
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinazoline-Schiff base conjugates: in silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins
Mostafa A.
Mansour
 a,
Asmaa M.
AboulMagd
a and
Hamdy M.
Abdel-Rahman
*ab
a,
Asmaa M.
AboulMagd
a and
Hamdy M.
Abdel-Rahman
*ab
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University (NUB), Beni-Suef, Egypt. E-mail: hamdy.abdelrahman@nub.edu.eg
bMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
First published on 15th September 2020
Abstract
The 2019 coronavirus (COVID-19) pandemic is spreading worldwide, with a dramatic increase in death without any effective therapeutic treatment available up to now. We previously reported quinazoline-trihydroxyphenyl Schiff base conjugates as phosphodiesterase 4B (PDE 4B) inhibitors (an enzyme that plays an essential role in the early stages of COVID-19 pneumonia). Additionally, the structural similarity between these conjugates and identified anti-severe acute respiratory syndrome (SARS)-coronavirus (CoV)-2 flavonoids inspired us to in silico study their possible binding interactions with essential SARS-CoV-2 proteins. Thus, this study provides an insight into the potential bindings between quinazoline-Schiff base conjugates and SARS-CoV-2 proteins, including spike glycoprotein (SGp), main protease (Mpro) and RNA-dependent RNA polymerase (RdRp), to offer an opportunity to find an effective therapy. Besides this, based on the role that COVID-19 plays in iron dysmetabolism, the conjugate trihydroxyphenyl moiety should be reconsidered as an iron chelator. Moreover, molecular dynamics simulations of quinazoline derivative Ic bound to the mentioned targets were carried out. Finally, ADMET calculations were performed for the studied compounds to predict their pharmacokinetic profiles.
1. Introduction
Since the emergence of severe acute respiratory syndrome (SARS) coronavirus (CoV) in Wuhan, China at the end of 2019, extensive studies on the pathogenicity and transmissibility of the causative virus, SARS-CoV-2, have been discussed.1 COVID-19 is considered the seventh coronavirus known to infect humans and cause severe diseases.2 Coronaviruses are enveloped structures, their RNA nucleic acid is positive and single-stranded, meaning that they can infect both humans and animals, especially cattle.3 The virus releases a S-glycoprotein on its surface that is responsible for its attachment to a host receptor, named angiotensin-converting enzyme 2 (ACE2), during cell entry.4 The sequencing of the SARS-CoV-2 genome revealed that it encodes for 16–17 non-structural proteins. Two proteases, namely papain-like protease (PLpro) and 3-chymotrypsin-like protease (3CLpro), which are known as Mpro, are essential in both maturation and infectivity of the virus.5–10 Additionally, RNA-dependent RNA polymerase (RdRp) allows the viral genome to be copied into the new RNA strand using the host cell's machinery11–15 (Fig. 1).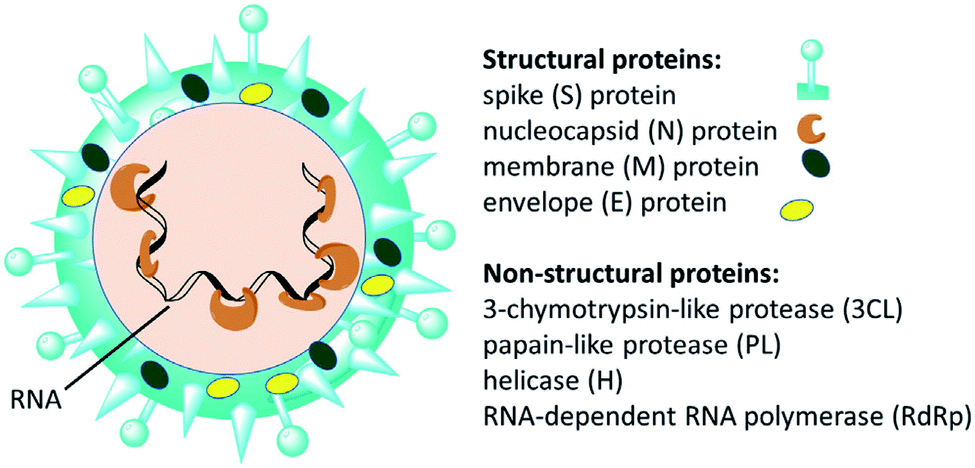 | ||
| Fig. 1 Scheme of SARS-CoV-2 and some of its molecular protein targets.6 | ||
A recent literature survey highlighted that two possible viral pathophysiological mechanisms exist. The first one deals with interaction with the hemoglobin molecule, through receptors present on erythrocyte precursors, such as CD147 (also known as Basigin or EMMPRIN) or cluster of differentiation 26 (CD26), causing COVID-19. In contrast, the other mechanism is the induction of a ferroportin blockage through hepcidin-mimetic action of a viral spike protein.16,17 Regarding iron dysmetabolism in SARS-CoV-2, a noticeable similarity between the hepcidin protein and SARS-CoV-2 spike glycoprotein cytoplasmic tail amino acid sequence has been reported.18 SARS-CoV-2 has evolved to use a variety of host proteases, including cathepsin L, cathepsin B, trypsin, factor X, elastase, furin, and TMPRSS2 (transmembrane protease serine 2), to facilitate cell entry following receptor binding. It has been revealed that circulating Ang II levels are markedly increased in COVID-19 patients, providing a direct connection between tissue ACE2 down regulation with SARS-CoV-2 via blocking of the receptor binding domain (RBD) of the viral S-protein.19 Hepcidin is considered as the master regulator of iron metabolism, interacting with ferroportin to increase the iron entrance inside the cells.20 In terms of COVID-19, significant iron metabolism dysregulation may occur, accompanied by hyperferritinemia and ultimately ferroptosis.21 Whether the main pathological viral infection begins in the lungs, resulting in general anemic hypoxia or the opposite, iron dysmetabolism, is a matter of future research due to its role in multi-organ disease and hypoxia (Fig. 2).
