
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of green fluorescent carbon dots from carbon nano-onions and graphene oxide†
Alessia Ventrellaab,
Adalberto Camisasca a,
Antonella Fontanab and
Silvia Giordani*a
a,
Antonella Fontanab and
Silvia Giordani*a
aSchool of Chemical Science, Dublin City University (DCU), Glasnevin, Dublin 9, Ireland. E-mail: silvia.giordani@dcu.ie
bDepartment of Pharmacy, G. D'Annunzio University, Chieti, Italy
First published on 5th October 2020
Abstract
In recent years, carbon dots (CDs) have triggered considerable interest due to their intriguing tunable photoluminescence properties. In this work, we report the synthesis of green-emitting CDs from two different carbon sources, namely carbon nano-onions and graphene oxide. We also investigate the effects of the two starting materials on the physico-chemical properties of the as-synthesised CDs. Our results show that both CDs exhibit remarkable emission properties and different fluorescence behaviour, which is attributed to the differences in size, surface defects, as well as the presence of different surface functional groups. Moreover, we propose an innovative, low-cost and time-saving method for the recovery of CDs from solution by acetone-mediated precipitation. We demonstrate that this methodology can rival the common dialysis-based purification approach; it shows excellent photostability, and the CD fluorescent properties are retained. Our work paves the way for the use of these particles for biomedical applications by exploiting their interesting fluorescent features as well as their oxygen-enriched surface for further functionalization strategies.
1. Introduction
Nanoparticles have been intensively investigated in the last decades in the biomedical field, in particular for drug delivery1,2 and theranostic3 applications. Among them, carbon dots (CDs), a zero-dimensional fluorescent carbon nanomaterial,4 have received increasing attention due to their remarkable electronic and tunable photoluminescence properties.5,6 CDs, firstly reported by Xu et al.7 in 2004, are quasi-spherical nanoparticles consisting of both graphitic and diamond-like sp3 carbon atoms, and have a typical size below 10 nm.8–10 The presence of carboxyl moieties on their surface gives excellent water solubility and allows for further functionalisation and surface passivation with various organic, polymeric, inorganic or biological materials.11 Moreover, CDs exhibit interesting electronic properties and, compared to traditional semiconductor quantum dots, they possess lower toxicity and excellent biocompatibility.8,12 Collectively, these features make CDs particularly suitable for bioimaging,13 biosensing,14 drug-delivery applications15 as well as in photocatalysis.16–18 The recent and growing interest in CDs is mainly due to their characteristic photoluminescent properties. The exact mechanism of the CDs photoluminescence has not yet been fully understood and remains a subject of current research and debate. Different factors that influence and regulate their fluorescence mechanism have been proposed, including size,19,20 shape,21 surface defects,11,22–24 as well as functional groups.25CDs are mainly produced by two different approaches: the “bottom-up” and “top-down” methods.17,26 Advantages and disadvantages of the different methodologies are schematically reported in Table S1.†
The “bottom-up” approach involves the assembly of small precursor molecules, such as polycyclic aromatic derivatives, into CDs. Typical synthetic strategies include thermal decomposition,27,28 hydrothermal synthesis29 and microwave-assisted hydrothermal (MAH) synthesis.30 The synthesis of CDs via these approaches typically involves the condensation of the precursor molecules into larger entities, which requires stepwise and complex synthetic procedures,31 generally associated with expensive reagents and inert atmosphere conditions.
The “top-down” approach exploits the cutting of big-sized carbon sources in smaller pieces and usually generates nanoparticles bearing oxygen functional groups that increase the water solubility and allow to passivate their surface.31 Typical synthetic strategies include chemical32,33 and electrochemical34 oxidation, hydrothermal35 and solvothermal36 treatments, microwave-assisted synthesis,37 arc discharge,7,38 laser ablation39,40 and ultrasonic synthesis.41 Among the several “top-down” synthetic routes, the chemical oxidation of carbon nanomaterials (CNMs) such as carbon nanotubes,42 graphene and related materials,43,44 carbon fibers19 and fullerenes,45 is one of the most common approaches for the synthesis of highly fluorescent CDs. The main advantages of this methodology over the others is its cost-effectiveness due to the low cost of the starting materials and reagents, ease of implementation as it doesn't require special instrumentation and the possibility to potentially use any carbon-containing materials for the synthesis.
Generally, the size of CDs is typically less than 10 nm, but it can reach up to tens of nm, depending on the synthesis protocol used.46,47
In this work, we employed two different CNMs, for the synthesis of CDs via chemical oxidation, namely graphene oxide (GO) and carbon-nano onions (CNOs).
GO is the oxidised derivative of graphene,48,49 with which shares some interesting properties, such as high mechanical strength50 and large surface area,51 while exhibiting excellent dispersibility in water, due to the presence of several hydrophilic terminal groups,52 and remarkable biocompatibility.53,54 GO is commonly employed as a carbon source to produce CDs because of its low cost and ease of processability and mass production.44 Several synthetic protocols are utilised for their production, including chemical oxidation,44,55,56 solvothermal,57 hydrothermal58 or ultrasonic method.59
CNOs, firstly reported by Iijima in 1980,60 are multi-shell fullerenes structured by concentric shells of carbon atoms.61 CNOs show great promise in different applicative fields, such as biology62 and electronics.63 In particular, several reports have shown that CNOs are biocompatible both in vitro64,65 and in vivo.66,67 They have also been used as a starting material to produce CDs, by exploiting both chemical oxidation68,69 and laser ablation70 strategies.
The CNM-derived CDs were characterised by UV-Vis and fluorescence spectroscopies, ζ-potential analysis, and atomic force microscopy (AFM) in order to investigate and evaluate the effects of the starting material on the physico-chemical properties of the CDs.
In addition, we proposed an innovative acetone-based purification process for the recovery of the CDs as an efficient alternative to the conventional dialysis approach.
