishihiraki-cho 34-4, Sakyou-ku, Kyoto 606-8103, Japan
First published on 30th September 2020
Abstract
The reaction of O2 with a Ru13@Pt42 core–shell particle consisting of a Ru13 core and a Pt42 shell was theoretically investigated in comparison with Pt55. The O2 binding energy with Pt55 is larger than that with Ru13@Pt42, and O–O bond cleavage occurs more easily with a smaller activation barrier (Ea) on Pt55 than on Ru13@Pt42. Protonation to the Pt42 surface followed by one-electron reduction leads to the formation of an H atom on the surface with considerable exothermicity. The H atom reacts with the adsorbed O2 molecule to afford an OOH species with a larger Ea value on Pt55 than on Ru13@Pt42. An OOH species is also formed by protonation of the adsorbed O2 molecule, followed by one-electron reduction, with a large exothermicity in both Pt55 and Ru13@Pt42. O–OH bond cleavage occurs with a smaller Ea on Pt55 than on Ru13@Pt42. The lower reactivity of Ru13@Pt42 than that of Pt55 on the O–O and O–OH bond cleavages arises from the presence of lower energy in the d-valence band-top and d-band center in Ru13@Pt42 than in Pt55. The smaller Ea for OOH formation on Ru13@Pt42 than on Pt55 arises from weaker Ru13@Pt42–O2 and Ru13@Pt42–H bonds than the Pt55–O2 and Pt55–H bonds, respectively. The low-energy d-valence band-top is responsible for the weak Ru13@Pt42–O and Ru13@Pt42–OH bonds. Thus, the low-energy d-valence band-top and d-band center are important properties of the Ru13@Pt42 particle.
1. Introduction
The proton exchange membrane fuel cell (PEMFC) is a promising candidate for a clean and sustainable energy source to cope with the growing energy consumption and related environmental concerns. Pt particles are used as a catalyst for the oxygen reduction reaction (ORR) at the PEMFC cathode because of their incomparable catalytic activity and stability in acidic solution, as discussed in recent reviews;1–6 we cite here reviews from the last 5 years because many reviews have been published. However, their limited availability on earth and high cost still remain major obstacles for the wide use of PEMFCs. One of the promising methods to solve this problem is the use of core–shell Pt particles (M@Pt) consisting of Pt for the shell and abundant metals (M) for the core, because Pt content can be reduced in the catalyst by the use of a M core but the Pt shell exhibits high catalytic activity and stability in acid solution, as reviewed in the last few years.7–14 Also, one can expect to improve the catalytic activity of the Pt shell by tuning the electronic structure of the Pt shell with the M core.Recently, core–shell Ru@Pt particles have been reported as excellent ORR catalysts.15–20 For instance, Adzic and co-workers demonstrated that the catalytic activity of Ru@Pt could be tuned by varying the Pt shell thickness; Ru@Pt2ML with two Pt layers was more active than Ru@PtxML (x = 1 and 3).15 Jackson and coworkers17,19 and Takimoto and coworkers18 reported that the catalytic activity of Ru@Pt for the ORR exceeded that of a commercial Pt electrode. Jackson and co-workers also interestingly obtained a volcano plot of the catalytic activity against the O binding energy, suggesting that both overly strong and overly weak O binding with the Pt surface is not good for ORR catalysts.17 However, the relation between electronic structure and origin of the O binding energy has been unclear.
Many theoretical ORR studies reported so far discuss the relation between electronic structure of the catalyst and ORR activity.21–39 In particular, the O–O bond activation has been theoretically investigated in many works, as discussed by a recent review40 and many works even after this review.41–46 However, the theoretical study of Ru@Pt has been limited so far; for instance, the O and OH-binding energies with Ru13@Pt42 and Pt55 have been theoretically investigated,47 but the O–O bond cleavage on Ru@Pt has not been investigated, despite the crucial importance of such O–O bond cleavage in the ORR. Considering that the theoretical study of nanoscale metal particles is still challenging and its development is needed even currently,48–50 the theoretical study of the O2 reaction on Ru@Pt and related metal particles is indispensable.
In this work, we theoretically investigated dioxygen (O2) adsorption and O–O bond cleavage by the Ru13@Pt42 particle in comparison with the Pt55 particle using DFT computations. In the O–O bond cleavage, two reaction courses are plausible; in one, the O–O bond of the adsorbed O2 molecule is cleaved. In the other reaction course, OOH species are formed on the surface, followed by O–OH bond cleavage, because it is likely that the OOH species are easily formed in the presence of excess protons and enough supply of electrons to the electrode. Our purposes here are to explore O2 adsorption, O–O bond cleavage, OOH formation, and O–OH bond cleavage, compare the reactivity between Ru13@Pt42 and Pt55, find important factor(s) determining the reactivities of Ru13@Pt42 and Pt55, and present a theoretical understanding of the differences between Ru13@Pt42 and Pt55. We believe that the theoretical findings on these issues are valuable for understanding the chemistry of nanometal particles.
2. Computational methods and models
Spin-polarized periodic DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP),51,52 where plane-wave basis sets were employed with an energy cutoff of 400 eV, and the projector-augmented-wave (PAW) pseudopotentials were used to represent core electrons. The PBE-D3 functional was employed in all calculations,53 where “D3” represents the dispersion correction proposed by Grimme's group.54,55 Ru@Pt and Pt particles were placed in a large supercell (25 Å × 25 Å × 25 Å) to ensure enough separation by vacuum. Geometry optimization of equilibrium structure was carried out in gas phase using the energy threshold of 0.0001 eV and force threshold of 0.01 eV Å−1. Optimization of transition state (TS) was carried out using the climing image nudged elastic band (CI-NEB) method with the VASP transition-state tools (VTST),56,57 in which thresholds for convergence were set to 0.0001 eV for energy and 0.02 eV Å−1 for force. To evaluate solvent effects, an implicit solvation model, which describes the effect of electrostatic interaction between solute and solvent, was employed as implemented in VASPsol,58,59 where optimized geometry in gas phase was used.Though Pt55 and Ru13@Pt42 particles are not very large compared to real catalysts, we employed these particles here as model nanoparticles because we have to optimize many intermediates and transition states. Also, the use of Pt55 and Ru13@Pt42 particles is not unreasonable, considering that subnanometer-scale metal particles with 55 metal atoms have been employed as model particle in theoretical studies of O2 adsorption and O–O bond cleavage46,60–63 and other catalytic reactions.64–69 The number “55” is a magic number for icosahedral (Ih) and cuboctahedral (Oh) structures. Here, we employed the Ih-like structure because the Ih structure is more stable than Oh in Ru13@Pt42; the relative stabilities of various spin states and comparison between core–shell and non-core–shell structures have been investigated recently.47 As shown in Scheme 1, the Pt42 surface of icosahedral Pt55 and Ru13@Pt42 consists of 20 triangular facets. Each facet has three different types of binding sites: top (t), bridge (b) and hollow (h). Adsorptions at these binding sites are classified as follows: (i) adsorption at the top site is denoted as t1 or t2, in which the adsorbate binds with one Pt atom at the edge or the vertex position. (ii) Adsorption in a bridging manner is denoted as b1 or b2, in which the adsorbate binds with two adjacent Pt atoms at either the edge and vertex positions or two adjacent edge positions. (iii) Adsorption at the hollow site is denoted as h1 or h2, in which the adsorbate binds at either the fcc-like position among three edge-Pt atoms or the hcp-like position among two edge-Pt atoms and one vertex-Pt atom. We explored all these possible adsorption sites.
