
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel rhodamine dye with large Stokes shifts by fusing the 1,4-diethylpiperazine moiety and its applications in fast detection of Cu2+†
Jin Gongab,
Chang Liua,
Xiaojie Jiaoa,
Song Hea,
Liancheng Zhaoab and
Xianshun Zeng *ab
*ab
aTianjin Key Laboratory for Photoelectric Materials and Devices, School of Materials Science & Engineering, Tianjin University of Technology, Tianjin, 300384, China. E-mail: xshzeng@tjut.edu.cn
bSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin, 150001, China
First published on 14th October 2020
Abstract
Rhodamine dyes were widely developed for designing probes due to their excellent photophysical properties and biocompatibility. However, traditional rhodamine dyes still bear major drawbacks of short emission wavelengths (<600 nm) and narrow Stokes shifts (<30 nm), which limit their biological imaging applications. Herein, we reported a novel mitochondria-targeted fluorescent dye JRQ with near-infrared (NIR) emission wavelength and improved Stokes shift (63 nm) by tuning the donor–acceptor–donor (D–A–D) character of the rhodamine skeleton. As expected, JRQ exhibited multiple excellent properties and could accumulate in mitochondria, and can therefore be used as a signal reporter for the design of fluorescent probes by taking advantage of the fluorescence controlled mechanism of the ring opening and closing chemical processes of the spirolactone platform. By using JRQ as a precursor, a highly sensitive fluorescent probe JRQN for the fast detection of mitochondrial Cu2+ ions was synthesized based on the Cu2+-triggered specific hydrolysis mechanism because mitochondria are an important reservoir of intracellular Cu2+. We expect that the Stokes shift increase of rhodamine dyes via tuning the donor–acceptor–donor (D–A–D) character of the rhodamine skeleton will provide a novel synthetic approach for the development of rhodamine dyes and expansion of their applications.
Introduction
Rhodamine derivatives were widely developed for designing probes due to their excellent photophysical properties and biocompatibility, such as tolerance to photobleaching, high quantum yields, and distinct photophysical advantages of ring-opening process.1 However, traditional rhodamine-based probes still bear some major drawbacks, such as short emission wavelengths (<600 nm) and narrow Stokes shifts (<30 nm), which are the main reasons for self-quenching, excitation wavelength interference and photodamage in living cells.2 Next generation applications, e.g. single-molecule analysis, demand fluorescent probes with NIR emission wavelength and large Stokes shifts to achieve precise imaging and accurate sensing.3 To data, several strategies have been introduced to increase the emission wavelength, for example, extending the π-conjugated system of rhodamine scaffolds and replacing the bridging oxygen atom with other atoms.4 However, these strategies cannot effectively improve the Stokes shifts. Therefore, it is of great significance to design and synthesize new rhodamine dyes with near-infrared (NIR) emission wavelengths and large Stokes shifts.Recently, it has been reported that altering the intramolecular charge transfer (ICT) properties of chromophoric skeleton by tuning electron-donating character of the chromophore is an efficient strategy to increase the absorption and emission wavelengths of rhodamine dyes.5 In pioneering works, the emission wavelengths of dyes were prominently red-shifted by fusing a piperazine in the xanthene skeleton as strong electron-donating group, increased with the Stokes shifts.6 For example, Zhang and coworkers demonstrated that the emission wavelengths of rhodamine dyes are prominently red-shifted by using the 1,4-diethyl-1,2,3,4-tetrahydroquinoxaline unit as a strong electron-donating group to modify the xanthene moiety, increased with the Stokes shifts.6a In 2018, Luck and Liu et al. reported the incorporation of a piperazine fused ring on the rhodamine scaffold, which also both increased the emission wavelengths and the Stokes shifts.6b These results have inspired our synthetic efforts aimed at the development of new rhodamine dyes with NIR emission wavelengths and improved Stokes shifts.
In this paper, we design of a new rhodamine dye with NIR emission wavelengths and larger Stokes shifts by introducing the electron-donating 1,4-diethylpiperazine moiety in rhodamine scaffold for tuning the D–A–D character of rhodamine dyes. In addition, the cationic xanthene moiety normally works as a functional group for targeting mitochondria,7 and increase the water solubility of the dye. As expected, JRQ exhibited multiple excellent properties and was capable of specifically distributing in mitochondria, which can be used as signal reporter for the design of fluorescence probes by taking advantage of the fluorescence controlled mechanism of the ring opening and closing chemical processes of the spirolactone platform. Furthermore, to demonstrate the utility of this dye for probe design, a highly sensitive and selective fluorescent probe JRQN for the specific detection of mitochondrial Cu2+ ions was synthesized based on the Cu2+-triggered specific hydrolysis mechanism because mitochondria are important reservoir of intracellular Cu2+.