2. Experimental
2.1 Materials
GO-V30 powder was purchased from Standard Graphene (Las Vegas, NV, USA). Pristine CNOs were synthesised through thermal annealing of detonation nanodiamonds following a previously reported protocol.65,71 Sulfuric acid (H2SO4, 95–98%) nitric acid (HNO3, ≥65%), acetone (≥99.5%) and sodium hydroxide (NaOH) pellets were purchased from Sigma-Aldrich. Qualitative filter paper was purchased from Fischer Scientific (Hampton, New Hampshire, USA). Polyethersulfone (PES) filters (0.20 μm pore size) were purchased from GVS Filter Technology (USA). Pur-A-Lyzer™ Mega Dialysis Kits with a capacity of 20 mL and molecular weight cut-off (MWCO) of 1 kDa were purchased from Sigma-Aldrich. All water utilised in this study was Ultrapure Milli-Q (electric resistance > 18.2 MΩ cm−1) water.2.2 Preparation of CDs
CDs were synthesised following a slightly modified protocol reported in the literature.44 50 mg of the starting material (CNOs or GO) was dispersed in 10 mL of a mixture of concentrated H2SO4 and HNO3 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), using an ice bath to limit the development of heat and fumes. After cooling, the reaction mixture underwent sonication at 37 kHz (Fischer Scientific Ultrasonic Bath FB15050 for CNOs and Elmasonic P60H for GO) for 2 h, to promote the fragmentation of the starting material. The solution was then left refluxed 100 °C for 3 h. After cooling down to room temperature, the solution was diluted with 20 mL of Milli-Q H2O and neutralised using NaOH. During the neutralisation process, the formation of salts, Na2SO4 and NaNO3, was promoted in ice-bath.
:1 (v/v), using an ice bath to limit the development of heat and fumes. After cooling, the reaction mixture underwent sonication at 37 kHz (Fischer Scientific Ultrasonic Bath FB15050 for CNOs and Elmasonic P60H for GO) for 2 h, to promote the fragmentation of the starting material. The solution was then left refluxed 100 °C for 3 h. After cooling down to room temperature, the solution was diluted with 20 mL of Milli-Q H2O and neutralised using NaOH. During the neutralisation process, the formation of salts, Na2SO4 and NaNO3, was promoted in ice-bath.
CDs were isolated from the solution through several vacuum filtrations. The final dark brown solution was filtered using a 0.20 μm pore size PES filter to get rid of the larger particles and residual starting material. To remove the excess of salt still present in the dispersion, 10 mL was dialysed against 1 L H2O for 24 h to yield CNO-CDs and GO-CDs. During dialysis, the dispersion was subject to constant stirring, and the medium was exchanged three times (after 2 h, 5 h and overnight).