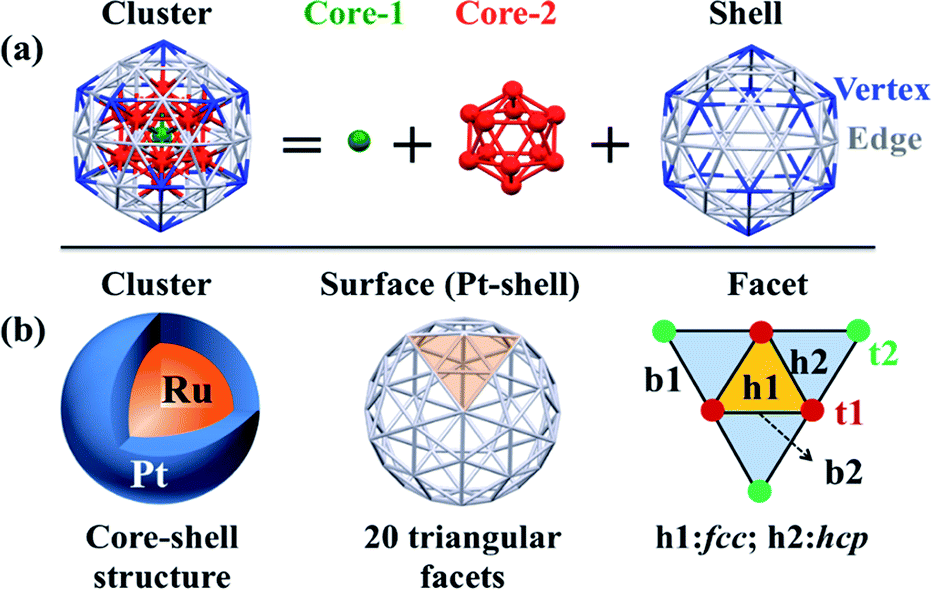 | ||
| Scheme 1 (a) Geometry of the icosahedron (Ih) 55-atom particle and (b) binding sites of the Pt42 shell of the Ih Ru13@Pt42 core–shell particle. | ||
3. Results and discussion
In this work, we firstly discuss O2 adsorption to Pt55 and Ru13@Pt42, followed by O–O bond cleavage, OOH formation from the adsorbed O2 molecule, and O–OH bond cleavage on the Pt55 and Ru13@Pt42 surfaces. Next, we show the differences in reactivity between Pt55 and Ru13@Pt42 in these reactions and elucidate the reasons for the differences. Lastly, we unveil the characteristic feature(s) of Ru13@Pt42 in comparison with Pt55.3.1 Dioxygen (O2) adsorption
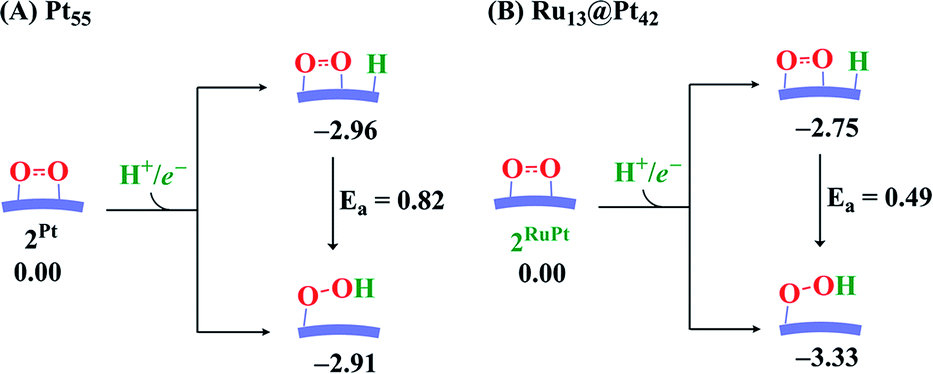
The binding energy Eb(O2) of dioxygen molecule (O2) increases (becomes more negative) following the order O2-η1/h1 < O2-η1/t1 < O2-η1/t2 < O2-μ2/b2 < O2-μ3/h2 < O2-μ3/h1 < O2-μ2/b1 for pure Pt55 and O2-η1/h1 < O2-μ2/b2 < O2-η1/t1 < O2-μ3/h2 < O2-μ3/h1 < O2-η1/t2 < O2-μ2/b1 for Ru13@Pt42, as shown in Fig. S1 in the ESI,† where h1, t1 etc., represent the adsorption site shown in Scheme 1 and “A-μ2/x” represents the interaction of adsorbate A with Pt at the x binding site in a μ2 manner, hereinafter. Obviously, O2 is preferentially adsorbed at the b1 site of both Pt55 and Ru13@Pt42 in a μ2-side-on manner (O2-μ2/b1). This is the most stable O2 adsorption structure. The coordination number of the surface Pt atom is one of the important factors for stabilization of O2 adsorption: because the coordination number of the vertex Pt atom is 6 but that of the edge Pt is 7, the O2 molecule tends to interact with the vertex Pt atom compared to the edge Pt atom. However, the O2 molecule cannot interact with two vertex Pt atoms because the vertex Pt is far from the neighboring vertex Pt. Thus, O2 interacts with one vertex Pt and one edge Pt in a bridging manner, as seen by the O2-μ2/b1 structure.In the most stable O2-μ2/b1-binding species (2), the O–O distance of Pt55(O2) 2Pt is moderately longer than that of Ru13@Pt42(O2) 2RuPt, and the Pt–O distance of 2Pt is moderately shorter than that of 2RuPt, as shown in Fig. 1. The Eb(O2-μ2/b1) value is −1.85 (−1.70) eV for Pt55 and −1.07 (−0.95) eV for Ru13@Pt42, as shown in Fig. 2, where the figures in parentheses represent the binding energy in gas phase. The larger binding energy of the O2 molecule with Pt55 than with Ru13@Pt42 is consistent with such geometrical features as the shorter Pt–O and longer O–O distances in 2Pt than in 2RuPt. The reasons for the stronger O2 adsorption with Pt55 than with Ru13@Pt42 are discussed below. It is also noted that solvation by water enhances O2 binding with these metal particles.
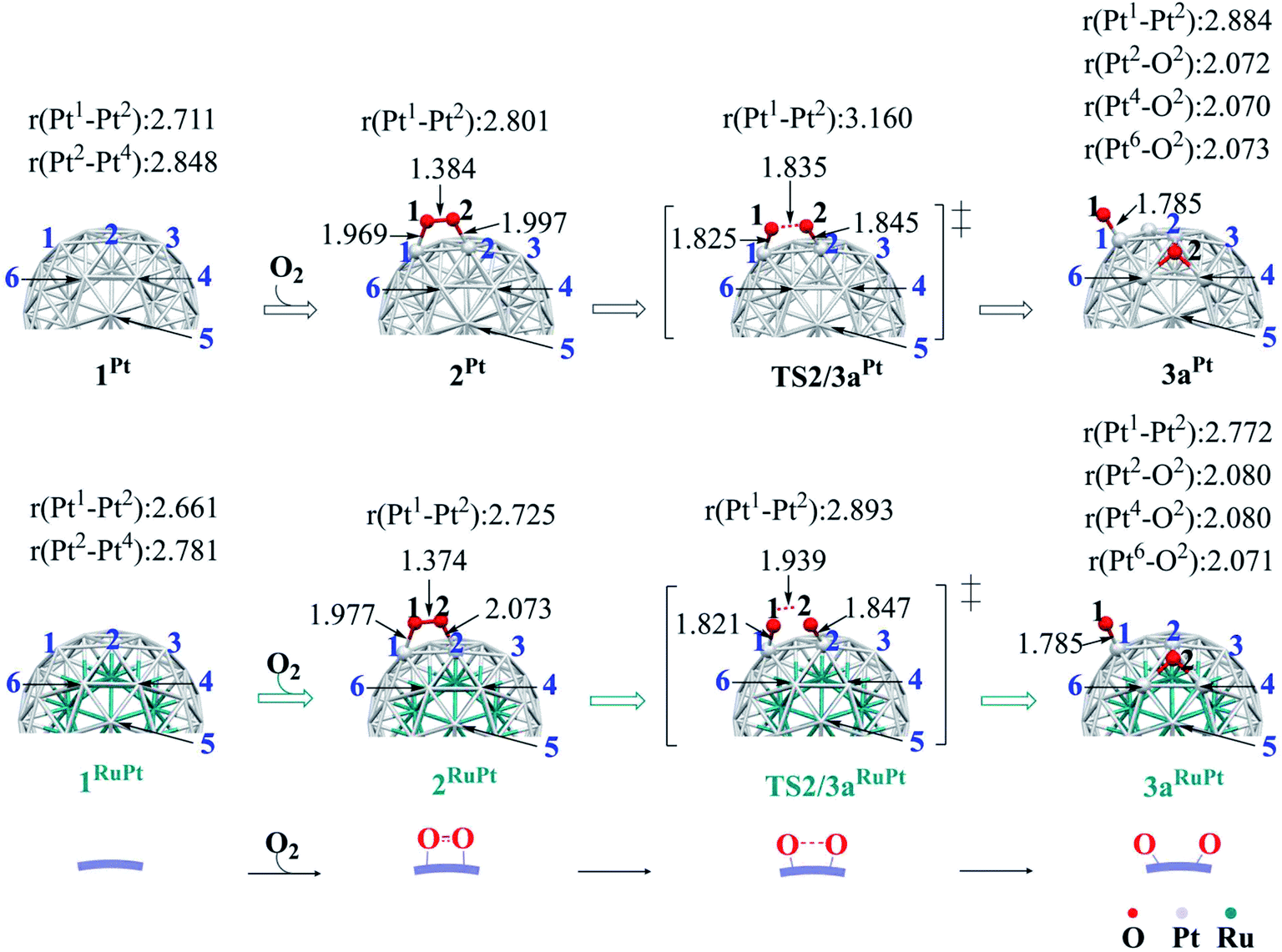 | ||
| Fig. 1 Geometry changes in O2 adsorption followed by O–O bond cleavage on Pt55 and Ru13@Pt42. Values represent bond distance in angstrom. | ||
 | ||
| Fig. 2 Energy changes in O2 adsorption followed by O–O bond cleavage and OOH formation, followed by O–OH bond cleavage, on (A) Pt55 and (B) Ru13@Pt42. Values represent energy (in eV) relative to Pt55 or Ru13@Pt42. In parentheses are values for energy in gas phase. | ||
3.2 O–O bond cleavage
Starting from 2Pt and 2RuPt, O–O bond cleavage occurs via transition states TS2/3aPt and TS2/3aRuPt to afford Pt55(O)2 3aPt and Ru13@Pt42(O)2 3aRuPt, respectively, as shown in Fig. 1. In TS2/3aPt, the O–O distance is considerably elongated to 1.835 Å, by 0.451 Å, and the Pt–O distances become shorter to 1.825 Å and 1.845 Å, by 0.144 and 0.152 Å (Fig. 1). In TS2/3aRuPt, the O–O distance is more elongated to 1.939 Å (by 0.565 Å) than in TS2/3aPt, indicating that TS2/3aPt is more reactant-like than TS2/3aRuPt. Consistent with the O–O bond elongation, the Pt–O distances become shorter in TS2/3a. Though they are almost the same between TS2/3aPt and TS2/3aRuPt, the average of Pt–O distances is moderately shorter in 3aPt than in 3aRuPt, suggesting the stronger binding energy of the O atom with Pt55 than with Ru13@Pt42, as discussed below. The activation barrier (Ea) relative to 2 is 0.31 (0.33) eV and 0.35 (0.49) eV for TS2/3aPt and TS2/3aRuPt, respectively, and the reaction energy (ΔE) relative to 2 is −0.90 (−0.83) eV and −0.95 (−0.85) eV, for 3aPt and 3aRuPt, as shown in