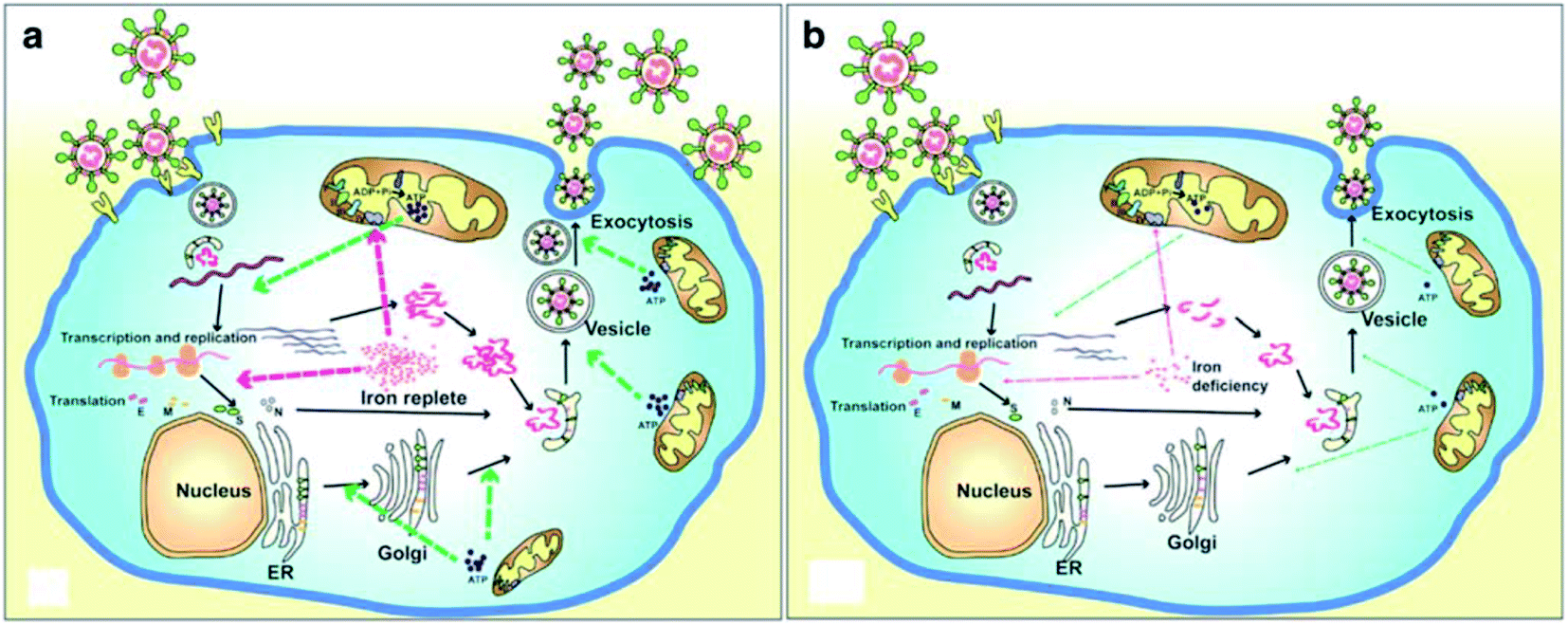 | ||
| Fig. 2 Life cycle of coronaviruses under iron replete and deficiency conditions. (a) Sufficient intracellular iron levels support coronavirus replication, whereas (b) iron deficiency undermines its replication process by interfering with viral transcription, translation, assembly, and exocytosis. CoVs enter into host cells via binding to various receptors and disassemble to release the viral genome and nucleocapsid.21 | ||
Although the hallmarks of COVID-19 have been studied in detail, until now there has been no specific drug used in its treatment, with its control mainly being achieved using known antiviral drugs combined with supportive care.22 Thus, many research groups worldwide began searching for drugs and/or molecules for rapid COVID-19 disease therapy from libraries of natural, synthetic or semi-synthetic derivatives. The main strategies for these drug discovery projects involve repurposing of approved drugs or using computational approaches such as molecular docking, virtual screening, simulations, etc., which can save time, money and facilitate the discovery of COVID-19 drugs.23
Remdesivir 1 (Fig. 3) was the first drug granted emergency use for the treatment of suspected or laboratory confirmed COVID-19 cases by the U.S. Food and Drug Administration (FDA). It is an antiviral nucleoside analog that was developed for treatment of general ribonucleic acid (RNA) viral infections. It works by inhibiting viral replication through competitively inhibiting viral RNA polymerase.24
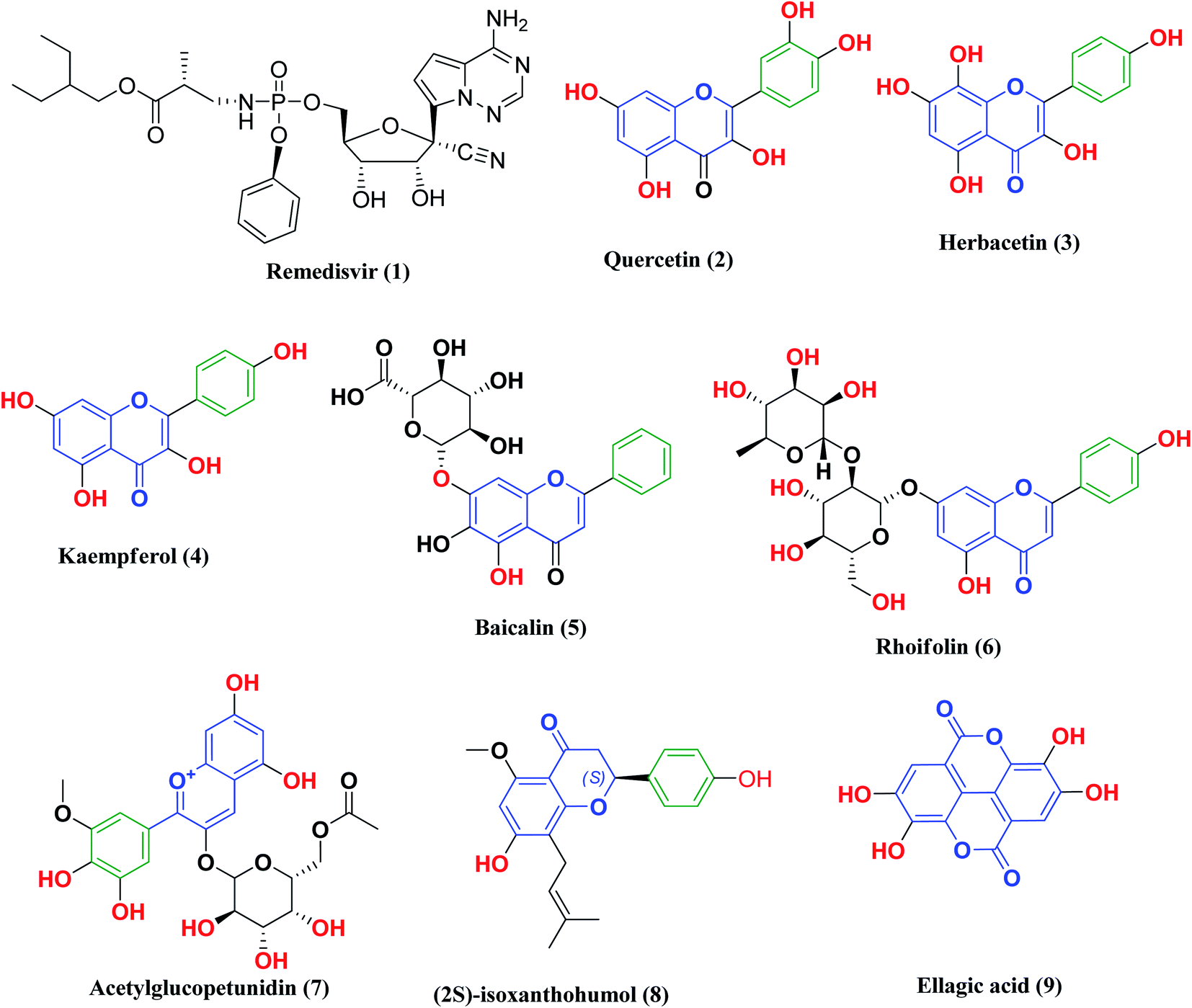 | ||
| Fig. 3 Remdesivir and reported polyphenolic compounds that are possible SARS-CoV-2 3CLpro inhibitors. | ||
Other research projects have focused on adjuvant therapies for the treatment of COVID-19, in particular iron chelation and phosphodiesterase 4 (PDE 4B) inhibitors. As previously mentioned, COVID-19 infections are characterized by iron dysmetabolism resulting in increasing intracellular iron, thus depriving iron supply to the virus represents a promising adjuvant therapy. For this reason, iron chelators such as deferoxamine or polyphenols have been screened for their effectiveness against SARS-CoV-2 viral infection.25 On the other hand, PDE4 inhibitors, which are one of the major PDE enzymes involved in cAMP-mediated regulation, play an important role in suppressing inflammatory cell functions that contribute to their anti-inflammatory actions in respiratory diseases such as chronic obstructive pulmonary disease (COPD) and asthma. Furthermore, the inhibition of PDE4 activity leads to smooth muscle relaxation and bronchodilation, which is useful for the treatment of asthma or COPD.26–29 Thus, selective PDE 4B inhibitors may be a promising option for treatment of the COVID-19 early hyperinflammatory state attributed to massive pro-inflammatory cytokine release.30
Likewise, many polyphenolic natural products such as flavonoids (2–6) have been in silico screened as a possible therapeutic treatment for COVID-19 disease as 3CLpro inhibitors (Fig. 3).31–33 In our previous study, three more phenolic compounds (7–9) were also screened against the SARS-CoV-2 main protease (Mpro) enzyme (Fig. 3). The structure–activity relationship analyses of the final top-hits presented in Fig. 3 reveal that they share similar binding modes in terms of hydrogen bonding and van der Waals interactions within the active site of Mpro.34
From all the above mentioned studies, we think that an effective protocol for the treatment of COVID-19 disease should include the following; (a) a drug that disrupts the life cycle of the virus at any stage of the viral replication cycle, such as blocking ACE2 receptors or inhibition of the SARS-CoV-2 main protease (Mpro); (b) an iron chelating agent to bind with the excess iron resulting from its dysmetabolism; (c) anti-inflammatory drugs as phosphodiesterase 4 inhibitors for the treatment of lung inflammation and necrosis.
