
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized pyridine in pyclen-based iron(III) complexes: evaluation of fundamental properties†
Magy A. Mekhail,
Kristof Pota ,
Timothy M. Schwartz and
Kayla N. Green*
,
Timothy M. Schwartz and
Kayla N. Green*
Department of Chemistry and Biochemistry, Texas Christian University, 2950 W. Bowie, Fort Worth, TX 76129, USA. E-mail: kayla.green@tcu.edu
First published on 26th August 2020
Abstract
The use of tetra-aza pyridinophanes is of increasing interest in the fields of bioinorganic modeling, catalysis, and imaging. However, a full study of how modifications to the pyridyl moiety affect the characteristics of the daughter metal complexes, has not been explored. In this study, six tetra-aza macrocyclic ligands were metalated with Fe(III) and were characterized for the first time. The pyridyl functional groups studied include: 4-hydroxyl (L1), 4-H (L2), 4-chloro (L3), 4-trifluoromethyl (L4), 4-nitrile (L5), and 4-nitro (L6) modified pyridyl on a pyclen base structure. The resulting iron complexes were characterized by X-ray diffraction analysis, cyclic voltammetry, and metal-binding affinities (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β) were determined. Analysis of these results indicate that such functionalizations introduce a handle by which electrochemical properties and thermodynamic stability of daughter complexes with transition metal ions can be tuned, which in turn, could potentially impact the reactivity of these complexes in future studies.
β) were determined. Analysis of these results indicate that such functionalizations introduce a handle by which electrochemical properties and thermodynamic stability of daughter complexes with transition metal ions can be tuned, which in turn, could potentially impact the reactivity of these complexes in future studies.
Introduction
Porphyrins, corrins, and chlorins are examples of macrocyclic compounds found in nature with vital roles in many animals (hemoglobin) and vegetation (chlorophyll).1 The reactions carried out by these systems are highly selective and take place under mild experimental conditions.1 The prospect of using model compounds to emulate such species in catalysis and medicine has been a driving force for efforts focused on the synthesis and modification of macrocyclic ligands dating back to 1926, with the first total synthesis of etioporphyrin-III and octamethylporphyrin.2 A large library of nitrogen-containing macrocycles has been developed in response to the increasing interest in the study and use of macrocyclic ligands, a class of which are pyridinophanes, also known as pyridine containing macrocycles.3–5 The unique attributes of introducing a pyridine moiety into macrocycles was recognized early on. For example, pyclen (1,4,7,10-tetra-aza-2,6-pyridinophane, Fig. 1) has conformational rigidity and decreased basicity compared to cyclen.6 Therefore, the inclusion of pyridine provides an avenue through which thermodynamic properties and complexation kinetics can be modulated.3–5,7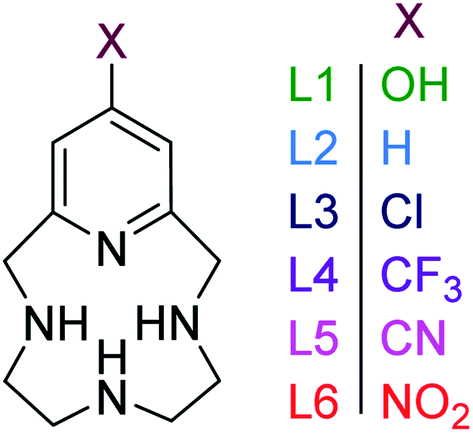 | ||
