
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of fluoro-substituted spiro-isoxazolines as potential anti-viral and anti-cancer agents†
Prasanta Das *a,
Sarah Booneb,
Dipanwita Mitrab,
Lindsay Turnerc,
Ritesh Tandonb,
Drazen Raucherc and
Ashton T. Hamme II*a
*a,
Sarah Booneb,
Dipanwita Mitrab,
Lindsay Turnerc,
Ritesh Tandonb,
Drazen Raucherc and
Ashton T. Hamme II*a
aDepartment of Chemistry and Biochemistry, Jackson State University, Jackson, Mississippi 39217, USA. E-mail: prasanta.das@jsums.edu; Tel: +1-601-203-5149
bDepartment of Microbiology and Immunology, University of Mississippi Medical Center, Jackson, Mississippi 39216, USA
cDepartment of Cellular and Molecular Biology, University of Mississippi Medical Center, Jackson, Mississippi 39216, USA
First published on 17th August 2020
Abstract
Electrophilic fluorine-mediated dearomative spirocyclization has been developed to synthesize a range of fluoro-substituted spiro-isoxazoline ethers and lactones. The in vitro biological assays of synthesized compounds were probed for anti-viral activity against human cytomegalovirus (HCMV) and cytotoxicity against glioblastomas (GBM6) and triple negative breast cancer (MDA MB 231). Interestingly, compounds 4d and 4n showed significant activity against HCMV (IC50 ∼ 10 μM), while 4l and 5f revealed the highest cytotoxicity with IC50 = 36 to 80 μM. The synthetic efficacy and biological relevance offer an opportunity to further drug-discovery development of fluoro-spiro-isoxazolines as novel anti-viral and anti-cancer agents.
Introduction
Spiro-heterocycles are prevalent in plant and animal domains and have become the cornerstone of pharmaceutical and medicinal chemistry.1 While their conformations, chirality, and structural implications are suitable for biological systems, a diverse group of spiro-heterocycles has widely been used in the drug-discovery process.1 Among heterocycles, the significance of isoxazole2 has been driven by a broader biological spectrum, including analgesic,3 anti-bacterial,4 anti-depressant,5 anti-cancer,6 and anti-herpetic7 of activities; it is worth mentioning that the synthesis of isoxazoles and isoxazolines is therefore receiving attention from organic chemists.2c In this context, the spiro-architecture represents a potential avenue for a diverse range of siblings8 for further synthetic and biological exploration. Moreover, the unique spiro-isoxazoline core, found in bromotyrosine derived marine natural products, delivering a wide range of biological activities,9,10 has attracted much synthetic attention. Due to their outstanding biological profile in combination with an intriguing molecular structure, we have focussed our research interest toward the synthesis of spiro-isoxazoline natural products and its congeners as potential leads for anti-cancer, anti-HCMV, and anti-GBM6 agents.11It is worth mentioning that the introduction of fluorine in a biologically active molecules can dramatically modify several parameters, such as acidity, basicity, and dipole moments, delivering its drug-like properties, such as lipophilicity, metabolic stability, and bioavailability of molecules.12 As a result, these unique properties of the fluorine atom make it relevant in pharmaceutical,12,13 agrochemical,12,14 and material sciences.15 Hence, inspired by the medicinal relevance of spiro-isoxazolines and fluorine, we herein report an electrophilic fluorine-mediated dearomative spirocyclization to various fluoro-spiro-isoxazolines and probe their biological activity (Scheme 1). The novelty of the current method includes the identity of effective fluoro-cyclization, the exploration of the chemistry for several fluoro-spiro-isoxazolines, and their anti-viral and anti-cancer activities (Scheme 1).
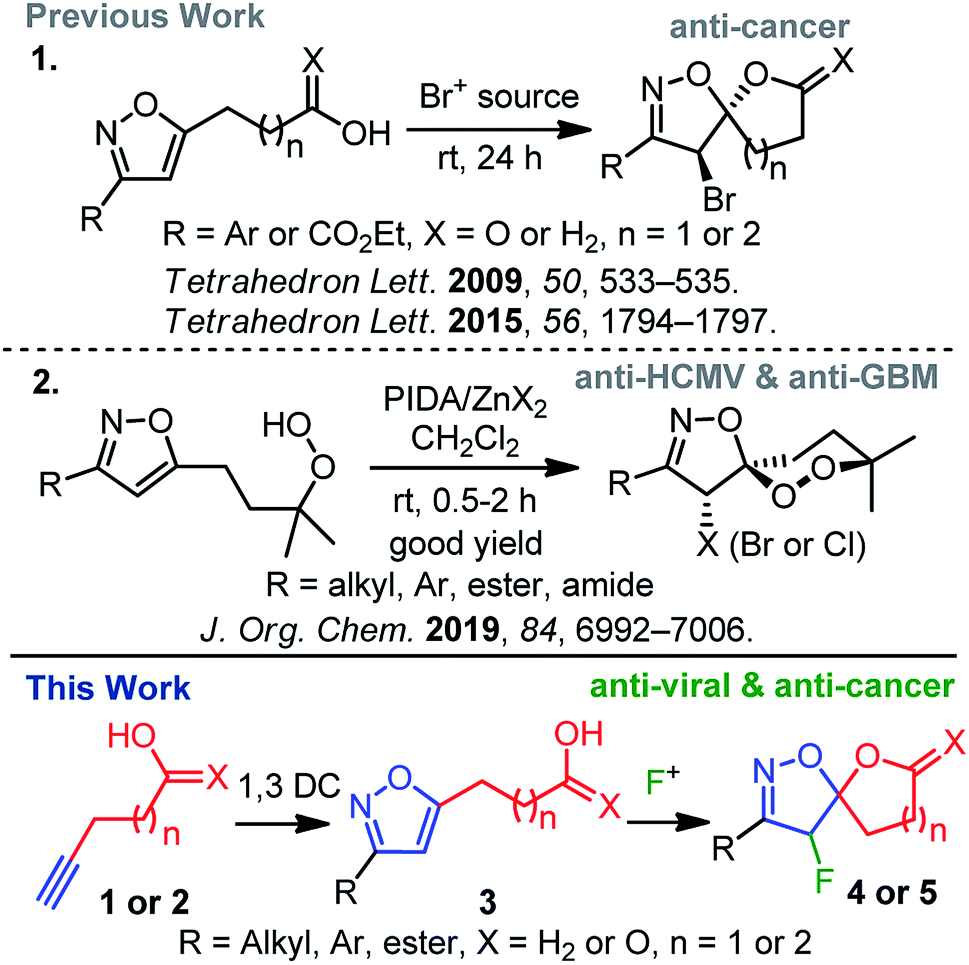 | ||
| Scheme 1 Synthesis of 4-halo-spiro-isoxazoline derivatives. | ||
Electrophilic fluorination has led to a powerful tool to access various fluorinated hetero- and carbocycles.16 Moreover, electrophilic dearomative-fluorination17 of aromatic compounds has received much attention since they produce 3D-molecular skeletons with two consecutive stereogenic centers. Though the mechanism of electrophilic fluorination remains controversial to date, isoxazole can act like a glycal system,16c,18 in which an oxonium ion mediated electrophilic fluorocyclization leads to the desired spirocyclization. If successful, this strategy would serve as a practical route to readily access a range of fluoro-spiro-isoxazolines in order to probe biological activity.
In the line with our continued interest in the synthesis of spiro-isoxazoline (Scheme 1),11 we herein report a two-step method that includes 1,3-dipolar cycloaddition and electrophilic fluoro-cyclization to access fluorinated variants of this important motifs. The effectiveness of this strategy is also examined using various substituted nitrile oxides and alkynes to obtain a diverse group.
Results and discussion
Chemistry
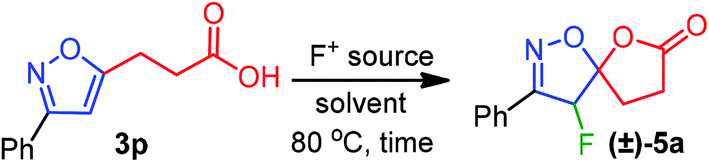
The fluorolactonization of isoxazoline 3p was selected as a model reaction to optimize the reaction conditions, including electrophilic fluorine sources, solvents, and temperatures (Table 1). As Selectfluor is inexpensive, commercially available, and widely used for electrophilic fluorocyclization, we chose to screen Selectfluor on 3p while using various protic, aprotic, and halogenated solvents at room temperature. However, since the role of the solvents was crucial for the desired transformation, none of the solvents were found suitable for the fluorolactonization at room temperature. To our surprise, as depicted in Table 1, when the temperature of the reaction was elevated to 80 °C, we observed remarkable changes in reactivity. However, the chlorinated solvents CH2Cl2, CHCl3, and ClCH2CH2Cl (DCE) were abortive to bring about the fluorolactonization, even at refluxed temperature. This prompted us to examine the reaction in aprotic polar and nonpolar solvents, such as THF, Et2O, acetone, and ethyl acetate (entry 5–7, Table 1). To our disappointment, the desired fluorolactonization was unsuccessful, and we recovered the substrate 3p, quantitatively. In mark contrast, when we performed the reaction in polar solvents, such as CH3CN and MeOH at 80 °C (entry 8 and 9, Table 1), a significant enhancement in reactivity was observed, and we were able to isolate 5a in 90% and 85% yields. Notably, when trifluoroethanol (TFE) was used as a fluorinated polar solvent, the desired reaction was completely absent (entry 10, Table 1). We speculated that due to the electronegativity of the trifluoromethyl group in TFE, the corresponding alcohol exhibits a stronger acidic character compared to methanol; thereby, plausible complexation with isoxazole, protonation of alkoxide or carboxylate anion was detrimental to reactivity. At this point, based on the reaction time, we chose CH3CN as our optimized solvent for the desired reaction. To further our investigation, we used two other fluorine sources that are also known for fluorination.19 In doing so, we performed a similar reaction but using 1.5 equiv. of N-fluorobenzenesulfonimide (NFSI) or N-fluoro-o-benzenedisulfonimide (NFOBS); unpredictably, no desired fluorolactonizations were observed (entries 11 and 12, Table 1). Therefore, we decided to use Selectfluor (1.5 equiv.) in CH3CN at 80 °C as an optimized reaction condition to achieve the desired transformation (Table 1).
|
||||
|---|---|---|---|---|
| Entry | F+-source | Solvent | Timeb (h) | Yieldc (%) |
| a General conditions: isoxazoline acid 3p (0.2 mmol, 1.0 equiv.) and F-source (1.5 equiv.) in 2 mL solvent was refluxed at 80 °C.b Time required for the reaction.c Based on isolated product after purification by chromatography. | ||||
| 1 | Selectfluor | CH2CI2 | 24 h | — |
| 2 | Selectfluor | CHCI3 | 24 h | — |
| 3 | Selectfluor | DCE | 24 | 10 |
| 4 | Selectfluor | THF | 12 h | — |
| 5 | Selectfluor | Ether | 24 | — |
| 6 | Selectfluor | Ethyl acetate | 24 h | — |
| 7 | Selectfluor | Acetone | 24 h | — |
| 8 | Selectfluor | CH3CN | 24 h | 90 |
| 9 | Selectfluor | MeOH | 24 h | 85 |
| 10 | Selectfluor | CF3CH2OH | 24 h | 10 |
| 11 | NFSI | CH3CN | 24 h | — |
| 12 | NFOBS | CH3CN | 24 h | — |
 |
||||
A plausible mechanistic pathway has been proposed in Scheme 2. Under the refluxed condition, Selectfluor could dearomatize the isoxazole ring via an oxonium-ion mediated electrophilic fluorination on C4–C5 double bond, regioselectivity, leading to a reactive intermediate A. Subsequently, the neighboring group participation of the carboxylate anion to the intermediate A produced the fluoro-spiro-isoxazoline (Scheme 2).