The purification of CDs (Fig. S1†) has been achieved through acetone-driven precipitation. Specifically, after the filtration with the PES filter, 10 mL of the sample was mixed with the same volume of acetone, resulting in the precipitation of CDs. The precipitate was further washed with acetone several times and dried. The final product (AP-CNO-CDs) was a brown powder (Fig. S1E†).
2.3 Characterisation of CDs
UV-Vis spectrophotometry was carried out by using a UV-1800 SHIMADZU spectrophotometer and a Varian Cary 100 spectrophotometer. The fluorescence properties were analysed using a PerkinElmer LS55 Fluorescence Spectrometer and a JASCO FP-6500. A quartz cuvette with an optical path length of 1 cm was used for all spectroscopic measurements. ζ-Potential analysis was carried out using a 90 Plus/BI-MAS Zeta Plus multiangle particle size analyser; the data reported are an average of three independent measurements.Atomic Force Microscopy (AFM) analyses were performed on a Multimode 8 Bruker AFM microscope coupled with a Nanoscope V controller and a commercial silicon tip (RTESPA 300; resonance of 300 kHz; nominal elastic constant of 40 N m−1) by using the ScanAsyst™ in air mode with a scan size of 3 μm. The samples were prepared by depositing 20 μL of the solutions on a silicon wafer support drying in an oven at 40 °C.
3. Result and discussion
CDs were synthesised via chemical oxidation of two different CNMs, namely CNOs and GO, to yield CNO-CDs and GO-CDs, respectively. Fig. 1 shows the schematisation of the methodology employed for the synthesis and purification.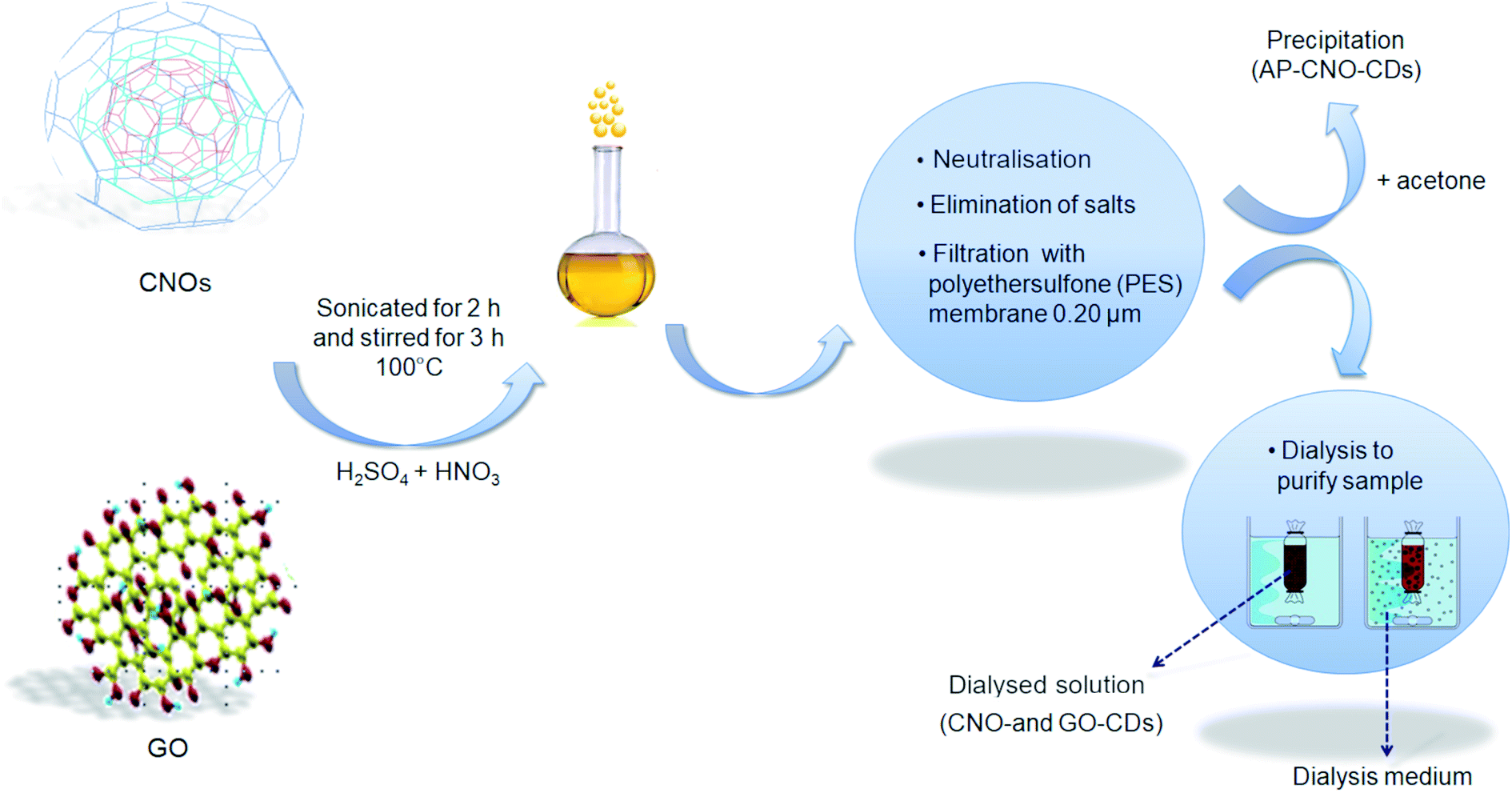 | ||
| Fig. 1 Schematic representation of the synthesis of CDs from CNOs and GO. | ||
Different techniques were used for the characterization of the as-produced materials. The absorption properties of the as-synthesised CDs were investigated by UV-Vis spectroscopy (Fig. 2A and B). Both CDs display high absorption in the UV region with a tail extending to the visible range. In particular, a peak at 230 nm as well as a shoulder at 265 nm can be observed. These absorption features are attributed to the π → π* transitions of the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and to the n → π* transitions of CO and COOH groups.5,44,72
C bond, and to the n → π* transitions of CO and COOH groups.5,44,72
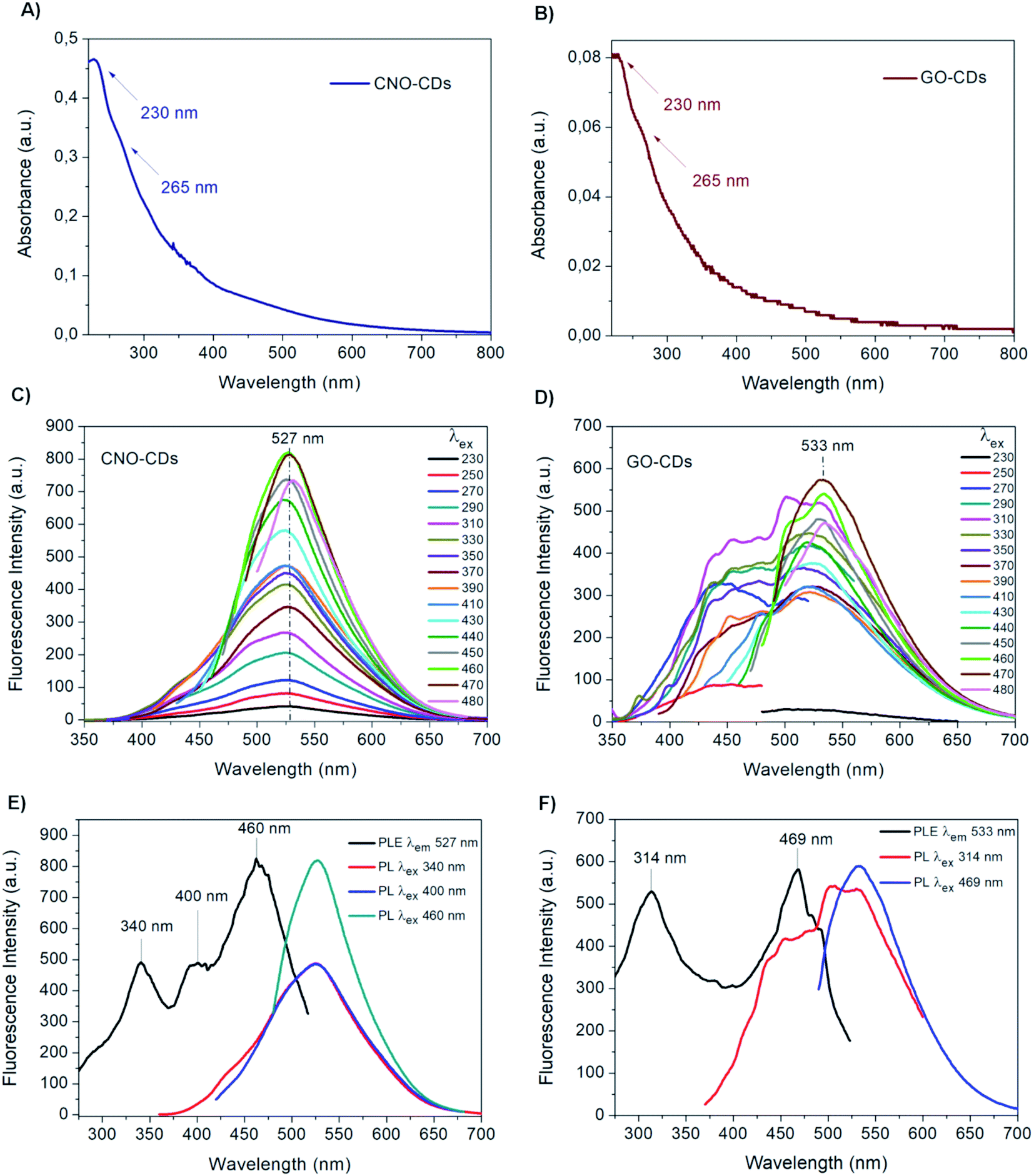 | ||
| Fig. 2 UV-Vis spectra of the 1:10 diluted aqueous solutions of (A) CNO-CDs and (B) GO-CDs. Fluorescence emission spectra of (C) CNO-CDs and (D) GO-CDs at an increasing excitation wavelength (from 230 to 480 nm). Fluorescence excitation spectra of (E) CNO-CDs and (F) GO-CDs. | ||
Although the UV-Vis spectra of both CDs have similar features, the absorption intensity of CNO-CDs is higher than that of GO-CDs, indicating a higher concentration of the CNO-CDs solution, and thus, higher reaction yield. In particular, approx. 5.5 times higher yield is observed for CNO-derived CDs compared to those derived from GO, despite an equal quantity of the respective starting materials.