| Fig. 1 Pyridinophane macrocycles investigated in this study with different functional groups on the 4-position of the pyridine ring. | ||
Congeners of pyclen have been widely studied to tune their properties and reactivity.8 For example, it was observed that 12- and 13-membered cavities cannot accommodate planar coordination compared to 14-membered py-2,3-cyclam, which significantly affects the electrochemical properties of the complex and leads to the stabilization of rather uncommon metal oxidation states.9,10 Introduction of pendant arms, amine functionalization on the macrocycle, and differing numbers of coordinating atoms are all examples of other modifications that have been studied to tune reactivity of pyridinophanes to achieve changes to geometric rigidity, electrochemical properties, and coordination environments, respectively.11–16 As a result of these modifications, pyridinophanes have been successfully used as mimics of biological systems,17 scaffolds for magnetic resonance imaging,18 and ligands for catalysis.12,19–21
The impact of the functionalization of the pyridine moiety, which has not been evaluated in pyridinophanes to date, adds a new method of tuning the reactivity of the ligand and the redox activity of a metal chelated to the pyridinophane. Such modifications are anticipated to control the characteristics and reactivity of daughter metal complexes.22 However, pyridinophanes (PyN3) with functionalized pyridine rings have been scarcely reported to date, due to synthetic barriers. Our group has reported L1 (Fig. 1), with an –OH in the 4-position of the pyridine ring, as a potential therapeutic for neurodegenerative diseases.3,23 The former is a daughter molecule to the unsubstituted 12-membered pyridinophane pyclen, henceforth referred to as L2.24,25 Metal complexes derived from L2 and modifications to the C–C linkers have been extensively explored, as noted above.8,25–27 The 4-OMe substituted pyridinophane was studied as a precursor to a scaffold used for iron-catalyzed oxidation chemistry.26 The 4-Br substituted pyridinophane was developed as an intermediate for synthesis of molecules explored for biomedical imaging.28 Finally, our group has recently synthesized a series of ligands with electron-withdrawing groups (EWG) (L3–L6) to diversify pyridinophane chemistry and allow the development of this library.29
Here we report the impact of modified pyridine moieties on the iron(III) metal complexes using ligands L1–L6 (Fig. 1). The functional groups range from electron-donating (ED) (L1 (OH)) to electron-withdrawing (EW) (L3 (Cl) < L4 (CF3) < L5 (CN) < L6 (NO2)) and will be compared to the unsubstituted L2 (H). Such modifications are hypothesized to introduce a handle by which electrochemical properties of daughter complexes with transition metal ions can be tuned. The iron(III) complexes were characterized by X-ray diffraction analysis, cyclic voltammetry, and metal-binding affinities.