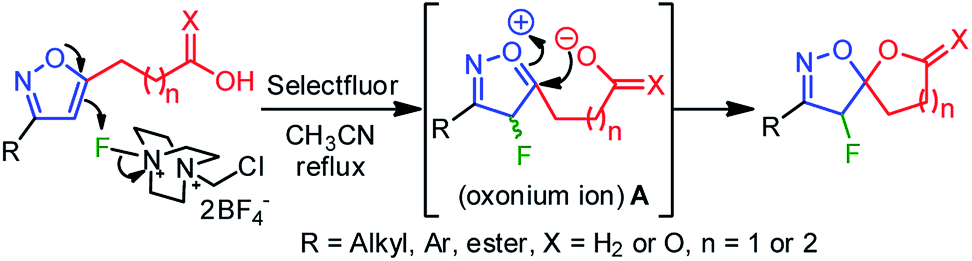 | ||
| Scheme 2 Plausible mechanism for 4-fluoro-spiro-isoxazolines. | ||
Having optimized conditions at hand, we proceeded to explore the synthetic compatibility, first by introducing various substituents on the isoxazole ring, and then, by evaluating the feasibility of five and six-membered ether and lactone ring formation. The desired precursors 3(a–z), possessing an isoxazole and pendant alcohol or acid, were initially synthesized by using a 1,3-dipolar cycloaddition reaction between alkynol 1 or alkynoic acid 2 and 1,3-dipole precursors B (Table 2).11a
|
||||
|---|---|---|---|---|
| Entry | Alkyne | R | 3(a–z) | Yieldc (%) |
| a General conditions 1: alkynol 1 (1.0 equiv.), hydroximoyl chloride (1.2 equiv.), triethylamine (1.5 equiv.) in CH2Cl2 (10 mL) at rt for 24 h or alkynoic acid 2 (1 equiv.), hydroximoyl chloride (2 equiv.), and K2CO3 (2.5 equiv.) in deionized water (1 M) at rt for 24 h.b General conditions 2: alkynoic acid 2b (1.0 equiv.), ethylnitroacetate (2.5 equiv.), DABCO (0.2 equiv.), ethanol (10 mL) at 80 °C in sealed vessel for 3 days.c Based on isolated product after purification by chromatography. | ||||
| 1 | 1a, n = 1, x = H2 | Ph | 3a | 96 |
| 2 | 1a, n = 1, x = H2 | 4-Me-Ph | 3b | 83 |
| 3 | 1a, n = 1, x = H2 | 4-F-Ph | 3c | 95 |
| 4 | 1a, n = 1, x = H2 | 4-CI-Ph | 3d | 95 |
| 5 | 1a, n = 1, x = H2 | 4-Br-Ph | 3e | 95 |
| 6 | 1a, n = 1, x = H2 | 2,6-Di-CI-Ph | 3f | 96 |
| 7 | 1a, n = 1, x = H2 | 4-CF3-Ph | 3g | 94 |
| 8 | 1a, n = 1, x = H2 | 4-NO2-Ph | 3h | 80 |
| 9 | 1b, n = 2, x = H2 | Ph | 3i | 92 |
| 10 | 1b, n = 2, x = H2 | 4-F-Ph | 3j | 94 |
| 11 | 1b, n = 2, x = H2 | 4-CI-Ph | 3k | 92 |
| 12 | 1b, n = 2, x = H2 | 4-Br-Ph | 3l | 93 |
| 13 | 1b, n = 2, x = H2 | 2,6-Di-CI-Ph | 3m | 94 |
| 14 | 1b, n = 2, x = H2 | 4-CF3-Ph | 3n | 95 |
| 15 | 1b, n = 2, x = H2 | 4-NO2-Ph | 3o | 78 |
| 16 | 2a, n = 1, x = O | Ph | 3p | 96 |
| 17 | 2a, n = 1, x = O | 4-Me-Ph | 3q | 89 |
| 18 | 2a, n = 1, x = O | 4-F-Ph | 3r | 80 |
| 19 | 2a, n = 1, x = O | 4-CI-Ph | 3s | 82 |
| 20 | 2a, n = 1, x = O | 4-Br-Ph | 3t | 85 |
| 21 | 2a, n = 1, x = O | 2,6-Di-CI-Ph | 3u | 80 |
| 22 | 2a, n = 1, x = O | 4-CF3-Ph | 3v | 92 |
| 23 | 2a, n = 1, x = O | Me | 3w | 76 |
| 24 | 2a, n = 2, x = O | n-Pr | 3x | 78 |
| 25 | 2a, n = 2, x = O | 2,6-Di-CI-Ph | 3y | 80 |
| 26b | 2a, n = 2, x = O | CO2Et | 3z | 82 |
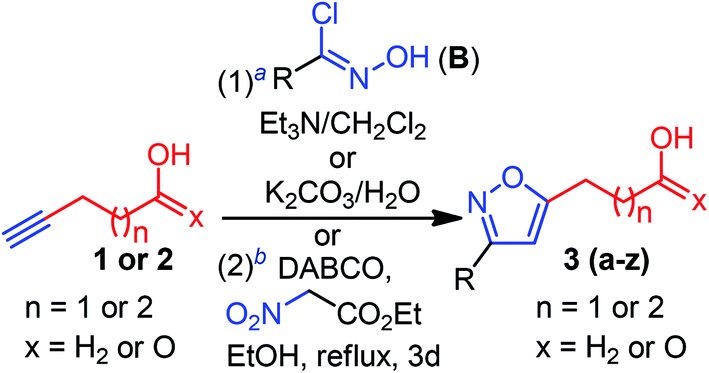
Although the substrate scope for the desired fluorocyclization was examined at the final stage, various functionalities were introduced by using several 1,3-dipole precursors B (Table 2). Therefore, as illustrated in Table 2, electron-donating, electron-withdrawing, and halo-functionality were introduced on an aromatic system; a couple of alkyl functionalities were also incorporated to introduce hydrophobic nature to the molecule for biological investigation. The 1,3-dipolar reaction was performed between 1 or 2 and in situ generated nitrile oxide from the corresponding chloro-oxime B upon treatment with Et3N/CH2Cl2.11a As summarized in Table 2, a range of substituted isoxazoles 3(a–z), comprising pendant alcohol 1 and acid 2 of different chain lengths, were successfully synthesized in high yields (Table 2). To determine the ester functionality that could facilitate obtaining an acid and amide functional group, we conducted a base-catalyzed 1,3-dipolar cycloaddition reaction, using ethyl-nitroacetate and alkynoic acid 2, under sealed tube refluxed condition, which provided the desired product 3z in 82% yield (Table 2).11a It is worth mentioning that the 1,3-dipolar cycloaddtion produced the 3,5-disubstituted isoxazole as a major isomers; however, 3,4-disubstituted isoxazole was insignificant to be isolated.
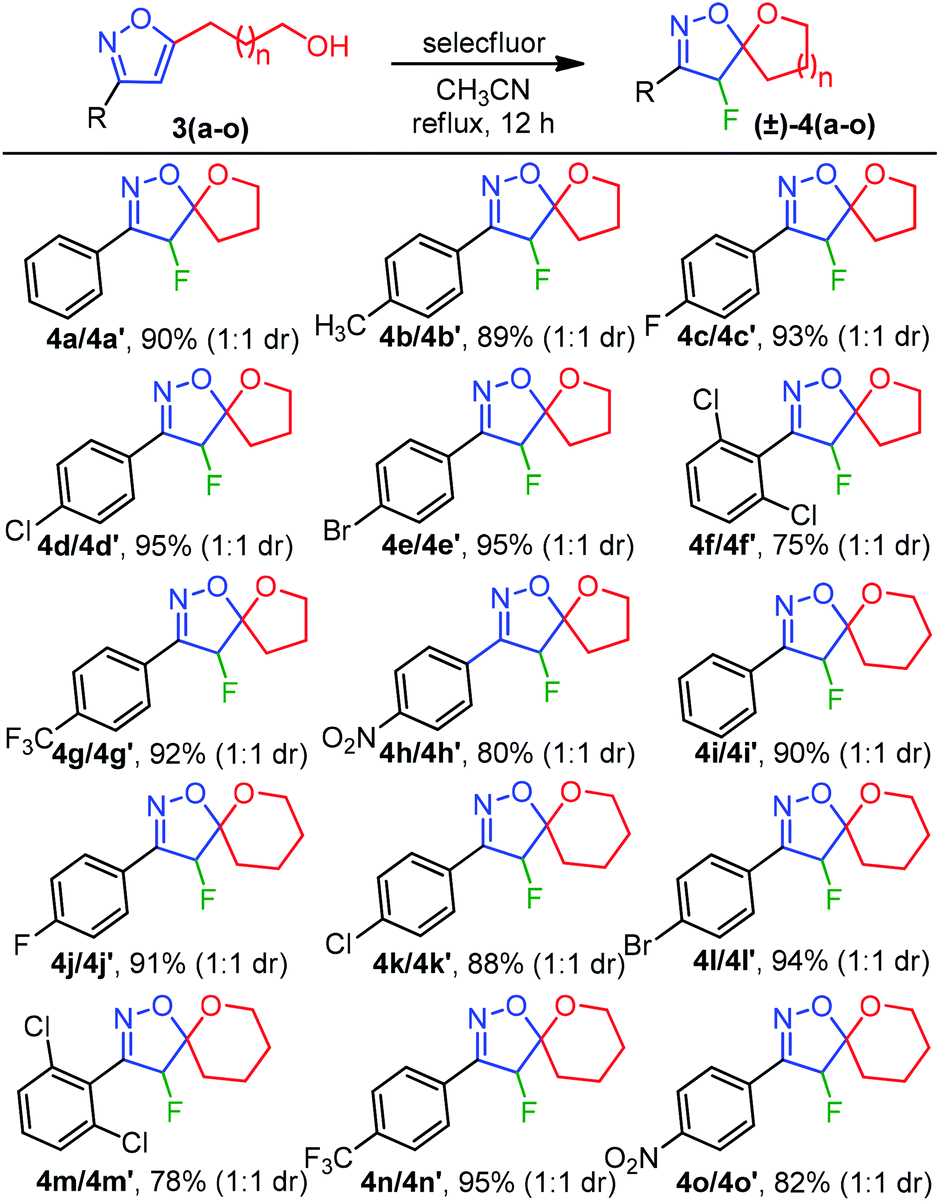
To evaluate the feasibility of five and six-membered ether ring formations, the list of alcohols 3(a–o) from Table 2 was executed for the fluorocyclization (Table 3), following our optimized reaction condition. As summarized in Table 3, the electron-donating effect of 4-Me was tolerated under our standard reaction conditions to afford 4b in 89% yield. Next, for the halo substitutions, 4-F-Ph, 4-Cl-Ph, 4-Br-Ph, and 4-CF3-Ph afforded 4(c–e) and 4g in 93%, 95%, 95%, and 92% isolated yields, respectively (Table 3). A similar pattern of reactivity was also observed for a six-membered etherification 4(i–l) and 4n with high yields. On the other hand, the spirocyclization of 2,6-dichloro substituted isoxazoles 3f and 3m were also successful to produce 4f and 4m, albeit with decreased efficiencies (75% and 78%) due to the steric environment generated between 2,6-di-Cl and F-atoms. The electrophilic fluoro-cyclization was also found to be effective in the presence of an electron-withdrawing group such as NO2, producing the desired cyclized products 4h and 4o in 80% and 82% yields, respectively. Therefore, the reaction conditions were found to be well tolerated for a wide range of functionalities leading to (±)-4-fluoro-spiro-isoxazoline-ether 4(a–o) (Table 3). Importantly, the diastereomers were isolated with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio with an identical chemical shifts (δ) in NMR for most cases. However, a well-defined chemical shift for the CHF proton was observed for few diastereomers.
