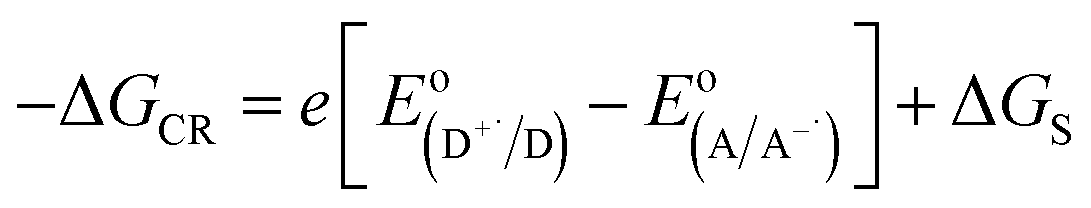DOI:
10.1039/D0RA06195F
(Paper)
RSC Adv., 2020,
10, 31720-31729
Solution-phase molecular recognition of an azafullerene-quinoline dyad by a face-to-face porphyrin-dimer tweezer†
Received
7th July 2020
, Accepted 10th August 2020
First published on 27th August 2020
Abstract
A face-to-face porphyrin dimer, (H2P)2 “porphyrin tweezer”, was explored as a photo- and redox-responsive host for the molecular recognition of an azafullerene (C59N) derivative bearing an amphoteric pentafluoroquinoline (FQ) domain. The intramolecular electronic coupling between the FQ substituent and the C59N cage, within the newly synthesized C59N-FQ dyad was evaluated, while the neutral and protonated form of the covalently attached FQ moiety were utilized as recognition motifs for the (H2P)2 tweezer. Complementary photophysical and electrochemical techniques were applied to investigate the electronic communication between the porphyrin-dimer (H2P)2 tweezer and the azafullerene cage as mediated by the FQ unit.
Introduction
Azafullerene C59N1,2 is the most explored heterofullerene3 participating in light harvesting nanoarchitectures.4 Starting from the parent azafullerene dimer (C59N)2, a series of monoadducts can be prepared via the C59N+ intermediate species.5 In this respect, several C59N-based dyads have been realized by the covalent incorporation of arenes,6 ferrocene,7 phthalocyanine,8 corole,9 perylene10 and oligophenylenevinylene11 derivatives onto the C59N cage. Apart from the mono-adducts, C59N has also a rich chemistry towards multi-addition patterns.12–17 Notably, we recently reported the tether-directed regioselective synthesis of an equatorial-face bis-adduct of C59N, as single isomer, based on a two-step mono-addition reaction sequence employing cyclo-[2]-octylmalonate.18 In contrast, the supramolecular chemistry of C59N is less explored. Recent advances on the chemistry of carbon nanobelts19–22 empowered the realization of supramolecular assemblies in solution driven by the molecular recognition of the C59N cage by [10]-cycloparaphenylene ([10]CPP) nanohoops through π–π interactions at the concave–convex interface.23,24 Furthermore, the shielding of the labile C59N˙ radical was enabled by supramolecular complexation with [10]CPP rings.25 In addition, the supramolecular chemistry of functionalized C59N cages is currently limited to derivatives owing monodentate nitrogen ligands suitable for coordination to the metal center of light harvesting molecules such as zinc-phthalocyanines26 and zinc-porphyrins.27,28
Quinolines are amongst heteroatom semiconducting organic species that have gained increased scientific attention due to their easy and versatile synthesis,29 excellent thermal and mechanical properties, but more importantly due to their interesting photonic, electronic, and conductive characteristics.30 The reverse protonation on the imino group31 allows their use as pH tunable sensors,32,33 while their ability to complex a variety of metal ions or organic conjugates reveals their applicability also as selective fluorescent chemosensors.34 Moreover, the incorporation of electron withdrawing units onto quinolines allows further modulation of their optical and electronic properties. To this end, the incorporation of a perfluorophenyl group has proven to be a valuable addition onto the quinoline core, since it significantly lowers their LUMO levels. Moreover, the presence of the halogen atoms enabled their straightforward transformation to azides, allowing the direct incorporation on the sp2 skeleton of carbon nanostructures via cycloaddition reactions.35–37 More specific, (tetrafluoro)azidophenyl-quinolines have been employed for the functionalization of carbon nanotubes,35,36 C60 (ref. 36 and 37) and phenyl-C61-butyric acid methyl ester (PCBM).37 Up to date, C60 and PCBM are the only fullerene cages functionalized by pentafluorophenyl-quinolines. In this work, we demonstrate the chemical incorporation of a pentafluorophenyl-quinoline (FQ) derivative on the azafullerene C59N cage, towards a molecular recognition motif for the development of host–guest supramolecular assemblies featuring a hairpin-like face-to-face dimer porphyrin (H2P)2.
The quest for light harvesting antennas owing face-to-face porphyrins macrocycles was fueled by exploration of nature-mimicking molecular photosynthetic approaches.38 P700, the electron donor unit of photosystem I, is considered to consist of two chlorophyll species (a and a′) axially coordinated by a histidine residual, resembling a face-to-face supramolecular porphyrin dimer.39 Progress in artificial photosynthetic systems has met the expansion of fullerene chemistry, generating an interface where natural-inspired electron donors have been coupled to synthetic fullerene electron acceptors.40,41 Porphyrin “tweezers” or “jaws” have attracted significant interest due to their versatile function as electron donor host-molecules and the simple, in general, chemistry required for their preparation.42,43 Up to date, porphyrin tweezers have been efficiently employed as hosts for the molecular recognition of small electron withdrawing compounds44 and fullerenes.45–50
The molecular recognition of acridine, with molecular structure resembling that of quinoline, due to strong electrostatic interactions by a metal-free porphyrin tweezer has been accomplished.51,52 However, the recognition of pristine C60 fullerene by this type of metal-free (H2P)2 tweezers is weak,46 requiring high excess of C60 in order to track detectable changes in the absorption features of the tweezer, especially of the Soret band arising from the S0 → S2 optical transition of (H2P)2. Steric hindrance most likely restricts those species from efficiently approaching each other. Therefore, the quinoline heterocycle present within the C59N-FQ dyad is, herein, exploited as flexible nanodomain capable of being sandwiched by the face-to-face porphyrin rings of the (H2P)2 tweezer. This, allows the development of effective electronic communication between C59N and (H2P)2 and mediated by FQ within the so-formed (H2P)2/C59N-FQ assemblies. Moreover, we prepare the protonated form of C59N-FQ, namely C59N-FQH+, benefited by the weak basic nature of the porphyrin heterocycles for empowering the development of electrostatic interactions between the individual species. Synergistic weak π–π interactions, electrostatic forces and weak hydrogen bonding between pentafluorophenyl-quinoline and the (H2P)2 macrocycle collectively stabilize the host–guest architecture. We perform complementary spectroscopic and electrochemical techniques to unveil and evaluate host–guest interactions in those nano-architectures. Specifically, electronic absorption, steady-state and time-resolved photoluminescence spectroscopy aided the assessment of the optical properties of (H2P)2/C59N-FQ assemblies at the ground and excited states, while the redox properties were screened by electrochemistry.
Results and discussion
The pentafluorophenyl-quinoline derivative, 6-(4-hydroxylphenyl)-(2-perfluorophenyl)-4-phenyl-quinoline (FQ),53 is chemically introduced on a pre-modified C59N cage bearing a –COOH unit.5,8 For the covalent functionalization of the C59N cage with the quinoline derivative, the synthetic route shown in Scheme 1a is followed. Briefly, (C59N)2 is initially treated with di-(tert)butyl malonate diester at elevated temperature in the presence of oxygen, in order to proceed the nucleophilic attack of the malonate to the in situ generated C59N+ intermediate,5 followed by thermally-induced decarboxylation to afford C59N-COOH.8 The latter compound is used for the incorporation of the 6-(4-hydroxylphenyl)-(2-perfluorophenyl)-4-phenyl-quinoline53 via carbodiimide-mediated condensation yielding the C59N-FQ dyad. 1H, 13C and 19F NMR, and IR spectroscopy, together with ESI mass spectrometry, ascertained the structure of C59N-FQ. In the 1H NMR spectrum of C59N-FQ (ESI, Fig. S1†) the characteristic signal at 5.07 ppm owed to the methylene protons next to the azafullerene cage, accompanied by the thirteen aromatic protons due to the FQ core in the region 7.3–8.3 ppm, is evident. In the 13C NMR spectrum of C59N-FQ, the typical signal at 168.2 ppm for the carbonyl ester is identified (ESI, Fig. S2†). Complementary, 19F NMR assays reveal three distinct signals at −124, −153 and −161 ppm, due to the pentafluorophenyl ring within FQ (ESI, Fig. S3†). The formation of the ester bond in C59N-FQ is further validated by ATR-IR spectroscopy, where the characteristic vibration of the ester carbonyl at 1758 cm−1, accompanied by the absence of the band centred at 1711 cm−1 owed to the precursor carboxylic acid unit (ESI, Fig. S4†), is evolved. Ultimately, solid proof for the formation of C59N-FQ is delivered by registering the corresponding [M+1]+ ion at 1227.17 amu by ESI mass spectrometry (ESI, Fig. S5†). The high solubility of C59N-FQ in common organic solvents, i.e. toluene, dichloromethane, chloroform and benzonitrile, enables not only performing a proper spectroscopic and electrochemical examination in liquid media, but also proceeding with the supramolecular integration of the porphyrin tweezer (Scheme 1b) yielding the (H2P)2/C59N-FQ nanoarchitecture (Scheme 1c).