Fig. 2, where a negative ΔE value represents exothermicity. The smaller Ea for the O–O bond cleavage on Pt55 than on Ru13@Pt42 is consistent with the more reactant-like TS2/3aPt than TS2/3aRuPt. The moderately smaller ΔE in the Pt55 case than in the Ru13@Pt42 case is seemingly inconsistent with the smaller Ea of the former case than in the latter. But, this is not unreasonable because the O2 binding energy with Pt55 is overly larger than that with Ru13@Pt42. It is noted that the Ea is smaller and ΔE is more negative in water than in gas phase, because CT is generally enhanced by polar solvents.The short Pt–Pt distance of the surface has been discussed as one important factor for high catalytic activity.70–73 The surface Pt1–Pt2 distance becomes longer by the O2 adsorption and the O–O bond cleavage in both Pt55 and Ru13@Pt42 (Fig. 1), suggesting that the short Pt–Pt distance of the surface is not beneficial to these processes. The other important factor is flexibility of the Pt surface. Actually, the energy destabilization of Pt55 is much smaller than that of Ru13@Pt42 when surface Pt–Pt distance is elongated; it is 1.0 kcal mol−1 in Pt55 and 7.5 kcal mol−1 in Ru13@Pt42 for the Pt–Pt elongation by 0.4 Å, where we employed rather arbitrarily the elongation of 0.4 Å because the Pt–Pt distance is elongated by about 0.3–0.4 Å at TS2/3aPt of the O–O bond cleavage on Pt55. These results suggest that the longer Pt–Pt distance and the larger flexibility of Pt55 than those of Ru13@Pt42 are favorable for O2 adsorption and O–O bond cleavage; in other words, the discussion that short Pt–Pt distance is good for high catalytic activity is not useful when these processes are rate-determining. In addition, it should be noted that the flexibility of the Pt surface is a crucially important factor besides Pt–Pt distance.
3.3 OOH formation followed by O–OH bond cleavage
In ORR, it is likely that the proton is adsorbed easily to the cathode surface because protons exist in excess in solution. Also, electrons are always supplied to the cathode. These features suggest that H species is formed on the cathode surface. Actually, the reactions of adsorbed H atoms with oxygen-containing species were discussed in recent works,25,74–76 which indicate they correspond to the Langmuir–Hinshelwood pathway. First, we investigated the formation of OOH species from the adsorbed O2 molecule and H atom on the surface; the adsorption sites of OOH and H are shown in Fig. S3, Tables S2 and S3 in the ESI.†The H+/e− addition occurs with significant exothermicity in 2Pt and 2RuPt to afford Pt55(O2)(H1) 3bPt and Ru13@Pt42(O2)(H1) 3bRuPt, as shown in Fig. 2, where one-half of ΔE of the eq. 2H+ + 2e− → H2, was taken as the energy of H+/e−; it is noted that this step can be tuned experimentally by the cell voltage. In 3bPt and 3bRuPt, H1 takes the position bridging two Pt atoms, as shown in Fig. 3 and 4. Starting from 3bPt and 3bRuPt, the H1 reacts with the adsorbed O2 via transition states TS3/4bPt and TS3/4bRuPt to afford an OOH species adsorbed on the Pt surface Pt55(OOH) 4bPt and Ru13@Pt42(OOH) 4bRuPt, respectively (Fig. 3 and 4). In the transition state, the H1 is approaching the O2, keeping a bonding interaction with one Pt, and simultaneously, the O2 is leaving the Pt42 surface. Though the Pt–O2 distance is very long in TS3/4bPt, the O2–H1 distance is still long (1.414 Å), and the O1–O2 distance is moderately elongated. In TS3/4bRuPt, the O2–H1 distance is much longer (2.006 Å) than that of TS3/4bPt, suggesting that TS3/4bRuPt is more reactant-like than TS3/4bPt, in contrast to the more product-like TS2/3aRuPt than TS2/3aPt. This contrast is reasonable according to the Hammond rule because the O–O bond cleavage occurs with smaller Ea (relative to 2Pt), but the OOH formation occurs with larger Ea on Pt55 than on Ru13@Pt42. In 4bPt and 4bRuPt, the O1–O2 distance is 1.451 Å and 1.456 Å, respectively, which is moderately shorter than that (1.471 Å, the PBE-D3-optimized value) of free HOOH. The surface Pt1–Pt2 distance becomes longer in this reaction, suggesting that the short Pt–Pt distance of the Pt surface is not favorable for this step, either.
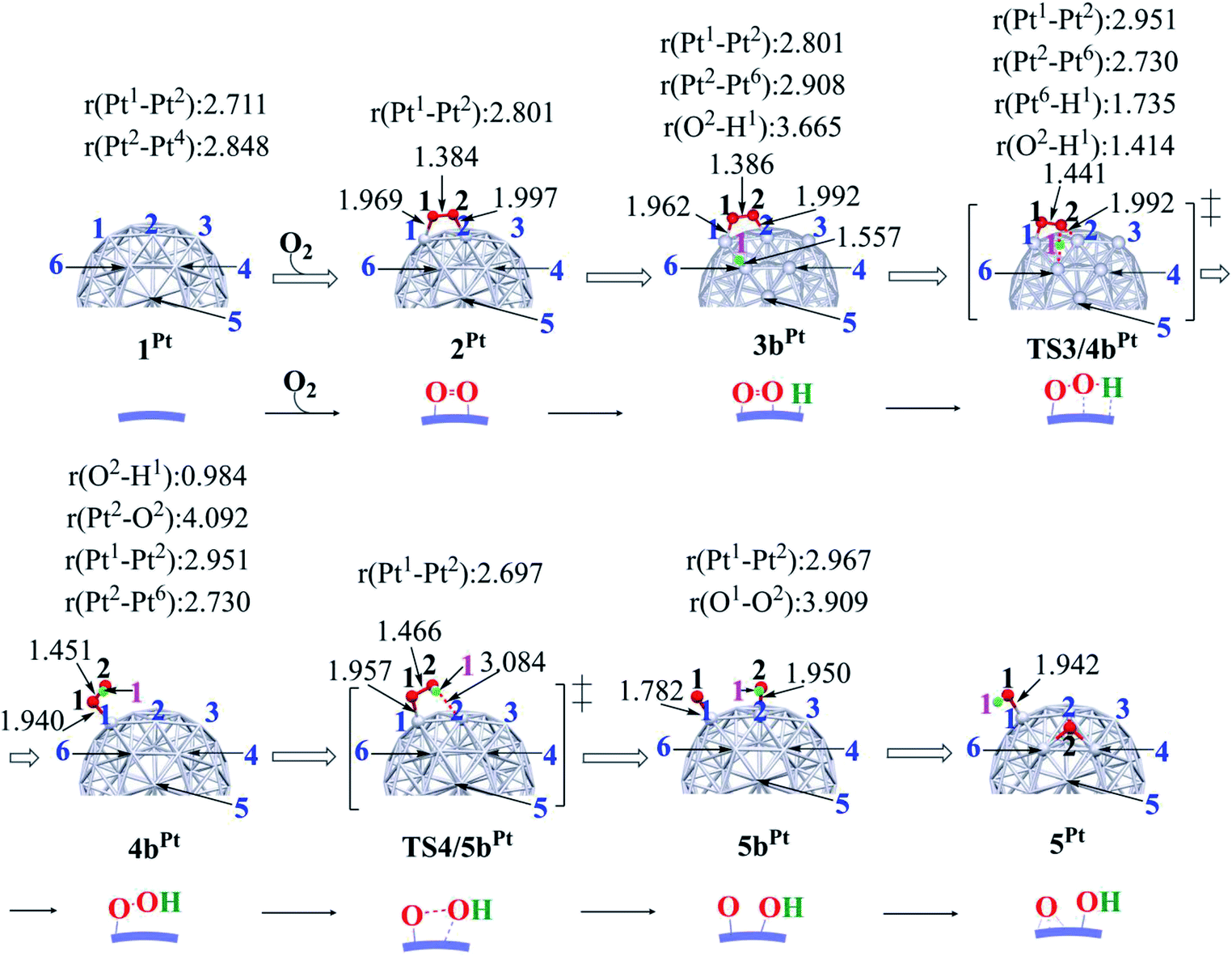 | ||