:1 diastereomeric ratio with an identical chemical shifts (δ) in NMR for most cases. However, a well-defined chemical shift for the CHF proton was observed for few diastereomers.
| a Reaction conditions: isoxazole alcohol 3(a–o) (0.2 mmol), Selectfluor (0.3 mmol, 1.5 equiv.), CH3CN (2 mL) at 80 °C for 12 h, product isolated after purification by chromatography. |
|---|
 |
Inspired by this success, we next explored the scope of isoxazole acids 3 to afford about five and six-membered fluoro-spiro-isoxazoline-lactones. As depicted in Table 4, regardless of various substitution on the phenyl ring and electronic properties, the reaction proceeded smoothly and provided the desired products 5(a–k) in excellent yields (Table 4). The direct alkyl substituents (Me and Pr) on the isoxazoline also endured the reaction conditions and resulted 5h and 5i in 82% and 80% yield, respectively (Table 4).
| a Reaction conditions: isoxazole acid 3(p–z) (0.2 mmol), Selectfluor (0.3 mmol, 1.5 equiv.), CH3CN (2 mL) at reflux for 12 h, product isolated after purification by chromatography. |
|---|
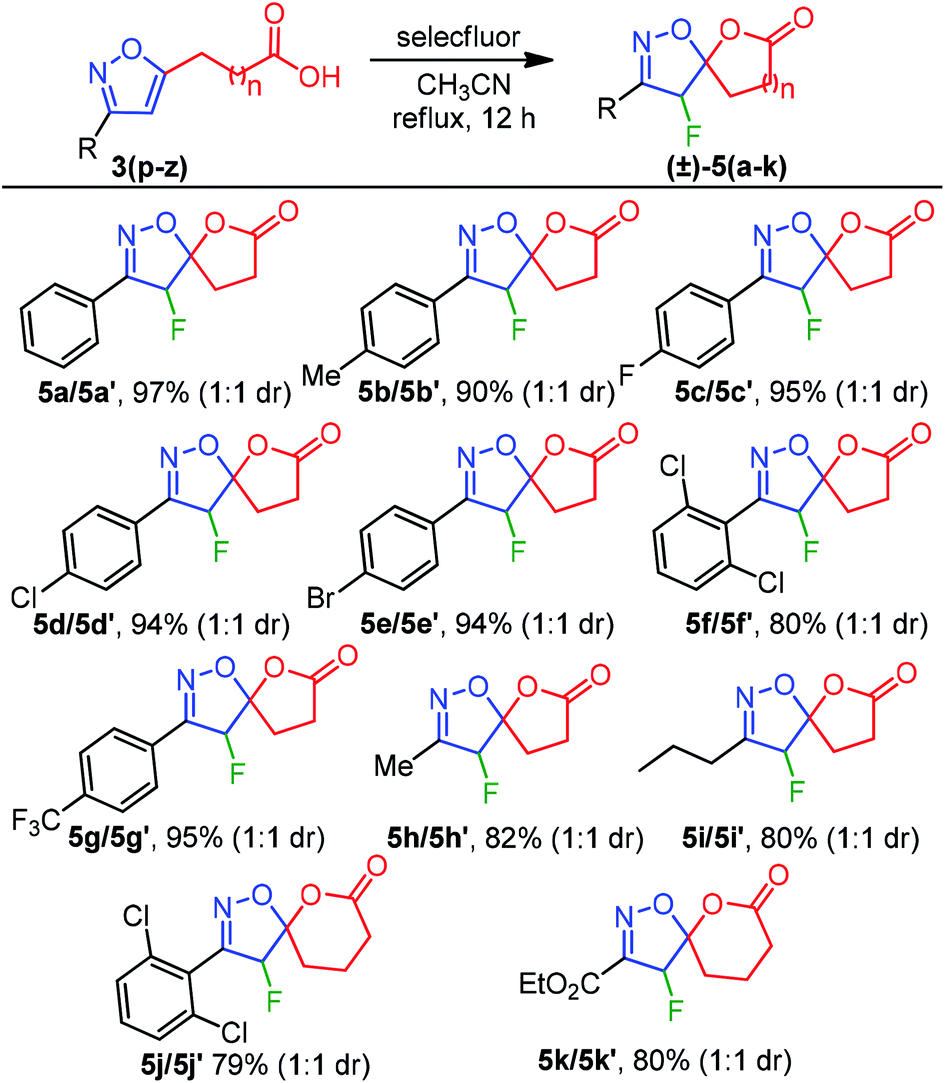 |
Furthermore, an ester functionality that is directly connected to an isoxazoline system also exhibited good compatibility to provide 5k in 80% yield. To our surprise, spiro-lactones 5(a–k) were separated well, which delivered a dr 1:1 (Table 4).
Biology
Having synthesized diverse fluoro-containing spiro-ethers and -lactones in Tables 3 and 4, we were interested in evaluating their anti-viral and anti-cancer activity.Next, we determined the half maximal inhibitory concentration (IC50) for the five compounds that showed promise in the preliminary screening against HCMV. Based on the number of GFP+ cells present in infected cells pre-treated with serial dilutions of test compounds, the IC50 for the compounds 4o, 4i, 4n, and 4d was calculated as 2.54 mM, 11.2 mM, 10.47 μM, and 9.47 μM, respectively (Fig. 1). IC50 for ganciclovir (GCV), a clinically approved drug for HCMV treatment, was calculated as 4.96 μM, which is very close to the reported IC50 of 5.2 μM,21 proving the validity of this assay. Since IC50 values are inversely proportional to the potency of a compound, this data showed that compounds 4d and 4n are the most effective inhibitors of HCMV in cell culture among the compounds tested (Fig. 1).
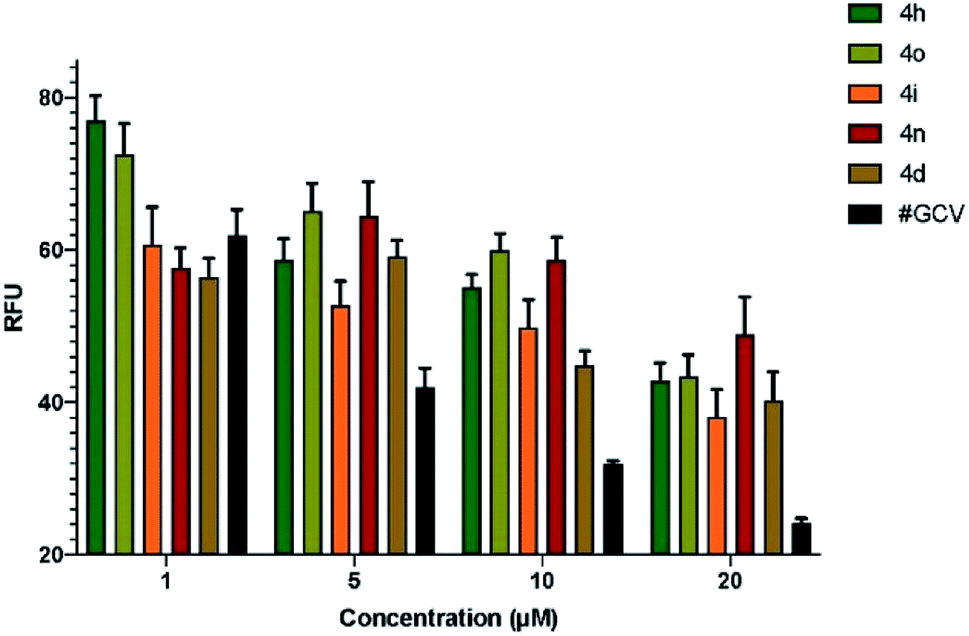 | ||
| Fig. 1 Determination of IC50 of the test compounds. Confluent HFF cells were pre-treated with serial dilutions (0–20 μM) of test compounds 4h, 4o, 4i, 4n, and 4d. As a control, cells were pre-treated with ganciclovir (GCV) followed by infection with HCMV (Towne-BAC-GFP) strain at a MOI of 3.0. At 5 days post infection, cells were fixed in 3.7% formaldehyde, and relative fluorescence units (RFU) were quantified using a microwell image cytometer (Celigo, Nexcelom Bioscience LLC, Lawrence, MA). Estimation of IC50 for the compounds 4o, 4i, 4n, and 4d as 2.54 mM, 11.2 mM, 10.47 μM, and 9.47 μM, respectively, is based on non-linear curve fitting on transformed data in Prism GraphPad software. Error bars represent standard error of mean (SEM) from three independent experiments. IC50 for GCV was 4.96 μM, which is close to the reported value of 5.2 μM.21 | ||
The compounds 4o and 4i showed virus inhibition in this assay; however, much more concentration is required, which may be impractical to attain physiologically.
Finally, we determined the possible cytotoxicity of the test compounds on HFF cells (Fig. 2). Cells were treated with the test compounds (4d and 4n) at 2× the IC50 concentrations or left untreated (UT). Cells were then either infected (I) with HCMV at MOI of 3.0 or left uninfected (UI), and cell viability at the end point was quantified by trypan blue exclusion assay.22 As presented in Fig. 2, cell viability for all tested compounds remained near 100%. Most importantly, the compounds 4n and 4d did not show significant cytotoxicity even when maintained on cells for a period of 5 days.
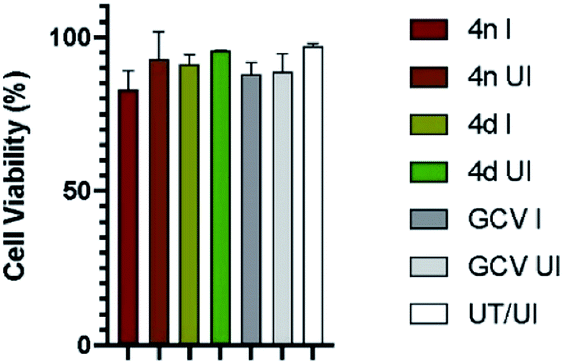 | ||
| Fig. 2 Effect of the test compounds on cell viability. HFF-1 cells were treated with the test compounds at 2× the IC50 concentrations or untreated (UT). Cells were either infected (I) with HCMV at MOI of 3.0 or left uninfected (UI). Cell viability was quantified by trypan blue exclusion assay at day 5 post infection. Error bars represent standard error of mean (SEM) from three independent experiments. | ||
Based on the biological assay, it is depicted that spiro-lactones 5(a–k) were more inactive against HCMV than the corresponding ether derivatives. Likewise, the phenyl and electron-donating groups on fluoro-spiro derivatives did not show an acceptable range of inhibition against HCMV replication. It is also imperative to highlight that compounds (4h, 4o, 4i, 4n, and 4d) containing electron-withdrawing groups such as NO2, –Cl, and –CF3 were primarily found to have potential anti-HCMV properties. The Ph and 4-NO2-Ph substituted fluoro-isoxazoline 4o and 4i proved to be moderately active against HCMV with IC50 2.54 mM and 11.2 mM. Interestingly, replacement of functionality to halo substituents such as the 4-Cl-Ph and 4-CF3-Ph in 4d and 4n lead to substantial increase in potency (IC50 values 9.47 μM and 10.47 μM). Although, their potency is comparatively lower than that of the control compound ganciclovir (GCV), considering widespread resistance against GCV in clinical strains of HCMV, and the well-known side effects such as neutropenia, the compounds 4d, and 4n may be developed into potential future drugs for HCMV.23–25
 | ||
| Fig. 3 GBM6 were plated at 1000 cells per well on 96-well format. After 24 hours, the cells were treated at 100 μM concentration of each given compound. The MTT assay was performed 72 hours after treatment and the absorbance (570 nm) data from each experimental group (n = 3) was normalized to control average, bars are S.E.M. | ||
The IC50 value was evaluated using an increased concentration (0.8–100 μM) for 4l and 5f. Survival percentages were plotted in Fig. 4 and IC50 values predicted based on non-linear regression curve fit, using Graphpad 8. These values are shown in Table 5.
 | ||
| Fig. 4 GBM6 were plated at 1000 cells per well on 96-well format. After 24 hours, the cells were treated with a range of concentrations (0.8–100 μM). MTT assay was performed 72 hours after treatment. n = 3, bars are S.E.M. | ||
| 4l | 5f | |
|---|---|---|
| IC50 in GBM6 | 36.08 | 43.21 |
| 95% C.I. in GBM6 | 23.21 to 55.06 | 35.69 to 52.38 |
| IC50 in MDA MB 231 | 68.18 | 79.80 |
| 95% C.I. in MDA MB 231 | 64.27 to 72.25 | 75.58 to 84.95 |
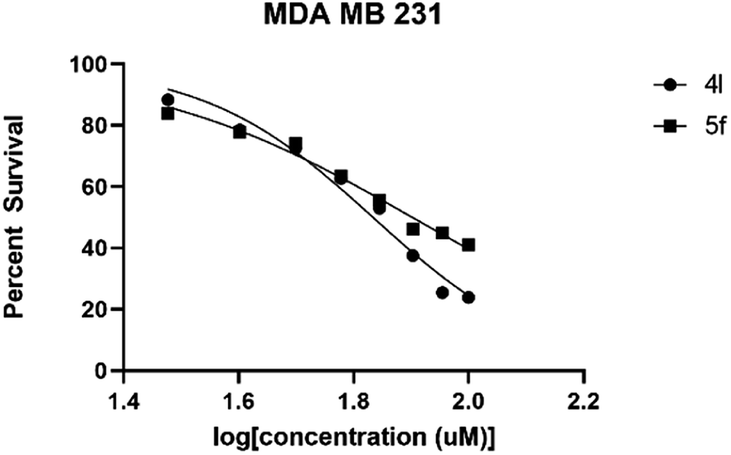 | ||
| Fig. 5 MDA MB 231 were plated at 2000 cells per well on 96-well format. After 24 hours, the cells were treated with a range of concentrations. MTT assay was performed 72 hours after treatment. n = 6, bars are S.E.M. | ||
As depicted in Table 5, compound 4l shows better activity than 5f against both GBM6 and MDA MB 231 cells suggesting that spiro-ether is more effective to show cytotoxicity than the corresponding spiro-lactone. The Br substitution on 4l, situated at the periphery of the molecular geometry, shows anti-GBM activity; however, the dichloro substitutions at 2,6-position are away from the periphery shows anti-MDA MB 231 activity.