Results and discussion
Molecular synthesis
The dye JRQ and probe JRQN were synthesized according to the procedures in Scheme 1. The intermediate compounds 4-methoxy-1,2-phenylenediamine (A), 1,4-diethyl-6-methoxy-1,2,3,4-tetrahydroquinoxaline (B) and 2-(2-hydroxyjulolidine) benzoic acid (C) were synthesized according to the literature,6a,8 and the detailed reaction reagents and conditions were depicted in Scheme 1. By the reaction of 2-(4-diethylamino-2-hydroxybenzoyl)benzoic acid with B, the dye JRQ was obtained in 76% yield. Followed by the reaction of the dye JRQ with hydrazine hydrate in methanol, the probe JRQN was obtained in 53% yield. The structures of the new compounds JRQ and JRQN were characterized by spectroscopic techniques, for example, 1H NMR, 13C NMR and HRMS, and the spectra were presented in the ESI† in detail.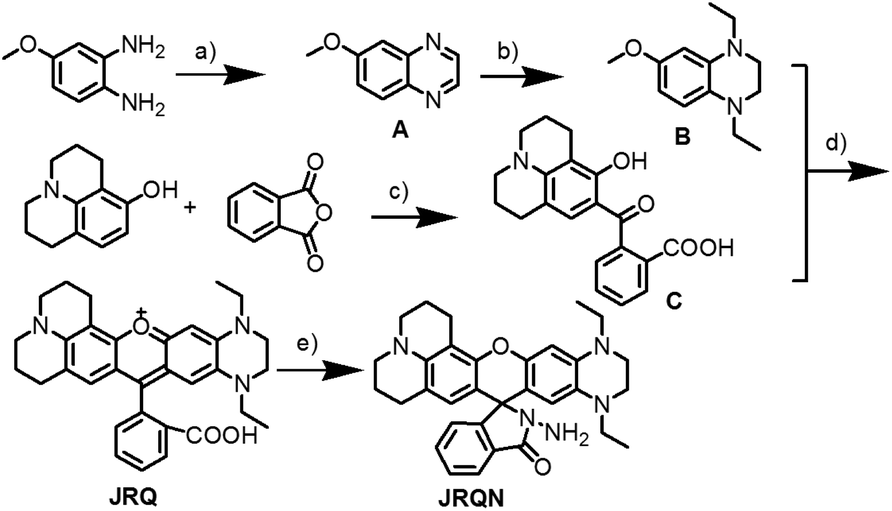 | ||
| Scheme 1 Synthesis of dye JRQ and probe JRQN. Reagents and conditions: (a) succinaldehyde, CH3CN, 60 °C, overnight; (b) NaBH4, toluene, AcOH, 5 °C to reflux, 5 h; (c) toluene, reflux, overnight; (d) CH3SO3H, 80 °C, 8 h; (e) NH2NH2, MeOH, 50 °C, 6 h. | ||
Characterization of JRQ
In order to verify whether JRQ meets our original design intentions, a detailed evaluation of the photophysical properties of the dye JRQ in distinct solvents (CH3CN, DMSO, DCM, EtOH, and H2O) was conducted. Surprisingly, JRQ showed the maxima absorption bands between 583 nm and 590 nm (ε ≥ 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 M−1 cm−1), and the maxima emission peaks of JRQ range from 654 nm to 668 nm in all test solvents (Table S1 and Fig. S1†). Comparatively, JRQ showed longer emission and larger Stokes shift than traditional rhodamine dyes in ethanol (Table 1). The maximum fluorescence emission of the dye was remarkably red-shifted ca. 61 nm (rhodamine 101), 86 nm (rhodamine B), and 100 nm (rhodamine 6G), respectively, which may be attributed to the excited intramolecular charge transfer (ICT). Meanwhile, the Stokes shift of JRQ was calculated to be 63 nm in ethanol, which was significantly larger than rhodamine 101 (30 nm), rhodamine B (26 nm), and rhodamine 6G (28 nm), respectively. It can be rationalized that the change of the electron-donating 1,4-diethylpiperazine group in the asymmetric xanthene fluorophore can regulate the donor–acceptor–donor (D–A–D) character of the chromophore. The donor–acceptor system in plane from the electron-donating 1,4-diethylpiperazine group to the electron-withdrawing group in JRQ can lead to large changes in the energy difference between the ground and the excited state, thereby producing an obviously increased Stokes shifts.9 Furthermore, the quantum yields of JRQ range from 5% to 54% in these solvents (Table S1†). Collectively, it proved that JRQ was a novel NIR fluorescence reagent with large Stokes shift, achieving original design intentions.
200 M−1 cm−1), and the maxima emission peaks of JRQ range from 654 nm to 668 nm in all test solvents (Table S1 and Fig. S1†). Comparatively, JRQ showed longer emission and larger Stokes shift than traditional rhodamine dyes in ethanol (Table 1). The maximum fluorescence emission of the dye was remarkably red-shifted ca. 61 nm (rhodamine 101), 86 nm (rhodamine B), and 100 nm (rhodamine 6G), respectively, which may be attributed to the excited intramolecular charge transfer (ICT). Meanwhile, the Stokes shift of JRQ was calculated to be 63 nm in ethanol, which was significantly larger than rhodamine 101 (30 nm), rhodamine B (26 nm), and rhodamine 6G (28 nm), respectively. It can be rationalized that the change of the electron-donating 1,4-diethylpiperazine group in the asymmetric xanthene fluorophore can regulate the donor–acceptor–donor (D–A–D) character of the chromophore. The donor–acceptor system in plane from the electron-donating 1,4-diethylpiperazine group to the electron-withdrawing group in JRQ can lead to large changes in the energy difference between the ground and the excited state, thereby producing an obviously increased Stokes shifts.9 Furthermore, the quantum yields of JRQ range from 5% to 54% in these solvents (Table S1†). Collectively, it proved that JRQ was a novel NIR fluorescence reagent with large Stokes shift, achieving original design intentions.