| Fig. 3 Geometry changes in OOH formation through the reaction between adsorbed O2 molecule and H atom, followed by O–OH bond cleavage on Pt55 particle. Numbers represent bond distance in angstrom. | ||
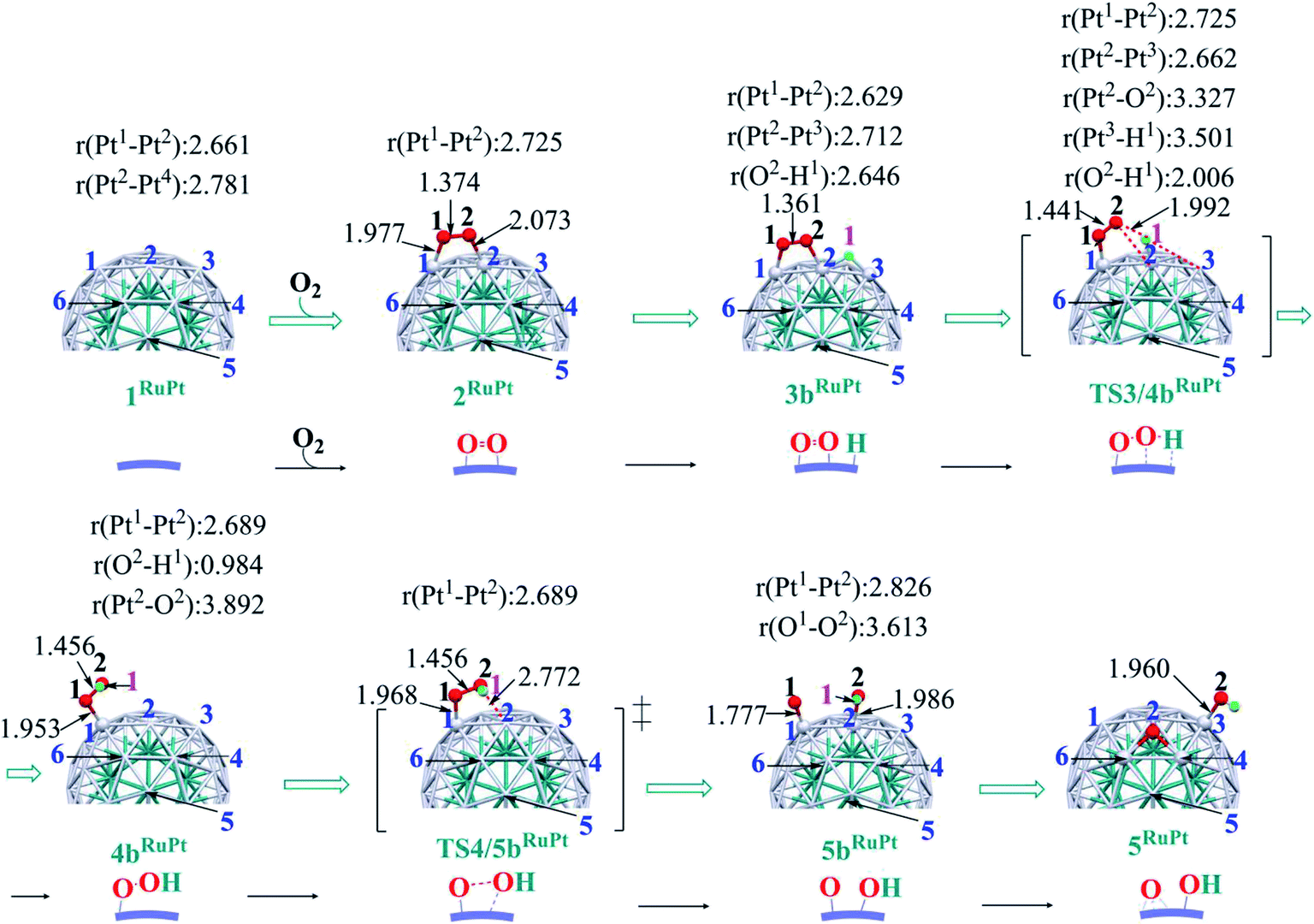 | ||
| Fig. 4 Geometry changes in OOH formation through the reaction between adsorbed O2 molecule and H atom, followed by O–OH bond cleavage on Ru13@Pt42 particle. Numbers represent bond distance in angstrom. | ||
We explored here another OOH formation pathway in which adsorbed O2 molecule undergoes protonation followed by one-electron reduction, as proposed by several works.77–79 This reaction corresponds to the Eley–Rideal pathway. We compared the energy change in this pathway with that of the Langmuir–Hinshelwood pathway, as shown in Scheme 2. In the Pt55 case, the energy changes differ little between these two pathways, suggesting that the OOH formation occurs via both pathways. In Ru13@Pt42, the H+/e− addition to the adsorbed O2 molecule is more exothermic than that to the Pt42 surface (Scheme 2), indicating that the Eley–Rideal pathway is more favorable than the Langmuir–Hinshelwood pathway from the viewpoint of reaction energy. Also, Scheme 2 strongly suggests that the OOH formation occurs more easily on Ru13@Pt42 via the Eley–Rideal pathway than that on Pt55 via both the Langmuir–Hinshelwood and Eley–Rideal pathways. Here, we need to mention that the reaction pathway significantly depends on the coverage of Pt surface by O2 molecules; the Langmuir–Hinshelwood pathway preferentially occurs at low coverage. On the other hand, the Eley–Rideal pathway preferentially occurs at high coverage from the viewpoint of possibility, while the Eley–Rideal pathway becomes less easy at high coverage than at low coverage, from the viewpoint of reactivity of adsorbed O2 molecule, because the adsorbed O2 molecule becomes less negatively charged at high coverage. The mechanism of OOH formation significantly depends on reaction conditions, which must be investigated carefully in the near future. In both pathways, it is reasonably concluded that OOH formation is an easy process on Pt55 and Ru13@Pt42 particles.
 | ||
| Scheme 2 Comparison of energy change between H+/e− addition to the Pt42 surface (the Langmuir–Hinshelwood pathway) and the adsorbed O2 molecule (the Eley–Rideal pathway). | ||
Starting from 4bPt and 4bRuPt, the O–OH bond is cleaved through transition states TS4/5bPt and TS4/5bRuPt to afford Pt55(O)(OH) 5bPt and Ru13@Pt42(O)(OH) 5bRuPt, respectively (Fig. 3 and 4). In the transition state, the O2 is approaching the Pt2, while the Pt2–O2 distance is still long, and the O1–O2 distance changes little from those in 4bPt and 4bRuPt, indicating that the transition state is reactant-like. These transition states differ little from each other except for moderately different Pt–O2 and O1–O2 distances.
The OOH formation from adsorbed O2 and H species occurs with a smaller Ea of 0.49 (0.50) eV on Ru13@Pt41 than on Pt55 (Ea = 0.82 (0.80) eV), as shown in Fig. 2. The smaller Ea in the reaction on Ru13@Pt42 than on Pt55 is consistent with the more reactant-like TS3/4bRuPt than TS3/4bPt. The O–OH bond cleavage occurs with a very small Ea on both Pt55 (Ea = 0.12 (0.12) eV) and Ru13@Pt41 (Ea = 0.17 (∼0) eV; Fig. 2). The very small Ea for the O–OH bond cleavage is consistent with the reactant-like transition states TS4/5bPt and TS4/5bRuPt. Though the Ea is moderately smaller in the reaction on Pt55 than on Ru13@Pt42, the difference is small, and therefore, the geometry of TS4/5bPt differs little from that of TS4/5bRuPt.
It should be noted that the O–OH bond cleavage occurs with a smaller Ea than the O–O bond cleavage of the adsorbed O2 molecule on both Pt55 and Ru13@Pt42. This is not surprising because the O–O bond of the adsorbed O2 molecule is weaker than the original O–O double bond of free O2 molecule, as shown by the elongated O–O bond, but still stronger than the O–O single bond of the OOH species. Another result to be noted is that the Ea of the OOH formation from adsorbed O2 and H species is larger in the Pt55 case than in the Ru13@Pt42 case, but the Ea for O–O bond cleavage is smaller in the Pt55 case than in the Ru13@Pt42 case. These findings are discussed below in more detail on the basis of electronic structure.