Conclusion
In conclusion, we have developed an efficient and practical route to access various 4-fluoro-substituted spiro-isoxazolines. The efficacy of this strategy mainly relies on 1,3-dipolar cycloaddition and electrophilic fluoro-etherification or -lactonization mediated by commercially available Selectfluor. The protocol has been shown to be useful for accessing spirocyclic compound comprising alkyl, aromatic, and ester substituted isooxazoline and five and six membered cyclic-ethers and lactones. Additionally, few synthetic compounds showed selective and moderate anti-HCMV, anti-GBM6, and anti-MDA MB 231 activity. The structural implication along with biological activity validates that the fluoro-spiro-isoxazolines have the potential to serve as inhibitors of HCMV infection as well as anti-cancer agents.Experimental section
Materials and methods
The reactions mentioned below are performed in non-inert atmosphere using HPLC grade CH2Cl2, commercial grade EtOH, anhydrous DMF, and deionized water as solvent. All reagents were used as supplied without prior purification unless otherwise stated. The progress of the reaction is monitored by analytical thin-layer chromatography comprised of 60 Å silica gel medium with layer-thickness 250 μm and visualized on 254 nm light, or KMnO4/Na2CO3/NaOH mixture and subsequent development with either no or gentle heating. Purifications by flash column chromatography were performed using flash silica gel (60 Å, 0.060–0.200 mm) with the indicated eluent.
Biological assay for antiviral activity against human cytomegalovirus (HCMV)
Biological assay for cytotoxicity against glioblastomas (GBM6)
Biological assay for MDA MB 231 breast carcinoma cells
General procedure for 1,3-dipolar cycloaddition of the alkynol (condition 1)
A solution of the alkynol (1 equiv.) and the hydroximoyl chloride (1.2 equiv.) in 10 mL of dichloromethane was treated with triethylamine (1.5 equiv.). The reaction mixture was stirred at rt for 24 h until complete consumption of starting material as monitored by TLC analysis. After the reaction was complete, a minimum amount of silica gel was added, and the solvent was evaporated under reduced pressure. The crude products were purified by column chromatography over silica gel using hexanes/ethyl acetate as eluent to provide the desired products.:1 ethyl acetate:hexanes); mp: 48–50 °C; IR νmax 3261, 3018, 2922, 2871, 2853, 1606, 1430, 1062, 1040, 798 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.65 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 6.28 (s, 1H), 3.71 (t, J = 6.2 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.00–1.95 (m, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 173.4, 162.4, 140.0, 129.6 (2C), 126.6 (2C), 126.3, 99.1, 61.4, 30.3, 23.2, 21.4; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H15NO2Na: 240.0995; found 240.0995.:1 ethyl acetate:hexanes); mp: 70–72 °C; IR νmax 3241, 3122, 2931, 2870, 1612, 1524, 1432, 1236, 1035, 842, cm−1; 1H NMR (500 MHz, CDCl3): δ 7.76–7.72 (m, 2H), 7.11 (t, J = 8.7 Hz, 2H), 6.27 (s, 1H), 3.72 (t, J = 6.2 Hz, 2H), 2.90 (t, J = 7.6 Hz, 2H), 2.26 (br s, 1H, OH), 1.98 (tt, J = 7.3, 6.2 Hz, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 173.7, 163.7 (d, J = 249.6 Hz), 161.5, 128.6 (d, J = 8.4 Hz, 2C), 125.4 (d, J = 3.4 Hz), 115.9 (d, J = 21.8 Hz, 2C), 99.0, 61.4, 30.3, 23.2; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H12Cl2FNO2Na: 244.0744; found 244.0743.:1 hexanes/ethyl acetate); mp 80–82 °C; IR νmax 3240, 3150, 2931, 2873, 1605, 1426, 1061, 1041, 833, 801 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.63 (d, J = 8.6 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 6.29 (s, 1H), 3.73 (t, J = 6.2 Hz, 2H), 2.91 (t, J = 7.6 Hz, 2H), 2.07 (br s, 1H, OH), 1.98 (dd, J = 14.3, 6.9 Hz, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 173.8, 161.5, 132.1 (2C), 128.2 (2C), 128.1, 124.1, 99.0, 61.4, 30.2, 23.2; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H12BrNO2Na: 303.9943; found 303.9942.:1 ethyl acetate:hexanes); mp: 50–52 °C; IR νmax 331, 2955, 2936, 2874, 1603, 1438, 1462, 1324, 1164, 1111, 1063, 848 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.90 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 9.1 Hz, 2H), 6.37 (s, 1H), 3.75 (t, J = 6.1 Hz, 2H), 2.95 (t, J = 7.6 Hz, 2H), 2.04–1.99 (m, 2H), 1.80 (br s, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ 174.2, 161.3, 132.6, 131.7 (q, J = 32.7 Hz), 127.1 (d, J = 8.2 Hz, 2C), 125.8 (q, J = 3.8 Hz, 2C), 123.8 (q, J = 272.3 Hz), 99.2, 61.4, 30.2, 23.2; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H12F3NO2Na: 294.0712; found 294.0711.:1 ethyl acetate:hexanes); IR νmax 3350, 3130, 2960, 2928, 2850, 1590, 1514, 1460, 1340, 1175, 1105, 1003, cm−1; 1H NMR (500 MHz, CDCl3): δ 8.30 (d, J = 8.7 Hz, 2H), 7.96 (d, J = 8.7 Hz, 2H), 6.41 (s, 1H), 3.76 (t, J = 5.9 Hz, 2H), 2.97 (t, J = 7.6 Hz, 2H), 2.05–2.00 (m, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 174.6, 160.6, 148.5, 135.4, 126.6 (2C), 124.2 (2C), 99.3, 61.4, 30.2, 23.2; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H12N2O4Na: 271.0689; found 271.0688.:1 ethyl acetate:hexanes); mp: 58–60 °C; IR νmax 3443, 3405, 3132, 2940, 2929, 2861, 1606, 1591, 1524, 1433, 1346, 1231, 910, 842 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.74–7.72 (m, 2H), 7.10 (t, J = 8.9 Hz, 2H), 6.24 (s, 1H), 3.66 (t, J = 6.4 Hz, 2H), 2.79 (t, J = 7.8 Hz, 2H), 2.61 (br s, 1H OH), 1.83–1.77 (m, 2H), 1.66–1.623 (m, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 174.1, 163.6 (d, J = 249.7 Hz), 161.4, 128.6 (d, J = 8.4 Hz, 2C), 125.4 (d, J = 3.3 Hz), 115.9 (d, J = 21.8 Hz, 2C), 98.9, 62.0, 31.8, 26.5, 23.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H14FNO2Na: 258.0900; found 258.0900.:3 ethyl acetate:hexanes); mp: 60–62 °C; IR νmax 3468, 3132, 2945, 2923, 2853, 1601, 1417, 1343, 1054, 902, 818, 721, 518 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.62 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 6.26 (s, 1H), 3.66 (t, J = 6.4 Hz, 2H), 2.8 (t, J = 7.6 Hz, 2H), 2.12 (br s, 1H, OH), 1.81 (dt, J = 15.4, 7.6 Hz, 2H), 1.67–1.61 (m, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 174.2, 161.4, 132.1 (2C), 128.2 (2C), 128.1, 124.1, 98.8, 62.1, 31.8, 26.5, 23.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H14BrNO2Na: 318.0100; found 318.0099.:3 ethyl acetate:hexanes); mp: 58–60 °C; IR νmax 3400, 3137, 2947, 2922, 2871, 1601, 1461, 1321, 1175, 1123, 1111, 1060, 1015, 912, 851 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.88 (d, 8.3 Hz, 2H), 7.68 (d. J = 8.3 Hz, 2H), 6.34 (s, 1H), 3.69 (t, J = 6.4 Hz, 2H), 2.84 (t, J = 7.6 Hz, 2H), 1.97 (br s, 1H, OH), 1.97–1.81 (m, 2H), 1.66 (dt, J = 13.3, 6.4 Hz, 2H); 13C{1H}NMR (125 MHz, CDCl3): δ 174.5, 161.2, 132.7, 131.6 (q, J = 32.6 Hz), 127.0 (2C), 125.8 (q, J = 3.8 Hz, 2C), 123.8 (q, J = 272.2 Hz), 99.0, 62.1, 31.8, 26.5, 23.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C14H14F3NO2Na: 308.0868; found 308.0867.:1 ethyl acetate:hexanes); mp: 62–64 °C; IR νmax 3349, 3132, 2955, 2923, 2854, 1593, 1514, 1460, 1342, 1175, 1105, 1003, 862, 809 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.31 (d, J = 8.9 Hz, 2H), 7.97 (d, J = 8.9 Hz, 2H), 6.4 (s, 1H), 3.72 (t, J = 6.3 Hz, 2H), 2.89 (t, J = 7.6 Hz, 2H), 1.88 (dt, J = 15.4, 7.6 Hz, 2H), 1.69 (dt, J = 13.5, 6.4 Hz, 2H), 1.34 (br s, 1H, OH); 13C{1H}NMR (125 MHz, CDCl3): δ 175.0, 160.5, 148.5, 135.4, 127.6 (2C), 124.1 (2C), 99.2, 62.2, 31.8, 26.5, 23.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H14N2O4Na: 285.0845; found 285.0844.General procedure for 1,3-dipolar cycloaddition of the alkynoic acid (condition 1).
Deionized water (10 mL) was added into a round-bottomed flask containing the hydroximoyl chloride (2 equiv.) with stirring. The alkynoic acid 2a (1 equiv.) was added into this mixture. Potassium carbonate (2.5 equiv.) was then added in small portions. The reaction mixture was stirred at rt until the reaction was complete as evidenced by TLC/NMR analysis. The reaction mixture was acidified with 4 N HCl and treated with diethyl ether and water (1:1, 20 mL). Sodium hydroxide was added to this mixture until the mixture was basic (litmus paper). The mixture was extracted with diethyl ether (3 × 10 mL), and 4 M HCl was then added to the aqueous layer until it was acidic (litmus paper). The acidified aqueous layer was extracted with ethyl acetate (3 × 15 mL), and the resulting organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure to provide the crude product, which was purified via column chromatography over silica gel using an appropriate hexanes–ethyl acetate ratio as an eluent system.
:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 162–164 °C; IR νmax 3122, 2912, 2856, 1693, 1601, 1421, 1243, 1206, 951, 809 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.74 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 6.64 (s, 1H), 3.11 (t, J = 7.4 Hz, 2H), 2.80 (d, J = 7.4 Hz, 2H), 2.37 (s, 3H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 172.7, 172.3, 162.0, 139.8, 129.5 (2C), 126.7, 126.4 (2C), 98.9, 30.9, 21.9, 20.4; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H13NO3Na: 254.0787; found 254.0787.:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 160–162 °C; IR νmax 3117, 2927, 2848, 1691, 1605, 1589, 1524, 1428, 1233, 1204, 1233, 1204, 1156, 903, 815 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.92 (dd, J = 8.4, 5.6 Hz, 2H), 7.27 (t, J = 8.7 Hz, 2H), 6.69 (s, 1H), 3.12 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 173.1, 172.2, 163.6 (d, J = 247.4 Hz), 161.2, 128.7 (d, J = 8.6 Hz, 2C), 125.9 (d, J = 2.8 Hz), 115.8 (d, J = 22.0 Hz, 2C), 99.0, 30.8, 21.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H10FNO3Na: 258.0537; found 258.0536.:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 180–182 °C; IR νmax 3120, 2928, 2849, 1690, 1600, 1590, 1525, 1425, 1230, 1200, 1155, 900 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.82 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.6 Hz, 2H), 6.72 (s, 1H), 3.13 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 173.3, 172.2, 161.2, 132.1 (2C), 128.7, 128.4 (2C), 123.5, 99.0, 30.8, 21.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H10BrNO3Na: 317.9736; found 317.9736.:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 173–175 °C; IR νmax 3129, 2929, 2851, 1702, 1599, 1441, 1320, 1166, 1133, 1062, 929, 846, 815 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 8.1 (d, J = 8.0 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 6.82 (s, 1H), 3.15 (t, J = 6.3 Hz, 2H), 2.83 (t, J = 6.4 Hz, 2H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 173.7, 172.3, 161.0, 133.3, 130.9 (q, J = 32.3 Hz), 127.2 (2C), 125.9 (d, J = 3.5 Hz, 2C), 124.2 (q, J = 271.4 Hz), 99.3, 30.8, 21.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H10F3NO3Na: 308.0505; found 308.0504.:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 80–82 °C; IR νmax 3129, 3033, 2993, 2927, 2866, 1690, 1600, 1436, 1412, 1309, 1196, 1001, 924, 881, 827, 671 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 6.01 (s, 1H), 3.00 (t, J = 7.4 Hz, 2H), 2.71 (t, J = 7.4 Hz, 2H), 2.2 (s, 3H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 172.3, 171.6, 159.4, 101.5, 30.8, 21.7, 10.3; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C7H9NO3Na: 178.0474; found 178.0474.:1); Rf 0.4 (1:1 ethyl acetate:hexanes); mp: 98–100 °C; IR νmax 3124, 2958, 2931, 2871, 1690, 1598, 1431, 1414, 1309, 1227, 1215, 1000, 924, 828, 878 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 6.07 (s, 1H), 3.01 (t, J = 7.4 Hz, 2H), 2.72 (t, J = 7.4 Hz, 2H), 2.56 (t, J = 7.5 Hz, 2H), 1.69–1.61 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H}NMR (125 MHz, CD3COCD3): δ 172.3, 171.5, 163.4, 100.4, 30.9, 27.6, 21.8, 21.3, 13.1; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C9H13NO3Na: 206.0787; found 206.0789.General procedure for fluoro-etherification 4(a–o)
A solution of isoxazole alcohol 3(a–o) (0.2 mmol) and Selectfluor (0.11 g, 0.3 mmol) in CH3CN (2 mL) was refluxed under N2 for 24 h. Then the reaction mixture was cooled, and water was added and extracted with ethyl acetate. The organic phase was washed several times with water and finally with brine. The combined organic layer was dried over anhydrous MgSO4 and filtered. The filtrate was evaporated under reduced pressure to give the crude product which was purified over silica gel column chromatography using hexane/ethyl acetate (9:1) as eluent.
:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3063, 2990, 2957, 2926, 1447, 1375, 1275, 1260, 1060, 1039, 907, 750 cm−1; 1H NMR (500 MHz, CDCl3): δ4a/4a′ 7.8–7.76 (m, 2H), 7.44 (d, J = 3.5 Hz, 3H), 5.76 (d, J = 53.2 Hz, 1H), 4.17 (dd, J = 14.8, 7.4 Hz, 1H), 4.09 (dd, J = 14.8, 7.4 Hz, 1H), 2.52–2.45 (m, 1H), 2.34–2.22 (m, 2H), 2.13–2.05 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4a/4a′ 162.2 (d, J = 16.0 Hz), 130.6, 128.9 (2C), 127.8, 126.7 (2C), 116.2 (d, J = 28.3 Hz), 96.7 (d, J = 187.6 Hz), 69.7, 28.8 (d, J = 5.6 Hz), 23.9; 19F NMR (376 MHz, CDCl3): δ4a −185.4 (d, J = 53.1 Hz); 19F NMR (376 MHz, CDCl3): δ4a′ −201.6 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H12FNO2Na: 244.0744; found 244.0745.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3036, 2987, 2957, 2922, 2894, 1611, 1456, 1372, 1275, 1260, 1059, 036, 906, 782 cm−1; 1H NMR (500 MHz, CDCl3): δ4b/4b′ 7.66 (d, J = 7.5 Hz, 2H), 7.24 (d, J = 7.9 Hz, 2H), 5.74 (d, J = 53.2 Hz, 1H), 4.16 (dd, J = 14.9, 7.3 Hz, 1H), 4.08 (dd, J = 14.3 Hz, 7.4 Hz, 1H), 2.50–2.44 (m, 1H), 2.39 (s, 3H), 2.32–2.19 (m, 2H), 2.13–2.04 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4b/4b′ 155.6 (d, J = 15 Hz), 141.0, 129.6 (2C), 126.6 (2C), 125.0, 116.0 (d, J = 27.8 Hz), 96.8 (d, J = 187.6 Hz), 69.6, 28.8 (d, J = 5.4 Hz), 23.9, 21.5; 19F NMR (376 MHz, CDCl3): δ4b −185.52 (d, J = 53.2 Hz); 19F NMR (376 MHz, CDCl3): δ4b′ −201.7 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H14FNO2Na: 258.0900; found 258.0901.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 2990, 2962, 2896, 1602, 1512, 1415, 1371, 1229, 1059, 1033, 907, 887 cm−1; 1H NMR (500 MHz, CDCl3): δ4c 7.76 (dd, J = 8.0, 5.8 Hz, 2H), 7.13 (t, J = 8.5 Hz, 2H), 5.73 (d, J = 53.3 Hz, 1H), 4.17 (dd, J = 14.6, 7.4 Hz, 1H), 4.1 (dd, J = 14.2, 7.5 Hz, 1H), 2.50–2.44 (m, 1H), 2.33–2.20 (m, 2H), 2.14–2.05 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4c 164.0 (d, J = 251.6 Hz), 154.7 (d, J = 15.1 Hz), 128.7 (d, J = 8.6 Hz, 2C), 124.1 (d, J = 3.4 Hz), 116.3 (d, J = 27.7 Hz), 116.1 (d, J = 22.1 Hz, 2C), 96.7 (d, J = 187.9 Hz), 69.7, 28.8 (d, J = 5.5 Hz), 23.9; 19F NMR (376 MHz, CDCl3): δ4c −108 (m), −185.8 (d, J = 53.2 Hz); 1H NMR (500 MHz, CDCl3): δ4c′ 7.79 (ddd, J = 8.8, 5.3, 1.1 Hz, 2H), 7.13 (t, J = 8.7 Hz, 2H), 5.86 (d, J = 52.6 Hz, 1H), 4.28–4.22 (m, 1H), 4.13–4.09 (m, 1H), 2.47–2.41 (m, 1H), 2.35–2.22 (m, 2H), 2.17–2.09 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4c′ 164.2 (d, J = 251.9 Hz), 155.3 (d, J = 16.2 Hz), 128.9 (d, J = 8.6 Hz, 2C), 123.9 (d, J = 2.2 Hz), 116.1 (d, J = 22.1 Hz, 2C), 112.7 (d, J = 13.1 Hz), 93.6 (d, J = 201.7 Hz), 70.1, 34.1, 24.2; 19F NMR (376 MHz, CDCl3): δ4c′ −108 (tt, J = 8.4, 5.3 Hz), −202.5 (d, J = 52.6 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H11F2NO2Na: 262.0650; found 262.0651.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 2987, 2957, 2851, 2922, 2896, 2597, 1494, 1405, 1355, 1275, 1091, 1062, 1014, 833 cm−1; 1H NMR (500 MHz, CDCl3): δ4d 7.72 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 5.72 (d, J = 53.3 Hz, 1H), 4.20–4.15 (m, 1H), 4.10 (dd, J = 14.9, 7.5 Hz, 1H), 2.55–2.41 (m, 2H), 2.31–2.20 (m, 1H), 2.13–2.05 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4d 156.9, 136.7, 129.2 (2C), 128.2 (2C), 125.8, 110.0, 96.5 (d, J = 187.9 Hz), 70.2, 31.2, 23.9; 19F NMR (376 MHz, CDCl3): δ4d′ −185.9 (d, J = 52.6 Hz); 1H NMR (500 MHz, CDCl3): δ4d′ 7.71 (d, J = 8.7 Hz, 2H), 7.41 (d, J = 8.7 Hz, 2H), 5.86 (d, J = 52.6 Hz, 1H), 4.33–4.26 (m, 1H), 4.14–4.09 (m, 1H), 2.47–2.42 (m, 1H), 2.35–2.27 (m, 2H), 2.18–2.12 (m, 1H); 19F NMR (376 MHz, CDCl3): δ4d′ −202.3 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H11ClFNO2Na: 278.0354; found 278.0356.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3005, 2987, 2960, 2927, 2899, 2858, 1594, 1491, 1457, 1275, 1260, 1067, 912, 754 cm−1; 1H NMR (500 MHz, CDCl3): δ4e 7.66–7.62 (m, 2H), 7.58–7.56 (m, 2H), 5.72 (d, J = 53.3 Hz, 1H), 4.23–4.14 (m, 1H), 4.13–4.07 (m, 1H), 2.56–2.51 (m, 1H), 2.33–2.20 (m, 2H), 2.13–2.06 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4e 157.1 (d, J = 16.5 Hz), 132.2 (2C), 132.1, 128.1 (2C), 125.1, 101.6 (d, J = 23.1 Hz), 96.4 (d, J = 188.1 Hz), 69.8, 28.9 (d, J = 5.6 Hz), 23.9; 19F NMR (376 MHz, CDCl3): δ4e −186.1 (d, J = 53.8 Hz); 1H NMR (500 MHz, CDCl3): δ4e′ 7.66 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 6.8 Hz, 2H), 5.86 (d, J = 52.7 Hz, 1H), 4.25–4.22 (m, 1H), 4.18–4.10 (m, 1H), 2.47–2.43 (m, 1H), 2.36–2.22 (m, 2H), 2.17–2.08 (m, 1H); 19F NMR (376 MHz, CDCl3): δ4e′ −190.4 (d, J = 52.2 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H11BrFNO2Na: 321.9849; found 321.9851.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 2988, 2956, 2850, 2920, 2890, 2595, 1494, 1400, 1350, 1272, 1090, 1060, 1010, 833 cm−1; 1H NMR (500 MHz, CDCl3): δ4f/4f′ 7.40 (dd, J = 7.7, 3.3 Hz, 2H), 7.31 (ddd, J = 8.9, 7.4, 2.7 Hz, 1H), 6.13 (d, J = 10.1 Hz, 1H), 4.23 (t, J = 6.3 Hz, 1H), 3.21 (t, J = 7.5 Hz, 1H), 2.95 (t, J = 7.5 Hz, 1H), 2.84 (t, J = 7.5 Hz, 1H), 2.36–2.31 (m, 1H), 2.17–2.11 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4f/4f′ 158.8, 135.5 (2C), 130.9, 128.3, 128.2 (2C), 102.8 (d, J = 12.42 Hz), 97.3 (d, J = 154.9 Hz), 63.6, 31.7, 23.6; 19F NMR (376 MHz, CDCl3): δ4f −187.2 (d, J = 52.7 Hz); 19F NMR (376 MHz, CDCl3): δ4f′ −202.4 (d, J = 53.4 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H10Cl2FNO2Na: 311.9965; found 311.9967.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3005, 2954, 2928, 2850, 1460, 1325, 1270, 1265, 1170, 1130, 1070, cm−1; 1H NMR (500 MHz, CDCl3): δ4g 7.89 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H), 5.77 (d, J = 53.3 Hz, 1H), 4.19 (dd, J = 14.6, 7.3 Hz, 1H), 4.12 (dd, J = 14.5, 7.4 Hz, 1H), 2.53–2.46 (m, 1H), 2.35–2.30 (m, 1H), 2.29–2.22 (m, 1H), 2.15–2.08 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4g 154.6 (d, J = 15.2 Hz), 131.2, 128.2, 126.9 (2C), 125.9 (dd, J = 7.5, 3.9 Hz, 2C), 123.7 (q, J = 276.6 Hz), 116.8 (d, J = 27.8 Hz), 96.2 (d, J = 188.1 Hz), 69.9, 28.9 (d, J = 5.6 Hz), 23.9; 1H NMR (500 MHz, CDCl3): δ4g′ 7.90 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 5.94 (d, J = 52.2 Hz, 1H), 3.00–2.85 (m, 3H), 2.79–2.73 (m, 1H), 2.62–2.57 (m, 1H), 2.36–2.31 (m, 1H); 19F NMR (376 MHz, CDCl3): δ4g −62.9 (s), −191.6 (d, J = 52.4 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C26H20F8N2O4Na: 599.1187; found 599.1200.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3008, 2987, 2950, 2921, 2852, 1607, 1518, 1458, 1345, 1275, 1260, 1055, 914, 853 cm−1; 1H NMR (500 MHz, CDCl3): δ4h/4h′ 8.30 (d, J = 8.9 Hz, 2H), 7.94 (d, J = 8.0 Hz, 2H), 5.79 (d, J = 53.3 Hz, 1H), 4.20 (dd, J = 14.5, 7.4 Hz, 1H), 4.13 (dd, J = 14.7, 7.5 Hz, 1H), 2.53–2.48 (m, 1H), 2.37–2.25 (m, 2H), 2.17–2.12 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4h/4h′ 167.6, 148.7, 133.8, 127.4 (2C), 124.2 (2C), 117.2 (d, J = 28.0 Hz), 95.9 (d, J = 188.3 Hz), 70.1, 28.9 (d, J = 5.5 Hz), 23.8; 19F NMR (376 MHz, CDCl3): δ4h −186.1 (d, J = 53.9 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H11FN2O4Na: 289.0595; found 289.0594.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3061, 3003, 2954, 2886, 2853, 1446, 1377, 1275, 1260, 1203, 905, 889, 764, 750 cm−1; 1H NMR (500 MHz, CDCl3): δ4i/4i′ 7.78–7.76 (m, 2H), 7.43 (dd, J = 5.2, 1,8 Hz, 3H), 5.5 (d, J = 53.1 Hz, 1H). 4.08 (td, J = 12.3, 2.7 Hz, 1H), 3.76 (dd, J = 11.4, 4.4 Hz, 1H), 2.03–1.89 (m, 4H), 1.81–1.74 (m, 1H), 1.69–1.66 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4i/4i′ 155.9 (d, J = 15.2 Hz), 130.6, 130.9 (2C), 127.8, 126.7 (2C), 107.4 (d, J = 26.4 Hz), 98.01 (d, J = 188.8 Hz), 63.6, 26.2 (d, J = 7.3 Hz), 24.8, 18.6 (d, J = 2.2 Hz); 19F NMR (376 MHz, CDCl3): δ4i −185.5 (d, J = 53.2 Hz); 19F NMR (376 MHz, CDCl3): δ4i′ −201.8 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H14FNO2Na: 258.0900; found 258.0901.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3003, 2950, 2927, 2889, 2851, 1603, 1513, 1375, 1233, 1160, 1078, 0146, 905, 838, 750 cm−1; 1H NMR (500 MHz, CDCl3): δ4j/4j′ 7.76 (d, J = 7.8, 5.5 Hz, 2H), 7.12 (t, J = 8.6 Hz, 2H), 5.51 (d, J = 53.2 Hz, 1H), 4.08 (td, J = 12.1, 2.7 Hz, 1H), 3.77 (dd, J = 11.4, 4.3 Hz, 1H), 2.03–1.89 (m, 4H), 1.82–1.73 (m, 1H), 1.69–1.67 (br m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4j/4j′ 164.0 (d, J = 251.6 Hz), 155.0 (d, J = 14.1 Hz), 128.7 (d, J = 8.6 Hz, 2C), 124.1 (d, J = 3.4 Hz), 116.1 (d, J = 22.1 Hz, 2C), 107.5 (d, J = 26.3 Hz), 98.0 (d, J = 188.8 Hz), 63.6, 26.1 (d, J = 7.2 Hz), 24.8, 18.6 (d, J = 2.2 Hz); 19F NMR (376 MHz, CDCl3): δ4j −109.1 (m), −186.0 (d, J = 53.2 Hz); 19F NMR (376 MHz, CDCl3): δ4j′ −112.3 (m), −206.2 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H13F2NO2Na: 276.0806; found 276.0807.