We previously reported the design and synthesis of a class of 3H-quinazolin-4-one/Schiff base conjugates and evaluated them in terms of their PDE 4B activity.35 Based on the above findings, as the most active derivatives of this series were trihydroxyphenyl Schiff bases, we thought that these compounds would be ideal drugs for the treatment of COVID-19 (Fig. 4). Thus, herein we perform molecular modeling studies on the quinazolinone-Schiff base conjugates against three essential target proteins of the SARS-CoV-2 virus and predict their pharmacokinetics properties for rapid drug discovery.
 | ||
| Fig. 4 Structures and rationale for the design of 2-phenylquinazolin-4(3H)one-trihydroxyphenyl Schiff base conjugates for COVID-19 therapy. | ||
2. Experimental
2.1. Chemistry
The synthesis of the target substituted 2-phenylquinazoline derivatives was performed according to previously reported work.35–37 Details of the synthetic procedures as well as full characterization of all compounds have been reported.35–37 Briefly, the general methods for the synthesis of compounds Ia–d are discussed below. Previous reports suggested the E configuration of compounds Ia–d.26 Furthermore, both Z and E conformers energies were minimized and the calculated total energy of compound Ic was 117.266 kcal mol−1, which is lower if it is in E configuration (Table 1).| Isomer | E isomer | Z isomer |
|---|---|---|
| a Total energy = EStr + EAng + EStb + EOop + ETor + EVdw + EEle. | ||
| 2D |
|

|
| 3D minimized |

|
|
| Total energy | 117.266 | 149.139 |
| E Str (bond stretch energy) | 8.122 | 7.595 |
| E Ang (bond angle bend energy) | 21.979 | 42.065 |
| E Stb (stretch-bend energy) | 1.966 | 2.621 |
| E Oop (out-of-plane energy) | 0.295 | 0.417 |
| E Tor (torsion energy) | 11.316 | 15.691 |
| E Vdw (van der Waals energy) | 71.813 | 68.254 |
| E Ele (electrostatics energy) | 1.774 | 12.497 |
Then, a mixture of the (un)substituted 2-phenyl-4H-benzo[d][1,3]oxazin-4-one intermediates (0.1 mol) and hydrazine hydrate 40% (0.1 mol) was refluxed in ethyl alcohol for 4 h. The reaction was monitored with thin layer chromatography (TLC), then poured on crushed ice, filtered, and washed many times with distilled water, dried in an oven (70–80 °C), and then recrystallized from ethanol (96%) to give a fragile solid mass of 3-amino-2-(un)substituted phenylquinazolin-4(3H)-one.
Finally, a mixture of the 3-amino-2-(un)substituted phenylquinazolin-4(3H)-one intermediates (0.001 mol) and 2,3,4-trihydroxybenzaldehyde (0.001 mol) was refluxed in absolute ethanol with a few drops of glacial acetic acid for 5 h. The reaction was monitored with TLC until completion of the reaction was reached. Then, the solution was cooled and the solid was precipitated, filtered off, dried, and recrystallized from ethanol.
2.2. PDE 4 inhibition assay
PDE assay was performed according to the reported method and detailed results were discussed in our previous work.35,37 HT-29 cells were harvested from 90–95% confluent 10 cm dishes, and diluted in growth media. The appropriate concentrations of cells were prepared to add 1250 cells in a volume of 25 μL per well in tissue culture treated Griener 384-well microplates, and were then incubated overnight at 37 °C. Experimental compounds were previously diluted to a concentration of 50 mM in DMSO. Concentrated dosing solutions of twice the final concentration were prepared by dilution of the DMSO stocks by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 in growth media. Two-fold serial dilutions were then prepared using a medium containing 0.2% DMSO to maintain a constant concentration of the vehicle throughout the concentration range of the compound. Each of these was added in equal volume (25 μL), to four wells on the cell assay plate, along with the vehicle control. Cells were incubated with the compounds for a further 72 h.