3.4 Electronic process in O2 adsorption, O–O bond cleavage, OOH formation, and O–OH bond cleavage
As shown in Table 1, the O2 moiety is negatively charged in Pt55(O2) 2Pt and Ru13@Pt42(O2) 2RuPt, because O2 adsorption occurs with charge-transfer (CT) from metal particle to O2 molecule. This CT is enhanced by the polar solvent (water), as expected. Notably, the O2 molecule is more negatively charged in 2RuPt than in 2Pt. Seemingly, this result is inconsistent with the larger adsorption energy of the O2 molecule with Pt55 than with Ru13@Pt42, which is discussed below. In the O–O bond cleavage (2 → 3a), both O1 and O2 atoms become much more negatively charged, as well known. A moderately larger CT occurs unexpectedly in Ru13@Pt42 than in Pt55, despite the larger Ea in Ru13@Pt42 than in Pt55.| 2 | TS2/3a | 3a | |
|---|---|---|---|
| a A positive value represents positive atomic charge, and vice versa.b Values in parentheses represent the Bader charge in gas phase.c A positive value represents the increase in charge transfer from the metal particle to O1 and O2 atoms. | |||
| Pt55 | |||
| O1 | −0.305 (−0.273)b | −0.496 (−0.434) | −0.610 (−0.509) |
| O2 | −0.300 (−0.259) | −0.480 (−0.406) | −0.706 (−0.684) |
| Pt55 | +0.605 (+0.532) | +0.976 (+0.840) | +1.316 (1.193) |
| Δ(CT)c | 0.0 (0.0) | 0.371 (0.308) | 0.711 (0.661) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Ru13@Pt42 | |||
| O1 | −0.326 (−0.289) | −0.519 (−0.445) | −0.640 (−0.536) |
| O2 | −0.320 (−0.268) | −0.506 (−0.427) | −0.719 (−0.689) |
| Ru13@Pt42 | +0.646 (+0.557) | +1.025 (0.872) | +1.359 (+1.225) |
| Δ(CT)c | 0.0 (0.0) | 0.379 (0.315) | 0.713 (0.668) |
| 3b | TS3/4b | 4b | TS4/5b | 5b | |
|---|---|---|---|---|---|
| a Values in parentheses represent the Bader charge in gas phase. | |||||
| Pt55 | |||||
| O1 | −0.295 (−0.263)a | −0.246 (−0.218) | −0.371 (−0.339) | −0.669 (−0.630) | −1.016 (−0.935) |
| O2 | −0.289 (−0.250) | −0.252 (−0.197) | −0.624 (−0.575) | −0.384 (−0.349) | −0.615 (−0.511) |
| H | −0.035 (−0.037) | 0.198 (0.204) | 0.683 (0.609) | 0.727 (0.653) | 0.676 (0.610) |
| Pt55 | 0.619 (0.550) | 0.300 (0.211) | 0.312 (0.305) | 0.326 (0.326) | 0.955 (0.836) |
|
|||||
| Ru13@Pt42 | |||||
| O1 | −0.312 (−0.261) | −0.260 (−0.235) | −0.401 (−0.367) | −0.945 (−0.671) | −1.182 (−1.138) |
| O2 | −0.278 (−0.245) | −0.210 (−0.140) | −0.800 (−0.728) | −0.401 (−0.367) | −0.708 (−0.680) |
| H | 0.008 (0.007) | 0.069 (0.083) | 0.872 (0.782) | 1.000 (0.675) | 0.822 (0.789) |
| Ru13@Pt42 | 0.582 (0.499) | 0.401 (0.292) | 0.329 (0.313) | 0.346 (0.363) | 1.068 (1.029) |
In the OOH formation via the reaction between the adsorbed O2 molecule and H atom (3b → TS3/4b → 4b), the H atom becomes more positively charged, the O1 and O2 atoms become more negatively charged, and the positive charges of Pt55 and Ru13@Pt42 decrease. However, these population changes are not simple. The positive charges of Pt55 and Ru13@Pt42 decrease when going from 3b to TS3/4b but change little after TS3/4b, suggesting that the CT from O2 and H to the metal particle mostly occurs in step 3b → TS3/4b but little after TS3/4b. In this 3b → TS3/4b step, the H atomic charge becomes considerably positive, but the O1 and O2 atomic charges moderately change, suggesting that the H atom mainly participates in the CT to the metal particle. As it goes from TS3/4b to 4b, the O1 becomes more negatively charged, the O2 is much more negatively charged, and the H atom becomes much more positively charged. Because the CT occurs little to metal particle in this step (TS3/4b → 4b), as discussed above, the change in electron distribution mainly occurs in the O2H moiety, suggesting that the Oδ−–Hδ+ polarization becomes strong in this step. It is noted that the positive charges of Pt55 and Ru13@Pt42 change to a lesser extent in this OOH formation than in the O2 adsorption, O–O bond cleavage, and O–OH bond cleavage, as shown in Tables 1 and 2. These features suggest that not only CT but also some other factors play important roles in this OOH formation, as discussed below.
In the O1–O2H bond cleavage (4b → TS4/5b → 5b), the O2H group becomes considerably positive at TS4/5b and then returns to moderately positive at 5b, while the negative charge of the O1 atom and the positive charges of Pt55 and Ru13@Pt42 increase when going from 4b to 5b. These population changes indicate that this step occurs with CT from the metal particle to the OOH moiety. These population changes resemble those by the oxidative addition in organometallic chemistry.80 This is reasonable because the σ-bond cleavage needs CT from the metal to the σ*-antibonding orbital. Because the CT deeply relates to the electronic structure of the metal particle, the next task is to elucidate the electronic structures of Pt55 and Ru13@Pt42.
3.5 Electronic structures of Pt55 and Ru13@Pt42, M–X bond energy (M = Pt55 and Ru13@Pt42; X = H, O, OH, and OOH), and their relation to O2 activation
The 5d-valence band-top and d-band center of the Pt42 shell are calculated at higher energy in Pt55 than in Ru13@Pt42, but the d-conduction band-bottom of the Pt42 shell is calculated at lower energy in Pt55 than in Ru13@Pt42 (Fig. 5A and B), where the d-band center was calculated using d-valence bands and the DOS energy was corrected according to Baldereschi and coworkers.81 We checked if the box size for periodic calculation influences little the Fermi level after the correction, as shown in Table S4 in the ESI (page S10†), and also we wish to note the 5d-valence band-top energy differs moderately from that of our previous work,47 because of the different computation method, as explained in the ESI (pages S11 to S12†). The O2 adsorption decreases the density of the d-valence band-top and that of the d-conduction band-bottom in both Pt55 and Ru13@Pt42, as shown by Fig. 5A, B and C, D, indicating that the d-valence band-top mainly participates in the CT from the Pt42 shell to the O2, and the d-conduction