| Dyes | λex (nm) | λem (nm) | Δλ (nm) |
|---|---|---|---|
| Rhodamine 101 | 564 | 594 | 30 |
| Rhodamine B | 543 | 569 | 26 |
| Rhodamine 6G | 527 | 555 | 28 |
| JRQ | 592 | 655 | 63 |
Fluorescent imaging of JRQ
The excellent photophysical properties, for example, NIR emission wavelengths and improved Stokes shifts, encouraged us to examine the potential applications of JRQ in living cells. Before the imaging experiments in living cells, the well-established MTT assay was adopted to study cytotoxicity of the aforementioned dye at varying concentrations (0, 1, 3, 5, 10, and 20 μM). As shown in Fig. S2,† more than 85% of cells survived after treatment with various concentrations of JRQ (0, 1, 3, 5, 10, and 20 μM) for 24 h, indicated that the dye has fairly low toxicity to living cells. Then imaging experiments of JRQ in living cells were carried out. HeLa cells were incubated with JRQ for 30 min, and then washed three times with PBS for removing excess reagents in the growth medium. As shown in Fig. S3,† strong red fluorescence signals were observed after treatment with JRQ (1 μm) for 30 min, indicating that the dye was membrane permeability and could be used for imaging in living cells.By carefully observing the overlap images, we found that the distribution of the red signal in the cells unevenly fill the entire cytoplasm, but concentrated in some regions, indicating that JRQ likely possessed organelles localization properties. According to the literature, the xanthene dyes leads to the proactive accumulation in mitochondria,7 therefore, JRQ was likely to accumulate in mitochondria. To verify the speculated organelles localization properties, the co-localization experiments through co-incubating the HeLa cells with commercial targeting reagents Mito-Tracker Green (Mito), Golgi-Tracker Green (Golgi), ER-Tracker Green (ER), and Lyso-Tracker Green (Lyso), respectively, were conducted. As expected, highlight yellow pixels on overlapping images were observed in Fig. 1a, and a high overlap coefficient (0.92) and Pearson's coefficient (0.91) could be calculated from the intensity correlation plot, suggesting that JRQ was mainly distributed in mitochondria. In contrast, red fluorescence signals of JRQ and green fluorescence signals of other trackers (Lyso, ER, or Golgi) were distinctly distributed in different regions, and low overlap coefficients and Pearson's coefficients were observed (Table S2†), indicating that JRQ does not accumulate in these organelles. Co-localization experiments confirmed that JRQ can be used as an effective mitochondrial targeting reagent.
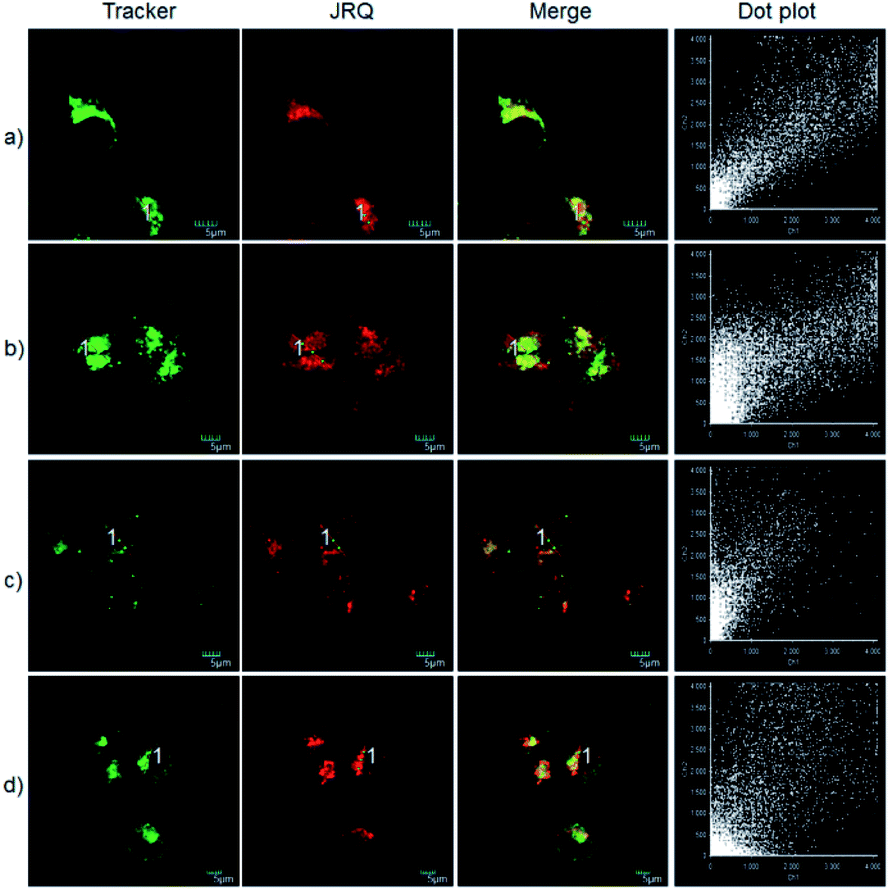 | ||
| Fig. 1 Co-localization experiments with HeLa cells stained with JRQ (1 μM) and other organelle-targeting Trackers (200 nM). (a) Costained with Mito-Tracker Green; (b) costained with Lyso-Tracker Green, (c) costained with ER-Tracker Green; (d) costained with Golgi-Tracker Green. Red channel: λex = 559 nm, λem = 618–718 nm; green channel: λex = 488 nm, λem = 520–541 nm. | ||
Recognition properties of JRQN
Copper is a redox-active metal that has a wide range of applications in the chemical and electronics industries, and plays an indispensable role in various physiological processes as enzyme cofactors and signal messengers, including cellular respiration, neurotransmitter synthesis and metabolism, maintenance of the immune system, redox process, bone health, etc.10 As a cofactor for the respiratory enzyme cytochrome c oxidase, mitochondria are important reservoir of intracellular copper.11 Therefore, humans must supplement their normal intake by means of food, and the average daily copper intake of adults should not exceed 10–12 mg.12 It has been proved that abnormal levels of copper (Cu2+) in the body can cause diseases, for example, Alzheimer's disease, coronary heart disease, Wilson's disease, arthritis, brain dysfunction, anemia, Menkes disease and vitiligo.13 Consequently, real-time sensing Cu2+ levels in mitochondria is important for early detection and prevention of