Results and discussion
Synthesis and X-ray crystallography of iron(III) complexes
Complexation was achieved by mixing equimolar ratios of iron(III) triflate with each ligand (L1–L6) in methanol and a minimal amount of water. X-ray quality crystals of FeL1 (orange), FeL2, FeL3, FeL4 and FeL5 (yellow), and FeL6 (brown) were obtained through slow evaporation of methanol solutions and used for the analyses described herein. The perchlorate salts of FeL1 and FeL2 were previously reported, but triflate salts were produced for consistency in this study.30 The results from X-ray diffraction analysis show that each complex adopts a six-coordinate, distorted octahedral geometry (N(2)–Fe–N(4) = 84–87°; N(1)–Fe–N(3) = 147–148°) (Table 1 and Fig. 2). The coordination sphere consists of four nitrogen donor atoms from the ligand set: two axial and two equatorial (cis). Likewise, two cis-coordinated chlorine atoms occupy the remaining equatorial positions around the iron(III) center. A line of symmetry was also observed for FeL5 and FeL6 through atoms N(2), Fe(1), and N(4), along with the functional group on the pyridine ring.| Bond angle (°)/length (Å) | FeL1 (
|
FeL2 (
|
FeL3 (
|
FeL4 (
|
FeL5 (
|
FeL6 (
|
|---|---|---|---|---|---|---|
| Cl(1)–Fe–Cl(2) | 92.06(3) | 98.37(14) | 100.084(19) | 100.34(2) | 92.83(3) | 99.95(13) |
| N(4)–Fe–N(2) | 87.23(8) | 85.21(4) | 84.45(6) | 84.52(7) | 85.94(14) | 84.4(4) |
| N(3)–Fe–N(1) | 148.45(8) | 147.08(5) | 147.19(6) | 147.11(8) | 147.32(15) | 147.1(4) |
| Fe–N(1) | 2.178(2) | 2.178(11) | 2.1719(11) | 2.1630(14) | 2.170(3) | 2.169(17) |
| Fe–N(2) | 2.156(2) | 2.197(12) | 2.2010(15) | 2.199(2) | 2.157(4) | 2.199(2) |
| Fe–N(3) | 2.175(2) | 2.158(11) | 2.1719(11) | 2.1630(14) | 2.170(3) | 2.169(7) |
| Fe–N(4) | 2.065(2) | 2.114(11) | 2.1163(15) | 2.127(2) | 2.105(4) | 2.131(2) |






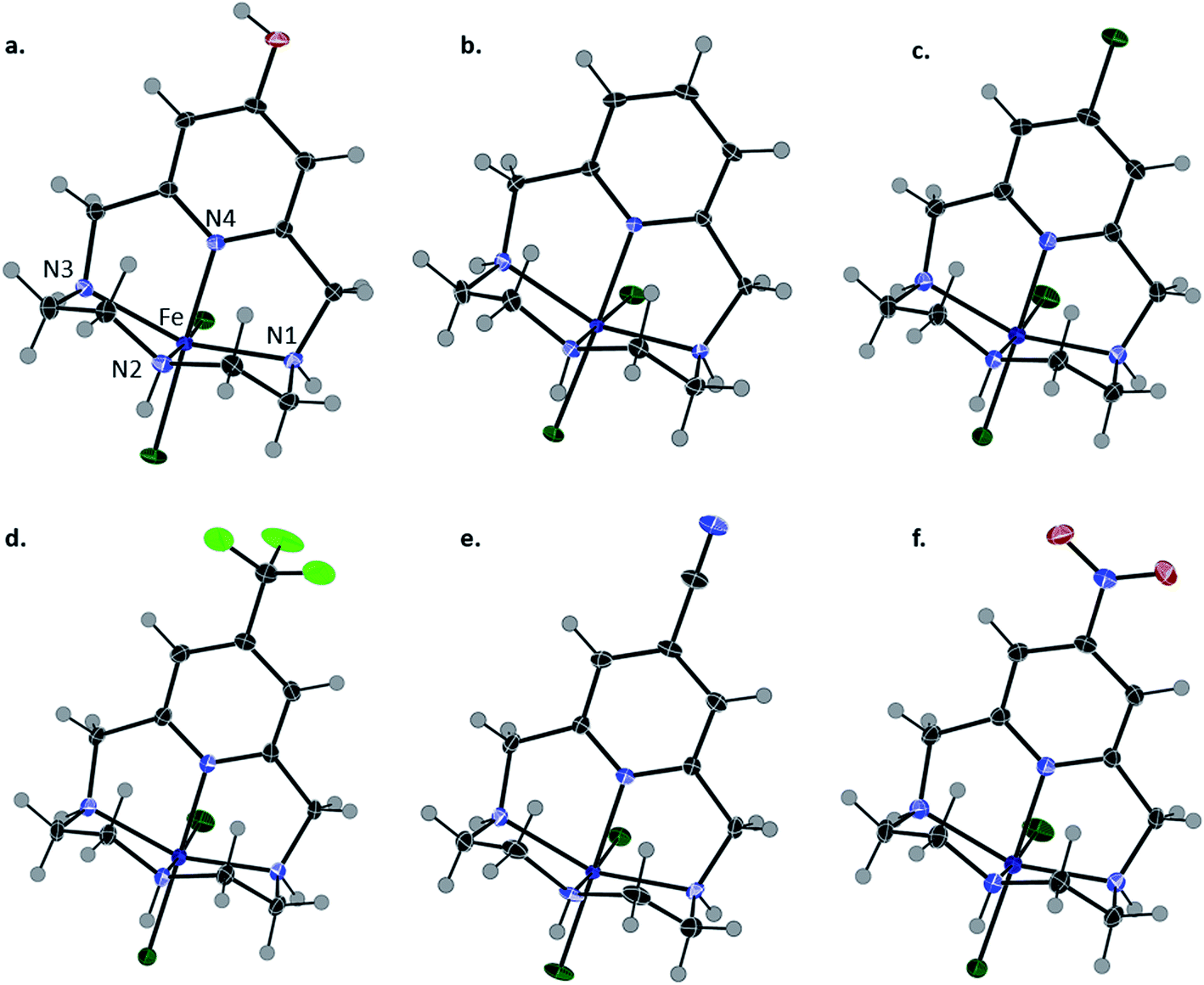 | ||
| Fig. 2 Solid-state structures of (a) FeL1 (OH), (b) FeL2 (H), (c) FeL3 (Cl), (d) FeL4 (CF3), (e) FeL5 (CN), and (f) FeL6 (NO2). Thermal ellipsoids are presented at 50%. Counterions are omitted for clarity. | ||
The Fe–Npyridine (Fe–N(4)) bond is most likely to be affected by the functionalization of the pyridine ring and was consistently observed to be the smallest of the Fe–N bonds in each complex. Within the series, the shortest Fe–N(4) bond distance (2.065(2)) was observed with FeL1 (largest EDG character), while the longest (2.131(2)) with FeL6 (largest EWG character). Compared to FeL1 and FeL2, the more electron withdrawing groups weaken the Fe–Npyridine bond as a result of the decreased localized electron density on the pyridine nitrogen. These results reveal that modifications to the pyridine moiety of the ligand provide subtle changes to the geometry of the complex.
Formation constants
Potentiometric titrations were used to identify correlations between functionalization of the pyridine ring in 5L1–L6 and stability constants (logβ) with iron(II) (Tables 2 and S1†). The formation of Fe(OH)3(s) and Fe2O3(s) at lower pH values precludes potentiometric titrations in the presence of Fe(III). Therefore, the stability of the Fe(III) species of L1–L5 was calculated using eqn (1).31 The binding affinity towards both iron(II) and iron(III) was observed to generally decrease with increased EW character. L1 (ED) was expected to have a larger stability constant in comparison to L2, however the observed data reveals otherwise. As previously observed, this ligand is observed in both enol and keto forms in solution (the ratio between the two is unknown).6
β) of the ML species formed, Fe(II)/(III)




The presence of the keto form lowers the formation constant of the iron complex because the non-bonding electron pair from the pyridine N-atom, which would be donated to the metal center, is incorporated as part of the delocalized seven-center eight-electron system. To further support this, the thermodynamic stability with iron(II) decreased as the EW character of the pyridine substitution increased, with the exception of L4 (Table 2). The observed preference of the electron donating ligand with ferric iron and the electron withdrawing ligands with ferrous iron is perhaps the most obvious evidence that functionalizing the pyridine ring can affect the complexes as a whole. Specifically, the iron(III) complexes derived from L1 and L2 were more thermodynamically favorable, compared to the iron(II) congeners. In contrast, larger stability constants were observed between ligands containing EWGs (L3–L5) with iron(II) as result of the ligands being electron deficient and, therefore, favoring the lower valent iron cation. Poor solubility of L6 (NO2) precluded potentiometric studies. Preliminary results of equilibrium studies of L4 with other metals reveal the same complexation trends between the divalent and trivalent iron and are the focus of an ongoing study.
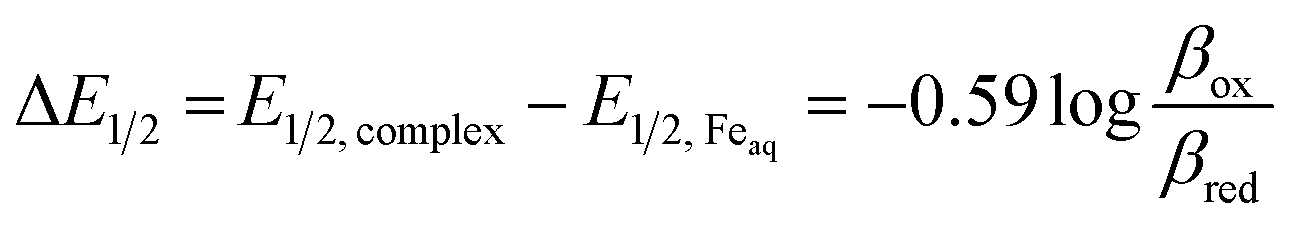 | (1) |
Electrochemistry
Cyclic voltammetry was used to further evaluate the impact of pyridine substitution on the electrochemical behavior of FeL1–FeL6. The cyclic voltammograms show that the iron(III/II) redox couple ranges between −470 mV and −360 mV for the series (Fig. 3). The results indicate that the observed iron(III/II) redox processes are quasi-reversible at 100 mV s−1 and diffusion controlled (Table 3), which is consistent with similar complexes in literature.20,30,32 The FeL1 (E1/2 = −462 mV) complex has the most negative half-cell potential followed by FeL2 (E1/2 = −453 mV). The potentials of the remaining complexes are more positive (favoring reduction) than FeL1 and FeL2, consistent with the pyridine functionalizations, and the most positive value observed with FeL6 (E1/2 = −372 mV). The trend observed is an indication that functionalization of the pyridine ring on the ligand can tune the electrochemical properties of the metal and potentially control reactivity. It is also important to point out that the trend observed with the electrochemical behavior of the iron complexes paralleled the thermodynamic stability determined for the metal complexes. The FeL4 complex was the exception to the trend observed with both the potentiometric and the electrochemical studies, suggesting there are more factors in play than simply the Hammett parameters derived from substituted benzoic acids.33 Interestingly, DFT analysis with a range of basis sets and functionals, including B3LYP,34,35 B3PW91,35,36 aug-cc-pvdz,37–39 aug-cc-pvtz,37–39 lanL2DZ,40 6-31++g(d,p),41–43 6-31++g(d′,p′),44 and 6-311++g(d,p),41,45 was not able to model the solid-state structures or other properties observed. This suggests that more is at play than simple changes to electronics. Such analysis is ongoing.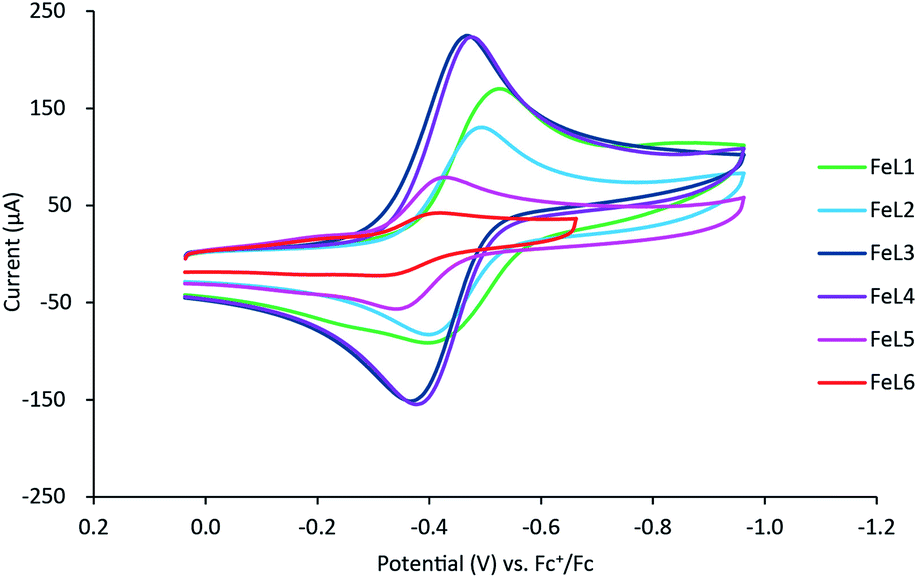 | ||
| Fig. 3 Cyclic voltammogram overlay of the iron(III/II) redox couple of FeL1–FeL6 in DMF (3–4 mM) containing [Bu4N][BF4] (100 mM) as the supporting electrolyte at a scan rate of 100 mV s−1. The potential values presented here have been normalized to the half-wave potential of the Fc+/Fc redox couple (E1/2 = 0.00 V). | ||
| Complexes | EPa (mV) | EPc (mV) | ΔE (mV) | E1/2 (mV) |
|---|---|---|---|---|
FeL1 ( ) ) |
−398 | −526 | 128 | −462 |
FeL2 ( ) ) |
−404 | −502 | 98 | −453 |
FeL3 ( ) ) |
−365 | −466 | 95 | −416 |
FeL4 ( ) ) |
−377 | −476 | 99 | −427 |
FeL5 ( ) ) |
−347 | −432 | 85 | −388 |
FeL6 ( ) ) |
−330 | −414 | 84 | −372 |
Additional oxidation (∼−1.5 mV) and reduction (−1.8 to −0.8 mV) events were also observed within the full experimental window of solutions containing FeL1–FeL6, which were hypothesized to be ligand-based. Therefore, cyclic voltammetry was conducted for ligands L1–L6 as well. The redox potentials observed are known to be a result of the pyridine ring.46 In these studies, reduction events were observed, in addition to the positive ligand-based oxidation reported previously with L1 and L2 (Fig. 4).30,47 Similar trends to the iron(III) complexes were observed for the reduction events in the ligands, where the more EWG character to the pyridine ring correlated to more positive reduction waves. It should be noted that the reduction wave of L6 (Epc = −783 mV) is significantly more positive compared to the other ligands (Table S14†), almost overlapping with the redox window of iron complexes themselves. Such activity is unique and potentially worthwhile to explore in possible further applications.
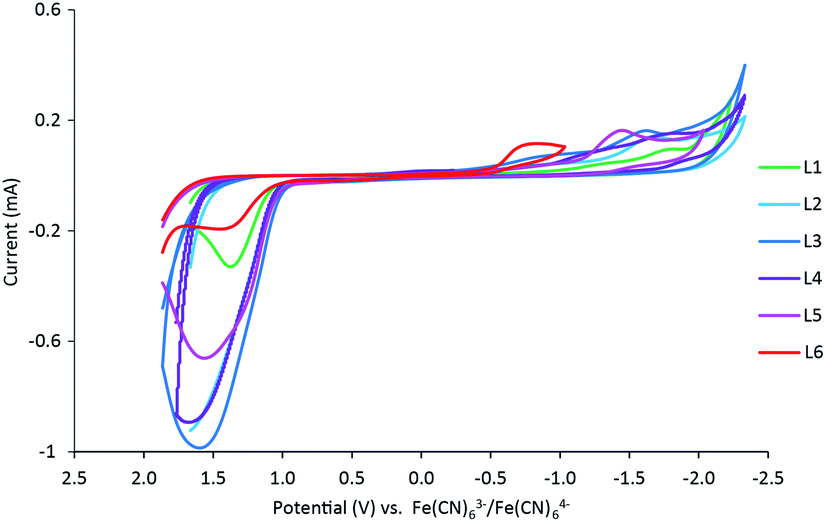 | ||
| Fig. 4 Cyclic voltammograms obtained for ligands L1–L6 in 3 mL of 0.1 M NaCl(aq) solution at a scan rate of 100 mV s−1 normalized to [Fe(CN)6]3−/[Fe(CN)6]4− (E1/2 = 0.00 V). | ||
Experimental
General methods
Elemental analysis was performed by Canadian Microanalytical Service Ltd. L1–L6 were synthesized using previously published procedures and isolated as water soluble trihydrochloride salts.3,23,29Potentiometric methods
The concentration of each ligand, as well as stability constants of the iron(II) complexes, were determined via pH-potentiometric titrations. A Metrohm 888 Titrando equipped with a Metrohm 6.0234.100 combined electrode was used to measure the pH in the titration experiments. For the calibration of the electrode, KH-phthalate (pH 4.008) and borax (pH 9.177) buffers were used.48,49 The calculation of [H+] from the measured pH values was performed with the use of the method proposed by Irving et al. by titrating a 0.02 M HCl solution with a standardized NaOH solution (0.2 M).50 The differences between the measured and calculated pH values were used to obtain the [H+] concentrations from the pH-data collected in the titrations. The ion product of water was determined from the same experiment in the pH range 11.40–12.00. The ionic strength in the titrated and thermostat controlled (at 25 °C) samples of 6.00 mL was kept constant and set to 0.15 M NaCl. The samples were stirred by a magnetic stirbar and kept under inert gas atmosphere (N2) to avoid the effect of CO2. The protonation constants of the ligands were obtained from previously reported data.29 The stability constants of the metal complexes were determined using the direct pH-potentiometric method by titrating samples with 1:1 metal-to-ligand ratios (the number of data pairs were between 100–250), allowing 1 min for sample equilibration to occur. The protonation and stability constants ((logβ) = [ML]/([M][L])) were calculated from the titration data with the PSEQUAD program.51
Synthesis of iron complexes
X-ray crystallography
Crystal diffraction data were collected at 100 K on a Bruker D8 Quest Diffractometer. Data collection, frame integration, data reduction (multi-scan), and structure determination were carried out using APEX2 software.52 Structural refinements were performed with XSHELL (v 6.3.1), by the full-matrix least-squares method.53 All non-hydrogen atoms were refined using anisotropic thermal parameters, while the hydrogen atoms were treated as mixed. The ORTEP molecular plots (50%) were produced using APEX2 (Version 2014.9-0).Electrochemistry
Cyclic voltammetry experiments for the Fe(III) complexes (FeL1–FeL6) used 3–4 mM complex and 100 mM [Bu4N][BF4] as the supporting electrolyte in DMF. The electrochemical cell was composed of a working glassy carbon electrode, a Pt auxiliary electrode, and a silver wire as the reference electrode. The potential values presented here have been normalized to the half-wave potential of the Fc+/Fc = 0.00 mV. Cyclic voltammetry experiments for ligands L1–L6 used 3–4 mM of ligand and 100 mM NaCl as the supporting electrolyte in water. The electrochemical cell was composed of a working glassy carbon electrode, a Pt auxiliary electrode, and Ag/AgCl as the reference electrode. The potential values presented here have been normalized to the half-wave potential of the [Fe(CN)6]3−/[Fe(CN)6]4− = 0.00 mV.Conclusions
Through this study, a library of 4-substituted pyridinophanes, ranging from electron donating to electron withdrawing, was metalated with iron(III), studied using single crystal X-ray diffraction, electrochemistry, and potentiometric titrations to study the impact of 4-substitution of the pyridine ring on the iron complex. Single crystal X-ray diffraction revealed that the substitution on the pyridine play only a small role in impacting coordination geometry. Cyclic voltammograms revealed that reduction is favored (more positive potentials) in case of ligands with more EW character. The binding stability constants supported the electrochemistry results in revealing that the ligands with EWGs favored Fe(II) over Fe(III). The effect of the substitution observed in the conducted studies points towards a new method of tuning the properties and potentially the reactivity of the iron complex.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Institutes of Health (R15GM123463). The authors are also grateful for generous financial support from TCU Andrews Institute of Mathematics & Science Education and Cambridge Isotope Laboratories. The authors acknowledge the TCU High-Performance Computing Center for providing HPC resources.Notes and references
- E. C. Constable, Coordination Chemistry of Macrocyclic Compounds, Oxford University Press, 1999 Search PubMed.
- J. K. H. Fischer, Justus Liebigs Ann. Chem., 1926, 448, 178–193 CrossRef.
- K. M. Lincoln, M. E. Offutt, T. D. Hayden, R. E. Saunders and K. N. Green, Inorg. Chem., 2014, 53, 1406–1416 CrossRef CAS PubMed.
- F. Dioury, S. Sambou, E. Guene, M. Sabatou, C. Ferroud, A. Guy and M. Port, Tetrahedron, 2007, 63, 204–214 CrossRef CAS.
- R. M. Nunes, R. Delgado, M. F. Cabral, J. Costa, P. Brandao, V. Felix and B. J. Goodfellow, Dalton Trans., 2007, 4536–4545, 10.1039/b710122h.
- K. N. Green, K. Pota, G. Tircso, R. A. Gogolak, O. Kinsinger, C. Davda, K. Blain, S. M. Brewer, P. Gonzalez, H. M. Johnston and G. Akkaraju, Dalton Trans., 2019, 48, 12430–12439 RSC.
- K. P. Guerra, R. Delgado, M. G. B. Drew and V. Felix, Dalton Trans., 2006, 4124–4133, 10.1039/b603484e.
- S. Xu, L. Bucinsky, M. Breza, J. Krzystek, C. H. Chen, M. Pink, J. Telser and J. M. Smith, Inorg. Chem., 2017, 56, 14315–14325 CrossRef CAS PubMed.
- M. Rezaeivala and H. Keypour, Coord. Chem. Rev., 2014, 280, 203–253 CrossRef CAS.
- A. M. Herrera, G. V. Kalayda, J. S. Disch, R. P. Wikstrom, I. V. Korendovych, R. J. Staples, C. F. Campana, A. Y. Nazarenko, T. E. Haas and E. V. Rybak-Akimova, Dalton Trans., 2003, 4482–4492, 10.1039/b308557k.
- B. Verdejo, A. Ferrer, S. Blasco, C. E. Castillo, J. Gonzalez, J. Latorre, M. A. Manez, M. Garcia Basallote, C. Soriano and E. Garcia-Espana, Inorg. Chem., 2007, 46, 5707–5719 CrossRef CAS PubMed.
- S. Taktak, W. H. Ye, A. M. Herrera and E. V. Rybak-Akimova, Inorg. Chem., 2007, 46, 2929–2942 CrossRef CAS PubMed.
- W. H. Ye, R. J. Staples and E. V. Rybak-Akimova, J. Inorg. Biochem., 2012, 115, 1–12 CrossRef CAS PubMed.
- W. H. Ye, D. M. Ho, S. Friedle, T. D. Palluccio and E. V. Rybak-Akimova, Inorg. Chem., 2012, 51, 5006–5021 CrossRef CAS PubMed.
- J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. Garcia-Espana, M. G. Basallote, E. Munck, L. Que, A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15842 CrossRef CAS PubMed.
- J. R. Khusnutdinova, J. Luo, N. P. Rath and L. M. Mirica, Inorg. Chem., 2013, 52, 3920–3932 CrossRef CAS PubMed.
- S. G. Rzuczek, D. S. Pilch, A. Liu, L. Liu, E. J. LaVoie and J. E. Rice, J. Med. Chem., 2010, 53, 3632–3644 CrossRef CAS PubMed.
- B. Drahos, J. Kotek, I. Cisarova, P. Hermann, L. Helm, I. Lukes and E. Toth, Inorg. Chem., 2011, 50, 12785–12801 CrossRef CAS PubMed.
- G. Tseberlidis, D. Intrieri and A. Caselli, Eur. J. Inorg. Chem., 2017, 3589–3603, DOI:10.1002/ejic.201700633.
- S. M. Brewer, K. R. Wilson, D. G. Jones, E. W. Reinheimer, S. J. Archibald, T. J. Prior, M. A. Ayala, A. L. Foster, T. J. Hubin and K. N. Green, Inorg. Chem., 2018, 57, 8890–8902 CrossRef CAS PubMed.
- J. Wen, S. Qin, L. F. Ma, L. A. Dong, J. Zhang, S. S. Liu, Y. S. Duan, S. Y. Chen, C. W. Hu and X. Q. Yu, Org. Lett., 2010, 12, 2694–2697 CrossRef CAS PubMed.
- L. Chiang, K. Herasymchuk, F. Thomas and T. Storr, Inorg. Chem., 2015, 54, 5970–5980 CrossRef CAS PubMed.
- K. M. Lincoln, P. Gonzalez, T. E. Richardson, D. A. Julovich, R. Saunders, J. W. Simpkins and K. N. Green, Chem. Commun., 2013, 49, 2712–2714 RSC.
- H. Stetter, W. Frank and R. Mertens, Tetrahedron, 1981, 37, 767–772 CrossRef CAS.
- J. Costa and R. Delgado, Inorg. Chem., 1993, 32, 5257–5265 CrossRef CAS.
- R. X. Fan, J. Serrano-Plana, W. N. Oloo, A. Draksharapu, E. Delgado-Pinar, A. Company, V. Martin-Diaconescu, M. Borrell, J. Lloret-Fillol, E. Garcia-Espana, Y. S. Guo, E. L. Bominaar, L. Que, M. Costas and E. Munck, J. Am. Chem. Soc., 2018, 140, 3916–3928 CrossRef CAS PubMed.
- G. P. Shkil and R. S. Sagitullin, Khim. Geterotsikl., 1998, 579–602 Search PubMed.
- S. Aime, M. Botta, L. Frullano, S. G. Crich, G. B. Giovenzana, R. Pagliarin, G. Palmisano and M. Sisti, Chem.–Eur. J., 1999, 5, 1253–1260 CrossRef CAS.
- A. Yepremyan, M. A. Mekhail, B. P. Niebuhr, K. Pota, N. Sadagopan, T. M. Schwartz and K. N. Green, J. Org. Chem., 2020, 85, 4988–4998 CrossRef CAS PubMed.
- S. M. Brewer, P. M. Palacios, H. M. Johnston, B. S. Pierce and K. N. Green, Inorg. Chim. Acta, 2018, 478, 139–147 CrossRef CAS.
- M. Botta, M. Ravera, A. Barge, M. Bottaro and D. Osella, Dalton Trans., 2003, 1628–1633, 10.1039/b211533f.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. A. Lewis, K. C. MacLeod, B. Q. Mercado and P. L. Holland, Chem. Commun., 2014, 50, 11114–11117 RSC.
- P. Murray, L. Jack, E. J. L. McInnes and L. J. Yellowlees, Dalton Trans., 2010, 39, 4179–4185 RSC.
- G. G. Manov, N. J. Delollis, P. W. Lindvall and S. F. Acree, J. Res. Natl. Bur. Stand., 1946, 36, 543–558 CrossRef CAS.
- G. G. Manov, N. J. Delollis and S. F. Acree, J. Res. Natl. Bur. Stand., 1945, 34, 115–127 CrossRef CAS.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- D. Domyati, S. L. Hope, R. Latifi, M. D. Hearns and L. Tahsini, Inorg. Chem., 2016, 55, 11685–11693 CrossRef CAS PubMed.
- Bruker Advanced X-ray Solutions, APEX2, Version 2014.9-0, Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- Bruker Advanced X-ray Solutions, SADABS, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1977733, 1984171–1984175. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05756h |
| This journal is © The Royal Society of Chemistry 2020 |