:500 in growth media. Two-fold serial dilutions were then prepared using a medium containing 0.2% DMSO to maintain a constant concentration of the vehicle throughout the concentration range of the compound. Each of these was added in equal volume (25 μL), to four wells on the cell assay plate, along with the vehicle control. Cells were incubated with the compounds for a further 72 h.
At the end of the treatment period, assay plates were allowed to cool to room temperature for 10 min prior to the addition of 25 μL per well of Promega CellTiterGlo reagent, followed by an additional 10 min of incubation at room temperature. The resulting luminescence was quantitated using Molecular Devices Spectramax Paradigm apparatus. The percentage growth inhibition was calculated as follows, where 100% represents the viability of the vehicle treated control samples:

Thus, on average, the growth inhibition of the vehicle control samples equals zero.
The potency of compounds was determined using a non-linear dose response algorithm (4-parameter logistic fit) with the GraphPad Prism 5 software.
2.3. Docking study
To study the protein–ligand interactions, compounds were sketched using Marvin sketch powered by Chem-Axon and then transferred to a Molecular Operating Environment (MOE) platform to undergo energy optimization for each compound using the MMFF94x forcefield (with the gradient set to root mean square (RMS) 0.1 kcal mol−1). The crystallographic 3D protein structures of SARS-CoV-2 RdRp in a complex with cofactors (PDB ID: 6M71), the crystal structure of the SARS-CoV-2 spike receptor-binding domain bound to ACE2 (PDB ID: 6M0J) and the crystal structure of the SARS-CoV-2 main protease Mpro in a complex with Z1220452176 (PDB ID: 5R7Z) were obtained from the RSCB protein data bank (http://www.rscb.org). Molecular docking was conducted using the MOE 2020.0101 package. Visualization and generation of the 3D figures were performed using PyMOL 2.4 software.2.4. Molecular dynamics simulations
The conformational stability of the protein–ligand interactions was evaluated using molecular dynamics (MD) simulations analysis performed using MOE software. In this work, the MD of the ligands were studied using Nosé–Poincaré–Andersen (NPA), which is the most precise and the most sensitive. In MD calculations, an AMBER10:EHT force field, sphere shape, water as a solvent, six margins and the deletion of solvent molecules at a distance of greater than 4 Å were selected to optimize the system.2.5. Prediction of the pharmacokinetic properties and toxicological properties using ADMET
For the calculation of the pharmacokinetic properties of the compounds under study, online pkCSM pharmacokinetics prediction properties were used.38 The following properties, (i) absorption, Lipinski's rule of five, water solubility, Caco-2 permeability, intestinal absorption (human), skin permeability and P-glycoprotein interactions, (ii) distribution: VDss, Fu, Log BB and CNS permeability, (iii) metabolism and (iv) excretion were selected.Also, online pkCSM pharmacokinetics were used to predict the toxicity of the molecules, including skin sensitization, hepatotoxicity, etc. The results obtained were analyzed and compared with the reference values of the pkCSM pharmacokinetics prediction properties.39
3. Results and discussion
3.1. PDE 4 inhibition assay
Compounds Ia–d were screened in vitro for their inhibition of PDE 4B in our previous study using rolipram, a positive reference drug,37 and also, the mentioned compounds were evaluated for their anticancer activity.40 The screening was performed at 10 μM in triplicate and the results revealed that all compounds have significant inhibitory activity (% inhibition = 56, 85, and 89, respectively). Structure activity correlation showed that 2,3,4-trihydroxy moiety that served as a hydrogen bond donor may be responsible for the PDE 4B inhibitory activity of the compounds. Additionally, the presence of a 7-Cl at the quinazolinone ring in compound Id further improved the inhibitory effect (% inhibition = 89). These results may suggest the possibility of repurposing these synthesized quinazolinone derivatives as multi-target inhibitors against coronavirus.3.2. Docking results
The crystal structure of the SARS-CoV-2 spike receptor-binding domain bound with ACE2 (PDB ID: 6M0J) contains two chains, where chain A represents the angiotensin-converting enzyme 2 structure of homo sapiens and chain E is the spike receptor binding domain.46
Recently, a library of ligands comprising 30 phytochemicals from medicinal plants was prepared for docking. The best compound, silybin (flavonolignan), scored −10.572 kcal mol−1 with a Silybin-S spike glycoprotein complex forming hydrogen bonds with residues His34, Arg403, Tyr453, Ser494 and Lys353 present in this pocket.47

Compound Ic exhibits a minimum binding energy −16.8866 kcal mol−1 by targeting His34 and Ser49 via hydrogen bonding, π–π and π–H interactions (Table 2). Furthermore, compound Ia shows significant binding interactions to SGp chain E; a spike glycoprotein that plays a vital role in attaching the virus to the human receptor ACE2 cell membrane. Thus, blockage of SGp reduces the binding of the virus to the host. Also, compound Ib exhibits hydrogen bonding and hydrophobic interactions with His34 and Arg403 amino acid residues in the same binding site as the ligand (Fig. 5). These interactions seem to inhibit the functions of SGp, thus achieving blockage of the SGp functional protein suppresses the activity of binding to the host. It is worth mentioning that ACE2 is responsible for altering the sensitivity of both the substrate and the inhibitor by binding to the cavity of chain B. Additionally, both π–alkyl and van der Waals interactions can reduce the binding capabilities of the virus, which enables target compounds with antioxidant activity and thus more inhibitory action. Compound Ic has a methoxy group that can be hydrolyzed inside the body, therefore its metabolite was redocked to predict its inhibitory activity. The results revealed that the metabolite reserve the initial π–π interactions with the His34 amino acid residue. Moreover, it binds via a hydrogen bond donor with Ala387, with a binding energy of −12.1711 kcal mol−1.
| Compound | Crystal structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2 (PDB ID: 6M0J) | ||||
|---|---|---|---|---|---|
| Affinity, kcal mol−1 | Distance (in Å) from main residue | Functional group | Interaction | ||
| Ia | −15.8821 | 2.54 | His34 | Phenyl ring | Pi–cation |
| 2.14 | Arg403 | N of Schiff base | H–acceptor | ||
| 3.07 | Ala387 | Phenolic OH | H–donor | ||
| Ib | −16.3852 | 2.82 | His34 | Phenolic OH | H–donor |
| 2.93 | Arg403 | Quinazoline ring | Pi–cation | ||
| Ic | −16.8866 | 2.19 | His34 | Cyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
H–acceptor |
| 2.21 | His34 | N of Schiff base | H–acceptor | ||
| 3.86 | His34 | Phenyl ring | Pi–pi | ||
| 3.03 | Ser494 | Quinazoline ring | Pi–H | ||
| Id | −16.3282 | 2.91 | His34 | Phenolic OH | H–donor |
| 2.71 | Arg403 | N of Schiff base | H–acceptor | ||
| Ic metabolite | −12.1711 | 2.35 | His34 | Quinazoline ring | Pi–H |
| 2.24 | His34 | Quinazoline ring | Pi–cation | ||
| 3.02 | Ala387 | Phenolic OH | H–donor | ||
 | ||
| Fig. 5 (a) A 2D animated structure showing the binding interactions between compound Ib and SGp and (b) a 3D representation showing the position of compound Ib within the cavity of SGp. | ||
Interestingly, the Schiff moiety formed interactions with His34 or Arg403 residues as a hydrogen bond acceptor and this may serve as a useful pharmacophoric feature for designing more active agents.