band-bottom mainly participates in the reverse CT from the O2 to the Pt42 shell. The higher energy d-valence band-top and the lower energy d-conduction band-bottom induce stronger CT from Pt55 to O2 than that from Ru13@Pt42 to O2 and, also, stronger CT from O2 to Pt55 than that from O2 to Ru13@Pt42, respectively. Consequently, the binding energy of O2 molecule with Pt55 is larger than that with Ru13@Pt42, as discussed above, but the O2 moiety is less negatively charged in Pt55(O2) than in Ru13@Pt42(O2); see Table 1. As shown in Fig. 5C and D, the d-valence band-top and d-band center exist at higher energy in Pt55(O2) than in Ru13@Pt42(O2). The higher energy d-valence band-top and d-band center in Pt55(O2) than in Ru13@Pt42(O2) are the origin of the smaller Ea value of the O–O bond cleavage on Pt55 than on Ru13@Pt42. Considering these results, we mainly employ the d-valence band-top and the conduction band-bottom for discussion; these DOSs correspond to HOMO and LUMO in the frontier orbital theory in molecular theory. Also, we used the d-band center for discussion because it is an important property representing the electronic structure of metal particles.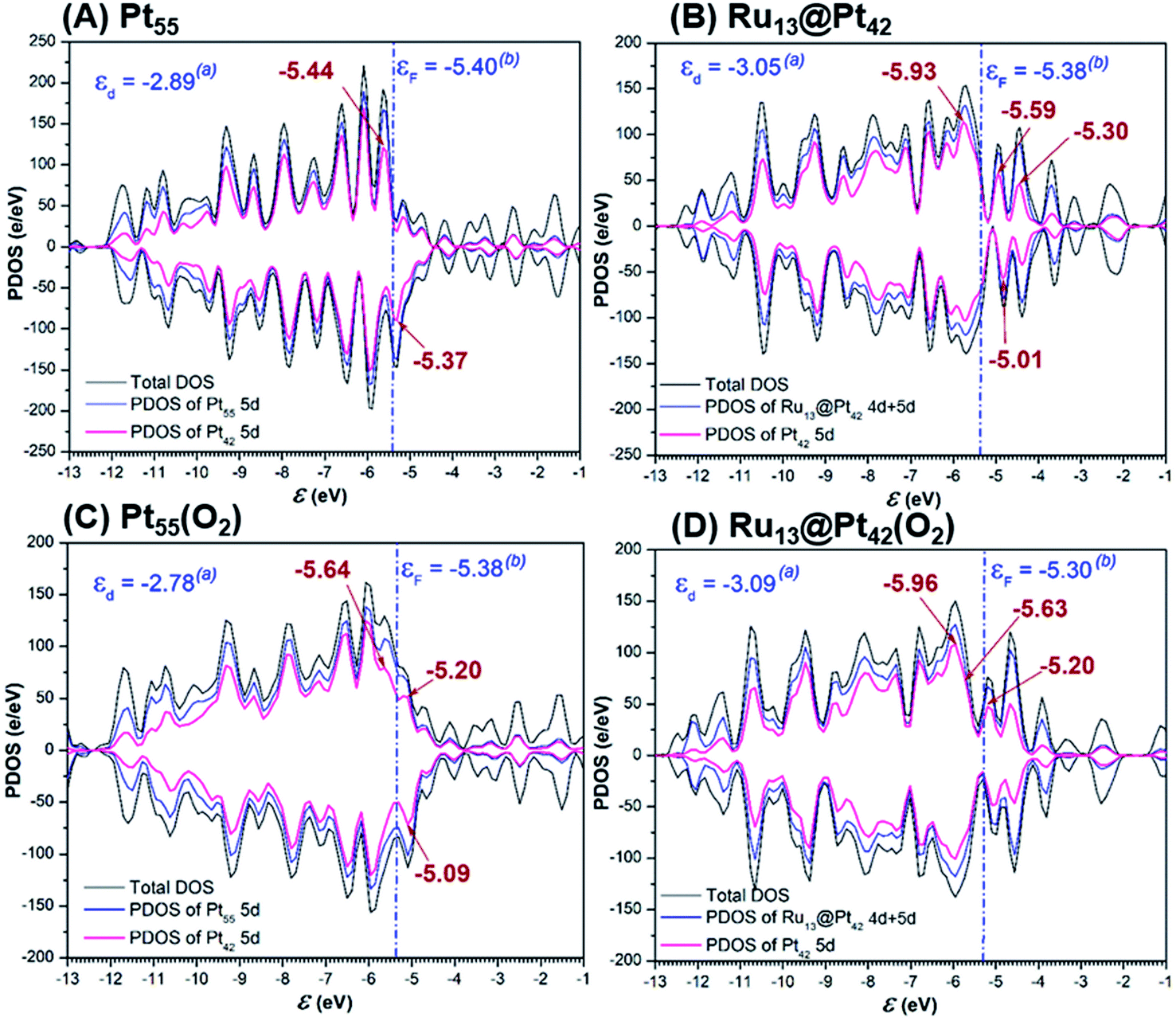 | ||
| Fig. 5 Density of states (DOS), partial density of states (PDOS) of total d of whole particle and 5d of the Pt42 shell in (A) Pt55, (B) Ru13@Pt42, (C) Pt55(O2), and (D) Ru13@Pt42(O2). | ||
In OOH formation, on the other hand, the charge distribution changes to a lesser extent than in the O–O bond cleavage, as mentioned above, but the Ea value is considerably different between Pt55 and Ru13@Pt42. This result suggests that some different factor plays an important role in this reaction. One plausible factor is bond dissociation energy (BDE). In OOH formation, M–O2 and M–H bonds are broken and M–(OOH) and O–H bonds are formed, where M represents Pt55 and Ru13@Pt42. Because the O–H bond formation is common in both Pt55 and Ru13@Pt42 cases, we focus here on M–O2, M–H, and M–(OOH) bonds. As shown in Scheme 3, the Pt55–(O2) and Pt55–H bonds are stronger than the Ru13@Pt42–(O2) and Ru13@Pt42–H bonds, respectively, in the reactant side, while the Pt55–(OOH) bond is stronger than the Ru13@Pt42–(OOH) bond in the product side. Therefore, two strong Pt55–(O2) and Pt55–H bonds (the sum of BDEs = 4.48 eV) are converted to one strong Pt55–(OOH) bond (BDE = 1.96 eV) in the OOH formation on Pt55, where the energy loss is 2.52 eV. In the OOH formation on Ru13@Pt42, on the other hand, two weaker Ru13@Pt42–(O2) and Ru13@Pt42–H bonds (the sum of BDEs = 3.52 eV) are converted to one weaker Ru13@Pt42–(OOH) bond (BDE = 1.55 eV), where the energy loss is 1.71 eV. Apparently, the reaction occurs more easily on Ru13@Pt42 than on Pt55 because of the smaller energy loss in the reaction by the former than by the latter. These results lead us to the conclusion that the stronger Pt55–(O2) and Pt55–H bonds than Ru13@Pt42–(O2) and Ru13@Pt42–H bonds, respectively, are reasons why OOH formation from adsorbed O2 and H needs a larger Ea on Pt55 than on Ru13@Pt42.
 | ||
| Scheme 3 Bond energy changes (in eV) in OOH formation followed by O–OH bond cleavage on Pt55 and Ru13@Pt42. | ||
Because the O–OH bond cleavage needs CT from the metal particle to the OOH moiety, the higher energy d-valence band-top and d-band center of Pt55 than those of Ru13@Pt42 are the origin of the smaller Ea on Pt55 than on Ru13@Pt42. In addition, the bond energies relating to this O–OH bond cleavage provide clear understanding of the larger reactivity of Pt55 than that of Ru13@Pt42, as follows: the Pt55–(OOH) bond is stronger than the Ru13@Pt42–(OOH) bond, as shown in Scheme 3, and the Pt55–O and Pt55–(OH) bonds are stronger than the Ru13@Pt42–O and Ru13@Pt42–(OH) bonds, respectively. This means that one stronger bond is broken, but two stronger bonds are formed in the O–OH bond cleavage by Pt55 than by Ru13@Pt42. Thus, Pt55 is more reactive for this reaction than Ru13@Pt42.
As discussed above, the stronger Pt55–(O2) and Pt55–X bonds (X = H, O, OH, and OOH) than the Ru13@Pt42–(O2) and Ru13@Pt42–X bonds, respectively, are responsible for the reactivity difference in OOH formation and O–OH bond cleavage between Pt55 and Ru13@Pt42. Also, it has been supposed that the overly strong binding energy of oxygen-containing species with the Pt electrode is unfavorable for ORR activity.17 Thus, it is of considerable importance to discuss the Pt55–X and Ru13@Pt42–X bond energies and determining factor of these bond energies. Because the Pt55–(O2) and Ru13@Pt42–(O2) bond energies were discussed above in terms of the d-valence band-top and d-band center energies, we focus here on Pt55–X and Ru13@Pt42–X bond energies. These bonds are neither pure ionic nor pure covalent, but they are understood to be strongly polarized covalent bonds. Polarized covalent bond energy Ecov (A–B) is approximately represented by eqn (1) on the basis of simple Hückel MO theory;82
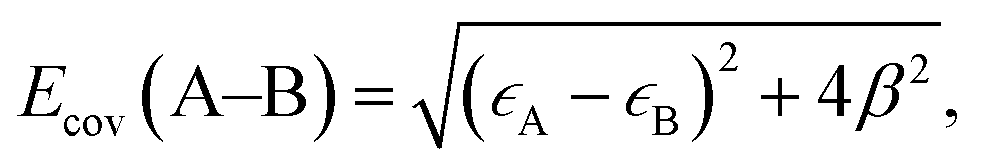 | (1) |
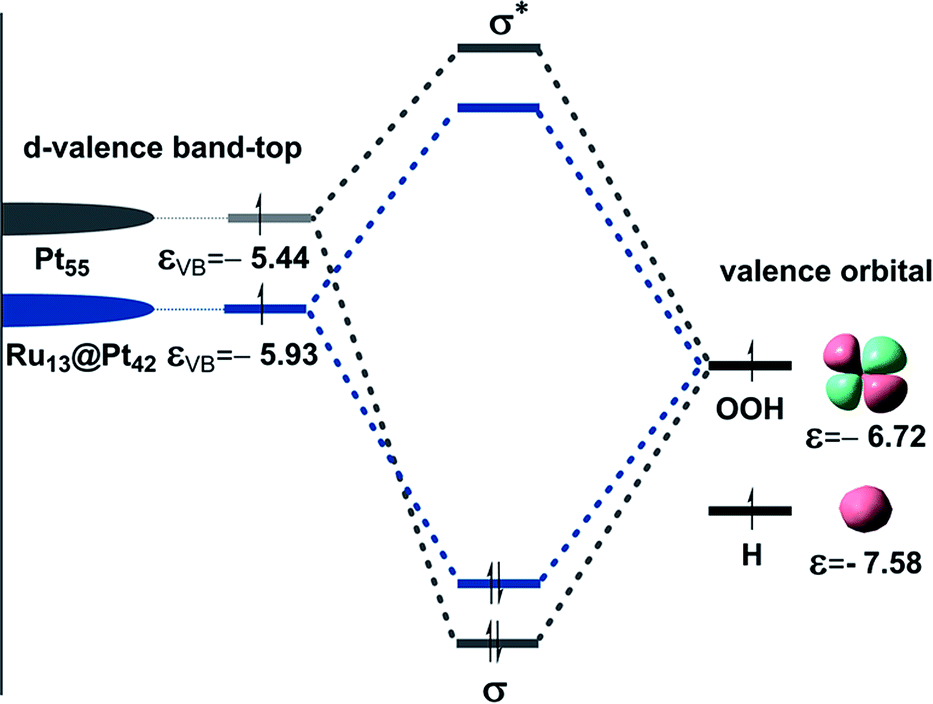 | ||
| Scheme 4 Schematic representation of orbital interaction between X (X = H or OOH) species and the Pt42 shell of Pt55 or Ru13@Pt42 particle. | ||
It should be clearly concluded that the higher energy d valence band-top of Pt55 than that of Ru13@Pt42 is the origin of the stronger Pt55–X bond than the Ru13@Pt42–X bond. The higher energy d valence band-top of Pt55 than that of Ru13@Pt42 is also responsible for the larger O2 adsorption energy to Pt55 than to Ru13@Pt42 and smaller Ea values of the O–O and O–OH bond cleavages on Pt55 than on Ru13@Pt42. Thus, one of the important characteristic features of Ru13@Pt42 is the presence of the d-valence band-top of the Pt42 shell at lower energy than that of Pt55; here we wish to mention that higher energy d-valence band-top relates to higher energy d-band center in many cases, indicating that the d-band center is also useful for discussion.