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 2985, 2958, 2850, 2925, 2898, 2595, 1490, 1400, 1352, 1276, 1093, 1060, 1010, 833 cm−1; 1H NMR (500 MHz, CDCl3): δ4k/4k′ 7.70 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 5.51 (d, J = 53.2 Hz, 1H), 4.07 (td, J = 12.3, 2.7 Hz, 1H), 3.77 (dd, J = 11.4, 4.4 Hz, 1H), 2.02–1.89 (m, 4H), 1.82–1.73 (m, 1H), 1.69–1.67 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4k/4k′ 155.1 (d, J = 15.1 Hz), 136.6, 129.2 (2C), 128.0 (2C), 126.4, 107.7 (d, J = 26.3 Hz), 97.8 (d, J = 188.9 Hz), 63.7, 26.1 (d, J = 7.3 Hz), 24.8, 18.6 (d, J = 2.1 Hz); 19F NMR (376 MHz, CDCl3): δ4k −185.6 (d, J = 53.6 Hz); 19F NMR (376 MHz, CDCl3): δ4k′ −192.7 (d, J = 53.2 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H10Cl2FNO3·H2ONa: 358.0019; found 358.0021.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3010, 2950, 2927, 2886, 2851, 1588, 1492, 1401, 1267, 1079, 1046, 1010, 908, 878, 764 cm−1; 1H NMR (500 MHz, CDCl3): δ4l/4l′ 7.63 (d, J = 7.7 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 5.5 (d, J = 53.2 Hz, 1H), 4.10–4.04 (m, 1H), 3.78–3.75 (m, 1H), 2.02–1.91 (m, 4H), 1.81–1.72 (m, 1H), 1.69–1.67 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4l/4l′ 155.37 (d, J = 15.1 Hz), 132.3 (2C), 128.8, 128.3 (2C), 125.1, 107.8 (d, J = 26.2 Hz), 97.9 (d, J = 188.9 Hz), 63.8, 26.3 (d, J = 7.3 Hz), 24.9, 18.7 (d, J = 2.1 Hz); 19F NMR (376 MHz, CDCl3): δ4l −190.5 (d, J = 52.1 Hz); 19F NMR (376 MHz, CDCl3): δ4l′ −206.0 (d, J = 52.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H13BrFNO2Na: 336.0005; found 336.0006.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3000, 2955, 2925, 2853, 1578, 1560, 1430, 1333, 1275, 1196, 876, 780 cm−1; 1H NMR (500 MHz, CDCl3): δ4m/4m′ 7.41–7.39 (m, 2H), 7.33 (dd, J = 8.9, 7.1 Hz, 1H), 5.54 (d, J = 53.2 Hz, 1H), 4.14 (td, J = 12.2, 2.7 Hz, 1H), 3.84 (dd, J = 11.6, 4.5 Hz, 1H), 2.06–1.89 (m, 4H), 1.82–1.76 (m, 1H), 1.72–1.68 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4m/4m′ 153.1 (d, J = 16.2 Hz), 135.9, 131.5 (2C), 128.4, 128.3 (2C), 108.0 (d, J = 25.8 Hz), 99.1 (d, J = 191.1 Hz), 63.9, 26.2 (d, J = 7.6 Hz), 24.8, 18.6 (d, J = 2.1 Hz); 19F NMR (376 MHz, CDCl3): δ4m −188.1 (d, J = 53.5 Hz); 19F NMR (376 MHz, CDCl3): δ4m′ −193.3 (d, J = 53.7 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H12Cl2FNO2Na: 326.0121; found 326.0120.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3003, 2952, 2924, 2853, 1459, 1324, 1275, 1260, 1169, 1130, 1070, 879, 764 cm−1; 1H NMR (500 MHz, CDCl3): δ4n/4n′ 7.88 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 5.55 (d, J = 53.2 Hz, 1H), 4.11–4.05 (m, 1H), 3.78 (dd, J = 11.5, 4.2 Hz, 1H), 2.04–1.91 (m, 4H), 1.80–1.74 (m, 1H), 1.71–1.67 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4n/4n′ 156.2 (d, J = 15.1 Hz), 130.9 (q, J = 31.6 Hz), 128.8 (d, J = 3.0 Hz), 126.9 (2C), 125.87 (dd, J = 6.7, 3.4 Hz, 2C), 123.7 (q, J = 265.0 Hz), 108.1 (d, J = 25.9 Hz), 97.6 (d, J = 189.9 Hz), 63.7, 29.7, 26.1 (d, J = 7.2 Hz), 18.5 (d, J = 2.2 Hz); 19F NMR (376 MHz, CDCl3): δ4n′ −62.8 (s), −191.4 (d, J = 52.3 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H10Cl2FNO3·H2ONa: 358.0019; found 358.0021.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); 1H NMR (500 MHz, CDCl3): δ4o/4o′ 8.30 (d, J = 8.8 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H), 5.56 (d, J = 53.3 Hz, 1H) 4.11–4.06 (td, J = 12.2, 2.7 Hz, 1H), 3.80 (dd, J = 11.3, 4.2 Hz, 1H), 2.05–1.92 (m, 4H), 1.84–1.79 (m, 1H), 1.72–1.69 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ4o/4o′ 154.5 (d, J = 15.2 Hz), 148.7, 133.4, 127.5 (2C), 124.2 (2C), 108.6 (d, J = 26.2 Hz), 97.2 (d, J = 189.4 Hz), 63.9, 26.0 (d, J = 7.3 Hz), 24.7, 18.5 (d, J = 1.9 Hz); 19F NMR (376 MHz, CDCl3): δ4o −185.7 (d, J = 54.0 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H13Cl2FN2O4Na: 303.0751; found 303.0752.General procedure for fluoro-lactonization 5(a–k)
A solution of isoxazole acid 3(p–z) (0.2 mmol) and Selectfluor (0.11 g, 0.3 mmol) in CH3CN (2 mL) was refluxed under N2 for 24 h. Then, the reaction mixture was cooled, and water was added and extracted with ethyl acetate. The organic phase was washed several times with water and finally with brine. The combined organic layers were dried over anhydrous MgSO4 and filtered. The filtrate was evaporated under reduced pressure to give the crude product which was purified over silica gel column chromatography using hexane/ethyl acetate (4:1) as eluent.
:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 2987, 2950, 2924, 2848, 1798, 1448, 1370, 1185, 1108, 1012, 880, 768 cm−1; 1H NMR (500 MHz, CDCl3): δ5a 7.77 (d, J = 7.8 Hz, 2H), 7.51–7.47 (m, 3H), 5.93 (d, J = 52.2 Hz, 1H), 2.98–2.83 (m, 2H), 2.76–2.70 (m, 1H), 2.59–2.54 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5a 173.5, 156.1 (d, J = 14.8 Hz), 131.5, 129.2 (2C), 127.0 (2C), 126.4, 112.7 (d, J = 30.1 Hz), 96.8 (d, J = 191.8 Hz), 27.1 (d, J = 1.7 Hz), 24.8 (d, J = 7.1 Hz); 19F NMR (376 MHz, CDCl3): δ5a −190.2 (d, J = 51.6 Hz); 1H NMR (500 MHz, CDCl3): δ5a′ 7.79 (d, J = 7.4 Hz, 2H), 7.49 (dt, J = 14.6, 6.8 Hz, 3H), 6.08 (d, J = 51.4 Hz, 1H), 2.95–2.85 (m, 1H), 2.77–2.71 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5a′ 173.0, 157.1 (d, J = 16.4 Hz), 137.7, 129.0 (2C), 127.1 (d, J = 2.1 HZ, 2C), 126.1 (d, J = 2.0 Hz), 108.9 (d, J = 14.0 Hz), 95.7 (d, J = 207.5 Hz), 28.7, 27.7; 19F NMR (376 MHz, CDCl3): δ5a′ −205.9 (d, J = 51.4 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H10FNO3Na: 258.0537; found 258.0536.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); mp: 112–114 °C; IR νmax 3028, 2990, 2955, 2922, 2848, 1795, 1608, 1452, 1371, 1278, 1247, 1176, 1128, 1005, 873, 820 cm−1; 1H NMR (500 MHz, CDCl3): δ5b 7.65 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 5.91 (d, J = 52.2 Hz, 1H), 2.98–2.82 (m, 2H), 2.75–2.70 (m, 1H), 2.59–2.53 (m, 1H), 2.41 (s, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5b 173.6, 156.0 (d, J = 14.7 Hz), 142.1, 129.9 (2C), 125.9 (2C), 123.5, 112.6 (d, J = 30.1 Hz), 96.9 (d, J = 191.8 Hz), 27.1 (d, J = 1.8 Hz), 24.8 (d, J = 7.0 Hz), 21.6; 19F NMR (376 MHz, CDCl3): δ5b −190.1 (d, J = 51.9 Hz); 1H NMR (500 MHz, CDCl3): δ5b′ 7.67 (d, J = 7.1 Hz, 2H), 7.26 (d, J = 7.6 Hz, 2H), 6.1 (d, J = 51.5 Hz, 1H), 2.93–2.86 (m, 1H), 2.79–2.70 (m, 3H), 2.40 (s, 3H); 19F NMR (376 MHz, CDCl3): δ5b′ −205.8 (d, J = 51.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H12FNO3Na: 272.0693; found 272.0692.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3076, 3013, 2952, 2924, 2852, 1796, 1602, 1513, 1416, 1376, 1232, 1022, 882 cm−1; 1H NMR (500 MHz, CDCl3): δ5c 7.77 (dd, J = 8.0, 5.5 Hz, 2H), 7.16 (t, J = 8.5 Hz, 2H), 5.90 (d, J = 52.5 Hz, 1H), 2.90 (ddt, J = 22.6, 19.4, 9.5 Hz, 2H), 2.73 (ddd, J = 16.4, 9.2, 2.9 Hz, 1H), 2.59–2.54 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5c 173.4, 164.5 (d, J = 253.3 Hz), 155.2 (d, J = 14.0 Hz), 129.2 (d, J = 8.7 Hz, 2C), 122.6 (d, J = 3.4 Hz), 116.5 (d, J = 22.2 Hz, 2C), 112.7 (d, J = 30.1 Hz), 96.7 (d, J = 192.0 Hz), 27.1 (d, J = 1.8 Hz), 24.8 (d, J = 7.0 Hz); 19F NMR (376 MHz, CDCl3): δ5c −107.1 (m), −190.4 (d, J = 52.2 Hz); 1H NMR (500 MHz, CDCl3): δ5c′ 7.80 (dd, J = 7.7, 5.4 Hz, 2H), 7.16 (t, J = 8.6 Hz, 2H), 6.1 (d, J = 51.5 Hz, 1H), 2.95–2.85 (m, 1H), 2.79–2.71 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5c′ 172.9, 164.7 (d, J = 253.5 Hz), 156.1 (d, J = 16.4 Hz), 129.3 (d, J = 8.4 Hz, 2C), 122.4 (d, J = 1.8 Hz), 116.4 (d, J = 22.1 Hz, 2C), 108.9 (d, J = 14.2 HZ), 95.6 (d, J = 207.5 Hz), 28.6, 27.7; 19F NMR (376 MHz, CDCl3): δ5c′ −106.7 (m), −206.2 (d, J = 51.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H9F2NO3Na: 276.0443; found 276.0445.