diseases. However, up to now, mitochondria-targeted NIR fluorescent probes of rhodamine derivatives with large Stokes shifts for detection of Cu2+ were rarely described.14 Therefore, it is of great significance to design and synthesize new NIR fluorescent probes with large Stokes shifts, for sensing Cu2+ in mitochondria.To demonstrate the utility of this dye for probe design, a highly sensitive fluorescent probe JRQN for the fast detection of mitochondrial Cu2+ ions was synthesized based on the Cu2+-triggered specific hydrolysis mechanism because mitochondria are important reservoir of intracellular Cu2+.15 With JRQN in hand, the different metalated analogues were evaluated, in order to study the influence of the metals on the absorption and emission spectrum of the probe. As shown in the Fig. 2a, upon addition of 10 equiv. of Cu2+, the intensity of the fluorescence emission spectrum at 678 nm was significantly enhanced, which could be attributed to the formation of large π–π conjugated system through Cu2+-triggered ring-opening and hydrolysis cascade reaction.15 In contrast, no significant change in the fluorescence emission spectrum was observed after addition of 10 equiv. other metal cations (Ag+, Al3+, Ca2+, Fe2+, Cd2+, Co2+, Cr3+, Fe3+, Mg2+, K+, Li+, Mn2+, Pd2+, Na+, Ni2+, and Zn2+). Meanwhile, the same trend was also exhibited in the absorption spectrum (Fig. S4†). Upon addition of 10 equiv. of Cu2+, the absorption intensity of JRQN at 594 nm was significantly enhanced. In contrast, no obvious responses of absorption spectrum were observed after addition of various other metal cations under the same conditions. In addition, the probe can also respond to high concentration of Hg2+ (Fig. S5†). Considering that the cells do not contain Hg2+, the recognition of Cu2+ in the living cells will not be affected. Accordingly, intense absorption (λabs = 594 nm) and fluorescence emission (λem = 678 nm) appear, indicating the formation of highly fluorescent ring-opening form after the reaction of JRQN and Cu2+. The Stokes shift of JRQN was found to be 84 nm, which was obviously larger than those of conventional rhodamine dyes (<30 nm).16 The results revealed that JRQN can function as a highly selective fluorescence probe for Cu2+. To verify the speculated mechanism, HRMS was conducted to analyze the change of molecular weight of JRQN after the reaction with Cu2+. As shown in Fig. S6,† an intense peak at m/z 508.2600 corresponding to JRQ was distinct after the reaction of JRQN with Cu2+, revealing that the open-ring and hydrolysis cascade reaction process was triggered and the carboxylic acid compound JRQ was constructed.
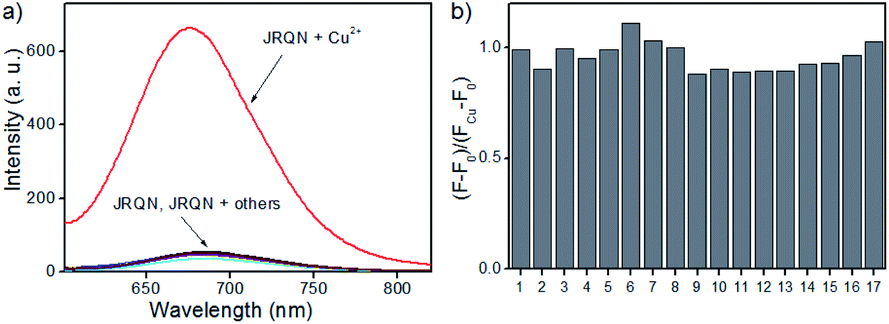 | ||
| Fig. 2 (a) Fluorescence emission responses of JRQN (10 μM) upon addition of different species; (b) fluorescence ratio ((F − F0)/(FCu − F0)) changes of JRQN in the presence of 100 μM Cu2+ and different species. Metal cations (100 μM): (1) Ag+; (2) Al3+; (3) Ca2+ (10 mM); (4) Cd2+; (5) Co2+; (6) Cr3+; (7) Cu2+; (8) Fe2+; (9) Fe3+; (10) Pd2+; (11) K+ (10 mM); (12) Li+ (10 mM); (13) Mg2+ (10 mM); (14) Mn2+; (15) Na+; (16) Ni2+; (17) Zn2+. The conditions: HEPES buffer (10 mM, pH = 7.4, containing 20% CH3CN), λex = 580 nm, slit = 5/5 nm. | ||
Subsequently, competitive experiments were conducted to assess whether the interfering metal ions showed interference with the detection of Cu2+. It was investigated that the response signal of JRQN towards Cu2+ in the presence of 10 equiv. coexistent potentially interfering metal ions (Ag+, Al3+, Ca2+, Fe2+, Cd2+, Co2+, Cr3+, Fe3+, Mg2+, K+, Li+, Mn2+, Pd2+, Na+, Ni2+, and Zn2+). As shown in Fig. 2b, all of the tested foreign metal ions had minor or no interference with signal response to Cu2+ and JRQN exhibited obvious response even in the presence of interfering metal species, revealing that interfering metal species had minor or no interference to the selective recognition properties of JRQN. Therefore, results of competition experiments further confirming the specificity of JRQN toward Cu2+.