4 Conclusions
O2 adsorption followed by O–O bond cleavage and OOH formation followed by O–OH bond cleavage on Pt55 and Ru13@Pt42 particles were investigated using DFT computations, and comparisons were made between Pt55 and Ru13@Pt42. Several important findings are summarized as follows: (i) O2 is preferentially adsorbed to the vertex Pt and the neighboring edge Pt atoms in a bridging μ2-side-on manner. (ii) The O2 adsorption energy with the Pt42 shell is larger in Pt55 than in Ru13@Pt42. (iii) The O–O bond cleavage occurs with a smaller Ea on Pt55 than on Ru13@Pt42. (iv) The OOH formation from the adsorbed O2 molecule and H atom occurs with a smaller Ea on Ru13@Pt42 than on Pt55. The CT occurs much less in this reaction than in the O–O bond cleavage. The stronger Pt55–(O2) and Pt55–H bonds than the Ru13@Pt42–(O2) and Ru13@Pt42–H bonds, respectively, are the origin of the larger Ea on Pt55 than on Ru13@Pt42. (v) The OOH formation via H+/e− addition to the adsorbed O2 molecule also occurs easily in Pt55 similarly to the reaction between adsorbed O2 molecule and H atom, but more easily in Ru13@Pt42. And, (vi) the O–OH bond cleavage occurs more easily with much smaller Ea than the O–O bond cleavage of the adsorbed O2 molecule.The abovementioned differences between Pt55 and Ru13@Pt42 are understood on the basis of the PDOS of these metal particles. The d-valence band-top and d-band center of the Pt42 shell are calculated at higher energy in Pt55 than in Ru13@Pt42, but the d-conduction band-bottom of the Pt42 shell is at lower energy in Pt55 than in Ru13@Pt42. Accordingly, the O2 molecule is adsorbed to Pt55 more strongly than to Ru13@Pt42, because the CT from the Pt42 shell to O2 and the reverse CT from the O2 to the Pt42 shell are more strongly formed with Pt55 than with Ru13@Pt42. Because the O–O bond cleavage needs CT from the metal particle to the O2 moiety, the presence of d-valence band-top at high energy is favorable. Consequently, Pt55 is more reactive than Ru13@Pt42. On the other hand, the reactivity for OOH formation from adsorbed O2 and H depends on the M–(O2) and M–H bond energies, as follows: because the Pt55–(O2) and Pt55–H bonds are stronger than the Ru13@Pt42–(O2) and Ru13@Pt42–H bonds, respectively, OOH formation on Pt55 needs a larger Ea than that on Ru13@Pt42.
The binding energy of oxygen-containing species with Pt-based electrode has been discussed as an important factor for ORR activity. Also, the above discussion suggests that the bond energy is an important property for understanding reactions on the Pt-based electrode. We explored the Pt55–X (X = H, O, OH, and OOH) and Ru13@Pt42–X bond energies and found that the Ru13@Pt42–X bond is weaker than the Pt55–X, and the lower energy d-valence band-top of Ru13@Pt42 than that of Pt55 is the origin of the weaker Ru13@Pt42–X bond than the Pt55–X bond. It is clearly concluded that the lower energy of the d-valence band-top of Ru13@Pt42 than that of Pt55 is one of the important characteristic features of Ru13@Pt42 in comparison to Pt55.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was performed in part with support from the Ministry of Education, Culture, Science, Sports, and Technology (Japan) through the Element Strategy Initiative for Catalysts and Batteries (ESICB) in Kyoto University and from the Ministry of Economy, Trade, and Industry though the New Energy and Industrial Technology Development Organization (NEDO) through a project JPNP16010. We wish to thank the computational center at the Institute for Molecular Science (IMS; Okazaki, Japan) and High Performance Computation Infrastructure (HPCI; Kobe, Japan) for use of their computers.Notes and references
- M. E. Scofield, H. Q. Liu and S. S. Wong, Chem. Soc. Rev., 2015, 44, 5836–5860 RSC.
- H. L. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- Y. J. Wang, B. Z. Fang, H. Li, X. T. T. Bi and H. J. Wang, Prog. Mater. Sci., 2016, 82, 445–498 CrossRef CAS.
- S. Sui, X. Y. Wang, X. T. Zhou, Y. H. Su, S. Riffatc and C. J. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- J. P. Lai and S. J. Guo, Small, 2017, 13, 1702156 CrossRef.
- T. Asset, R. Chattot, M. Fontana, B. Mercier-Guyon, N. Job, L. Dubau and F. Maillard, ChemPhysChem, 2018, 19, 1552–1567 CrossRef CAS.
- K. Singh, E. B. Tetteh, H. Y. Lee, T. H. Kang and J. S. Yu, ACS Catal., 2019, 9, 8622–8645 CrossRef CAS.
- O. Lori and L. Elbaz, ChemCatChem, 2020, 12, 3434–3446 CrossRef CAS.
- M. Sharma, N. Jung and S. J. Yoo, Chem. Mater., 2018, 30, 2–24 CrossRef CAS.
- S. T. Hunt and Y. Roman-Leshkov, Acc. Chem. Res., 2018, 51, 1054–1062 CrossRef CAS.
- B. W. Zhang, H. L. Yang, Y. X. Wang, S. X. Dou and H. K. Liu, Adv. Energy Mater., 2018, 8, 1703597 CrossRef.
- N. K. Chaudhari, Y. Hong, B. Kim, S. I. Choi and K. Lee, J. Mater. Chem. A, 2019, 7, 17183–17203 RSC.
- X. F. Ren, Q. Y. Lv, L. F. Liu, B. H. Liu, Y. R. Wang, A. M. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- K. Sasaki, K. A. Kuttiyiel and R. R. Adzic, Curr. Opin. Electrochem., 2020, 21, 368–375 CrossRef CAS.
- L. J. Yang, M. B. Vukmirovic, D. Su, K. Sasaki, J. A. Herron, M. Mavrikakis, S. J. Liao and R. R. Adzic, J. Phys. Chem. C, 2013, 117, 1748–1753 CrossRef CAS.
- A. Jackson, V. Viswanathan, A. J. Forman, J. K. Nørskov and T. F. Jaramillo, ECS Trans., 2013, 58, 929–936 CrossRef.
- A. Jackson, V. Viswanathan, A. J. Forman, A. H. Larsen, J. K. Nørskov and T. F. Jaramillo, ChemElectroChem, 2014, 1, 67–71 CrossRef.
- D. Takimoto, T. Ohnishi, J. Nutariya, Z. R. Shen, Y. Ayato, D. Mochizuki, A. Demortiere, A. Boulineau and W. Sugimoto, J. Catal., 2017, 345, 207–215 CrossRef CAS.
- A. Jackson, A. L. Strickler, D. Higgins and T. F. Jaramillo, Nanomaterials, 2018, 8, 38 CrossRef.
- J. S. Zou, M. Wu, S. L. Ning, L. Huang, X. W. Kang and S. W. Chen, ACS Sustainable Chem. Eng., 2019, 7, 9007–9016 CrossRef CAS.
- Y. Sha, T. H. Yu, Y. Liu, B. V. Merinov and W. A. Goddard III, J. Phys. Chem. Lett., 2010, 1, 856–861 CrossRef CAS.
- T. H. Yu, Y. Sha, B. V. Merinov and W. A. Goddard III, J. Phys. Chem. C, 2010, 114, 11527–11533 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov, P. Shirvanian and W. A. Goddard III, J. Phys. Chem. Lett., 2011, 2, 572–576 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov, P. Shirvanian and W. A. Goddard III, J. Phys. Chem. C, 2012, 116, 21334–21342 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov and W. A. Goddard III, ACS Catal., 2014, 4, 1189–1197 CrossRef CAS.