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3099, 2957, 2922, 2848, 1798, 1608, 1446, 1275, 1123, 1024, 880, 747 cm−1; 1H NMR (500 MHz, CDCl3): δ5d 7.70 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 5.95 (d, J = 52.1 Hz, 1H), 2.98–2.83 (m, 1H), 2.77–2.69 (m, 1H), 2.59–2.54 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5d 173.4, 155.23 (d, J = 14.9 Hz), 137.7, 129.5 (2C), 128.2 (2C), 124.8, 112.8 (d, J = 30.2 Hz), 96.56 (d, J = 192.1 Hz), 27.02 (d, J = 1.87 Hz), 24.81 (d, J = 7.1 Hz); 19F NMR (376 MHz, CDCl3): δ5d −190.4 (d, J = 52.6 Hz); 1H NMR (500 MHz, CDCl3): δ5d′ 7.73 (d, J = 8.5 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H), 6.1 (d, J = 51.5 Hz, 1H), 2.95–2.86 (m, 1H), 2.78–2.71 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5d′ 172.8, 156.2 (d, J = 16.5 Hz), 137.9, 129.4 (2C), 128.3 (d, J = 2.2 Hz, 2C), 124.6 (d, J = 2.2 Hz), 109.0 (d, J = 14.1 Hz), 95.5 (d, J = 207.7 Hz), 28.7, 27.7; 19F NMR (376 MHz, CDCl3): δ5d′ −206.4 (d, J = 51.4 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H9ClFNO3Na: 292.0147; found 292.0146.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3025, 2957, 2927, 2848, 1793, 1591, 1487, 1412, 1269, 1134, 1009, 886 cm−1; 1H NMR (500 MHz, CDCl3): δ5e 7.66–7.60 (m, 4H), 5.89 (d, J = 52.3 Hz, 1H), 3.00–2.83 (m, 2H), 2.77–2.69 (m, 1H), 2.60–2.54 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5e 173.5, 155.3 (d, J = 15.9 Hz), 132.5, 132.3 (2C), 128.4 (2C), 126.1, 112.8 (d, J = 30.1 Hz), 96.5 (d, J = 192.1 Hz), 27.0 (d, J = 1.8 Hz), 24.8 (d, J = 7.1 Hz); 19F NMR (376 MHz, CDCl3): δ5e −190.4 (d, J = 52.4 Hz); 1H NMR (500 MHz, CDCl3): δ5e′ 7.66 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 6.1 (d, J = 51.5 Hz, 1H), 2.95–2.89 (m, 1H), 2.76–2.74 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5e′ 172.8, 156.3 (d, J = 16.7 Hz), 132.4 (2C), 131.7 (d, J = 29.1 Hz), 128.4 (d, J = 2.3 Hz, 2C), 126.4, 109.0 (d, J = 14.2 Hz), 95.5 (d, J = 207.6 Hz) 28.7, 27.6; 19F NMR (376 MHz, CDCl3): δ5e′ −206.4 (d, J = 51.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H9BrFNO3Na: 335.9642; found 335.9645.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3010, 2955, 2924, 2854, 1710, 1560, 1432, 1275, 1260, 1194 cm−1; 1H NMR (500 MHz, CDCl3): δ5f 7.48–7.37 (m, 3H), 5.89 (d, J = 52.1 Hz, 1H), 3.00–2.91 (m, 1H), 2.90–2.81 (m, 1H), 2.78–2.71 (m, 1H), 2.64–2.59 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5f 173.5, 153.5 (d, J = 16.2 Hz), 135.8, 132.2 (2C), 128.4 (2C), 125.2, 112.8 (d, J = 29.8 Hz), 97.6 (d, J = 94.4 Hz), 27.0 (d, J = 1.8 Hz), 24.8 (d, J = 7.1 Hz); 19F NMR (376 MHz, CDCl3): δ5f −187.6 (d, J = 50.5 Hz); 1H NMR (500 MHz, CDCl3): δ5f′ 7.48–7.37 (m, 3H), 6.1 (d, J = 51.2 Hz, 1H), 2.98–2.88 (m, 1H), 2.83–2.71 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5f′ 176.1, 153.6, 135.7, 132.3 (2C), 128.3 (2C), 116.2, 109.1 (d, J = 13.2 Hz), 95.7 (d, J = 209.4 Hz), 28.8, 27.6; 19F NMR (376 MHz, CDCl3): δ5f′ −191.5 (d, J = 51.9 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C12H8Cl2FNO3Na: 325.9757; found 325.9757.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); mp: 130–132 °C; IR νmax 3005, 2955, 2921, 2851, 1798, 1416, 1325, 1269, 1163, 1115, 1025, 883 cm−1; 1H NMR (500 MHz, CDCl3): δ5g 7.90 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 5.94 (d, J = 52.2 Hz, 1H), 3.00–2.85 (m, 2H), 2.75 (d, J = 16.3, 9.1, 2.8 Hz, 1H), 2.62–2.57 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3): δ5g 173.2, 155.1 (d, J = 13.6 Hz), 133.1 (q, J = 32.1 Hz), 129.8, 127.3 (2C), 126.2 (d, J = 3.3 Hz, 2C), 125.5 (q, J = 272.6 Hz), 112.9 (d, J = 30.4 Hz), 96.3 (d, J = 192.4 Hz), 26.9, 24.8 (d, J = 7.2 Hz); 19F NMR (376 MHz, CDCl3): δ5g −63.1 (s), −190.5 (d, J = 52.2 Hz); 1H NMR (500 MHz, CDCl3): δ5g′ 7.92 (d, J = 8.3 Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 6.1 (d, J = 51.4 Hz, 1H), 2.97–2.88 (m, 1H), 2.79–2.73 (m, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5g′ 172.7, 156.0 (d, J = 16.9 Hz), 133.3 (dd, J = 63.7, 30.71 Hz), 129.5 (dd, J = 3.7, 2.3 Hz), 127.4 (d, J = 2.2 Hz, 2C), 126.0 (dd, J = 7.7, 3.9 Hz, 2C), 125.5 (q, J = 272.5 Hz), 109.1 (d, J = 13.9 Hz), 95.3 (d, J = 207 Hz) 28.7, 27.6; 19F NMR (376 MHz, CDCl3): δ5g′ −63.2 (s), −206.6 (d, J = 51.5 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H9ClF4NO3Na: 326.0411; found 326.0413.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3008, 2957, 2929, 2853, 1797, 1436,1 1391, 1260, 1182, 1123, 1033, 905, 839 cm−1; 1H NMR (500 MHz, CDCl3): δ5h 5.37 (d, J = 52.0 Hz, 1H), 2.88 (dt, J = 17.2, 9.5 Hz, 1H), 2.79–2.72 (m, 1H), 2.67 (ddd, J = 17.2, 9.8, 2.8 Hz, 1H), 2.45 (ddd, J = 13.4, 9.6, 2.8 Hz, 1H), 2.19 (d, J = 2.9 Hz, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5h 173.7, 154.8 (d, J = 15.1 Hz), 111.9 (d, J = 29.6 Hz), 98.6 (d, J = 191.2 Hz), 27.1, 24.7 (d, J = 6.8 Hz), 11.1; 19F NMR (376 MHz, CDCl3): δ5h −189.9 (d, J = 55.9 Hz); 1H NMR (500 MHz, CDCl3): δ5h′ 5.57 (d, J = 51.2 Hz, 1H), 2.87–2.79 (m, 1H), 2.71–2.63 (m, 3H), 2.14 (s, 3H); 13C{1H}NMR (125 MHz, CDCl3): δ5h′ 173.1, 157.6 (d, J = 17.7 Hz), 108.5 (d, J = 13.5 Hz), 97.1 (d, J = 206.1 Hz), 28.1, 27.7, 10.7; 19F NMR (376 MHz, CDCl3): δ5h′ −197.9 (d, J = 52.7 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C7H8FNO3Na: 196.0380; found 196.0380.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3008, 2965, 2937, 2876, 1796, 1713, 1447, 1275, 1177, 1042, 915, 764 cm−1; 1H NMR (500 MHz, CDCl3): δ5i 5.61 (d, J = 51.2 Hz, 1H), 2.87–2.79 (m, 2H), 2.66–2.63 (m, 2H), 2.53–2.41 (m, 2H), 1.77–1.64 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H}NMR (125 MHz, CDCl3) δ5i 173.2, 160.6 (d, J = 18.4 Hz), 108.4 (d, J = 13.7 Hz), 96.7 (d, J = 206.5 Hz), 28.1, 27.8, 27.5, 18.8, 13.7; 19F NMR (376 MHz, CDCl3): δ5i −188.8 (d, J = 52.2 Hz); 1H NMR (500 MHz, CDCl3): δ5i′ 5.4 (d, J = 51.9 Hz, 1H), 2.92–2.85 (m, 2H), 2.72–2.64 (m, 2H), 2.54–2.48 (m, 2H), 1.77–1.65 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H); 19F NMR (376 MHz, CDCl3): δ5i′ −192.7 (d, J = 50.7 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C9H12FNO3Na: 224.0693; found 224.0695.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3008, 2957, 2926, 2851, 1707, 1560, 1430, 1275, 1260, 1194, 1052, 782, 750 cm−1; 1H NMR (500 MHz, CDCl3): δ5j/5j′ 7.46–7.36 (m, 3H), 5.84 (d, J = 52.5 Hz, 1H), 2.91–2.85 (m, 1H), 2.75–2.62 (m, 2H), 2.35–2.24 (m, 2H), 2.09–2.03 (m, 1H); 13C{1H}NMR (125 MHz, CDCl3) δ5j/5j′ 168.0, 162.5 (d, J = 16.1 Hz), 135.9, 132.1 (2C), 128.4 (2C), 125.3, 110.6 (d, J = 26.9 Hz), 98.8 (d, J = 195.4 Hz), 29.3, 23.6 (d, J = 7.8 Hz), 15.4 (d, J = 1 Hz); 19F NMR (376 MHz, CDCl3): δ5j −187.7 (d, J = 52.3 Hz); 19F NMR (376 MHz, CDCl3): δ5j′ −191.5 (d, J = 52.0 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H10Cl2FNO3·H2ONa: 358.0019; found 358.0021.:1); Rf 0.4 (1:3 ethyl acetate:hexanes); IR νmax 3005, 2950, 2923, 2853, 1719, 1462, 1275, 1259, 1016, 925, 764 cm−1; 1H NMR (500 MHz, CDCl3): δ5k/5k′ 5.67 (d, J = 51.8 Hz, 1H), 4.43 (q, J = 7.2 Hz, 2H), 2.87–2.81 (m, 1H), 2.69–2.63 (m, 1H), 2.35–2.23 (m, 3H), 2.08–2.04 (m, 1H), 1.40 (t, J = 7.2 Hz, 3H); 13C{1H}NMR (125 MHz, CDCl3) δ5k/5k′ 167.1, 164.7, 160.5 (d, J = 16.0 Hz), 111.9 (d, J = 13.8), 95.1 (d, J = 193.7 Hz), 62.9, 29.3, 23.1 (d, J = 7.2 Hz), 15.5, 14.0; 19F NMR (376 MHz, CDCl3): δ5k −191.3 (d, J = 55.8 Hz); 19F NMR (376 MHz, CDCl3): δ5k′ −198.1 (d, J = 52.7 Hz); HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C10H12FNO3·H2ONa: 286.0697; found 286.0699.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by grants from the National Institute of General Medicinal Sciences (NIH/NIGMS) (2SC3GM094081-08) and partially supported by the National Science Foundation (HBCU-EiR) (1900127). The Analytical and NMR CORE Facilities were supported by NIH/NIMHD (G12MD007581).Notes and references
- (a) P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS; (b) M. Zaki, A. Oukhrib, M. Akssira and S. B. Raboin, RSC Adv., 2017, 7, 6523–6529 RSC; (c) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed; (d) T. Sengoku, A. Shirai, A. Takano, T. Inuzuka, M. Sakamoto, M. Takahashi and H. Yoda, J. Org. Chem., 2019, 84, 12532–12541 CrossRef CAS PubMed; (e) T. P. I. Saragi, T. Spehr, A. Siebert, T. F. Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed; (f) R. R. Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari and D. Sriram, J. Med. Chem., 2008, 51, 5731–5735 CrossRef CAS PubMed; (g) C.-Y. Chan, Y.-C. Wong, M.-Y. Chan, S.-H. Cheung, S.-K. So and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2016, 8, 24782–24792 CrossRef CAS PubMed.