In computational drug discovery, MD is a technique that sheds light on the allosteric binding site of the protein, and conformation of the ligand–protein complex. Also, it can simulate the conditions that are hard to perform under in wet experiments. Herein, compound Ic was subjected to MD simulations bound to the SARS-CoV-2 spike receptor-binding domain bound with ACE2. It can be seen from Fig. 6 that compound Ic retained its binding affinity and firmly bound to its respective binding site. The root mean square deviation (RMSD) value produced in the compound Ic complex was stable at 0.6 nm.
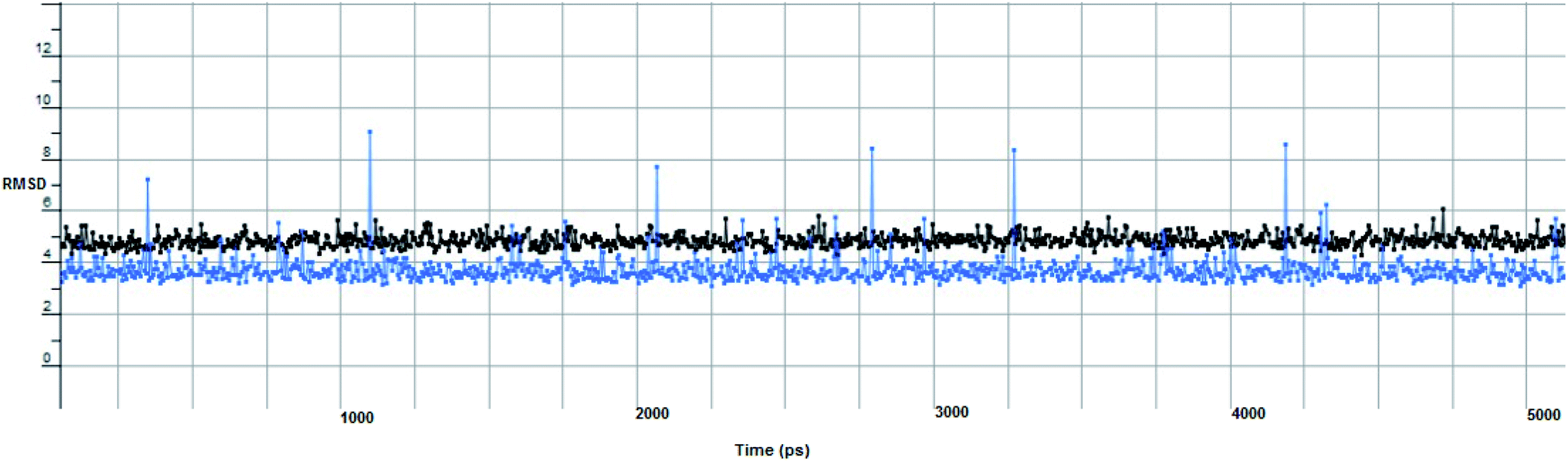 | ||
| Fig. 6 The RMSD curve from the molecular dynamics simulation of compound Ic. The x-axis represents the simulation time (in ps), while the y-axis represents the RMSD value (in nm). | ||
| Compound | Crystal structure of SARS-CoV-2 main protease in complex with Z1220452176 (PDB ID: 5R7Z) | ||||
|---|---|---|---|---|---|
| Affinity, kcal mol−1 | Distance (in Å) from main residue | Functional group | Interaction | ||
| Ia | −13.3909 | 2.42 | Glu166 | Phenolic OH | H–donor |
| 2.66 | His41 | Phenyl ring | H–pi | ||
| Ib | −12.7504 | 3.25 | Glu166 | Quinazoline ring | H–donor |
| 2.12 | Thr190 | Phenolic OH | Pi–H | ||
| Ic | −13.5962 | 2.44 | Glu166 | Phenolic OH | H–donor |
| 2.89 | Glu166 | Phenolic OH | H–donor | ||
| 3.17 | Gln189 | Quinazoline ring | Pi–H | ||
| 3.04 | Gln189 | Quinazoline ring | Pi–H | ||
| Id | −13.0212 | 2.83 | Glu166 | Phenolic OH | H–donor |
| 2.89 | Glu166 | Phenolic OH | H–donor | ||
| 2.99 | Gln189 | Phenyl ring | Pi–H | ||
| Ic metabolite | −10.3634 | 2.21 | Glu166 | Phenolic OH | H–donor |
| 3.12 | Gln189 | Phenyl ring | Pi–H | ||
Furthermore, two hydrogen bond donations to the main chain of Glu166 and π–H interactions to the Gln 189 amino acid, with a binding energy of −13.5962 kcal mol−1 (Fig. 7), were calculated. Also, the other tested compounds showed comparable binding interactions to main amino acids; Glu 166, Gln 189, His 41 and Thr 190; which seemed to be useful in the inhibition of the enzyme. So, a tri-hydroxy moiety alongside a quinazoline scaffold may serve as excellent binding features to inhibit this enzyme. Regarding the metabolite analogue of derivative Ic, it reserves the same hydrophobic and hydrogen bonding interaction with the Glu166 and Gln 189 amino acid residues, respectively. In the MD simulation, compound Ic retained its binding affinity and still firmly bound to the respective binding site. The average RMSD value of the ligand relative to the initial structure was around 0.7 Å and increased up to 3.9 Å without losing the key interactions and staying stable for the rest of the MD simulations (Fig. 8).
 | ||
| Fig. 7 (a) A 2D animated structure showing the binding interactions between compound Ic and Mpro and (b) a 3D representation showing the position of compound Ic within the cavity of Mpro. | ||
 | ||
| Fig. 8 The RMSD curve from the molecular dynamics simulation of compound Ic. The x-axis represents the simulation time (in ps), while the y-axis represents the RMSD value (in nm). | ||
The active site containing these amino acids was chosen using the site finder properties of MOE 2020.0101. All the target compounds bind at the RdRp functional sites, with a high docking score and the interaction details and the binding energy of the four quinazoline derivatives Ia–d at the active site of RdRp are summarized in Table 4.