In order to assess the sensitivity of JRQN towards Cu2+, continuous titration experiments were conducted by using fluorescence and UV/Vis spectroscopy. As shown in Fig. 3a, after the addition of increasing concentration of Cu2+ (0–105 μM), the fluorescent maximum at 678 nm remarkably increased, and eventually reached constant when the amount of Cu2+ reached 10 equiv. Subsequently, the linear fitting could be obtained between Cu2+ concentrations ranging from 0 to 30 μM and the fluorescence emission intensity at 678 nm with an excellent linear relationship (R = 0.9977) (Fig. 3b). Then, the detection limit of JRQN towards Cu2+ was obtained to be 29 nM based on the equation of 3σ/k, where k is the slope plotted from the fitting equation mentioned above and σ is the relative standard deviation of 10 times fluorescence measurements of the blank solution in parallel without Cu2+. The detection limit was far lower than the maximum allowable level of the World Health Organization (WHO) limit (30 μM)17 and the U.S. Environmental Protection Agency's (EPA) limit for drinking water (20 μM).18 At the same time, after the addition of increasing concentration of Cu2+ (0–10.5 equiv.), the absorption intensity of JRQN at 594 nm remarkably increased and reached the plateau when the amount of Cu2+ reached 5 equiv. (Fig. S7†). These results suggested that JRQN could be used as a practical probe for quantitative detection of Cu2+ in drinking water.
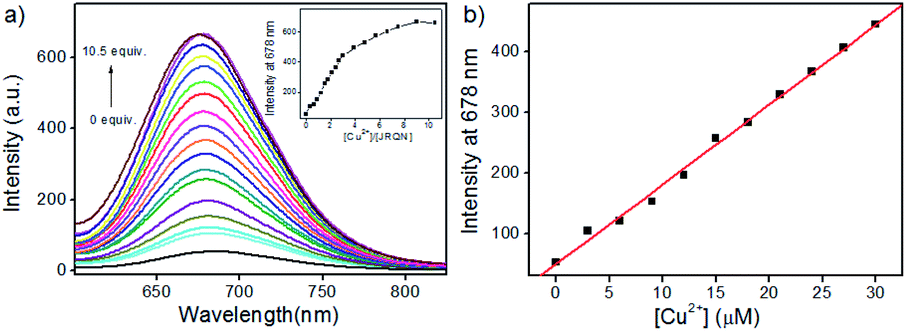 | ||
| Fig. 3 (a) The fluorescence changes of JRQN (10 μM) treated with increasing concentrations of Cu2+ (0–105 μM). Inset: the plot of the fluorescence intensities at 678 nm versus the equivalents of Cu2+; (b) the plot of the fluorescence intensities of JRQN at 678 nm versus the concentrations of Cu2+ (0–30 μM). The conditions: HEPES buffer (10 mM, pH = 7.4, containing 20% CH3CN), λex = 580 nm, slit = 5/5 nm. | ||
In order to evaluate whether JRQN was suitable for detecting Cu2+ in real time, time-dependent fluorescent responses of JRQN towards Cu2+ were evaluated by fluorescence spectroscopy at room temperature. The spectral data were recorded within 500 s in the absence and presence of Cu2+. As shown in Fig. 4a, in the absence of Cu2+, the fluorescence intensity of JRQN at 678 nm remained substantially constant, suggesting that the probe owned high photo-stability. In contrast, in the presence of Cu2+, the fluorescence emission intensity of JRQN at 678 nm rapidly increased and reached a plateau within about 50 s, implying that JRQN could be used as an effective candidate for monitoring Cu2+ in real-time. In addition, in order to evaluate whether JRQN was suitable for detecting Cu2+ in physiological environment and find a suitable pH span in which JRQN could selectively detect Cu2+ efficiently, the emission spectra of JRQN in the absence and presence of Cu2+ at different pH values were measured. As illustrated in Fig. 4b, in the absence of Cu2+, fluorescence intensity of JRQN at 678 nm remained substantially constant under a pH range of 6.0–10.0, while high fluorescence was observed under acidic conditions (4.0–5.0) for ring-opening induced by H+. In contrast, in the presence of Cu2+, the fluorescence intensity at 678 nm was obviously enhanced in the pH range of 5.0–10.0 for the ring-opening induced by Cu2+, implying that JRQN could be used for detecting Cu2+ under physiological conditions. Collectively, above results suggested that JRQN was ability to detect trace amount of Cu2+ in real-time in physiological environments.