- (a) A. Sysak and B. Obmińska-Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS PubMed; (b) T. M. V. D. Pinho e Melo, Curr. Org. Chem., 2005, 9, 925–958 CrossRef CAS; (c) C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Angew. Chem., 2016, 128, 4065–4069 CrossRef.
- (a) G. A. Habeeb, P. N. P. Rao and E. E. Kanus, J. Med. Chem., 2001, 44, 2921–2927 CrossRef PubMed; (b) G. Daidone, D. Raffa, B. Maggio, F. Plescia, V. M. C. Cutuli, N. G. Mangano and A. Caruso, Arch. Pharm., 1999, 332, 50–54 CrossRef CAS.
- P. Cali, L. Nærum, S. Mukhija and A. Hjelmencrantz, Bioorg. Med. Chem. Lett., 2004, 14, 5997–6000 CrossRef CAS PubMed.
- J. Liu, L.-F. Yu, J. B. Eaton, B. Caldarone, K. Cavino, C. Ruiz, M. Terry, A. Fedolak, D. Wang, A. Ghavami, D. A. Lowe, D. Brunner, R. J. Lukas and A. P. Kozikowski, J. Med. Chem., 2011, 54, 7280–7288 CrossRef CAS PubMed.
- (a) K. Kaur, V. Kumar, A. K. Sharma and G. K. Gupta, Eur. J. Med. Chem., 2014, 77, 121–133 CrossRef CAS PubMed; (b) R. K. Howe and B. R. Shelton, J. Org. Chem., 1990, 55, 4603–4607 CrossRef CAS; (c) R. S. Compagnone, R. Avila, A. I. Suárez, O. V. Abrams, H. R. Rangel, F. Arvelo, I. C. Piña and E. Merentes, J. Nat. Prod., 1999, 62, 1443–1444 CrossRef CAS PubMed; (d) J. P. Yong, C. Z. Lu and X. Y. Wu, MedChemComm, 2014, 7, 968–972 RSC; (e) R. M. Kumbhare, U. B. Kosurkar, M. Janaki Ramaiah, T. L. Dadmal, S. N. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 5424–5427 CrossRef CAS PubMed.
- (a) K. M. Dawood, H. Abdel-Gawad, H. A. Mohamed and F. A. Badria, Med. Chem. Res., 2011, 20, 912–919 CrossRef CAS; (b) Y.-S. Lee, S. M. Park and B. H. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 1126–1128 CrossRef CAS PubMed.
- (a) J. J. Harburn, N. P. Rath and C. D. Spilling, J. Org. Chem., 2005, 70, 6398–6403 CrossRef CAS PubMed; (b) G. M. Nicholas, L. L. Eckman and G. L. Newton, Bioorg. Med. Chem. Lett., 2003, 11, 601–608 CrossRef CAS; (c) C. Patrizia, D. Carmela and F. Ernesto, J. Nat. Prod., 1999, 62, 590–593 CrossRef PubMed; (d) M. Murakata, M. Tamura and O. Hoshino, J. Org. Chem., 1997, 62, 4428–4433 CrossRef CAS PubMed; (e) S. Bardhan, D. C. Schmitt and J. A. Porco, Org. Lett., 2006, 8, 927–930 CrossRef CAS PubMed; (f) E. McClendon, A. O. Omollo, E. J. Valente and A. T. Hamme, Tetrahedron Lett., 2009, 50, 533–535 CrossRef CAS PubMed; (g) E. D. Ellis, J. Xu, E. J. Valente and A. T. Hamme, Tetrahedron Lett., 2009, 50, 5516–5519 CrossRef CAS PubMed; (h) P. Savage, Curr. Org. Chem., 2010, 14, 1478–1499 CrossRef.
- (a) P. R. Bergquist and R. J. Wells, in Marine Natural Products–Chemical and Biological Perspectives, ed. P. J. Scheuer, Academic Press, New York, 1983, vol. 5, pp. 1–50 Search PubMed; (b) F. Hentschel and T. Lindel, Synthesis, 2010, 181–204 CAS; (c) R. S. Compagnone, R. Avila, A. I. Suárez, O. V. Abrams, H. R. Rangel, F. Arvelo, I. C. Piña and E. Merentes, J. Nat. Prod., 1999, 62, 1443–1444 CrossRef CAS PubMed; (d) R. D. Encarnacion, E. Sandoval, J. Mamstrom and C. Christophersen, J. Nat. Prod., 2000, 63, 874–875 CrossRef CAS PubMed; (e) G. M. Konig and A. D. Wright, Heterocycles, 1993, 36, 1351–1358 CrossRef; (f) K. Anake, W. Rawiwan, S. Pichai, A. M. S. Silva, G. Eaton and H. Werner, J. Biosci., 2001, 56, 1116–1119 Search PubMed; (g) X. Yang, R. A. Davis, M. S. Buchanan, S. Duffy, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2010, 73, 985–987 CrossRef CAS PubMed; (h) S. Tsukamoto, H. Kato, H. Hirota and N. Fusetani, Tetrahedron, 1996, 52, 181–8186 CrossRef; (i) K. T. Okamoto and J. Clardy, Tetrahedron Lett., 1987, 28, 4969–4972 CrossRef CAS; (j) X. Yang, R. A. Davis, M. S. Buchanan, S. Duffy, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2010, 73, 985–987 CrossRef CAS PubMed; (k) Y.-J. Lee, S. Han, H.-S. Lee, J. S. Kang, J. Yun, C. J. Sim, H. J. Shin and J. S. Lee, J. Nat. Prod., 2013, 76, 1731–1736 CrossRef CAS PubMed; (l) I. W. Mudianta, T. Skinner-Adams, K. T. Andrews, R. A. Davis, T. A. Hadi, P. Y. Hayes and M. J. Garson, J. Nat. Prod., 2012, 75, 2132–2143 CrossRef CAS PubMed.
- (a) D. M. Roll, C. W. J. Chang, P. J. Sheuer, G. A. Gray, J. N. Shoolery, G. K. Matsumoto, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1985, 107, 2916–2920 CrossRef CAS; (b) B. R. Copp and C. M. Ireland, J. Nat. Prod., 1992, 55, 822–823 CrossRef CAS PubMed; (c) T. Ichiba and P. J. Scheuer, J. Org. Chem., 1993, 58, 4149–4150 CrossRef CAS; (d) S. Liu, X. Fu, F. J. Schmitz and M. Kelly-Borges, J. Nat. Prod., 1997, 60, 614–615 CrossRef CAS PubMed; (e) X. Yang, R. A. Davis, M. S. Buchanan, S. Duffy, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2010, 73, 985–987 CrossRef CAS PubMed; (f) M. Xu, K. T. Andrews, G. W. Birrell, T. L. Tran, D. Camp, R. A. Davis and R. Quinn, Bioorg. Med. Chem. Lett., 2011, 21, 846–848 CrossRef CAS PubMed; (g) A. D. Wright, P. J. Schupp, J.-P. Schrör, A. Engemann, S. Rohde, D. Kelman, N. D. Voogd, A. Carroll and C. A. Motti, J. Nat. Prod., 2012, 75, 502–506 CrossRef CAS PubMed.
- (a) P. Das, A. O. Omollo, L. J. Sitole, E. McClendon, E. J. Valente, D. Raucher, L. R. Walker and A. T. Hamme II, Tetrahedron Lett., 2015, 56, 1794–1797 CrossRef CAS PubMed; (b) P. Das, M. H. Hasan, D. Mitra, R. Bollavarapu, E. J. Valente, R. Tandon, D. Raucher and A. T. Hamme II, J. Org. Chem., 2019, 84, 6992–7006 CrossRef CAS PubMed; (c) P. Das, E. J. Valente and A. T. Hamme II, Eur. J. Org. Chem., 2014, 2659–2663 CrossRef CAS PubMed; (d) P. Das and A. T. Hamme II, Eur. J. Org. Chem., 2015, 5159–5166 CrossRef CAS.
- (a) M. R. C. Gerstenberger and A. Haas, Angew. Chem., Int. Ed. Engl., 1981, 20, 647–667 CrossRef; (b) K. Mueller, C. Faehand and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef CAS PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (d) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (b) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef; (c) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (b) P. Jeschke, Pest Manag. Sci., 2010, 66, 10–27 CrossRef CAS PubMed; (c) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- (a) S. C. Wilkinson, R. Salmon and V. Gouverneur, Future Med. Chem., 2009, 1, 847–863 CrossRef CAS PubMed; (b) J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. V. Pidgeon, P. R. Moore, G. Sanford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796–9800 CrossRef CAS PubMed; (c) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed.
- (a) K. Sato, G. Sandford, Y. Konishi, N. Yanada, C. Toda, A. Tarui and M. Omote, Org. Biomol. Chem., 2019, 17, 2818–2823 RSC; (b) X.-W. Liang, Y. Cai and S.-L. You, Chin. J. Chem., 2018, 36, 925–928 CrossRef CAS; (c) K. Sato, G. Sandford, K. Shimizu, S. Akiyama, M. J. Lancashire, D. S. Yufit, A. Tarui, M. Omote, I. Kumadaki, S. Harusawa and A. Ando, Tetrahedron, 2016, 72, 1690–1698 CrossRef CAS; (d) J. Z. M. Fong, S. S. S. Choo, J.-A. Richard, M. V Garland, L. Guo, C. W. Johannes and T. M. Nguyen, Eur. J. Org. Chem., 2015, 995–1006 CrossRef CAS; (e) J. R. Breen, G. Sandford, B. Patel and J. Fray, Synlett, 2015, 26, 51–54 CAS; (f) M. Wang, X. Liu, L. Zhou, J. Zhu and X. Sun, Org. Biomol. Chem., 2015, 13, 3190–3193 RSC; (g) T. M. Nguyen, H. A. Duong, J.-A. Richard, C. W. Johannes, F. Pincheng, D. Kwong Jia Ye and E. L. Shuying, Chem. Commun., 2013, 49, 10602–10604 RSC; (h) O. Lozano, G. Blessley, T. Martinez del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105–8109 CrossRef CAS PubMed; (i) R. Lin, S. Ding, Z. Shi and N. Jiao, Org. Lett., 2011, 13, 4498–4501 CrossRef CAS PubMed.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- (a) F. A. Davis and W. Han, Tetrahedron Lett., 1991, 32, 1631–1634 CrossRef CAS; (b) F. A. Davis, W. Hart and C. K. Murphy, J. Org. Chem., 1995, 60, 4730–4737 CrossRef CAS; (c) M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed; (d) Q. Gu and E. Vessally, RSC Adv., 2020, 10, 16756–16768 RSC.
- W. Dunn, C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu and F. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 100, 14223–14228 CrossRef PubMed.
- D. J. Maggs and H. E. Clarke, Am. J. Vet. Res., 2004, 65, 399–403 CrossRef CAS PubMed.
- W. Strober, Curr. Protoc. Immunol., 2001, 21(1), A.3B.1–A.3B.2 Search PubMed , appendix 3, appendix 3B.
- R. R. Razonable, Curr. Opin. Organ Transplant., 2018, 23, 388–394 CrossRef CAS PubMed.
- T. E. Komatsu, A. Pikis, L. K. Naeger and P. R. Harrington, Antiviral Res., 2014, 101, 12–25 CrossRef CAS PubMed.
- M. A. Archer, T. M. Brechtel, L. E. Davis, R. C. Parmar, M. H. Hasan and R. Tandon, Sci. Rep., 2017, 7, 46069–46081 CrossRef CAS PubMed.
- H. Tada, O. Shiho, K. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157–165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C, and 19F NMR for synthesized compounds have been included. See DOI: 10.1039/d0ra06148d |
| This journal is © The Royal Society of Chemistry 2020 |