- A. Fortunelli, W. A. Goddard III, Y. Sha, T. H. Yu, L. Sementa, G. Barcaro and O. Andreussi, Angew. Chem., Int. Ed., 2014, 53, 6669–6672 CrossRef CAS.
- H. C. Tsai, T. H. Yu, Y. Sha, B. V. Merinov, P. W. Wu, S. Y. Chen and W. A. Goddard III, J. Phys. Chem. C, 2014, 118, 26703–26712 CrossRef CAS.
- A. Fortunelli, W. A. Goddard III, L. Sementa, G. Barcaro and F. R. Negreiros, Chem. Sci., 2016, 6, 3915–3925 RSC.
- A. Fortunelli, W. A. Goddard III, L. Sementa and G. Barcaro, Nanoscale, 2015, 7, 4514–4521 RSC.
- H. C. Tsai, Y. C. Hsieh, T. H. Yu, Y. J. Lee, Y. H. Wu, B. V. Merinov, P. W. Wu, S. Y. Chen, R. R. Adzic and W. A. Goddard III, ACS Catal., 2015, 5, 1568–1580 CrossRef CAS.
- L. Sementa, O. Andreussi, W. A. Goddard III and A. Fortunelli, Catal. Sci. Technol., 2016, 6, 6901–6909 RSC.
- E. Toyoda, R. Jinnouchi, T. Hatanaka, Y. Morimoto, K. Mitsuhara, A. Visikovskiy and Y. Kido, J. Phys. Chem. C, 2011, 115, 21236–21240 CrossRef CAS.
- X. Liu, L. Li, C. G. Meng and Y. Han, J. Phys. Chem. C, 2012, 116, 2710–2719 CrossRef CAS.
- L. Zhang, R. Iyyamperumal, D. F. Yancey, R. M. Crooks and G. Henkelman, ACS Nano, 2013, 7, 9168–9172 CrossRef CAS.
- Z. Chen, X. Zhang and G. Lu, J. Phys. Chem. C, 2017, 121, 1964–1973 CrossRef CAS.
- T. Ikeshoji and M. Otani, Phys. Chem. Chem. Phys., 2017, 19, 4447–4453 RSC.
- X. Wang, W. Huang, S. Liao and B. Li, Mater. Chem. Phys., 2018, 212, 378–384 CrossRef CAS.
- Y. Wang, Y. F. Li and T. Heine, J. Am. Chem. Soc., 2018, 140, 12732–12735 CrossRef CAS.
- J. Huang and M. Eikerling, Curr. Opin. Electrochem., 2019, 13, 157–165 CrossRef CAS.
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS.
- S. Liu and P. Liu, J. Electrochem. Soc., 2018, 165, J3090–J3094 CrossRef CAS.
- X. Wang, W. Huang, S. Liao and B. Li, Mater. Chem. Phys., 2018, 212, 378–384 CrossRef CAS.
- Y. Zhuang, J.-P. Chou, P.-Y. Liu, T.-Y. Chen, J.-J. Kai, A. Hu and H.-Y. T. Chen, J. Mater. Chem. A, 2018, 6, 23326–23335 RSC.
- A. S. Nair and B. Pathak, J. Phys. Chem. C, 2019, 123, 3634–3644 CrossRef CAS.
- C. Fu, C. Liu, T. Li, X. Zhang, F. Wang, J. Yang, Y. Jiang, P. Cui and H. Li, Comput. Mater. Sci., 2019, 170(12), 1092022 Search PubMed.
- Z. Zhao, H. Xu, Z. Feng, Y. Zhang, M. Cui, D. Cao and D. Cheng, Chem.–Eur. J., 2020, 26, 4128–4135 CAS.
- J. Lu, K. Ishimura and S. Sakaki, J. Phys. Chem. C, 2018, 122, 9081–9090 CrossRef CAS.
- F. Viñes, J. R. B. Gomesb and F. Illas, Chem. Soc. Rev., 2014, 43, 4922–4939 RSC.
- S. T. Hunt and Y. Roman-Leshkov, Acc. Chem. Res., 2018, 51, 1054–1062 CrossRef CAS.
- J. A. Trindell, Z. Y. Duan, G. Henkelman and R. M. Crooks, Chem. Rev., 2020, 120, 814–850 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- G. Stefan, E. Stephan and G. Lars, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef.
- G. Mills and H. Jónsson, Phys. Rev. Lett., 1994, 72, 1124–1127 CrossRef CAS.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef.
- M. Fishman, H. L. Zhuang, K. Mathew, W. Dirschka and R. G. Hennig, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 245402 CrossRef.
- D. J. Cheng and W. C. Wang, Nanoscale, 2012, 4, 2408–2415 RSC.
- J. H. Shin, J.-H. Choi, Y.-S. Bae and S.-C. Lee, Chem. Phys. Lett., 2014, 610, 86–90 CrossRef.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Nanoscale, 2014, 6, 1153–1165 RSC.
- S. Dobrin, Phys. Chem. Chem. Phys., 2012, 14, 12122–12129 RSC.
- D. Cheng, H. Xu and A. Fortunelli, J. Catal., 2014, 314, 47–55 CrossRef CAS.
- L. Ma and J. Akola, Phys. Chem. Chem. Phys., 2019, 21, 11351–11358 RSC.
- T. Morishita, T. Ueno, G. Panomsuwan, J. Hieda, M. A. Bratescu and N. Saito, RSC Adv., 2016, 6, 98091–98095 RSC.
- F. Deushi, A. Ishikawa and H. Nakai, J. Phys. Chem. C, 2017, 121, 15272–15281 CrossRef CAS.
- N. Takagi, K. Ishimura, R. Fukuda, M. Ehara and S. Sakak, J. Phys. Chem. A, 2019, 123, 7021–7033 CrossRef CAS.
- B. Zhu, M. Ehara and S. Sakaki, Phys. Chem. Chem. Phys., 2020, 22, 11783–11796 RSC.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS.
- Q. Jia, C. U. Segre, D. Ramaker, K. Caldwell, M. Trahan and S. Mukerjee, Electrochim. Acta, 2013, 88, 604–613 CrossRef CAS.
- Q. Jia, W. Liang, M. K. Bates, P. Mani, W. Lee and S. Mukerjee, ACS Nano, 2015, 9, 387–400 CrossRef CAS.
- A. B. Anderson, R. A. Sidik, J. Narayanasamy and P. Shiller, J. Phys. Chem. B, 2003, 107, 4618–4623 CrossRef CAS.
- Q. Liang, J. Yu and J. Lia, J. Chem. Phys., 2006, 125, 054701 CrossRef.
- A. S. Haile, W. Yohannes and Y. S. Mekonnen, RSC Adv., 2020, 10, 27346–27356 RSC.
- R. A. Sidik and A. B. Anderson, J. Electroanal. Chem., 2002, 528, 69–76 CrossRef CAS.
- Y. X. Wang and P. B. Balbuena, J. Phys. Chem. B, 2004, 108, 4376–4384 CrossRef CAS.
- Y. X. Wang and P. B. Balbuena, J. Phys. Chem. B, 2005, 109, 14896–14907 CrossRef CAS.
- W. Guan, F. B. Sayyed, G. Zeng and S. Sakaki, Inorg. Chem., 2014, 53, 6444–6457 CrossRef CAS.
- C. J. Fall, N. Binggeli and A. Baldereschi, J. Phys.: Condens. Matter, 1999, 11, 2689–2696 CrossRef CAS.
- S. Sakaki, B. Biswas and M. Sugimoto, Organometallics, 1998, 17, 1278–1289 CrossRef CAS.
- S. Sakaki, S. Kai and M. Sugimoto, Organometallics, 1999, 18, 4825–4837 CrossRef CAS.
- B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 1999, 18, 4015–4026 CrossRef CAS.
- M. Ray, Y. Nakao, H. Sato and S. Sakaki, Organometallics, 2007, 26, 4413–4423 CrossRef CAS.
- M. Ray, Y. Nakao, H. Sato, H. Sakaba and S. Sakaki, Organometallics, 2009, 28, 65–73 CrossRef CAS.
- R. L. Zhong and S. Sakaki, J. Am. Chem. Soc., 2019, 141, 9854–9866 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Possible adsorption structures of 2(O) species and (O)(OH) species (Scheme S1); total energies for Pt55 and Pt42Ru13 in different possible spin states using PBE-D3 method (Table S1); binding energies (Eb, in eV) for O2 and OOH species (Table S2); binding energies (Eb, in eV) for O, OH, and H/x-binding species (Table S3); optimized structures of O2-binding species (Fig. S1), 2O-binding species (Fig. S2), OOH-binding species (Fig. S3), (O)(OH)-binding species (Fig. S4) and Cartesian coordinates of important optimized species discussed in the main text. The effect of box size for periodic calculation on the Fermi level (εF, eV) and the d-valence band top (εVB_top, eV) energies (Table S4). d-Valence band top energy (in eV) calculated using several different functionals and basis sets (Table S5). See DOI: 10.1039/d0ra05738j |
| This journal is © The Royal Society of Chemistry 2020 |