The results show that all of the compounds have excellent affinity to the selected pocket, according to the affinity in kcal mol−1. The synthesized derivative Ib is encapsulated in the receptor cavity with a binding energy score of −13.8649 kcal mol−1. The binding site is located between the NiRAN domain and β-hairpin structure that polymerizes the 3′ end,49 and therefore it may interfere with the polymerizing activity. The mentioned derivative binds firmly to amino acids residues Thr556, Arg553, Arg555 (Fig. 9). Also, compound Id forms a non-covalent interaction with the divalent cationic residue of Asp618. The potent synthesized derivative was docked at the active site between the NSP12-NSP7 residues forms conventional hydrogen bonds to Asp452, and van der Waals interactions with residues Tyr619 and Lys621, including π–alkyl/π–πcontacts with residue Cys622 located in the RdRp tunnel structure. Notably, that tri-hydroxy moiety plays a significant role in these interactions as it forms hydrogen bond interactions with the Thr556 and Asp452 amino acid residues. All these interactions between the trihydroxy moiety and RdRp may serve as a crucial pharmacophore for further development of more potent target derivatives. Moreover, the interactions of the Ic metabolite analogue were examined and it was shown that it forms π–cation interactions with Arg 553 and Arg 555. The distances and energy bindings are presented in Table 4.
 | ||
| Fig. 9 (a) 2D animated structure showing the binding interactions between compound Ib and RdRp and (b) a 3D representation showing the position of compound Ib within the cavity of RdRp. | ||
| Compound | SARS-Cov-2 RNA-dependent RNA polymerase (PDB ID: 6M71) | ||||
|---|---|---|---|---|---|
| Affinity, kcal mol−1 | Distance (in Å) from main residue | Functional group | Interaction | ||
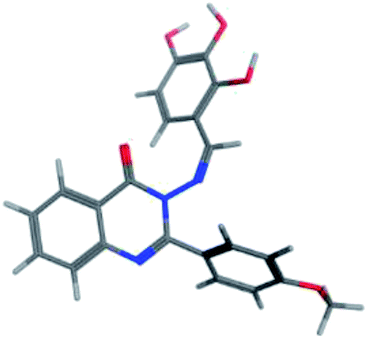
| Penciclovir | −14.5929 | 2.63 | Thr556 | –NH2 | H–donor |
| 2.71 | Thr556 | N of diazine | H–acceptor | ||
| 2.89 | Arg553 | Cyclic CO |
H–acceptor | ||
| 2.14 | Arg 555 | Cyclic CO |
H–acceptor | ||
| Arg555 | N of Diazole | H–acceptor | |||
| Ia | −14.0245 | 3.02 | Thr556 | Phenolic OH | H–donor |
| 2.99 | Thr556 | Phenolic OH | H–acceptor | ||
| 3.19 | Cys622 | Quinazoline ring | Pi–H | ||
| Ib | −13.8649 | 2.96 | Thr556 | Phenolic OH | H–donor |
| 2.70 | Thr556 | Phenolic OH | H–donor | ||
| 2.98 | Arg553 | Cyclic CO |
H–acceptor | ||
| 2.98 | Arg555 | Cyclic CO |
H–acceptor | ||
| Ic | −14.6145 | 2.93 | Cys622 | Cyclic CO |
H–acceptor |
| 2.94 | Asp452 | phenolic OH | H–donor | ||
| 3.09 | Lys621 | Quinazoline ring | Pi–H | ||
| 3.17 | Tyr619 | Quinazoline ring | Pi–H | ||
| Id | −12.9652 | 2.81 | Thr556 | Phenolic OH | H–donor |
| 2.73 | Lys621 | Quinazoline ring | H–donor | ||
| 3.18 | Asp618 | –Cl | Halogen bond | ||
| Ic metabolite | −14.5929 | 3.71 | Arg553 | –Phenyl ring | Pi–cation |
| 3.61 | Arg555 | –Phenyl ring | Pi–cation | ||
The proposed inhibitor Ic for SARS-CoV-2 identified as a result of molecular docking against RdRp was further investigated to determine its binding mode stability at the active site through MD simulations studies. The preliminary analysis performed on MD trajectory files was RMSD. The RMSD analysis was performed on the backbone atoms of the proteins (both in complex and free state) and average RMSD values obtained for ligand bound proteins (i.e. the inhibitor protein complex) were ∼0.8 Å, establishing their overall stability over the explored timescale, as shown in Fig. 10.
 | ||
| Fig. 10 The RMSD curve from the molecular dynamics simulation of compound Ic. The x-axis represents the simulation time (in ps), while the y-axis represents the RMSD value (in nm). | ||
3.3. Predication of the pharmacokinetic properties and toxicological properties via ADMET
In this study, the pharmacokinetic predictions of the quinazolinone Schiff base conjugates (Ia–d) were estimated. The results revealed that all the investigated molecules show significant values for oral absorption. Compound Ic shows the highest water solubility value among the other analogues. All of the evaluated compounds are predicted to have high cellular permeability, especially for intestinal cells (91.62–99.16%) (Table 5). Generally, it is known that an orally available molecule that satisfies both Lipinski's and Veber's rules has a balance between lipophilicity and hydrophilicity. As shown from Table 6, compounds Ia–d follow both Lipinski's and Veber's rules and thus qualify as possible drug-like molecules. Furthermore, the observed lipophilicities correlate negatively with the water solubility potentials of the target compound but have an association with the Caco2 permeability. This corresponds to the reported observation that there was no correlation between the lipophilicity and drug permeability measured using the human colon adenocarcinoma (Caco-2) cell line assay reported previously.53 Prediction of the distribution properties showed that compound Id shows the highest blood–brain barrier (BBB) permeability and unbound fraction among the other analogues (Table 5). The quinazoline derivatives were predicted to be substrates of P-glycoprotein, which is a member of the ATP-binding cassette transporter found primarily in epithelial cells. Thus, this can be explained by the ability of these compounds to modulate the physiological functions of P-glycoprotein in limiting the active uptake and distribution of these compounds. All of the synthesized compounds showed relatively high activity as they inhibit CYP2C19, CYP2C9, CYP2D6, and CYP3A4. This can be positively correlated to the lipophilicity of the compounds to metabolism related toxicity. These results show that these compounds could be involved in drug–drug interactions, and could initiate oxidative stress. It was observed that there were no remarkable difference in the parameters