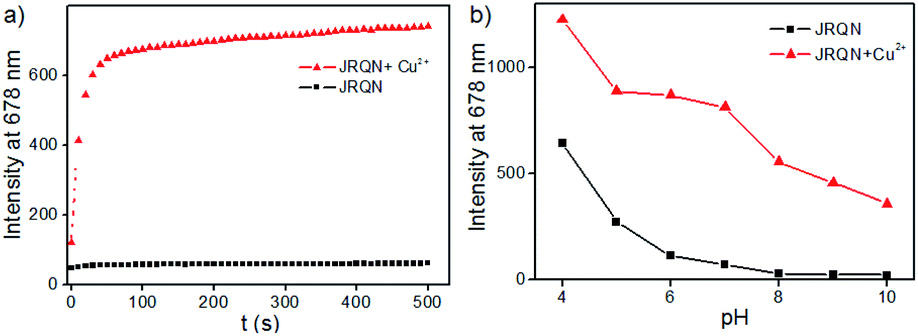 | ||
| Fig. 4 (a) Time-dependence fluorescence intensity of JRQN (10 μM) upon addition of 100 μM Cu2+; (b) fluorescence intensity of JRQN (10 μM) as a function of pH value in the absence/presence of Cu2+ (100 μM); the conditions: HEPES buffer (10 mM, pH = 7.4, containing 20% CH3CN), λex = 580 nm, slit = 5/5 nm. | ||
Fluorescent imaging of JRQN in HeLa cells
The spectral properties confirmed that JRQN had advantages of high selectivity, high sensitivity and fast response time for Cu2+ recognition, which encouraged us to examine the potential applications of JRQN for imaging Cu2+ in living cells. Before the imaging experiments in living cells, the well-established MTT assay was adopted to study cytotoxicity of the aforementioned probe at varying concentrations (0, 1, 3, 5, 10, and 20 μM). As shown in Fig. S8,† the viabilities of HeLa cells were not noticeably affected after incubation for 24 h, indicating low cytotoxicity of JRQN. Then, cell imaging of JRQN for monitoring of Cu2+ in living cells were carried out (Fig. 5). HeLa cells were incubated with JRQN for 30 min, and then washed three times with PBS for removing excess reagents in the growth medium. As shown in Fig. 5a, weak red fluorescence signals were observed in the red channel, implying the extremely low background of JRQN. In contrast, obvious enhancement fluorescence signals were observed in the red channel after treatment with Cu2+ (5 μm) for another 30 min under the same conditions (Fig. 5b). Compared with the controlled cells, the relative enhancement of fluorescence signals is about 5-fold (Fig. 5c). Results indicated that JRQN was membrane permeability and could be used for imaging Cu2+ in living cells.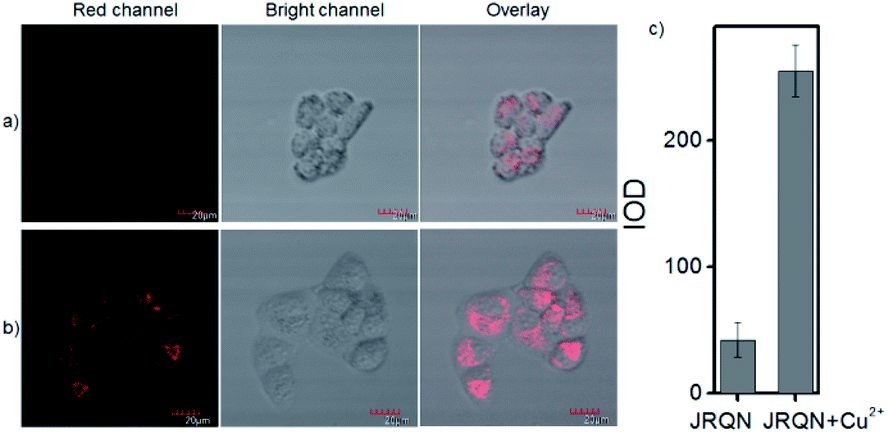 | ||
| Fig. 5 (a) Fluorescent images of HeLa cells incubated with RQS (1 μM) for 30 min; (b) and then further incubated with Cu2+ (5 μM) for 30 min; (c) histogram of fluorescence enhancement. λex = 559 nm, λem = 650–750 nm. | ||
By carefully observing the overlap images, we found that the distribution of the red signal in the cells unevenly fill the entire cytoplasm, but concentrated in some regions, indicating that JRQN likely possessed organelles localization properties. According to the literature, Cu2+ mainly accumulates in the mitochondria and lysosome of cells,19 and the xanthene dyes leads to the proactive accumulation in mitochondria,7 therefore, JRQN was likely to accumulate in one of the two organelles. To verify the speculated organelles localization properties, the co-localization experiments through co-incubating the HeLa cells with commercial targeting reagents Mito-Tracker Green (Mito), Golgi-Tracker Green (Golgi), ER-Tracker Green (ER), and Lyso-Tracker Green (Lyso), respectively, were conducted. As expected, highlight yellow pixels on overlapping images were observed in Fig. 6a, and a high overlap coefficient (0.93) and Pearson's coefficient (0.92) could be calculated from the intensity correlation plot, suggesting that JRQN was mainly distributed in mitochondria. In contrast, red fluorescence signals of JRQN and green fluorescence signals of other trackers (Lyso, ER, or Golgi) were distinctly distributed in different regions, and low overlap coefficients and Pearson's coefficients were obtained (Table S3†), indicating that JRQN does not accumulate in these organelles. Taken together, considering a series of excellent spectral properties and mitochondrial localization properties, the probe may be operated well in monitoring of Cu2+ concentration in the mitochondria for the relevant diseases.