The emission properties of CNO-CDs and GO-CDs were evaluated by fluorescence spectroscopy (Fig. 2C and D). The fluorescence spectra were acquired by increasing the excitation wavelength from 230 to 480 nm. Both CDs exhibit remarkable green fluorescent properties, with CNO-CDs displaying a much higher intensity. From the analysis of the emission spectra reported in Fig. 2C and D, a different behaviour is observed.
In particular, CNO-CDs show a constant emission maximum centred at 527 nm independently from the excitation wavelength, with the most intense band obtained when excited at 460 nm (Fig. 2C). Regardless of the synthetic procedure, CDs generally present excitation-dependent photoluminescence behaviour.35,73
Therefore, the excitation-independent emission properties shown by CNO-CDs make CNOs particularly intriguing as a starting material for CD synthesis.
Furthermore, the CNO-CDs green emission properties are similar to those reported by Zhang et al.,69 with a slightly red-shifted emission maxima (527 vs. 519 nm). Liu and Kim68 reported blue- and violet-emitting CDs produced through the chemical oxidation of CNOs, suggesting that our procedure is able to produce longer wavelength emitting CDs.
On the contrary, GO-CDs exhibit a excitation-dependent photoluminescence behaviour. The emission spectra, shown in Fig. 2D, can be resolved into two photoluminescent bands at shorter excitation wavelengths, showing an emission maxima located at 533 nm, while exciting at 470 nm. This behaviour is different to that of GO-CDs produced using a similar method by Tang et al.,44 showing a unique, constant emission band centred at 500 nm and the most intense band under an excitation of 295 nm.
Furthermore, from the comparison of the emission spectra of CNO- and GO-CDs (Fig. 2C and D), a red-shifted emission maximum is observed for the latter compared to that of CNO-CDs. This is attributed to the higher oxygen content on the GO-CDs surface.
Despite the same synthetic route should cause a similar degree of oxidation of the source material, GO, unlike pristine CNOs, exhibits a structure with a considerable amount of oxygen-containing groups, which likely leads to a final product richer in oxygen.
The surface oxidation state of CDs affects the overall electronic structure; as the degree of surface oxidation increases, surface defects increase, resulting in red-shifted emission.74
Also, the size of CDs can influence an emission red-shifting; the energy gap of the surface emissive sites, not only depends on the surface chemistry, but also on the π-electron system, as the extent of this conjugation influences the energy levels of the surface electronic states. This causes a lower band gap for π → π* transitions and therefore a smaller energy gap for surface states.6
In order to better understand and explain the different nature of the PL spectra of the two CNM-derived CDs, we deconvoluted the spectra, using a multi-Gaussian fit function (Fig. S2†). Fig. S2A and B† show the deconvoluted fluorescence spectra of CNO-CDs at two different wavelengths. In both cases, two peaks can be identified. The first peak at ca. 520 nm is an excitation-independent emission band attributed to fluorophore features of the CDs such as, for example, an abundance of conjugated carboxylic moieties on edges.75
The second band, which is an excitation-dependent peak, can be assigned to the carbogenic cores, quantum confinement effect (QCE), as well as to the corresponding edge structure variations of CDs with different sizes.75
On the other hand, the deconvoluted fluorescence spectra of GO-CDs, (Fig. S2C and D†), exhibit peaks that are excitation-dependent.
This suggests that the photoluminescent emission is due to the peculiarity of the carbonaceous core, the presence of defects and the co-existence of CDs with different sizes and diversified surface oxygen groups.76 In particular, even if both CDs consist of a carbon core, the GO-CDs surface is characterised by a large variety of O- and H-containing functional groups, much higher than those obtained from CNO-CDs, as discussed above. This greater amount of surface functional groups results in the existence of various energy levels between the π and π* states of CC, which may develop a series of emissive sites. Therefore, as the excitation wavelength changes, different surface state emissive sites become dominant.30,77
Fluorescence excitation analyses were performed to gain further information on the photoluminescent properties of the CDs. Fig. 2E and F report the excitation and the emission spectra at relevant excitation wavelengths for both samples.
In the excitation spectrum of CNO-CDs (Fig. 2E), three strong transitions occurred specifically at 340, 400 and 460 nm at the maximum emission wavelength of 527 nm, with the latter showing much higher intensity compared to the other peaks. For GO-CDs, as shown in Fig. 2F, two strong transitions with similar intensities are observed at 314 and 469 nm for an emission wavelength of 533 nm.
These results explain the different behaviour of the obtained CDs.78 While for CNO-CDs the luminescence depends primarily on the excitation-independent band, related to the surface functional groups, the optical properties of GO-CDs depend on the existence of states related to the carbogenic cores of the nanoparticles and states related to features of the graphene surface and edges. Therefore, the photoluminescence excitation data recorded confirmed the emission results.79
To analyse the stability of the synthesised CDs in aqueous solution, ζ-potential measurements were performed.
This technique allows to determinate the surface charge of nanoparticles in a colloidal solution.80 The ζ-potential of both CNM-derived CDs is presented in Table 1.
| Sample | ζ-Potential ± S.D. (mV) |
|---|---|
| CNO-CDs | −28.7 ± 0.7 |
| GO-CDs | −27.2 ± 0.9 |
The negative, superficial charge of the CDs suggests the presence of polar groups derived from the oxidation process, which tend to deprotonate in non-buffered aqueous dispersions. The presence of a negative charge on the surface of the CDs should aid the electrostatic repulsion between the nanoparticles and thus promote the stability of the dispersion. These ζ-potential values obtained are within the range associated with stable dispersions. Indeed, both CDs solutions were stable for months without any visible material sedimentation.
The morphology of the as-synthesised CDs has been investigated by AFM (Fig. 3). The results show the presence of individual CD particles as well as small aggregates <100 nm in size. The formation of these aggregates can be attributed to the drying process the sample underwent prior to the analysis. The AFM results suggest that GO-CDs (Fig. 3B) tend to form larger aggregates than CNO-CDs (Fig. 3A).
 | ||
| Fig. 3 AFM results for (A) CNO-CDs and (B) GO-CDs, showing, from left to right, topography, peak force error and height profile along with the lines evidenced in topography images. | ||
While AFM overestimates the width of the carbon dots due to the AFM probe interaction with the objects, the height of the dots can be extracted from the AFM images (Table S2†). In particular, CNO-CDs and GO-CDs exhibit an average size equal to 6.4 ± 1.8 and 3 ± 0.2 nm, respectively.
It should be noted that a small amount of residual salts, formed during the neutralisation process, is observed in the samples. However, their presence has negligible effects on the optical properties of CDs. Further workup could be carried out to ensure the complete removal of residual salts where necessary extending the dialysis time or changing the dialysis bags cut-off.
3.1 Optimisation of the purification protocol
Dialysis is a standard strategy employed for the purification of CDs. However, it has some limitations: it is expensive, typically requires long operational time and further steps, such as freeze-drying, are necessary to obtain a solid sample.For these reasons, we developed an alternative, quick and low-cost CD purification process involving an acetone-mediated liquid extraction.
It should be noted that this strategy worked only for the CNO-derived CDs due to their high yield and thus the high concentration of particles in solution.
The CNO-CDs precipitated by acetone (AP-CNO-CDs) were characterised through UV-Vis, fluorescence and Fourier transform infrared (FTIR) spectroscopy as well as AFM and the results have been compared to those of CNO-CDs.
The UV-Vis of both AP-CNO-CDs (Fig. 4A) and CNO-CDs (Fig. 2A) show identical absorption features, suggesting that the purification process doesn't affect the optical properties of the CDs.
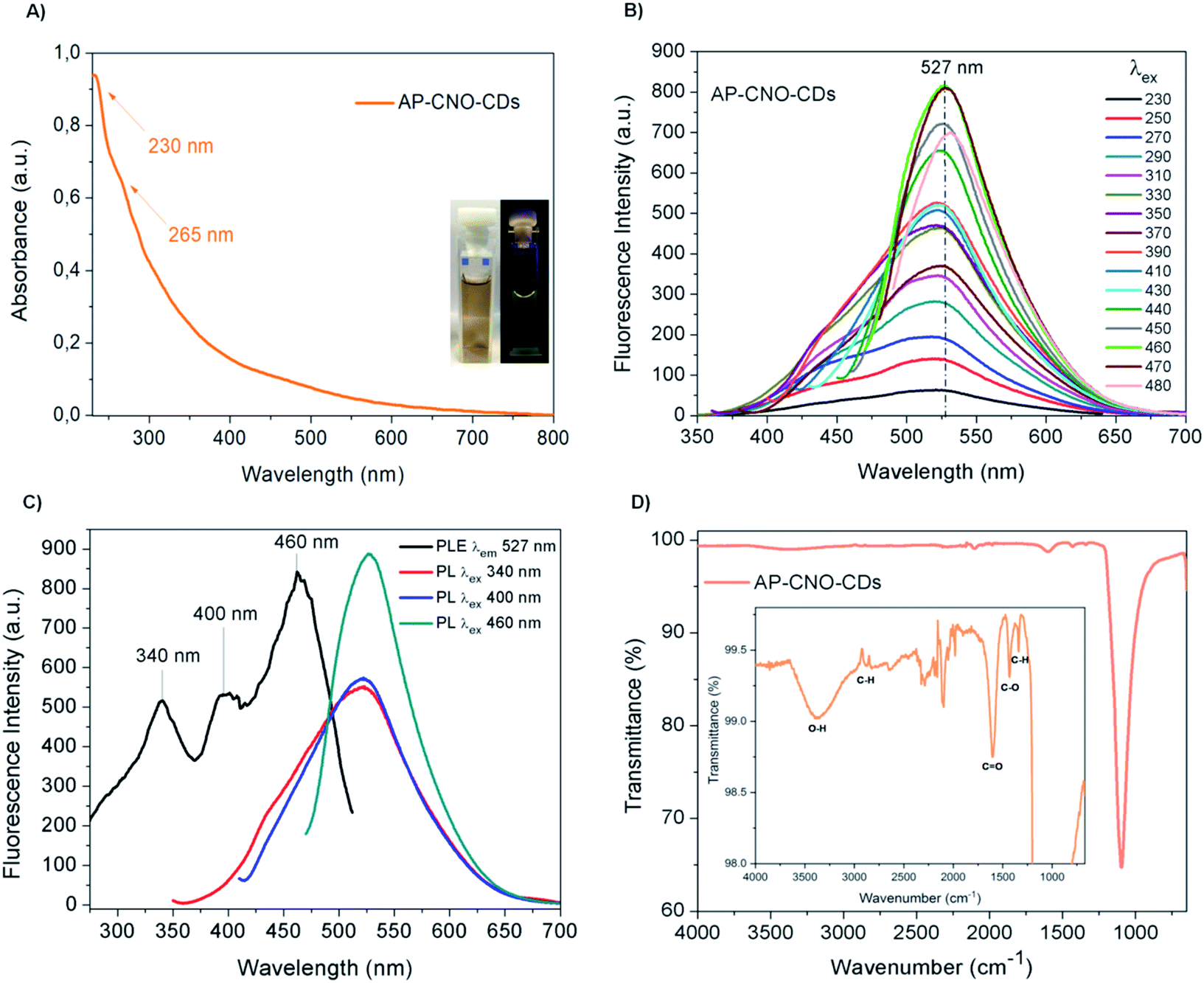 | ||
| Fig. 4 AP-CNO-CDs spectroscopic analyses: (A) UV-Vis spectrum at the concentration of 1 mg mL−1, the inset shows the aqueous solution of CDs irradiated under visible (left) and 365 nm UV light (right); (B) fluorescence spectra at a concentration of 5 mg mL−1 at different excitation wavelengths; (C) fluorescence excitation spectrum represented in black and the emission spectra of the relevant excitation wavelengths reported in other colours; (D) FTIR spectrum of AP-CNO-CDs, with a magnification of the most significant peaks. | ||
The fluorescence spectra of AP-CNO-CDs (Fig. 4B) and CNO-CDs (Fig. 2C) exhibit similar green emission profiles, with the emission maximum centred at 527 nm and identical transitions at 340, 400 and 460 nm in their respective excitation spectrum (Fig. 4C and 2E).
FTIR spectrum of AP-CNO-CDs (Fig. 4D) shows different peaks related to the presence of oxygen-containing functional groups, thus accounting for the excellent solubility in water.
The peaks at 1098 and 1607 cm−1 are representative of the stretching vibrations of C–O and CO; the peaks at 1344 and 1432 cm−1 are assigned to the bending vibrations of C–H and C–O; and peaks at 2950/2880 and 3384 cm−1 are attributed to the stretching vibrations of C–H and O–H groups, respectively.
We further investigated the photostability of the fluorescence under continuous irradiation of the sample for 100 min. AP-CNO-CDs show excellent photostability overtime, retaining over 94% of the initial intensity, suggesting an excellent resistance to photobleaching (Fig. S3†).
Further in line with the results for CNO-CDs, the AFM characterization of AP-CNO-CDs highlights the presence of a certain degree of aggregation (Fig. 5) and a small quantity of residual salts that may be removed by increasing the number of washes of the precipitate.
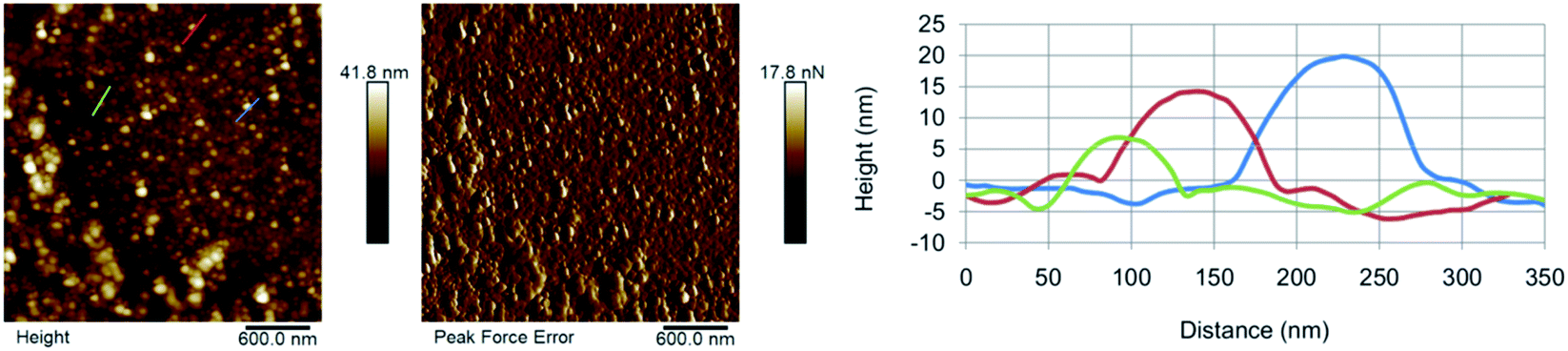 | ||
| Fig. 5 AFM micrographs of AP-CNO-CDs showing, from left to right, topography, peak force error and high profile along the lines evidenced in topography image. | ||
These results confirm that the herein outlined acetone-mediated purification step is a viable alternative makes to the standard dialysis approach. It has the benefits of being quick and low-cost, while showing no physicochemical differences.
4. Conclusions
In this work, we reported a simple, fast and low-cost method for the synthesis of carbon dots (CDs) through the chemical oxidation of two carbon nanomaterials, namely carbon nano-onions (CNOs) and graphene oxide (GO).The protocol developed allowed for the generation of green-emitting CDs with remarkable emission properties and different photoluminescence behaviour, depending on the carbon core and the content of oxygen-functional groups. In particular, CNO-derived CDs showed an excitation-independent fluorescence, which is generally difficult to achieve in CDs, and could represent a useful feature for bioimaging applications. The as-synthesised CDs exhibit excellent water dispersibility and long-term stability with no sedimentation of the material after 3 months.
Furthermore, a higher reaction yield was obtained by using CNOs as the starting material.
Moreover, we have proposed an interesting purification approach via acetone-mediated liquid extraction as an alternative to the standard dialysis process. Our strategy makes the purification process cheaper, faster and easier, while not affecting the optical properties of the CDs. In the recent years, CDs have shown to be a promising material in a number of bio-related applications. In particular, several studies have demonstrated the safety of the nanomaterial both in vitro and in vivo.46,81 Based on this, we are confident that the unique tunable fluorescence properties, as well as rich surface functional groups of our CDs, would make them suitable for biological applications such as bio-imaging agents and drug delivery nanocarriers.
In this regards, studies investigating the toxicological profile of these nanoparticles are on-going in our lab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The School of Chemical Sciences and the Nano Research Facility at Dublin City University (DCU) are gratefully acknowledged for financial support and access to instrumentation. Financial assistance from the University “G. d'Annunzio” in the form of doctoral funds to A. V., Fontana FAR 2018, Fontana FAR 2019 are also gratefully acknowledged. A. C. and S. G. wish to thank Istituto Italiano di Tecnologia (IIT) for access to the furnace for the synthesis of CNOs and Michał Bartkowski for his valuable proofreading.References
- N. Avramović, B. Mandić, A. Savić-Radojević and T. Simić, Pharmaceutics, 2020, 12, 298 CrossRef
.
- F. Oroojalian, F. Charbgoo, M. Hashemi, A. Amani, R. Yazdian-Robati, A. Mokhtarzadeh, M. Ramezani and M. R. Hamblin, J. Controlled Release, 2020, 321, 442–462 CrossRef CAS
- S. Indoria, V. Singh and M.-F. Hsieh, Int. J. Pharm., 2020, 582, 119314 CrossRef CAS
- B. Zhi, X. X. Yao, Y. Cui, G. Orr and C. L. Haynes, Nanoscale, 2019, 11, 20411–20428 RSC
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC
- L. Bao, C. Liu, Z. L. Zhang and D. W. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef CAS
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS
- A. P. Demchenko and M. O. Dekaliuk, Methods Appl. Fluoresc., 2013, 1, 042001 CrossRef
- S.-T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y.-P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS
- Y. Dong, J. Cai and Y. Chi, in Carbon Nanoparticles and Nanostructures, ed. N. Yang, X. Jiang and D.-W. Pang, Springer, Cham (Switzerland), 2016, pp. 161–238 Search PubMed
- Q. Li, T. Y. Ohulchanskyy, R. Liu, K. Koynov, D. Wu, A. Best, R. Kumar, A. Bonoiu and P. N. Prasad, J. Phys. Chem. C, 2010, 114, 12062–12068 CrossRef CAS
- K.-Q. Lu, Q. Quan, N. Zhang and Y.-J. Xu, J. Energy Chem., 2016, 25, 927–935 CrossRef
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC
- R. Wang, K.-Q. Lu, F. Zhang, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2018, 233, 11–18 CrossRef CAS
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J. J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS
- D. V. Melnikov and J. R. Chelikowsky, Phys. Rev. Lett., 2004, 92, 046802 CrossRef
- S. Kim, S. W. Hwang, M.-K. Kim, D. Y. Shin, D. H. Shin, C. O. Kim, S. B. Yang, J. H. Park, E. Hwang, S.-H. Choi, G. Ko, S. Sim, C. Sone, H. J. Choi, S. Bae and B. H. Hong, ACS Nano, 2012, 6, 8203–8208 CrossRef CAS
- J. Shen, Y. Zhu, C. Chen, X. Yang and C. Li, Chem. Commun., 2011, 47, 2580–2582 RSC
- J. Shen, Y. Zhu, X. Yang, J. Zong, J. Zhang and C. Li, New J. Chem., 2012, 36, 97–101 RSC
- L. Cao, M. J. Meziani, S. Sahu and Y.-P. Sun, Acc. Chem. Res., 2013, 46, 171–180 CrossRef CAS
- M. L. Liu, B. Bin Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC
- S. Huang, W. Li, P. Han, X. Zhou, J. Cheng, H. Wen and W. Xue, Anal. Methods, 2019, 11, 2240–2258 RSC
- B. Chen, F. Li, S. Li, W. Weng, H. Guo, T. Guo, X. Zhang, Y. Chen, T. Huang, X. Hong, S. You, Y. Lin, K. Zeng and S. Chen, Nanoscale, 2013, 5, 1967 RSC
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272 RSC
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS
- A. Kalluri, D. Debnath, B. Dharmadhikari and P. Patra, in Methods in Enzymology, ed. C. V. Kumar, Elsevier Inc., Philadelphia (USA), 1st edn, 2018, ch. 12, vol. 609, pp. 335–354 Search PubMed
- S. Hu, R. Tian, L. Wu, Q. Zhao, J. Yang, J. Liu and S. Cao, Chem.–Asian J., 2013, 8, 1035–1041 CrossRef CAS
- Z.-A. Qiao, Y. Wang, Y. Gao, H. Li, T. Dai, Y. Liu and Q. Huo, Chem. Commun., 2010, 46, 8812 RSC
- M. Liu, Y. Xu, F. Niu, J. J. Gooding and J. Liu, Analyst, 2016, 141, 2657–2664 RSC
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS
- S. Zhu, J. Zhang, X. Liu, B. Li, X. Wang, S. Tang, Q. Meng, Y. Li, C. Shi, R. Hu and B. Yang, RSC Adv., 2012, 2, 2717–2720 RSC
- L.-L. Li, J. Ji, R. Fei, C.-Z. Wang, Q. Lu, J.-R. Zhang, L.-P. Jiang and J.-J. Zhu, Adv. Funct. Mater., 2012, 22, 2971–2979 CrossRef CAS
- M. Bottini, C. Balasubramanian, M. I. Dawson, A. Bergamaschi, S. Bellucci and T. Mustelin, J. Phys. Chem. B, 2006, 110, 831–836 CrossRef CAS
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS
- X. Li, H. Wang, Y. Shimizu, A. Pyatenko, K. Kawaguchi and N. Koshizaki, Chem. Commun., 2011, 47, 932–934 RSC
- H. Li, X. He, Y. Liu, H. Yu, Z. Kang and S.-T. Lee, Mater. Res. Bull., 2011, 46, 147–151 CrossRef CAS
- D. Iannazzo, A. Pistone, M. Salamò, S. Galvagno, R. Romeo, S. V. Giofré, C. Branca, G. Visalli and A. Di Pietro, Int. J. Pharm., 2017, 518, 185–192 CrossRef CAS
- S. Zhu, J. Shao, Y. Song, X. Zhao, J. Du, L. Wang, H. Wang, K. Zhang, J. Zhang and B. Yang, Nanoscale, 2015, 7, 7927–7933 RSC
- D. Tang, J. Liu, X. Yan and L. Kang, RSC Adv., 2016, 6, 50609–50617 RSC
- C. K. Chua, Z. Sofer, P. Šimek, O. Jankovský, K. Klímová, S. Bakardjieva, Š. Hrdličková Kučková and M. Pumera, ACS Nano, 2015, 9, 2548–2555 CrossRef CAS
- F. Yuan, S. Li, Z. Fan, X. Meng, L. Fan and S. Yang, Nano Today, 2016, 11, 565–586 CrossRef CAS
- S. Sagbas and N. Sahiner, in Nanocarbon and its Composites: Preparation, Properties and Applications, ed. A. Khan, M. Jawaid, Inamuddin and A. M. A. Asiri, Woodhead Publishing-Elsevier, Cambridge (UK), 1st edn, 2019, ch. 22, pp. 651–676 Search PubMed
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS
- J. Kim, L. J. Cote and J. Huang, Acc. Chem. Res., 2012, 45, 1356–1364 CrossRef CAS
- R. J. Young, I. A. Kinloch, L. Gong and K. S. Novoselov, Compos. Sci. Technol., 2012, 72, 1459–1476 CrossRef CAS
- A. Trusek, E. Kijak and L. Granicka, Mater. Sci. Eng., C, 2020, 116, 111240 CrossRef CAS
- E. Campbell, M. T. Hasan, C. Pho, K. Callaghan, G. R. Akkaraju and A. V. Naumov, Sci. Rep., 2019, 9, 416 CrossRef CAS
- A. Di Crescenzo, S. Zara, C. Di Nisio, V. Ettorre, A. Ventrella, B. Zavan, P. Di Profio, A. Cataldi and A. Fontana, ACS Appl. Bio Mater., 2019, 2, 1643–1651 CrossRef CAS
- R. Di Carlo, S. Zara, A. Ventrella, G. Siani, T. Da Ros, G. Iezzi, A. Cataldi and A. Fontana, Nanomaterials, 2019, 9, 1–13 CrossRef
- Y. Zhang, H. Gao, J. Niu and B. Liu, New J. Chem., 2014, 38, 4970–4974 RSC
- B. Bali Prasad, A. Kumar and R. Singh, Biosens. Bioelectron., 2017, 94, 1–9 CrossRef CAS
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. Gao, H. Wei, H. Zhang, H. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC
- D. Pan, L. Guo, J. Zhang, C. Xi, Q. Xue, H. Huang, J. Li, Z. Zhang, W. Yu, Z. Chen, Z. Li and M. Wu, J. Mater. Chem., 2012, 22, 3314–3318 RSC
- Y. Zhu, G. Wang, H. Jiang, L. Chen and X. Zhang, Chem. Commun., 2015, 51, 948–951 RSC
- S. Iijima, J. Cryst. Growth, 1980, 50, 675–683 CrossRef CAS
- D. Ugarte, Nature, 1992, 359, 707–709 CrossRef CAS
- A. Camisasca and S. Giordani, Inorg. Chim. Acta, 2017, 468, 67–76 CrossRef CAS
- M. Zeiger, N. Jäckel, V. N. Mochalin and V. Presser, J. Mater. Chem. A, 2016, 4, 3172–3196 RSC
- M. d'Amora, A. Camisasca, A. Boarino, S. Arpicco and S. Giordani, Colloids Surf., B, 2020, 188, 110779 CrossRef
- S. Lettieri, A. Camisasca, M. D'Amora, A. Diaspro, T. Uchida, Y. Nakajima, K. Yanagisawa, T. Maekawa and S. Giordani, RSC Adv., 2017, 7, 45676–45681 RSC
- M. d'Amora, A. Camisasca, S. Lettieri and S. Giordani, Nanomaterials, 2017, 7, 414 CrossRef
- M. d'Amora, M. Rodio, J. Bartelmess, G. Sancataldo, R. Brescia, F. Cella Zanacchi, A. Diaspro and S. Giordani, Sci. Rep., 2016, 6, 33923 CrossRef
- Y. Liu and D. Y. Kim, Chem. Commun., 2015, 51, 4176–4179 RSC
- C. Zhang, J. Li, X. Zeng, Z. Yuan and N. Zhao, Nano Res., 2018, 11, 174–184 CrossRef CAS
- R. L. Calabro, D.-S. Yang and D. Y. Kim, J. Colloid Interface Sci., 2018, 527, 132–140 CrossRef CAS
- A. Camisasca, A. Sacco, R. Brescia and S. Giordani, ACS Appl. Nano Mater., 2018, 1, 5763–5773 CrossRef CAS
- L. Lin and S. Zhang, Chem. Commun., 2012, 48, 10177 RSC
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS
- L. Bao, Z. L. Zhang, Z. Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D. W. Pang, Adv. Mater., 2011, 23, 5801–5806 CrossRef CAS
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS
- S. Yang, J. Sun, X. Li, W. Zhou, Z. Wang, P. He, G. Ding, X. Xie, Z. Kang and M. Jiang, J. Mater. Chem. A, 2014, 2, 8660–8667 RSC
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CrossRef CAS
- A. S. Hassanien, R. A. Shedeed and N. K. Allam, J. Phys. Chem. C, 2016, 120, 21678–21684 CrossRef CAS
- S. K. Das, Y. Liu, S. Yeom, D. Y. Kim and C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef CAS
- A. Kumar and C. K. Dixit, in Advances in Nanomedicine for the Delivery of Therapeutic Nucleic Acids, ed. S. Nimesh, R. Chandra and N. Gupta, Woodhead Publishing-Elsevier, Cambridge (UK), 1st edn, 2017, ch. 3, pp. 43–58 Search PubMed
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Scheme of purification by acetone; fluorescence deconvolution spectra; data related to the topographic AFM analysis; and photostability fluorescence analysis. See DOI: 10.1039/d0ra06172g |
| This journal is © The Royal Society of Chemistry 2020 |