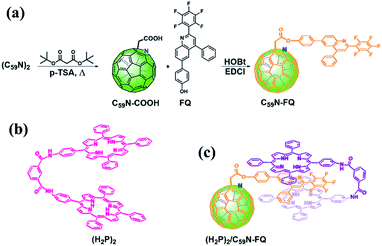 |
| | Scheme 1 (a) Synthetic procedure for the preparation of C59N-FQ. (b) Structure of the co-facial dimer porphyrin tweezer (H2P)2. (c) Illustration of the supramolecular complex (H2P)2/C59N-FQ. | |
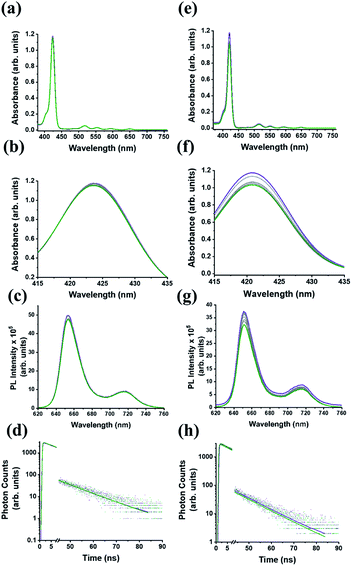
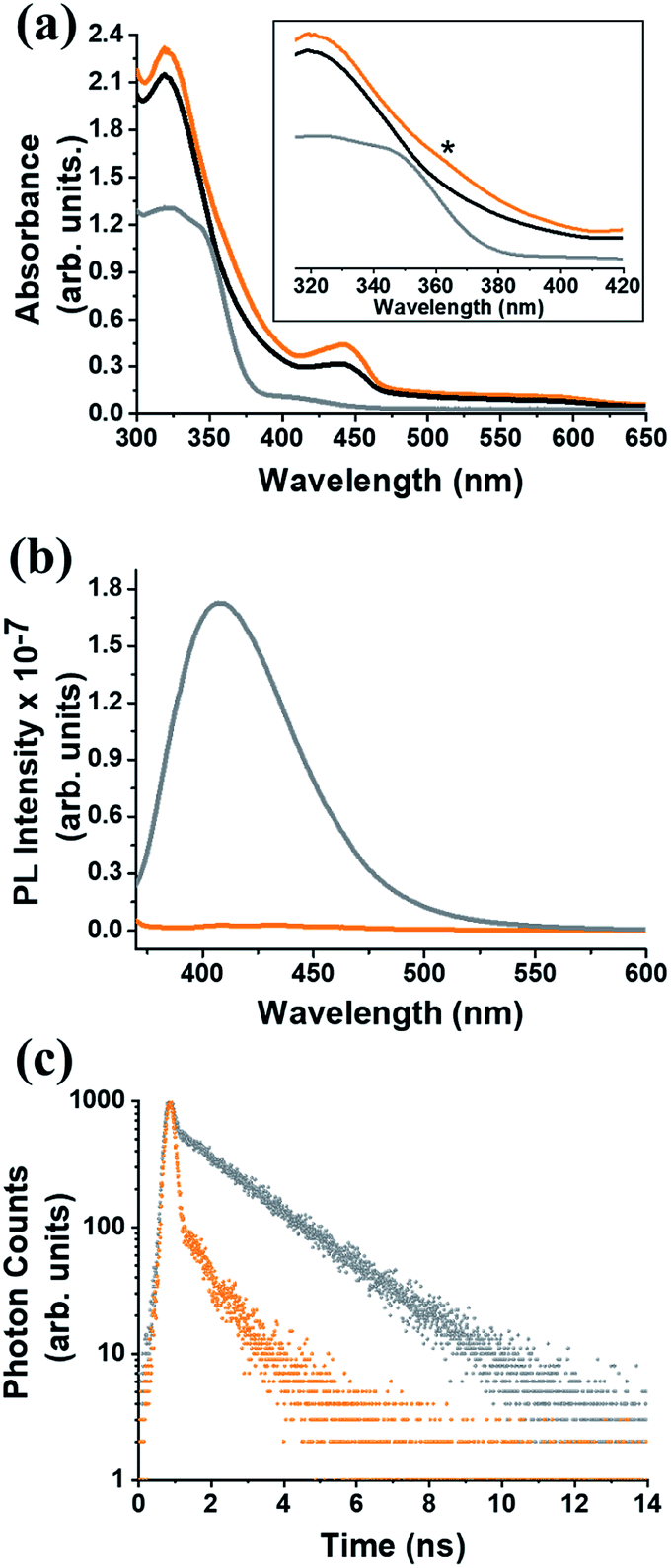
In the UV-Vis electronic absorption spectrum of C59N-FQ the main absorption features of (C59N)2 at 320, 440 and 600 nm, together with a new broad shoulder at 368 nm attributed to the attached FQ unit (Fig. 1a), are evident. Further, photoexcitation of C59N-FQ at 320 and 340 nm, where FQ shows maximum absorbance, reveals quantitative quenching of the FQ emission band centred at 401 nm (Fig. 1b), indicating strong electronic communication between the two species, C59N and FQ, at the excited state. This is further supported by the examination of the emission decay profile of C59N-FQ by the time-correlated-single-photon-counting (TCSPC) method, since a significantly shorter lifetime is recorded for the single excited state of the covalently attached FQ (∼76 ps) in the C59N-FQ dyad, as compared to the corresponding emission lifetime recorded for free FQ (1.9 ns), as shown in Fig. 1c. The fast deactivation of 1FQ* suggests energy and/or electron transfer phenomena to occur in the presence of the azafullerene cage.
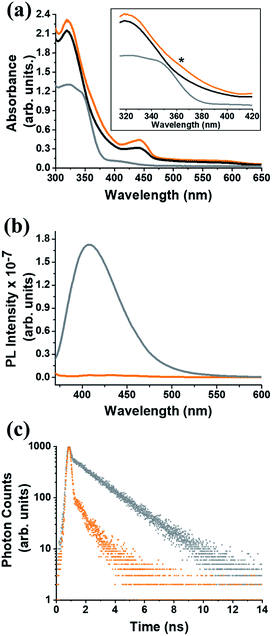 |
| | Fig. 1 (a) Electronic absorption UV-Vis spectra, (b) emission spectra at upon excitation at 340 nm, and (d) emission decay profiles (λexc 376 nm), for free FQ (gray), (C59N)2 (black) and C59N-FQ (orange). All measurements were performed in toluene solutions. Inset: zoom of the 310–420 nm region, where a new absorption feature (marked with an asterisk) at 360 nm was observed for C59N-FQ. | |
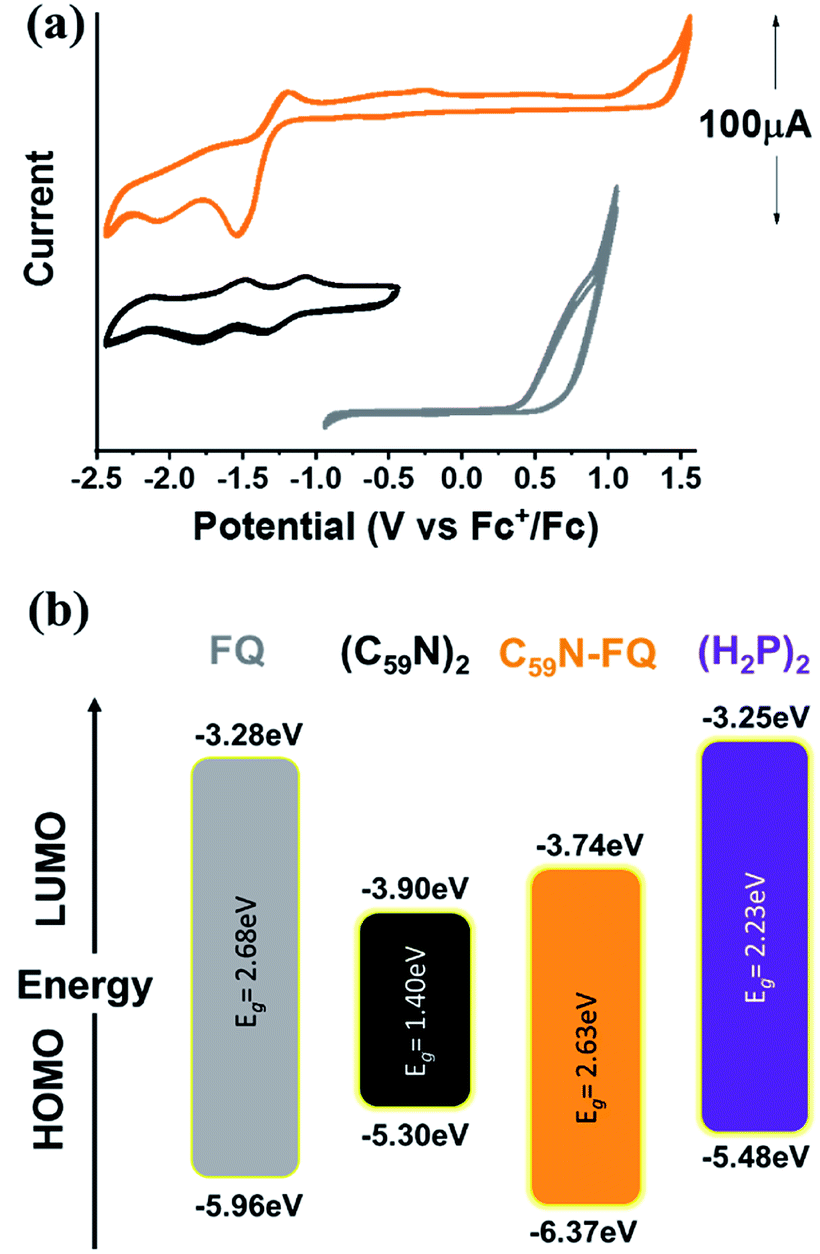
Insight into the redox properties of C59N-FQ is given by complementary cyclic voltammetry (CV) assays. As presented in the summarized data of Table 1, the covalent attachment of FQ on C59N anodically shifts the first oxidation potential E1ox by 0.41 V compared to that registered for free FQ (Fig. 2a), while cathodically shifts the first reduction potential E1red of functionalized C59N by 0.16 V compared to the value for (C59N)2. Based on the following equations (eqn (1) and (2)):54
| | |
HOMO = −(Eox vs. Fc+/Fc + 5.1) (eV)
| (1) |
| | |
LUMO = −(Ered vs. Fc+/Fc + 5.1) (eV)
| (2) |
and the energy gap of
FQ (ESI, Fig. S6
†) and
(C59N)2,
55 an energy level diagram is constructed (
Fig. 2b). The energy alignment of the individual
FQ and
(C59N)2 suggests that the
C59N-FQ is an electron donor–acceptor dyad, where
FQ is the donor and
C59N the acceptor. Accordingly, we may calculate the HOMO–LUMO energy levels of the
C59N-FQ dyad by the observed
E1ox and
E1red values, as −6.37 and −3.81 eV, respectively. Taking into consideration the HOMO–LUMO energy levels of the
(H2P)2 tweezer, the energy alignment of the supramolecular
(H2P)2/C59N-FQ host–guest assemblies was favorable towards a p–n junction, where the
(H2P)2 tweezer is the electron donor and the
C59N-FQ is the electron acceptor. In previous studies, analogous covalent dyads consisting of
FQ and the C
60 fullerene have been successfully integrated in organic solar cells as electron acceptors utilizing poly-(3-hexyl thiophene) (P3HT), a widely adopted electron donor polymer with similar to
(H2P)2 tweezer HOMO–LUMO energy levels, as electron donor.
36
Table 1 Redox potentials and energy gaps (Eg) for FQ, (C59N)2 and C59N-FQ
| Material |
Redox potentials (vs. Fc+/Fc) |
Eg (eV) |
| E1ox |
E1red |
E2red |
E3red |
| Optical gap calculated from the Tauc plot, ESI, Fig. S6. Optical gap value adopted from ref. 55. Electrochemical gap based on Fig. 2a. Electrochemical gap based on ESI, Fig. S10. |
| FQ |
+0.86 |
— |
— |
— |
2.68a |
| (C59N)2 |
— |
−1.20 |
−1.63 |
−2.20 |
1.40b |
| C59N-FQ |
+1.27 |
−1.36 |
−1.91 |
— |
2.63c |
| (H2P2) |
+0.38 |
−1.85 |
−2.14 |
— |
2.23d |
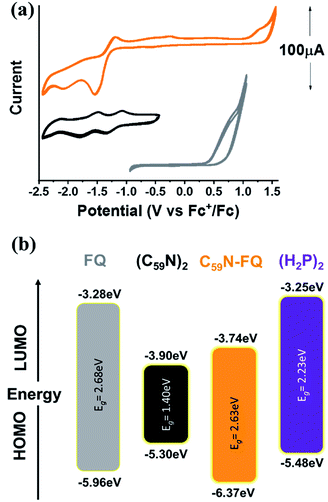 |
| | Fig. 2 (a) Cyclic voltammetry (CV) graphs for FQ (gray), (C59N)2 (black) and C59N-FQ (orange) vs. Fc+/Fc. CV curves recorded in N2 saturated toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (vol/vol 1:4) with 0.1 M TBAPF6 as electrolyte and 50 mV s−1 scan rate. (b) Energy level diagram constructed from the data presented in Table 1. :acetonitrile (vol/vol 1:4) with 0.1 M TBAPF6 as electrolyte and 50 mV s−1 scan rate. (b) Energy level diagram constructed from the data presented in Table 1. | |
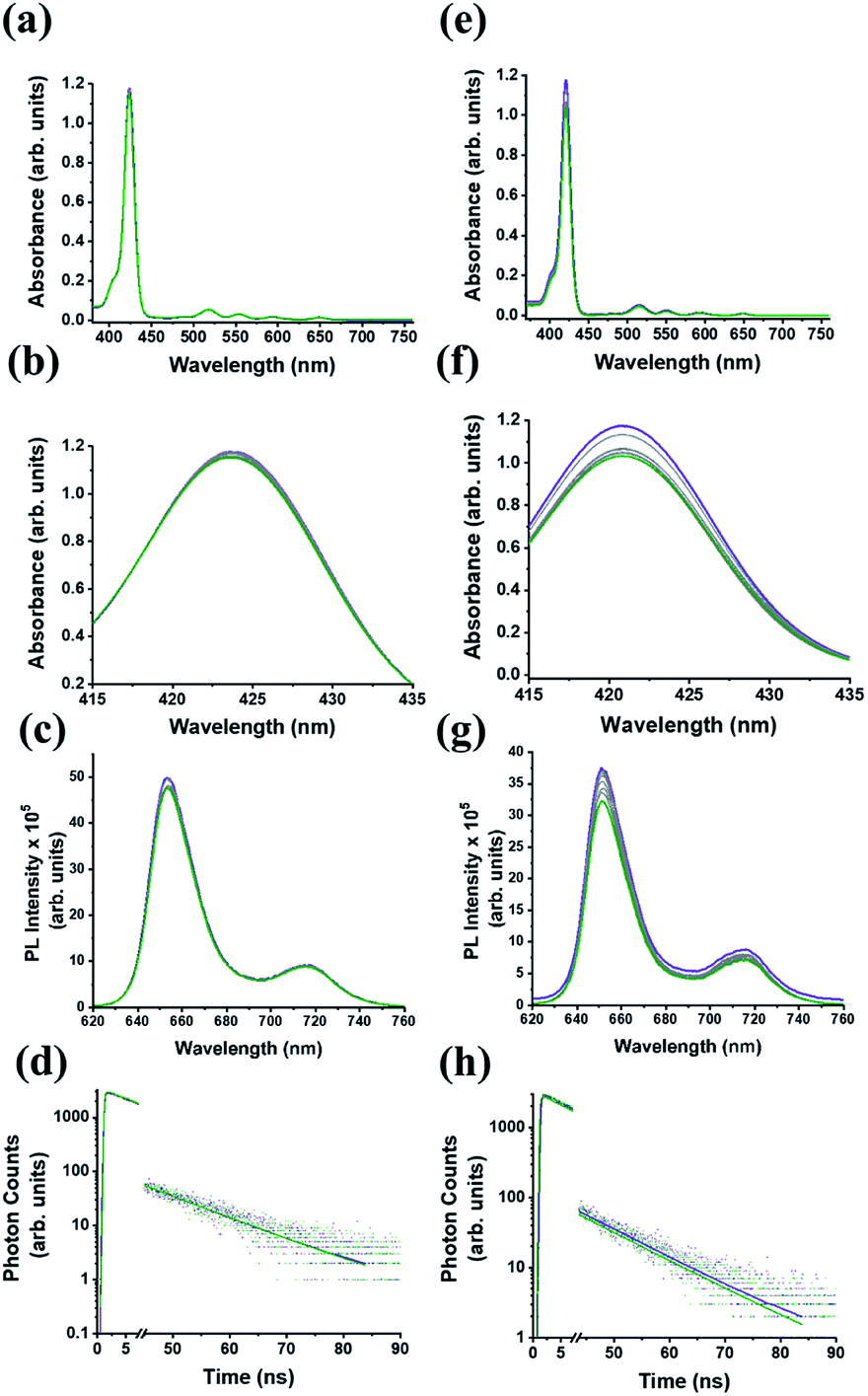
We firstly explore the complexation dynamics between C59N-FQ and (H2P)2 tweezer by UV-Vis electronic absorption, steady-state and time-resolved fluorescence emission spectroscopy. As depicted in Fig. 3a–d, upon incremental addition of C59N-FQ (0–2 equivalents) in a benzonitrile solution of (H2P)2, negligible changes are recorded in the UV-Vis spectra of (H2P)2, namely a 2% decrement of the absorption intensity of the Soret band at 420 nm and a 4% quenching of the emission intensity of (H2P)2 under excitation of the Soret band. Evidently, a weak interaction between the individual species occurs, which is also verified by the analysis of the time-resolved fluorescence spectra of (H2P)2 in the presence of 2 equivalents of C59N-FQ, where the decay trace is best fitted by a mono-exponential model giving a fluorescence lifetime of 10 ns, corresponding to that of non-complexed (H2P)2.
 |
| | Fig. 3 (a and e) UV-Vis electronic absorption spectra of (H2P)2 tweezer (purple) upon incremental additions of up to 2 equivalents (olive) of C59N-FQ and C59N-FQH+, respectively. (b and f) Zoom of the corresponding Soret band regions. In all spectra the absorbance of C59N-FQ and C59N-FQH+ is subtracted. (c and g) Steady-state fluorescence emission spectra of (H2P)2 tweezer (purple) upon excitation of the Soret band (λexc 420 nm) upon incremental additions of up to 2 equivalents (olive) of C59N-FQ and C59N-FQH+, respectively. (d and h) Emission decay profiles (λexc 440 nm, probed at 650 nm) of (H2P)2 tweezer (purple) upon incremental additions of up to 2 equivalents (olive) of C59N-FQ and C59N-FQH+, respectively. Dotted lines represent the decay trace, while solid lines represent the mathematical fitting of the decay spectra. All spectra were recorded in benzonitrile at 1 × 10−6 M concentration of the (H2P)2 tweezer. | |
Taking advantage of the low basicity of the quinoline domain, we next treat C59N-FQ with an excess of trifluoroacetic acid leading to the corresponding protonated C59N-FQH+ species. The protonation of C59N-FQ is monitored by FT-IR spectroscopy (ESI, Fig. S7†) and discrete spectra profiles are obtained, while the process is fully reversible upon addition of excess triethylamine. More specific, it is known that protonation of the quinoline nitrogen atom results to a shielding effect to the neighboring phenyl groups where the oxygen atom resides, as it was previously demonstrated by 1H NMR assays.53 The latter effect strongly affects the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the ester group connecting the C59N cage and the quinoline derivative. As a result, the CO stretching mode registered at 1758 cm−1 for C59N-FQ is shifted by 24 cm−1 for protonated C59N-FQH+ at 1734 cm−1.
O stretching vibration of the ester group connecting the C59N cage and the quinoline derivative. As a result, the CO stretching mode registered at 1758 cm−1 for C59N-FQ is shifted by 24 cm−1 for protonated C59N-FQH+ at 1734 cm−1.
Focusing on the impact of the protonated quinoline in the supramolecular complexation of C59N-FQH+ with the (H2P)2 tweezer, a series of titration assays are performed. Please note that NMR titration assays are hampered due to overlap of the signals of interest, namely the host molecules C59N-FQ and C59N-FQH+, contribute 1H-signals arising due to the FQ domain which overlap with the pyrrolic 1H of the porphyrin dimer (H2P)2 tweezer. In addition, 19F NMR and especially FAXS (fluorine chemical shift anisotropy and exchange for screening) and n-FABS (n-fluorine atoms for biochemical screening), methods developed for competitive binding assays in biosystems, require fluorinated fragments with strong sharp singlet signals, in contrast to the C–F groups of the pentafluorophenyl moiety which are strongly coupled resulting to complex splitting patterns as shown in Fig. S3.† Nevertheless, the electronic absorption spectrum of (H2P)2 upon incremental additions of C59N-FQH+ (up to 2 equivalents) reveal enhanced interactions as expressed by the suppression of the Soret band absorption by 15%, accompanied by 18% quenching to the fluorescence emission intensity of the (H2P)2 tweezer (Fig. 3e–g). Further, time-resolved fluorescence emission spectroscopy reveals the evolution of a new short-lived component (10% population, τ = 1 ns), which is attributed to the deactivation pathway of the photoexcited electron from the singlet excited state of (H2P)2 to C59N-FQH+ (Fig. 3h). These findings underscore the impact of the positively charged guests, in the form of protonated FQH+ domains, to the hosting ability of the co-facial metal-free porphyrin dimer and are fully in line with previously reported assemblies centered on acridinium cations.51,52
For the (H2P)2/C59N-FQ system, both the UV-Vis and PL emission changes during the titration assays were negligible suggesting the absence of notable interactions. In contrast, the changes observed in the UV-Vis and PL spectra for the (H2P)2/C59N-FQH+ system allowed us to obtain a rough estimation for the stoichiometry and the binding constant. In order to further validate the presence of complexation events between the C59N-FQH+ and the tweezer, we constructed the corresponding Job plot (ESI, Fig. S8a†) for unravelling the stoichiometry as well as the Hill plot (ESI, Fig. S8b†) to evaluate the binding constants. Specifically, the Job's plot for the (H2P)2/C59N-FQH+ system suggests the co-existence 1:1 and 1:2 stoichiometries,56 while from the Hill plot the binding constant was calculated to be 3.17 × 102 M. Moreover, from the Stern–Volmer plot derived from the fluorescence titration we find a KSV value of 2.6 × 104 M−1 (ESI, Fig. S8c†) and by taking into account the new short-lived component revealed form time-resolved fluorescence emission spectroscopy, a dynamic quenching mechanism is proposed. An estimation of the charge separation dynamics can be deduced by eqn (3) and (4):
| | |
kCS = (1/τ)(H2P)2/C2N-FQH+ − (1/τ)(H2P)2
| (3) |
| | |
ΦCS = kCS/(1/τ)(H2P)2/C2N-FQH+
| (4) |
where
τ is the fluorescence lifetime of the free
(H2P)2 in solution or the
(H2P)2 host within the supramolecular
(H2P)2/C59N-FQH+ host–guest assembly, allowing us to calculate the charge separation rate constant (
kCS) and the charge separation quantum yield (
ΦCS) for the
(H2P)2/C59N-FQH+ host–guest assembly, based on the fluorescence decay values recorded by TCSPC spectroscopy. The
kCS and
ΦCS values are found as high as 0.9 × 10
9 s
−1 and 0.9, respectively, suggesting a slow charge separation with high quantum yield.
A comprehensive examination of the UV-Vis spectrum of C59N-FQH+ reveals a slight broadening of the absorption spectra in the range of 300–380 nm, where both the azafullerene cage and the chemically attached protonated quinoline derivative absorb light. This feature is further accompanied by a weak band centred at 420 nm, as compared to the absorption profile of neutral C59N-FQ (Fig. 4a). In addition, the protonation of quinoline induces a more intense variation to the fluorescence emission profile of C59N-FQH+. It is known that quinoline derivatives are sensitive to charge delocalization and specifically to protonation of the nitrogen atom.57 Hence, the protonated quinoline domain impacts the emission spectrum of C59N-FQH+, since upon photoexcitation of the quinoline core (λexc 340 nm), a red-shifted and quenched emission band centered at 440 nm is observed (Fig. 4b). Moreover, an excitation-dependent emission spectrum is registered for C59N-FQH+ (ESI, Fig. S9†), in contrast to that derived from neutral C59N-FQ. As long as the emission spectra of C59N-FQH+ are found independent of the concentration, the presence of excimers is excluded.58 In general, protonation of quinolines lowers the energy of their singlet excited state and the absorption band drifts to lower energy.57 Therefore we assume that the properties of C59N-FQH+ are related to the photo-basicity of quinolines as described by the Förster cycle (Fig. 4c). Neutral quinolines show increased pKa values during photoexcitation 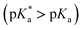 inducing proton capture at the singlet excited states.59 Considering that the complexation assays of C59N-FQH+ for hosting the (H2P)2 tweezer are performed in aprotic benzonitrile (cf. Fig. 3), dissociation to the neutral state by proton release to the solvent medium is blocked and therefore C59N-FQH+ remains charged under photoexcitation. Evidently, the tetrapyrrole core of the (H2P)2 tweezer, which is a weak base, favors the interactions with positively charged species, thus allowing electronic communication within the supramolecular assembly (H2P)2/C59N-FQH+ both at the ground and the excited state. It should be mentioned that protonation of metal-free (H2P)2 is not observed, since the corresponding absorption spectrum remains unaltered.
inducing proton capture at the singlet excited states.59 Considering that the complexation assays of C59N-FQH+ for hosting the (H2P)2 tweezer are performed in aprotic benzonitrile (cf. Fig. 3), dissociation to the neutral state by proton release to the solvent medium is blocked and therefore C59N-FQH+ remains charged under photoexcitation. Evidently, the tetrapyrrole core of the (H2P)2 tweezer, which is a weak base, favors the interactions with positively charged species, thus allowing electronic communication within the supramolecular assembly (H2P)2/C59N-FQH+ both at the ground and the excited state. It should be mentioned that protonation of metal-free (H2P)2 is not observed, since the corresponding absorption spectrum remains unaltered.
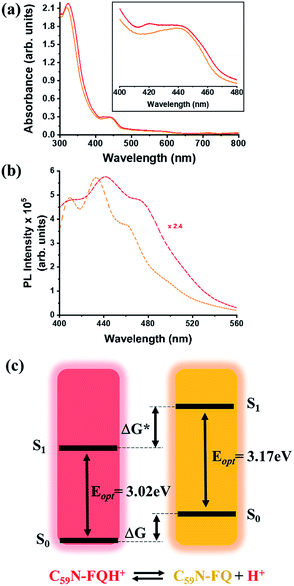 |
| | Fig. 4 (a) UV-Vis and (b) fluorescence emission spectra (λexc 340 nm) of C59N-FQ (orange) and C59N-FQH+ (red), acquired in benzonitrile. (c) Schematic illustration of the Förster cycle. | |
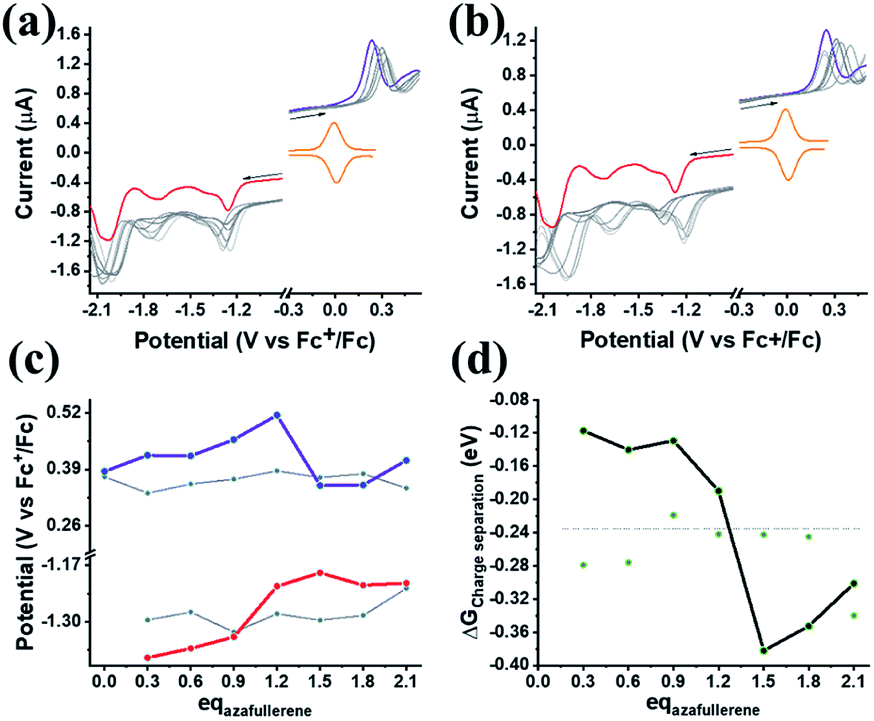
Finally, we explore the electronic communication between the (H2P)2 tweezer and the C59N-FQ derivatives into an electrochemical environment via differential pulse voltammetry (DPV) studies. DPV is selected as a sensitive electrochemical technique capable to the feasibility of the redox processes arose from the supramolecular assembly. Therefore, we monitor the redox features of (H2P)2 upon incremental additions of up to 2 equivalents of C59N-FQ and C59N-FQH+, respectively, as compared to the corresponding features of the individual (H2P)2, C59N-FQ and C59N-FQH+ species in nitrogen saturated solutions of 0.1 M TBAPF6 in benzonitrile. Firstly, we perform anodic and cathodic scans to address the redox potential of the individuals and evaluate the reversibility of the redox processes, while also to monitor the potent impact of the azafullerene derivatives to the oxidation and reduction properties of the (H2P)2 tweezer (ESI, Fig. S10†). The recorded DPV curves display strong interactions at working electrode interface between the host–guest species. Then, we focus at the HOMO level of the tweezer and the LUMO level of the quinoline-modified C59N cage at the neutral and charged state of the quinoline domain, namely we monitor the anodic wave of (H2P)2 tweezer oxidation and the cathodic wave of the first reduction of the azafullerene cage within C59N-FQ and C59N-FQH+. Prior each titration step, the system is allowed to reach equilibrium in nitrogen saturated benzonitrile solutions with 0.1 M TBAPF6 as the supporting electrolyte. These redox studies are presented in Fig. 5a and b. Analysis of the registered oxidation potentials for the (H2P)2 tweezer and the reduction potential of the pentafluoroquinoline-functionalized azafullerene cages show a slightly different redox response of (H2P)2/C59N-FQ and (H2P)2/C59N-FQH+ (Fig. 5c). Evidently, for the (H2P)2/C59N-FQ host–guest assemblies the oxidation potential of (H2P)2 tweezer and the first reduction potential of the azafullerene cage within C59N-FQ, remain almost unaltered during the different host–guest ratios. In contrast, the (H2P)2/C59N-FQH+ host–guest assemblies display more intense changes. More specific, in the presence of ∼1.5 eq. of C59N-FQH+ guest, the oxidation potential of (H2P)2 host display a cathodic shift by 132 mV, suggesting easier oxidation, accompanied by the anodic shift of the first reduction potential of the guest by 83 mV, indicating easier reduction. These observations are further probed by means of charge separation efficiency. The Gibbs energy of photoinduced electron transfer (ΔETG0) between the (H2P)2 host (electron donor) and the C59N-FQ or C59N-FQH+ guest (electron acceptor) can be approximated by eqn (5):60
| |
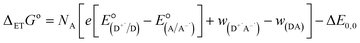 | (5) |
where
e is the elementary charge,
NA is the Avogadro constant,
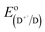
is the electrode potential of the donor radical cation resulting from the electron transfer,
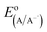
the electrode potential of the acceptor radical anion, while the Δ
E0,0 is the energy level of the singlet excited state of the electron donor (ESI, Fig. S11
†). The
w(D
+˙A
−˙) and
w(DA) are electrostatic work terms accounting for the effect of Coulombic interactions.
Eqn (5) is usually referred as the “Rehm–Weller relation”. The Gibbs energy for the ion pair separation (Δ
GCS) in benzonitrile is represented by
eqn (6), based on the Gibbs energy of charge recombination (Δ
GCR,
eqn (7)) and the static energy (Δ
Gs,
eqn (8)):
61,62| | |
−ΔGCS = ΔE0,0 − (−ΔGCR)
| (6) |
| |
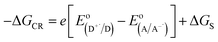 | (7) |
| | |
ΔGs = e2/4πε0[(1/Rhost + 1/Rguest − 1/Rhost–guest)(1/εS) − (1/Rhost + 1/Rguest)(1/εS)]
| (8) |
where
ε0 is the vacuum permittivity,
εS is the dielectric constant of the solvent used for the photophysical studies,
εR is the dielectric constant of the solvent used for the determination of redox potential,
Rguest is the radius of the guest molecule,
Rhost is the radius of the host molecule and
Rhost–guest is center-to-center distance between the host and guest components in the supramolecular assembly.
39
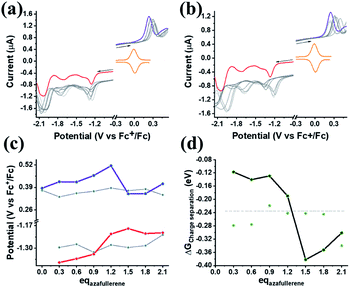 |
| | Fig. 5 (a and b) DPV curves recorded upon incremental additions of up to 2.1 equivalents (gray lines) of C59N-FQ and C59N-FQH+, respectively. Purple line represents the DPV oxidation wave of free (H2P)2, red line refers to the reduction wave of the azafullerene cage within C59N-FQ and C59N-FQH+, while orange line displays the Fc+/Fc redox pair employed as internal reference. All graphs recorded in N2-saturated solutions of 0.3 mM (H2P)2 in benzonitrile with 0.1 M TBAPF6 as the supporting electrolyte at room temperature with scan rate 0.01 V s−1. (c) Plots for the (H2P)2 oxidation potential (purple) and the first reduction potential for the azafullerene cage of C59N-FQH+ (red) versus the different host:guest ratios. The gray lines represent the corresponding plots in the presence of the C59N-FQ derivative. (d) Plots for the Gibbs energy of charge separation (ΔGCS) versus the host–guest ratio for the (H2P)2/C59N-FQ (gray) and (H2P)2/C59N-FQH+ (black) assemblies. The dotted line represents the level of the ΔGCS calculated via the redox potentials of the non-interacting individuals. | |
Based on eqn (6)–(8) we can estimate the variation of the ΔGCS value in respect to the host:guest ratio. As presented in Fig. 5d, for both (H2P)2/C59N-FQ and (H2P)2/C59N-FQH+ assemblies, the Gibbs energy of charge separation is negative, manifesting an exothermic process and thus a thermodynamically favored mechanism. Moreover, for the (H2P)2/C59N-FQH+ assembly we observe a sharp contrast in the region of 1.0–1.5 eq. of C59N-FQH+. These values correspond to a molar fraction of (H2P)2:C59N-FQH+ ranging between 1:1 to 1:1.5 (Xguest = 0.5–0.6) for the used solutions of 0.3 mM in benzonitrile, matching the stoichiometry calculated from the Job's plot for 1 × 10−6 M solutions in benzonitrile (ESI, Fig. S8a†). A maximum −ΔGCS of ∼−0.4 eV is registered in the presence of 1.5 eq. of C59N-FQH+ indicating that the formation of the host–guest complexes requires a slight excess of the guest molecules in order to be activated. Concluding, the redox behavior of the (H2P)2/C59N-FQ and (H2P)2/C59N-FQH+ mixtures complement the corresponding photophysical profiles, suggesting that the protonation of the quinoline domain induces enhanced interactions between the individuals and enables the formation of relatively stable host–guest complexes enabling the electronic communication of the porphyrin tweezer and the azafullerene cage.
Experimental
General
All reagents were used as received. The solvents were dried using standard techniques. Reactions were monitored by thin layer chromatography on silica gel 60 F254 aluminum plates. Products were isolated by column chromatography (silica gel 60, particle size 0.004–0.063 mm). FQ,53 porphyrin-dimer (H2P)2 tweezer45 and C59N-COOH5,8 were synthesized according to published procedures.
Instrumentation
1H, 13C and 19F Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance (Bruker BioSpin GmbH, Magnet Division, Karlsruhe, Germany) DPX 60013, 15092 and 56463 MHz spectrometer, respectively, in deuterated solvents containing TMS as internal standard. The chemical shifts are given in ppm relative to the appropriate solvent peak at standard reference. The resonance multiplicity is indicated as s (singlet), d (doublet), t (triplet), dd (doublet of doublets), td (triplet of doublets) and m (multiplet). Electrospray Ionization Mass Spectroscopy (ESI-MS) measurements were performed using an amaZon SL ion trap of Bruker Daltonics GmbH, Bremen Germany. UV-Vis spectra were recorded on a Perkin Elmer Lambda 19 spectrometer. Fluorescence spectra were recorded in toluene solutions on a Fluorolog-3 Jobin Yvon-Spex spectrofluorometer (model GL3-21) equipped with a UV-Vis detector (200–800 nm) operated at room temperature. TCSPC spectra recorded via the Horiba Fluorohub single photon counting controller. IR spectra were recorded on Bruker Equinox 55 FTIR spectrometer equipped with a Pike Miracle Ge ATR accessory. Electrochemistry studies were performed by using a standard three-electrode cell. Glassy carbon was used as a working electrode and platinum wire and platinum mesh were used as pseudo-reference and counter electrodes, respectively. A mixture of toluene/acetonitrile (4:1) was used as the solvent and ferrocene was employed as an internal reference for the CV studies. DPV studies were performed in benzonitrile. TBAPF6 (98%) was recrystallized three times from acetone and dried under vacuum at 100 °C before used as an electrolyte with 0.1 M concentration for the CV and DPV studies. Before each experiment, the cell was purged with high purity N2 for 15 min and the inert gas was turned to “blanket mode”. The working electrode was cleaned before each experiment (also in between the titration steps in DPV studies) through polishing with a cloth and 6, 3 and 1 mm diamond pastes. CV measurements were recorded by using an EG&G Princeton Applied Research potentiostat/galvanostat model VersaSTAT 4 instrument connected to a personal computer running the VersaStudio software. DPV measurements were recorded by AUTOLAB PGSTAT128N potentiostat/galvanostat instrument connected to a personal computer running the Nova 2.1 software.
Synthetic procedures
Synthesis of C59N-FQ. C59N-COOH (59 mg, 0.075 mmol) was mixed with FQ (104 mg, 0.225 mmol) in 100 mL dry CHCl3. Then, HOBt (33 mg, 0.247 mmol) and EDC·HCl (47 mg, 0.247 mmol) were also added and the mixture was stirred for 8 h. The reaction was monitored by TLC (SiO2) with CHCl3 as eluent and two greenish fullerene spots were evident. The solvent was removed by rotary evaporation and the residue was dissolved in CS2 and purified by column chromatography (SiO2) with toluene as eluent. Only one greenish fraction was eluted, evaporated to 2–3 mL and the product precipitated by addition of cold hexane. The solid collected by centrifugation (300 rpm for 5 min) and dried under vacuum. 1H NMR (CDCl3): 8.32 (1H, d, J = 4.35 Hz), 8.19 (1H, d, J = 0.2 Hz), 8.07 (1H, dd, J1 = 0.84 Hz, J2 = 0.87 Hz), 7.74 (2H, d, J = 4.29 Hz), 7.61–7.51 (8H, m) and 5.1 (2H, s) ppm. 13C NMR (CDCl3): 168.21 (CO), 155.23, 150.48, 149.76, 147.92, 147.67, 147.53, 147.42, 147.07, 146.77, 146.71, 146.43, 146.04, 146.27, 144.59, 144.53, 144.15, 143.30, 142.98, 142.29, 141.95, 141.60, 141.27, 141.24, 141.14, 139.89, 137.74, 137.31, 134.48, 130.85, 129.72, 129.58, 128.94, 128.91, 126.37, 124.50, 123.75, 123.47, 122.30 and 76.98 ppm. 19F NMR (CDCl3): −161 (2H, td, J1 = 4.17 Hz, J2 = 3.66 Hz, J3 = 4.35 Hz), −153 (1H, t, J = 11.04 Hz) and −142 (2H, dd, J1 = 4.17 Hz, J2 = 4.26 Hz) ppm. ESI-MS: calculated m/z for C88H15F5N2O2 [M + 1]: 1227.11; found: 1227.17.
Synthesis of C59N-FQH+. 10 mg of C59N-FQ were dissolved in 10 mL chloroform at room temperature and the was immersed into an ice-bath, followed by the dropwise addition of excess glacial acetic acid (∼1 mL) under stirring. The mixture was allowed to reach room temperature under stirring and then evaporated to dryness under vacuum. The solid residue was dissolved in a minimum amount of chloroform, cooled in the fridge and precipitated by addition of cold hexane. C59N-FQH+ was recovered by centrifugation, dried under vacuum and collected as brown powder (8 mg).
Conclusions
A new azafullerene (C59N) derivative covalently carrying a pentafluoroquinoline (FQ) was synthesized and fully characterized. The evidenced strong intramolecular electronic communication between the pentafluoroquinoline and azafullerene cage allowed us to exploit the quinoline domain as guest for hosting the (H2P)2 porphyrin tweezer. Interestingly, protonation of the quinoline heterocycle enables even stronger interactions, due to enhanced electrostatic attraction forces occurring between the (H2P)2 host and the protonated C59N-FQH+ guest species within the (H2P)2/C59N-FQH+ supramolecular nanoassembly. Photophysical studies revealed detectable host–guest interactions in (H2P)2/C59N-FQH+ represented by the slight decrement of the Soret band intensity of (H2P)2, complemented by partial quenching of the fluorescence emission of (H2P)2, in contrast to the corresponding (H2P)2/C59N-FQ where almost negligible differences were observed. Analysis of the time-resolved fluorescence spectra uncovered a new decay component of 1 ns for (H2P)2/C59N-FQH+, attributed to the deactivation of the singlet excited state of (H2P)2 to the azafullerene. In addition, differential pulse voltammetry titration assays displayed intense changes for the oxidation potential of (H2P)2 and the first reduction potential of the azafullerene cage within C59N-FQH+, suggesting easier oxidation of the host and easier reduction of the guest, respectively, in the range of 1.0–1.5 eq. of guest. Overall, the beneficial effect of the protonated quinoline domain in the formation of the host–guest supramolecular complexes was further confirmed by the calculated value of Gibbs free energy for charge separation ΔGCS. Here, it is the maximum value of around −0.4 eV registered for the (H2P)2/C59N-FQH+ assembly, which guarantees as an energetic favorable process the electronic communication between (H2P)2 and C59N. Collectively, we presented a very first investigation concerning the electronic interactions of C59N with molecular porphyrin-dimer (H2P)2 tweezers via a host–guest approach mediated by a versatile amphoteric heterocycle motif of a pentafluoroquinoline derivative conjugated on the azafullerene cage.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Partial financial support of this work by the project “Advanced Materials and Devices” (MIS 5002409), which is implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund) is acknowledged.
Notes and references
- J. C. Hummelen, M. Prato and F. Wudl, There Is a Hole in My Bucky, J. Am. Chem. Soc., 1995, 117, 7003–7004 CrossRef CAS.
- J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Isolation of the Heterofullerene C59N as Its Dimer (C59N)2, Science, 1995, 269, 1554 CrossRef CAS PubMed.
- O. Vostrowsky and A. Hirsch, Heterofullerenes, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed.
- G. Rotas and N. Tagmatarchis, Azafullerene C59N in Donor–Acceptor Dyads: Synthetic Approaches and Properties, Chem.–Eur. J., 2016, 22, 1206–1214 CrossRef CAS PubMed.
- F. Hauke and A. Hirsch, C59N+: a key intermediate in azaheterofullerene chemistry, Tetrahedron, 2001, 57, 3697–3708 CrossRef CAS.
- F. Hauke, S. Atalick, D. M. Guldi, J. Mack, L. T. Scott and A. Hirsch, Molecular satellite dishes: attaching parabolic and planar arenes to heterofullerenes, Chem. Commun., 2004, 766–767 RSC.
- F. Hauke, A. Hirsch, S.-G. Liu, L. Echegoyen, A. Swartz, C. Luo and D. M. Guldi, Evidence of Pronounced Electronic Coupling in a Directly Bonded Fullerene–Ferrocene Dyad, ChemPhysChem, 2002, 3, 195–205 CrossRef CAS PubMed.
- G. Rotas, J. Ranta, A. Efimov, M. Niemi, H. Lemmetyinen, N. Tkachenko and N. Tagmatarchis, Azafullerene C59N–Phthalocyanine Dyad: Synthesis, Characterisation and Photoinduced Electron Transfer, ChemPhysChem, 2012, 13, 1246–1254 CrossRef CAS PubMed.
- G. Rotas, G. Charalambidis, L. Glätzl, D. T. Gryko, A. Kahnt, A. G. Coutsolelos and N. Tagmatarchis, A corrole–azafullerene dyad: synthesis, characterization, electronic interactions and photoinduced charge separation, Chem. Commun., 2013, 49, 9128–9130 RSC.
- L. Martín-Gomis, G. Rotas, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi, N. Tagmatarchis and Á. Sastre-Santos, Does a nitrogen matter? Synthesis and photoinduced electron transfer of perylenediimide donors covalently linked to C59N and C60 acceptors, Nanoscale, 2015, 7, 7437–7444 RSC.
- G. Rotas, K. Stranius, N. Tkachenko and N. Tagmatarchis, Ultralong 20 Milliseconds Charge Separation Lifetime for Photoilluminated Oligophenylenevinylene–Azafullerene Systems, Adv. Funct. Mater., 2018, 28, 1702278 CrossRef.
- U. Reuther and A. Hirsch, Pyrrole-embedded [60]fullerenes, Chem. Commun., 1998, 1401–1402 RSC.
- N. B. Shustova, I. V. Kuvychko, A. A. Popov, M. von Delius, L. Dunsch, O. P. Anderson, A. Hirsch, S. H. Strauss and O. V. Boltalina, Nitrogen Directs Multiple Radical Additions to the 9,9′-Bi-1-aza(C60-Ih)[5,6]fullerene: X-ray Structure of 6,9,12,15,18-C59N(CF3)5, Angew. Chem., Int. Ed., 2011, 50, 5537–5540 CrossRef CAS.
- K. Y. Amsharov, J. Holzwarth, K. Roshchyna, D. I. Sharapa, F. Hampel and A. Hirsch, Synthesis, Structural Characterization, and Crystal Packing of the Elusive Pentachlorinated Azafullerene C59NCl5, Chem.–Eur. J., 2017, 23, 9014–9017 CrossRef CAS.
- R. Neubauer, F. W. Heinemann, F. Hampel, Y. Rubin and A. Hirsch, Pentaarylazafullerenes and their Triaryldihydro and Tetraarylmonohydro Precursors, Angew. Chem., Int. Ed., 2012, 51, 11722–11726 CrossRef CAS.
- R. Eigler, F. W. Heinemann and A. Hirsch, Hydro-aza-(C59N)fullerenes: Formation Mechanism and Hydrogen Substitution, Chem.–Eur. J., 2016, 22, 13575–13581 CrossRef CAS.
- F. Hauke and A. Hirsch, Regioselective formation of highly functionalised heterofullerenes: pentamalonates of RCN involving an octahedral addition pattern, Chem. Commun., 2001, 1316–1317 RSC.
- A. Stergiou, K. Asad, A. Kourtellaris, N. Chronakis and N. Tagmatarchis, Tether-Directed Regioselective Synthesis of an Equatorialface Bisadduct of Azafullerene Using Cyclo-[2]-octylmalonate, Chem.–Eur. J., 2019, 25, 5751–5756 CrossRef CAS PubMed.
- S. E. Lewis, Cycloparaphenylenes and related nanohoops, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- Y. Segawa, A. Yagi, K. Matsui and K. Itami, Design and Synthesis of Carbon Nanotube Segments, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed.
- D. Wu, W. Cheng, X. Ban and J. Xia, Cycloparaphenylenes (CPPs): An Overview of Synthesis, Properties, and Potential Applications, Asian J. Org. Chem., 2018, 7, 2161–2181 CrossRef CAS.
- Y. Xu and M. von Delius, The Supramolecular Chemistry of Strained Carbon Nanohoops, Angew. Chem., Int. Ed., 2020, 59, 559–573 CrossRef CAS PubMed.
- Y. Xu, B. Wang, R. Kaur, M. B. Minameyer, M. Bothe, T. Drewello, D. M. Guldi and M. von Delius, A Supramolecular [10]CPP Junction Enables Efficient Electron Transfer in Modular Porphyrin–[10]CPP⊃Fullerene Complexes, Angew. Chem., Int. Ed., 2018, 57, 11549–11553 CrossRef CAS PubMed.
- J. Rio, S. Beeck, G. Rotas, S. Ahles, D. Jacquemin, N. Tagmatarchis, C. Ewels and H. A. Wegner, Electronic Communication between two [10]cycloparaphenylenes and Bis(azafullerene) (C59N)2 Induced by Cooperative Complexation, Angew. Chem., Int. Ed., 2018, 57, 6930–6934 CrossRef CAS PubMed.
- A. Stergiou, J. Rio, J. H. Griwatz, D. Arčon, H. A. Wegner, C. P. Ewels and N. Tagmatarchis, A Long-Lived Azafullerenyl Radical Stabilized by Supramolecular Shielding with a [10]Cycloparaphenylene, Angew. Chem., Int. Ed., 2019, 58, 17745–17750 CrossRef CAS PubMed.
- G. Rotas, L. Martín-Gomis, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi, N. Tagmatarchis and Á. Sastre-Santos, Axially Substituted Silicon Phthalocyanine as Electron Donor in a Dyad and Triad with Azafullerene as Electron Acceptor for Photoinduced Charge Separation, Chem.–Eur. J., 2016, 22, 15137–15143 CrossRef CAS PubMed.
- F. Hauke, S. Atalick, D. M. Guldi and A. Hirsch, Covalently linked heterofullerene–porphyrin conjugates; new model systems for long-lived intramolecular charge separation, Tetrahedron, 2006, 62, 1923–1927 CrossRef CAS.
- F. Hauke, A. Swartz, D. M. Guldi and A. Hirsch, Supramolecular assembly of a quasi-linear heterofullerene–porphyrin dyad, J. Mater. Chem., 2002, 12, 2088–2094 RSC.
- J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. d. C. Carreiras and E. Soriano, Recent Advances in the Friedländer Reaction, Chem. Rev., 2009, 109, 2652–2671 CrossRef CAS PubMed.
- C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Blue-Light-Emitting Oligoquinolines: Synthesis, Properties, and High-Efficiency Blue-Light-Emitting Diodes, Adv. Funct. Mater., 2007, 17, 863–874 CrossRef CAS.
- S. P. Economopoulos, A. K. Andreopoulou, V. G. Gregoriou and J. K. Kallitsis, Synthesis and Optical Properties of New End-Functionalized Polyquinolines, Chem. Mater., 2005, 17, 1063–1071 CrossRef CAS.
- L. Lu and S. A. Jenekhe, Poly(vinyl diphenylquinoline): A New pH-Tunable Light-Emitting and Charge-Transport Polymer Synthesized by a Simple Modification of Polystyrene, Macromolecules, 2001, 34, 6249–6254 CrossRef CAS.
- I. Thivaios, S. Kakogianni and G. Bokias, A Library of Quinoline-Labeled Water-Soluble Copolymers with pH-Tunable Fluorescence Response in the Acidic pH Region, Macromolecules, 2016, 49, 3526–3534 CrossRef CAS.
- S. Yu, S. Wang, H. Yu, Y. Feng, S. Zhang, M. Zhu, H. Yin and X. Meng, A ratiometric two-photon fluorescent probe for hydrazine and its applications, Sens. Actuators, B, 2015, 220, 1338–1345 CrossRef CAS PubMed.
- A. A. Stefopoulos, S. N. Kourkouli, S. Economopoulos, F. Ravani, A. Andreopoulou, K. Papagelis, A. Siokou and J. K. Kallitsis, Polymer and Hybrid Electron Accepting Materials Based on a Semiconducting Perfluorophenylquinoline, Macromolecules, 2010, 43, 4827–4828 CrossRef CAS.
- L. Sygellou, S. Kakogianni, A. K. Andreopoulou, K. Theodosiou, G. Leftheriotis, J. K. Kallitsis and A. Siokou, Evaluation of the electronic properties of perfluorophenyl functionalized quinolines and their hybrids with carbon nanostructures, Phys. Chem. Chem. Phys., 2016, 18, 4154–4165 RSC.
- S. Kakogianni, S. N. Kourkouli, A. K. Andreopoulou and J. K. Kallitsis, A versatile approach for creating hybrid semiconducting polymer–fullerene architectures for organic electronics, J. Mater. Chem. A, 2014, 2, 8110–8117 RSC.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Molecular artificial photosynthesis, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- A. N. Webber and W. Lubitz, P700: the primary electron donor of photosystem I, Biochim. Biophys. Acta, Bioenerg., 2001, 1507, 61–79 CrossRef CAS.
- M. Rudolf, S. V. Kirner and D. M. Guldi, A multicomponent molecular approach to artificial photosynthesis – the role of fullerenes and endohedral metallofullerenes, Chem. Soc. Rev., 2016, 45, 612–630 RSC.
- V. Nikolaou, A. Charisiadis, C. Stangel and G. Charalambidis, Porphyrinoid–Fullerene Hybrids as Candidates in Artificial Photosynthetic Schemes, C, 2019, 5, 57 CAS.
- V. Valderrey, G. Aragay and P. Ballester, Porphyrin tweezer receptors: binding studies, conformational properties and applications, Coord. Chem. Rev., 2014, 258–259, 137–156 CrossRef CAS.
- M. Davor, Host–guest Studies of Bis-porphyrins, Curr. Org. Chem., 2012, 16, 829–851 CrossRef.
- T. Haino, T. Fujii and Y. Fukazawa, Guest Binding and New Self-Assembly of Bisporphyrins, J. Org. Chem., 2006, 71, 2572–2580 CrossRef CAS PubMed.
- G. Pagona, S. P. Economopoulos, T. Aono, Y. Miyata, H. Shinohara and N. Tagmatarchis, Molecular recognition of La@C82 endohedral metallofullerene by an isophthaloyl-bridged porphyrin dimer, Tetrahedron Lett., 2010, 51, 5896–5899 CrossRef CAS.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker and P. D. W. Boyd, Supramolecular Fullerene-Porphyrin Chemistry. Fullerene Complexation by Metalated “Jaws Porphyrin” Hosts, J. Am. Chem. Soc., 2002, 124, 6604–6612 CrossRef CAS PubMed.
- Z.-Y. Xiao, J.-L. Hou, X.-K. Jiang, Z.-T. Li and Z. Ma, Complexes between hydrogen bonded bisporphyrin tweezers and cholesterol-appended fullerenes as organogelators and liquid crystals, Tetrahedron, 2009, 65, 10182–10191 CrossRef CAS.
- M. Fathalla and J. Jayawickramarajah, Configurational Isomers of a Stilbene-Linked Bis(porphyrin) Tweezer: Synthesis and Fullerene-Binding Studies, Eur. J. Org. Chem., 2009, 2009, 6095–6099 CrossRef.
- M. Pérez Emilio and N. Martín, Molecular tweezers for fullerenes, Pure Appl. Chem., 2010, 82, 523 Search PubMed.
- R. M. K. Calderon, J. Valero, B. Grimm, J. de Mendoza and D. M. Guldi, Enhancing Molecular Recognition in Electron Donor–Acceptor Hybrids via Cooperativity, J. Am. Chem. Soc., 2014, 136, 11436–11443 CrossRef CAS PubMed.
- M. Tanaka, K. Ohkubo, C. P. Gros, R. Guilard and S. Fukuzumi, Persistent Electron-Transfer State of a π-Complex of Acridinium Ion Inserted between Porphyrin Rings of Cofacial Bisporphyrins, J. Am. Chem. Soc., 2006, 128, 14625–14633 CrossRef CAS PubMed.
- A. Chaudhary and S. P. Rath, Encapsulation of TCNQ and the Acridinium Ion within a Bisporphyrin Cavity: Synthesis,
Structure, and Photophysical and HOMO–LUMO-Gap-Mediated Electron-Transfer Properties, Chem.–Eur. J., 2012, 18, 7404–7417 CrossRef CAS PubMed.
- K. J. Kallitsis, R. Nannou, A. K. Andreopoulou, M. K. Daletou, D. Papaioannou, S. G. Neophytides and J. K. Kallitsis, Crosslinked wholly aromatic polyether membranes based on quinoline derivatives and their application in high temperature polymer electrolyte membrane fuel cells, J. Power Sources, 2018, 379, 144–154 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- S. Haffner, T. Pichler, M. Knupfer, B. Umlauf, R. Friedlein, M. S. Golden, J. Fink, M. Keshavarz-K, C. Bellavia-Lund, A. Sastre, J. C. Hummelen and F. Wudl, The electronic structure of (C59N)2 from high energy spectroscopy, Eur. Phys. J. B, 1998, 1, 11–17 CrossRef CAS.
- D. Brynn Hibbert and P. Thordarson, The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis, Chem. Commun., 2016, 52, 12792–12805 RSC.
- E. W. Driscoll, J. R. Hunt and J. M. Dawlaty, Photobasicity in Quinolines: Origin and Tunability via the Substituents' Hammett Parameters, J. Phys. Chem. Lett., 2016, 7, 2093–2099 CrossRef CAS PubMed.
- S. A. Jenekhe and X. L. Chen, Self-Assembled Aggregates of Rod-Coil Block Copolymers and Their Solubilization and Encapsulation of Fullerenes, Science, 1998, 279, 1903 CrossRef CAS PubMed.
- E. W. Driscoll, J. R. Hunt and J. M. Dawlaty, Proton Capture Dynamics in Quinoline Photobases: Substituent Effect and Involvement of Triplet States, J. Phys. Chem. A, 2017, 121, 7099–7107 CrossRef CAS PubMed.
- S. E. Braslavsky, A. M. Braum, A. E. Cassano, A. V. Emeline, M. I. Litter, L. Palmisano, V. N. Parmon and N. Serpone, Glossary of terms used in photocatalysis and radiation catalysis (IUPAC Recommendations 2011), Pure Appl. Chem., 2011, 83, 931–1014 CAS.
- M. Fujitsuka, A. Masuhara, H. Kasai, H. Oikawa, H. Nakanishi, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, Photoinduced Charge Separation and Recombination Processes in Fine Particles of Oligothiophene-C60 Dyad Molecules, J. Phys. Chem. B, 2001, 105, 9930–9934 CrossRef CAS.
- J. L. Delgado, M. E. El-Khouly, Y. Araki, M. J. Gómez-Escalonilla, P. de la Cruz, F. Oswald, O. Ito and F. Langa, Synthesis and photophysical properties of a [60]fullerene compound with dimethylaniline and ferrocene connected through a pyrazolino group: a study by laser flash photolysis, Phys. Chem. Chem. Phys., 2006, 8, 4104–4111 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and 19F NMR, FT-IR spectra and ESI-MS for C59N-FQ; FT-IR spectra of C59N-FQH+; Tauc plot of FQ, Job's, Hill and Stern–Volmer plots for (H2P)2/C59NFQH+; PL-spectra of C59N-FQH+; anodic and cathodic DPV graphs for (H2P)2/C59NFQ and (H2P)2/C59NFQH+; UV/Vis-PL cross-section spectra of (H2P)2. See DOI: 10.1039/d0ra06195f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Aikaterini K. Andreopoulou
*a,
Aikaterini K. Andreopoulou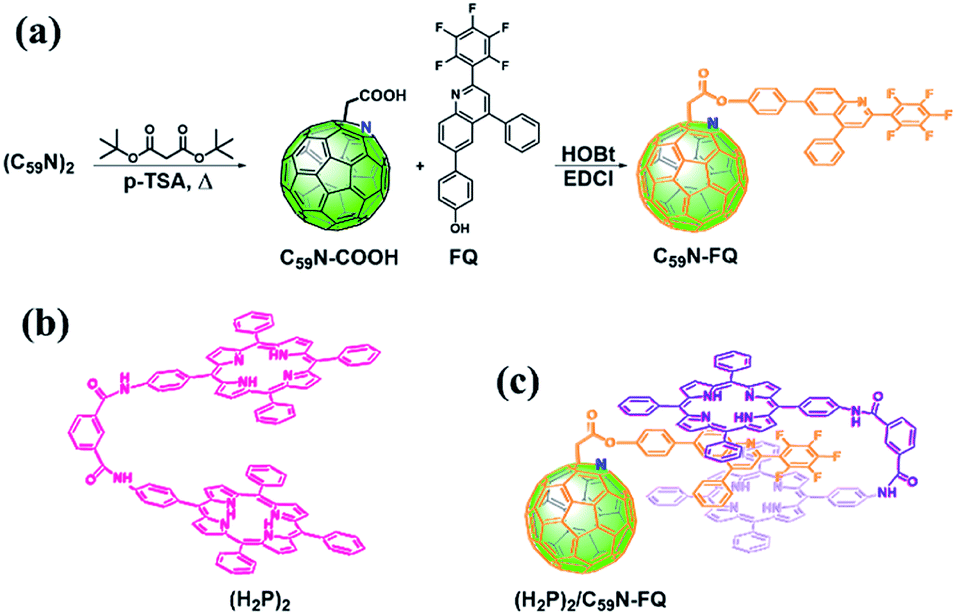
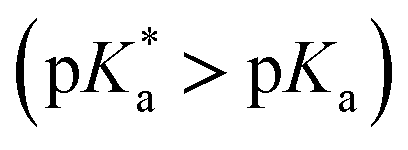 inducing proton capture at the singlet excited states.59 Considering that the complexation assays of C59N-FQH+ for hosting the (H2P)2 tweezer are performed in aprotic benzonitrile (cf. Fig. 3), dissociation to the neutral state by proton release to the solvent medium is blocked and therefore C59N-FQH+ remains charged under photoexcitation. Evidently, the tetrapyrrole core of the (H2P)2 tweezer, which is a weak base, favors the interactions with positively charged species, thus allowing electronic communication within the supramolecular assembly (H2P)2/C59N-FQH+ both at the ground and the excited state. It should be mentioned that protonation of metal-free (H2P)2 is not observed, since the corresponding absorption spectrum remains unaltered.
inducing proton capture at the singlet excited states.59 Considering that the complexation assays of C59N-FQH+ for hosting the (H2P)2 tweezer are performed in aprotic benzonitrile (cf. Fig. 3), dissociation to the neutral state by proton release to the solvent medium is blocked and therefore C59N-FQH+ remains charged under photoexcitation. Evidently, the tetrapyrrole core of the (H2P)2 tweezer, which is a weak base, favors the interactions with positively charged species, thus allowing electronic communication within the supramolecular assembly (H2P)2/C59N-FQH+ both at the ground and the excited state. It should be mentioned that protonation of metal-free (H2P)2 is not observed, since the corresponding absorption spectrum remains unaltered.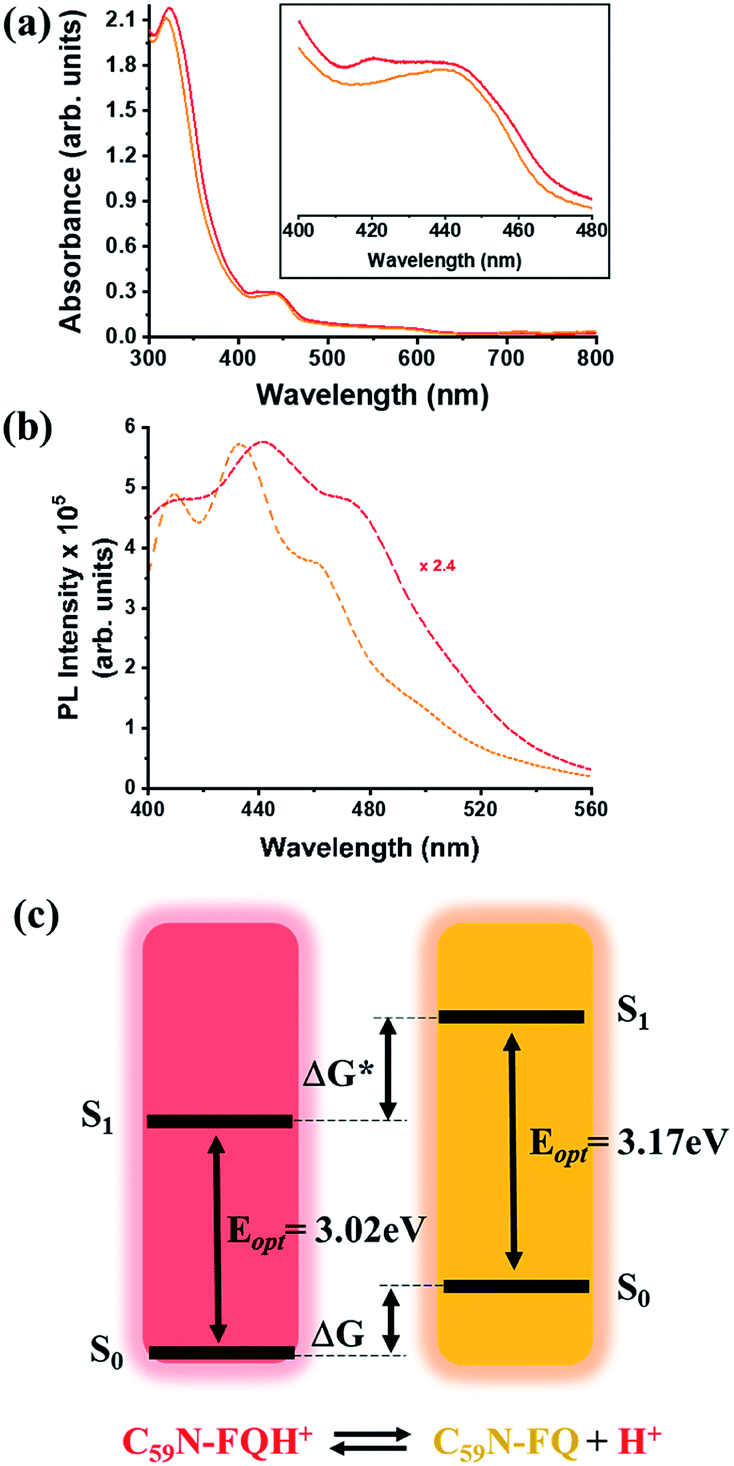
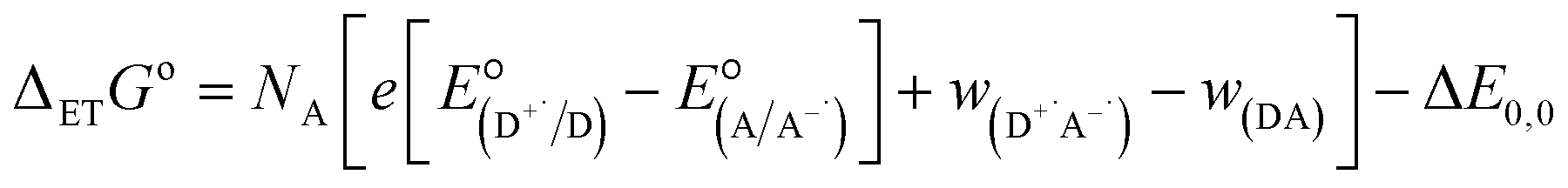
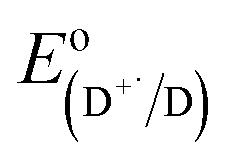 is the electrode potential of the donor radical cation resulting from the electron transfer,
is the electrode potential of the donor radical cation resulting from the electron transfer, 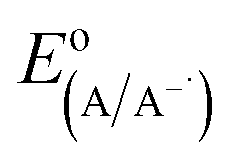 the electrode potential of the acceptor radical anion, while the ΔE0,0 is the energy level of the singlet excited state of the electron donor (ESI, Fig. S11†). The w(D+˙A−˙) and w(DA) are electrostatic work terms accounting for the effect of Coulombic interactions. Eqn (5) is usually referred as the “Rehm–Weller relation”. The Gibbs energy for the ion pair separation (ΔGCS) in benzonitrile is represented by eqn (6), based on the Gibbs energy of charge recombination (ΔGCR, eqn (7)) and the static energy (ΔGs, eqn (8)):61,62
the electrode potential of the acceptor radical anion, while the ΔE0,0 is the energy level of the singlet excited state of the electron donor (ESI, Fig. S11†). The w(D+˙A−˙) and w(DA) are electrostatic work terms accounting for the effect of Coulombic interactions. Eqn (5) is usually referred as the “Rehm–Weller relation”. The Gibbs energy for the ion pair separation (ΔGCS) in benzonitrile is represented by eqn (6), based on the Gibbs energy of charge recombination (ΔGCR, eqn (7)) and the static energy (ΔGs, eqn (8)):61,62