 | ||
| Fig. 6 Fluorescence imaging of the Cu2+-pretreated HeLa cells being costained with JRQN (1 μM) and other organelle-targeting Trackers (200 nM). Cells were incubated with probes at 37 °C for 30 min, and then treated with Cu2+ (5 μM) for another 30 min. (a) Costained with Mito-Tracker Green; (b) costained with Lyso-Tracker Green, (c) costained with ER-Tracker Green; (d) costained with Golgi-Tracker Green. Red channel: λex = 559 nm, λem = 650–750 nm; green channel: λex = 488 nm, λem = 511–534 nm. | ||
Conclusions
In summary, we reported a novel rhodamine dye (JRQ) with NIR emission wavelengths and larger Stokes shift by introducing the electron-donating 1,4-diethylpiperazine moiety in rhodamine scaffold for tuning the D–A–D character of rhodamine dyes. As expected, JRQ exhibited multiple excellent properties and was capable of specifically distributing in mitochondria, which can be used as signal reporter for the design of fluorescence probes by taking advantage of the fluorescence controlled mechanism of the ring opening and closing chemical processes of the spirolactone platform. Furthermore, to demonstrate the utility of this dye for probe design, a highly sensitive and selective fluorescent probe JRQN for the specific detection of mitochondrial Cu2+ ions was synthesized based on the Cu2+-triggered specific hydrolysis mechanism because mitochondria are important reservoir of intracellular Cu2+. JRQN was an outstanding “turn-on” fluorescence sensor with multiple excellent properties, and showed specifically distributing in mitochondrial regions, and thus detect Cu2+ in real-time there, which indicated that the probe can be used for monitoring intracellular mitochondria Cu2+ levels.Experimental
Instruments and reagents
All reagents and organic solvents were commercially available in analytical grade, and were used directly without further purification. Compounds structures were characterized by HRMS and NMR spectrometry. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were measured by Bruker spectrometer using tetramethylsilane (TMS) as an internal standard. Agilent 6510 Q-TOF LC/MS instrument (Agilent Technologies, Palo Alto, CA) was used to measure molecular mass. UV-2550 UV/Vis spectrophotometer (Hitachi Japan) and F-4600 fluorescence spectrophotometer (Hitachi Japan) were used to collect spectroscopic properties. The pH was measured using a FE 20/EL 20 pH meter (Mettler-Toledo Instruments (Shanghai) Co., Ltd.). Fluorescent imaging was carried out by Olympus FV 1000-IX81 laser scanning confocal imaging.Synthetic procedures
Cell culture and fluorescence imaging
HeLa cells for imaging were cultured in DMEM medium supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin (100 μg mL−1) in an atmosphere of 5% CO2 at 37 °C. HeLa cells were incubated with JRQ (1 μM) for 30 minutes. HeLa cells were incubated with JRQN (1 μM) for 30 minutes, and then Cu2+ (5 μM) was added and incubated for another 30 minutes. For co-localization experiments, RQN (1 μM) were incubated with 200 nM trackers (Mito-Tracker Green (Mito), Golgi-Tracker Green (Golgi), ER-Tracker Green (ER), and Lyso-Tracker Green (Lyso)) for 30 minutes, respectively; JRQN (1 μM) were incubated with 200 nM trackers (Mito-Tracker Green (Mito), Golgi-Tracker Green (Golgi), ER-Tracker Green (ER), and Lyso-Tracker Green (Lyso)) for 30 minutes, respectively, and then treated with Cu2+ (5 μM) for another 30 min. Fluorescence imaging was performed after washing the medium three times with PBS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the Natural Science Foundation of China (NNSFC 21907075; 21272172), and the Natural Science Foundation of Tianjin City (19JCZDJC32400; 18JCQNJC75900).Notes and references
- (a) M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC; (b) D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280 CrossRef CAS; (c) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef CAS; (d) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2012, 113, 192 CrossRef; (e) K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564 CrossRef CAS; (f) S. Cai, Y. Lu, S. He, F. Wei, L. Zhao and X. Zeng, Chem. Commun., 2013, 49, 822 RSC.
- (a) Z. Zhang and S. Achilefu, Org. Lett., 2004, 6, 2067 CrossRef CAS; (b) J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025 CrossRef CAS; (c) Q. Wang, X. Jiao, C. Liu, S. He, L. Zhao and X. Zeng, J. Mater. Chem. B, 2018, 6, 4096 RSC; (d) C. Liu, X. Jia, S. He, L. Zhao and X. Zeng, Org. Biomol. Chem., 2017, 15, 3947 RSC; (e) X. Jiao, C. Liu, Q. Wang, K. Huang, S. He, L. Zhao and X. Zeng, Anal. Chim. Acta, 2017, 969, 49 CrossRef CAS; (f) X. Wang, L. Cui, N. Zhou, W. Zhu, R. Wang, X. Qian and Y. Xu, Chem. Sci., 2013, 4, 2936 RSC.
- (a) Z. Q. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16 RSC; (b) R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123 CrossRef CAS; (c) A. N. Butkevich, G. Lukinavičius, E. D'Este and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 12378 CrossRef CAS; (d) L. Yuan, W. Y. Lin, K. B. Zheng, L. W. He and W. M. Huang, Chem. Soc. Rev., 2013, 42, 622 RSC.