related to metabolism and excretion among the four analogues, except for the unsubstituted Ia and methoxy Ic derivatives, which showed a higher total clearance relative to the others. In conclusion, in this study, pkCSM software was used to predict the toxicological properties of the target compounds. Furthermore, this software has a system that performs predictions on the type of toxicity that a compound presents, such as mutagenicity, hepatotoxicity, cardiotoxicity, and skin sensitization. Herein, the bacterial mutagenic potential of the quinazoline derivatives through Ames toxicity testing showed that two analogues, namely Ic and Id, could be considered as non-mutagenic agents. Yet, the toxicities of all of the compounds in T. pyriformis are high. Also, the investigated compounds were predicted for one of the important parameters regarding toxicity, which is cardiotoxicity, in the form of human ether-a-go-go-related gene I (hERG I), which was found to be at an acceptable level.| Property | Compound Ia | Compound Ib | Compound Ic | Compound Id | |
|---|---|---|---|---|---|
| Absorption | Water solubility (log mol L−1) | −3.602 | −3.515 | −3.955 | −3.603 |
| Caco2 permeability (logPapp in 10−6 cm s−1) |
0.024 | 0.091 | 0.064 | 0.014 | |
| Intestinal absorption (%) | 91.625 | 97.956 | 94.345 | 99.161 | |
| Skin permeability (logKp) |
−2.735 | −2.735 | −2.735 | −2.735 | |
| P-Glycoprotein substrate | Yes | Yes | Yes | Yes | |
| P-Glycoprotein I | Yes | Yes | Yes | Yes | |
| P-Glycoprotein II | Yes | Yes | Yes | Yes | |
| Distribution | VDss (log L kg−1) | −1.021 | −0.92 | −1.179 | −0.831 |
| Fraction unbound (Fu) | 0.087 | 0.137 | 0 | 0.14 | |
| BBB permeability (logBB) |
−1.1 | −1.327 | −1.41 | −1.5 | |
| CNS permeability (logPS) |
−2.339 | −3.14 | −3.371 | −3.068 | |
| Metabolism | CYP2D6 substrate | No | No | No | No |
| CYP3A4 substrate | Yes | Yes | Yes | Yes | |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | |
| CYP2D6 inhibitor | Yes | Yes | No | Yes | |
| CYP3A4 inhibitor | No | No | Yes | Yes | |
| Excretion | Total clearance (log ml mim−1 kg−1) | 0.385 | 0.239 | 0.325 | −0.1 |
| Renal OCT2 substrate | No | No | No | No | |
| Toxicity | AMES toxicity | Yes | Yes | No | No |
| Max. tolerated dose (log mg kg−1 day−1) | 0.44 | 0.492 | 0.343 | 0.495 | |
| hERG I inhibitor | No | No | No | No | |
| hERG II inhibitor | Yes | Yes | Yes | Yes | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 2.627 | 2.513 | 2.706 | 2.533 | |
| Oral rat chronic toxicity (LOAEL) (mol kg−1 bw day−1) | 3.527 | 2.595 | 3.504 | 2.541 | |
| Hepatotoxicity | Yes | Yes | Yes | Yes | |
| Skin sensitisation | No | No | No | No | |
| T. pyriformis toxicity (log μg L−1) | 0.287 | 0.286 | 0.286 | 0.286 | |
| Minnow toxicity (log mM) | 0.155 | 0.002 | −0.98 | −0.515 | |
| Property | Compound Ia | Compound Ib | Compound Ic | Compound Id |
|---|---|---|---|---|
| logP |
3.06 | 3.20 | 3.07 | 3.85 |
| Molecular weight | 373.37 | 391.36 | 403.39 | 425.80 |
| NORTB | 3 | 3 | 4 | 3 |
| H-bond acceptors | 7 | 7 | 8 | 7 |
| H-bond donor | 3 | 3 | 3 | 3 |
| No. of Lipinski's rule violations | 0 | 0 | 0 | 0 |
| TPSA | 158.65 | 162.82 | 170.13 | 173.120 |
4. Conclusions
Designing effective small-molecule therapeutics promises to be the fastest therapeutic way to stem the tide of the COVID-19 pandemic. Therefore, a series of previously synthesized quinazoline Schiff base conjugates designed as PDE 4B inhibitors was in silico examined for possible ability to stop SARS-CoV-2 viral infection via several mechanisms. Docking studies results revealed that the Schiff moiety may inhibit the spike glycoprotein, while the tri-hydroxy group alongside a quinazoline ring may serve as a good scaffold to inhibit both Mpro and RdRp enzymes. Compound Ib was found to fit the substrate binding site and presents strong interactions with the mentioned targets. The binding stability of compound 1c inside the pocket of the mentioned proteins with time was further validated through molecular dynamic simulations involving root mean square deviation. Furthermore, ADMET properties were calculated and showed satisfactory pharmacokinetic and toxicological properties. Therefore, combining the docking results, ADMET predictions and the biological activity of the studied quinazoline conjugates as PDE 4B inhibitors, we suggest that these compounds can be further studied as suitable therapeutic treatments for COVID-19.Conflicts of interest
No potential conflicts of interest are reported by the authors.Acknowledgements
The authors thank Nahda University in Beni Suef (NUB) for supporting this work.References
- S. Jiang, Z. Shi, Y. Shu, J. Song, G. F. Gao, W. Tan and D. Guo, Lancet, 2020, 395, 949 CrossRef CAS.
- V. M. Corman, D. Muth, D. Niemeyer and C. Drosten, Adv. Virus Res., 2018, 100, 163 CAS.
- T. P. Velavan and C. G. Meyer, Trop. Med. Int. Health, 2020, 25, 278 CrossRef CAS PubMed.
- P. Liu, W. Chen and J. P. Chen, Viruses, 2019, 11, 979 CrossRef CAS PubMed.
- M. Hagar, H. A. Ahmed, G. Aljohani and O. A. Alhaddad, Int. J. Mol. Sci., 2020, 21, 3922 CrossRef CAS.
- D. Gentile, V. Patamia, A. Scala, M. T. Sciortino, A. Piperno and A. Rescifina, Mar. Drugs, 2020, 18, 225 CrossRef CAS PubMed.
- O. O. Olubiyi, M. Olagunju, M. Keutmann, J. Loschwitz and B. Strodel, Molecules, 2020, 25, 3193 CrossRef CAS PubMed.
- S. T. Ngo, N. Q. A. Pham, L. T. Le, D.-H. Pham and V. V. Vu, J. Chem. Inf. Model., 2020 DOI:10.1021/acs.jcim.0c00491.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409 CrossRef CAS.
- W. R. Ferraz, R. A. Gomes, A. L. S Novaes and G. H. G. Trossini, Future Med. Chem., 2020 DOI:10.4155/fmc-2020-0165.
- M. S. Yu, J. Lee, J. M. Lee, Y. Kim, Y. W. Chin, J. G. Jee and Y. J. Jeong, Bioorg. Med. Chem. Lett., 2012, 22, 4049 CrossRef CAS PubMed.
- Y. Kumar, H. Singh and C. N. Patel, J. Infect. Public Health, 2020 DOI:10.1016/j.jiph.2020.06.016.