- (a) E. David, J. Lejeune, R. S. Pellet, J. Schulz, M. Lemaire and J. Chauvin, Tetrahedron Lett., 2008, 49, 1860 CrossRef CAS; (b) C. Liu, X. Jiao, Q. Wang, K. Huang, S. He, L. Zhao and X. Zeng, Chem. Commun., 2017, 53, 10727 RSC; (c) Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 5680 CrossRef CAS; (d) Q. Wang, K. Huang, S. Cai, C. Liu, X. Jiao, S. He, L. Zhao and X. Zeng, Org. Biomol. Chem., 2108, 16, 7609 RSC; (e) Q. Wang, X. Jiao, C. Liu, K. Huang, S. He, L. Zhao and X. Zeng, Org. Biomol. Chem., 2018, 16, 7163 RSC; (f) C. Liu, Q. Wang, X. Jiao, H. Yao, S. He, L. Zhao and X. Zeng, Dyes Pigm., 2019, 160, 989 CrossRef CAS; (g) W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713 CrossRef CAS.
- T. Ren, W. Xu, W. Zhang, X. Zhang, Z. Wang, Z. Xiang, L. Yuan and X. Zhang, J. Am. Chem. Soc., 2018, 140, 7716 CrossRef CAS.
- (a) Z. Tian, B. Tian and J. Zhang, Dyes Pigm., 2013, 99, 1132 CrossRef CAS; (b) H. Liu, Y. Zhang, S. Xia, M. Fang, W. Mazi, Y. Zeng, T. Johnston, A. Pap and R. L. Luck, Chem. Commun., 2018, 54, 7625 RSC; (c) J. Gong, C. Liu, X. Jiao, S. He, L. Zhao and X. Zeng, Spectrochim. Acta, Part A, 2020, 243, 118821 CrossRef CAS; (d) J. Gong, C. Liu, X. Jiao, S. He, L. Zhao and X. Zeng, RSC Adv., 2020, 10, 29536 RSC.
- Y. K. Kim, H. H. Ha, J. S. Lee, X. Bi, Y. H. Ahn, S. Hajar, J. J. Lee and Y. T. Chang, J. Am. Chem. Soc., 2010, 132, 576 CrossRef CAS.
- (a) E. A. Belyaeva, D. Dymkowska, M. R. Wieckowski and L. Wojtczak, Toxicol. Appl. Pharmacol., 2008, 231, 34 CrossRef CAS; (b) E. A. Belyaeva, T. V. Sokolova, L. V. Emelyanova and I. O. Zakharova, Sci. World J., 2012, 2012, 1 CrossRef.
- W. R. Vollmer and E. Birckner, J. Fluoresc., 1994, 4, 65 CrossRef.
- (a) G. E. Cartwright and M. M. Wintrobe, Am. J. Clin. Nutr., 1964, 14, 224 CrossRef CAS; (b) A. V. Davis and T. V. O'Halloran, Nat. Chem. Biol., 2008, 4, 148 CrossRef CAS; (c) S. Lutsenko, Curr. Opin. Chem. Biol., 2010, 14, 211 CrossRef CAS.
- (a) P. A. Cobine, F. Pierrel and D. R. Winge, Biochim. Biophys. Acta, 2006, 1763, 759 CrossRef CAS; (b) L. A. Abriata, L. Banci, I. Bertini, S. Ciofi-Baffoni, P. Gkazonis, G. A. Spyroulias, A. J. Vila and S. Wang, Nat. Chem. Biol., 2008, 4, 599 CrossRef CAS.
- R. Uauy, M. Olivares and M. Gonzalez, Am. J. Clin. Nutr., 1998, 67, 952S CrossRef CAS.
- (a) D. Y. Sasaki, D. R. Shnek, D. W. Pack and F. H. Arnold, Angew. Chem., 1995, 107, 994 CrossRef; (b) R. Krämer, Angew. Chem., 1998, 110, 804 CrossRef; (c) S. G. Kaler, Nat. Rev. Neurol., 2011, 7, 15 CrossRef CAS; (d) K. M. Davies, J. F. B. Mercer, N. Chen and K. L. Double, Clin. Sci., 2016, 130, 565 CrossRef.
- J. Gong, C. Liu, X. Jiao, S. He, L. C. Zhao and X. S. Zeng, J. Mater. Chem. B, 2020, 8, 2343 RSC.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386 CrossRef CAS.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC.
- Guidelines for drinking-water quality, World Health Organization Press, Geneva, 4th edn, 2011, p. 340 Search PubMed.
- US EPA, National Primary Drinking Water Regulations: EPA 812-Z-94-001, May 2009 Search PubMed.
- (a) Y. Li, Y. Zhao, W. Chan, Y. Wang, Q. You, C. Liu, J. Zheng, J. Li, S. Yang and R. Yang, Anal. Chem., 2015, 87, 584 CrossRef CAS; (b) M. Ren, B. Deng, J. Y. Wang, Z. R. Liu and W. Lin, J. Mater. Chem. B, 2015, 3, 6746 RSC; (c) A. Atkinson and D. R. Winge, Metal acquisition and availability in the mitochondria, Chem. Rev., 2009, 109, 4708 CrossRef CAS; (d) H. li, R. Zhang, C. Li, B. Huang, T. Yu, X. Huang, X. Zhang, F. Li, H. Zhou and Y. Tian, Org. Biomol. Chem., 2017, 15, 598 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05835a |
| This journal is © The Royal Society of Chemistry 2020 |