- S. Shahinshavali, K. A. Hossain, A. V. D. N. Kumar, A. G. Reddy, D. Kolli, A. Nakhi, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2020, 152336 CrossRef CAS PubMed.
- R. Alexpandi, J. F. De Mesquita, S. K. Pandian and A. V. Ravi, Front. Microbiol., 2020, 11, 1796 CrossRef.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907 CrossRef CAS.
- M. Dalamaga, I. Karampela and C. S. Mantzoros, Metabolism, 2020, 109, 154282 CrossRef CAS.
- Z. Varga, A. J. Flammer and P. Steiger, Lancet, 2020, 395, 1417 CrossRef CAS.
- J. Luan, Y. Lu, X. Jin and L. Zhang, Biochem. Biophys. Res. Commun., 2020, 526, 165 CrossRef CAS PubMed.
- M. Gheblawi, K. Wang, A. Viveiros, Q. Nguyen, J. C. Zhong, A. J. Turner, M. K. Raizada, M. B. Grant and G. Y. Oudit, Circ. Res., 2020, 126, 1457 CrossRef.
- T. Hirschhorn and B. R. Stockwell, Free Radic. Biol. Med., 2019, 133, 130 CrossRef CAS PubMed.
- W. Liu, S. Zhang, S. Nekhai and S. Liu, Curr. Clin. Microbiol. Rep., 2020, 7, 13 CrossRef.
- D. TangI, P. ComishID and R. Kang, PLoS Pathog., 2020, 16, 1008536 CrossRef.
- D. C. Hall and H.-F. Ji, Trav. Med. Infect. Dis., 2020, 35, 101646 CrossRef.
- R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Cent. Sci., 2020, 6, 672 CrossRef CAS.
- A. Cavezzi, E. Troiani and S. Corrao, Clin. Pract., 2020, 10, 1271 CrossRef.
- J. E. Phillips, Front. Pharmacol., 2020, 11, 259 CrossRef CAS.
- H. Li, J. Zuo and W. Tang, Front. Pharmacol., 2018, 9, 1048 CrossRef CAS.
- A. Kodimuthali, S. S. L. Jabaris and M. Pal, J. Med. Chem., 2008, 51, 5471 CrossRef CAS.
- R. Sunke, R. Bankala, B. Thirupataiah, E. V. V. Shivaji Ramarao, J. S. Kumar, H. M. Doss, R. Medishetti, P. Kulkarni, R. K. Kapavarapu, M. Rasool, J. Mudgal, J. E. Mathew, G. G. Shenoy, K. V. L. Parsa and M. Pal, Eur. J. Med. Chem., 2019, 174, 198–215 CrossRef CAS PubMed.
- M. Dalamaga, I. Karampela and C. S. Mantzoros, Metabolism, 2020, 109, 154282 CrossRef CAS.
- S. Jo, H. Kim, S. Kim, D. H. Shin and M. S. Kim, Chem. Biol. Drug Des., 2019, 94, 2023 CrossRef CAS.
- S. Jo, S. Kim, D. H. Shin and M. S. Kim, J. Enzyme Inhib. Med. Chem., 2020, 35, 145 CrossRef CAS.
- S. Vardhan and S. K. Sahoo, Comput. Biol. Med., 2020, 124, 103936 CrossRef CAS PubMed.
- A. M. Sayed, A. R. Khattab, A. M. AboulMagd, H. M. Hassan, M. E. Rateb, H. Zaid and U. R. Abdelmohsen, RSC Adv., 2020, 10, 19790 RSC.
- H. M. Abdel-Rahman, M. Abdel-Aziz, J. C. Canzoneri, B. D. Gary and G. A. Piazza, Arch. Pharm., 2014, 347, 650 CrossRef CAS PubMed.
- K. S. Kumar, S. Ganguly, R. Veerasamy and E. De Clercq, Eur. J. Med. Chem., 2010, 45, 5474 CrossRef CAS PubMed.
- M. Mansour, M. T. El-Saadi, N. H. Amin, J. Canzoneri, A. Keeton, G. Piazza and H. M. Abdel-Rahman, Egypt. J. Chem., 2020 DOI:10.21608/ejchem.2020.28992.2624.
- Pharmacokinetic properties, http://biosig.unimelb.edu.au/pkcsm/prediction.
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066 CrossRef CAS PubMed.
- S. F. El-Menshawe, O. M. Sayed, H. A. Abou Taleb, M. A. Saweris, D. M. Zaher and H. A. Omar, J. Drug Deliv. Sci. Technol., 2020, 101569 CrossRef CAS.
- W. Li, M. J. Moore, N. Vasilieva, J. Sui, S. K. Wong, M. A. Berne, M. Somasundaran, J. L. Sullivan, K. Luzuriaga and T. C. Greenough, Nature, 2003, 426, 450 CrossRef CAS PubMed.
- A. J. Turner, J. A. Hiscox and N. M. Hooper, Trends Pharmacol. Sci., 2004, 25, 291 CrossRef CAS.
- N. E. Clarke and A. J. Turner, Int. J. Hypertens., 2012, 2012, 307315 Search PubMed.
- T. Hashimoto, T. Perlot, A. Rehman, J. Trichereau, H. Ishiguro, M. Paolino, V. Sigl, T. Hanada, R. Hanada and S. Lipinski, Nature, 2012, 487, 477 CrossRef CAS.
- A. Wu, Y. Peng, B. Huang, X. Ding, X. Wang, P. Niu, J. Meng, Z. Zhu, Z. Zhang and J. Wang, Cell Host Microbe, 2020, 27, 325 CrossRef CAS.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan and X. Wang, Nature, 2020, 581, 215 CrossRef CAS PubMed.
- A. C. Walls, Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesle, Cell, 2020, 180, 281 CrossRef PubMed.
- S. Ekins, M. Mottin, P. R. Ramos, B. K. Sousa, B. J. Neves, D. H. Foil and A. C. Puhl, Drug Discov. Today, 2020, 25, 928 CrossRef CAS PubMed.
- Y. Gao, L. Yan, Y. Huang, F. Liu, Y. Zhao, L. Cao and J. Ge, Science, 2020, 368, 779 CrossRef CAS PubMed.
- W. Hao, J. A. Wojdyla, R. Zhao, R. Han, R. Das, I. Zlatev, M. Manoharan, M. Wang and S. Cui, PLoS Pathog., 2017, 13, e1006474 CrossRef PubMed.
- R. N. Kirchdoerfer and A. B. Ward, Nat. Commun., 2019, 10, 2342 CrossRef PubMed.
- M. Ahmad, A. Dwivedy, R. Mariadasse, S. Tiwari, J. Jeyakanthan and B. K. Biswal, ACS Omega, 2020 DOI:10.31219/osf.io/fjnzc.
- M. Yazdanian, S. L. Glynn, J. L. Wright and A. Hawi, Pharm. Res., 1998, 15, 1490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |