
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous TiO2 anatase films for enhanced photocatalytic activity under UV and visible light†
Olga M. Ishchenko *abf,
Guillaume Lamblina,
Jérôme Guillota,
Ingrid C. Infantec,
Maël Guennoud,
Noureddine Adjerouda,
Ioana Fechetebgh,
Francois Garinb,
Philippe Tureke and
Damien Lenoble*a
*abf,
Guillaume Lamblina,
Jérôme Guillota,
Ingrid C. Infantec,
Maël Guennoud,
Noureddine Adjerouda,
Ioana Fechetebgh,
Francois Garinb,
Philippe Tureke and
Damien Lenoble*a
aLuxembourg Institute of Science and Technology (LIST), Materials Research and Technology (MRT), 41 Rue du Brill, L-4422 Belvaux, Luxembourg. E-mail: olgaishchenk@gmail.com; damien.lenoble@list.lu
bInstitut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé-ICPEES, UMR 7515 CNRS, Université de Strasbourg, 25 Rue Becquerel, 67087 Strasbourg Cedex 2, France
cInstitut des Nanotechnologies de Lyon, CNRS UMR 5270, ECL, INSA, UCBL, CPE, Villeurbanne, France
dDepartment of Physics and Materials Science, University of Luxembourg, 41 Rue du Brill, L-4422 Belvaux, Luxembourg
eLaboratoire POMAM, Institut de Chimie de Strasbourg, UMR 7177, France
fTE-OX, 2 Rue Jean Rostand, 91400 Orsay, France
gICD-LASMIS, Université de Technologie de Troyes, Antenne de Nogent, Pôle Technologique de Sud Champagne, 26, Rue Lavoisier, Nogent, France
hNogent International Center for CVD Innovation – NICCI, LRC-CEA-ICD-LASMIS, Université de Troyes-Antenne de Nogent, Pôle Technologique Sud Champagne, 26, Rue Lavoisier, 52800 Nogent, France
First published on 16th October 2020
Abstract
Mesoporous TiO2 films with enhanced photocatalytic activity in both UV and visible wavelength ranges were developed through a non-conventional atomic layer deposition (ALD) process at room temperature. Deposition at such a low temperature promotes the accumulation of by-products in the amorphous TiO2 films, caused by the incomplete hydrolysis of the TiCl4 precursor. The additional thermal annealing induces the fast recrystallisation of amorphous films, as well as an in situ acidic treatment of TiO2. The interplay between the deposition parameters, such as purge time, the amount of structural defects introduced and the enhancement of the photocatalytic properties from different mesoporous films clearly shows that our easily upscalable non-conventional ALD process is of great industrial interest for environmental remediation and other photocatalytic applications, such as hydrogen production.
1. Introduction
In the context of growing concerns about environmental pollution and a need for substitutes to fossil resources, the interest in photocatalysis has recently been boosted by the use of different nanotechnological approaches.1 Photocatalysis is considered one of the most promising sustainable technologies for the environmental remediation of pollutants or for hydrogen production (by photocatalytic water splitting). Both technologies rely on the light activation of photocatalysts to either degrade pollutants, like complex organic compounds in aqueous or gas phases into simple CO2 and H2O, or produce hydrogen by splitting water into O2 and H2.The first water splitting property of a photocatalyst was shown by Honda–Fujishima on titanium dioxide by a light irradiation at a wavelength lower than 400 nm, which corresponds to the TiO2 band gap at 3.0 eV.2 Since then, a large number of investigations have been published over the last four decades. However, the high potential of the photocatalytic approach still attracts the interest of the scientific community. Typically, different binary oxides, namely TiO2, ZnO and SnO2, have been identified as photocatalytically active, but also chemically stable, biocompatible, non-toxic and low-cost solutions.3,4
Therefore, a significant amount of work has been dedicated to the understanding and improvement of the photocatalytic performances where TiO2 remains the leading material for environmental remediation purposes. The major problem of these materials is still the limited visible light absorption due to their “wide” band gap. In fact, the activation of the above-cited photocatalytic materials is only allowed for light with an energy higher than the band gap; meaning that only 3–4% of the solar spectrum can be used effectively for the photocatalytic reaction. Photocatalytic activation under visible light has hence become a major research challenge, which is still under intensive investigation by the research community. In addition, it is known that the short lifetime of photogenerated carriers, due to their fast recombinations, also reduces the overall efficiency of photocatalysts.
Currently, major research efforts are based on different strategies targeting band-gap engineering via doping, mainly with carbon, phosphorus, nitrogen, or the introduction of other defects in the photocatalyst lattice, on the fabrication of heterostructures with overlapping band-gap levels (ZnO/SnO2), or on the coupling of the metal oxide photocatalysts with plasmonic nanoparticles. This last approach is very promising when considering the spectral shift of the light absorption. Indeed, under visible light irradiation, metal nanoparticles induce the phenomenon of surface electron oscillations, known as surface plasmon resonance.5 The downside of this plasmonic approach is the possible covering of the photocatalytic surface with metal nanoparticles, which reduces the specific surface area of metal oxides.
Oxygen vacancies (VO) are specific defects of the crystalline lattice of TiO2, which could be present either in bulk or on the surface and result in the reduction of Ti4+ to Ti3+.6 The presence of Ti3+ defects, also known as self-doping defects, significantly enhances TiO2 absorption in the visible range, creating in-gap states. It explains the important increase in research interest into non-stoichiometric TiO2 in recent years.7–10 For example, oxygen vacancies in the TiO2 lattice can be introduced by high vacuum annealing or by the use of a reduction agent during the growth of TiO2. However, important drawbacks of these methods are their high cost and their critical (even hazardous) experimental conditions.10 Recently, Sasinska and co-workers achieved a highly Ti3+ doped titania film by performing a hydrogen plasma post-treatment of amorphous TiO2 films deposited by ALD.11 The hydrogen treated TiO2 films demonstrate a significant improvement in photocurrent density and light absorption in the visible range. It was also underlined that hydrogenated TiO2 undergoes a band-gap edge shift down to 1.5 eV, which renders the materials black.12 This “black TiO2” was initially reported by Chen et al.13 and launched a new wave of interest in this material. It is also worth noting that many published strategies for “black TiO2” material synthesis rely on hydrothermal approaches for Ti3+ generation.7,9,10,14,15 Ti3+ self-doped TiO2 powders synthesised by the hydrothermal approach confirm the significant enhancement of photocatalytic performances in the visible range. Another way to improve TiO2 photocatalytic activity is with acidic treatments by HCl or H2SO4. Such treatments likely introduce oxygen vacancies that improve TiO2 photocatalytic activity under UV and visible light.16,17 Yuan et al. suggested that a TiO2 chlorinated surface retains a very low concentration of Cl− ions (below the XPS detection limit), which may also generate chlorine radicals under visible light and participate in the photocatalytic reaction. However, authors also confirm that the amount of oxygen vacancies increases in samples with a higher chlorine content.17 Xu et al.18 and Li et al.19 used a chlorine precursor to introduce lattice distortion in the TiO2 matrix during a high-pressure synthesis. In this case, the TiO2 lattice distortion caused by Ti3+ defects in TiO2 is expected to play a role in the visible light photocatalytic activation. The use of ethylene glycol in their synthesis process, which is known to be a reduction agent and to generate hydrogen under elevated temperature and pressure conditions, contributes to the hydrogenation of TiO2, the increase of Ti3+ defect concentration and the significant increase in the photocatalytic activity.
TiO2 with introduced structural defects (VO, Ti3+) has gain additional asset to the already well known application of TiO2 for energy conversion and storage devices such as dye-sensitized solar cells20–22 and batteries.23,24 TiO2 has been recognized for its electrochemical properties as an anode for Li-ion batteries (LIBs) with a theoretical capacity of 335 mA h g−1.23,25 However, in practice, the use of TiO2 as anode is limited due to low electronic conductivity and ionic diffusivity. TiO2 with structural defects addresses this problematic and offers significantly higher electronic conductivity due to the important concentration of VO defects.23,24,26 Moreover, a recent theoretical study predicts the insertion of Na+ ions as a function of the anatase surface termination facets: this opens new perspectives for the alternative to LIBs, namely Na-ion batteries (NIBs).27 In the scope of the growing interest to the TiO2 with structural defects, the low-cost and upscalable fabrication approaches would enable the industrial implementation of new concepts. In this publication, we report on a novel fabrication approach of mesoporous TiO2 anatase coatings with significantly enhanced photocatalytic activity in both UV and visible ranges, using a non-conventional ALD regime. The basic principle of this regime relies on an important accumulation of hydrolysis by-products in the amorphous TiO2 films upon their growth at room temperature. The post-deposition annealing then completes the hydrolysis reaction within the film, which releases an acidic medium (HCl). The relatively high temperature of the annealing process enhances the volatilisation of acidic hydrolysis by-products trapped in the amorphous films, while their concomitant degassing leads to the formation of a mesoporous anatase film structure with structural defects that favour photocatalysis. Such films showed a great performance for the photocatalytic degradation of a large family of pollutants in the UV and visible light ranges.
2. Experimental section
2.1. ALD synthesis details
The TiO2 film deposition was carried out at room temperature (RT) in a commercial ALD reactor, the TFS 200 from Beneq, using TiCl4 (≥99.995%, from Sigma-Aldrich) and water as precursors for TiO2 synthesis by ALD. The ALD experimental conditions were optimised for both precursors with a pulse time of 0.2 s and purge time of 5 s (short) and 30 s (long). The reactor was pressurised and maintained at 3.5 mbar with a continuous nitrogen flow. The in situ control on the mass increment was carried out using quartz crystal microbalances (QCM) (Neyco 6 MHz). The amorphous TiO2 films deposited on Si (100) (Siegert) and on Si (100) with 40 nm of thermally-grown SiO2 were annealed in air at 600 °C for 3 h using a Nabertherm furnace N7/H with a heating ramp of 10 °C min−1. Samples were left to cool down to room temperature in the furnace naturally after annealing.To control the reproducibility of the systems two series of samples were deposited in the same ALD experimental conditions except the reactor pressure, which was decreased from 3.5 mbar (series 1) to 2.5 mbar (series 2) due to the freshly performed pump maintenance. In the configuration of the ALD reactor, the pump extraction flow cannot be controlled by a valve.
2.2. Structural and chemical analysis
The crystallinity of samples was characterised by X-ray diffraction (XRD) on a D8 Discover diffractometer from Bruker with a copper X-ray source at Kα1–2, operating in parallel beam configuration at grazing incidence with an angle of incidence of 0.5°. X-ray reflectometry (XRR). Experiments were carried out using a PANalytical X'Pert Pro MPD instrument equipped with a copper X-ray source at Kα1–2 to precisely determine TiO2 film thicknesses and densities for further determination of each sample mass. Measurements were performed in parallel beam configuration using a fixed divergent slit of 0.05 mm and a PIXcel line detector.The sample morphology was studied by scanning electron microscopy (SEM) on a dual beam Helios Nanolab™ 650 from FEI and atomic force microscopy (AFM) on a commercial AFM (Innova, Bruker Inc.).
The chemical analysis on as-deposited and annealed samples was carried out by X-ray photoelectron spectrometry (XPS) on an Axis Ultra DLD, from Kratos Analytical Ltd., using an X-ray source (Al Kα monochromated, E = 1486.6 eV) at 150 W, a pass energy of 20 eV for narrow spectra and a step size of 0.1 eV. An argon beam was used to sputter the sample surface and obtain the chemical profile in the depth. The analysed area was 300 μm × 700 μm for surface analysis and 110 μm in diameter for depth profiles. The XPS spectra were calibrated by placing the main Ti 2p3/2 peak at 458.8 eV and applying a constant shift to the other peaks. The relative amount of titanium-bonded oxygen (O–Ti) on the surface was calculated by subtracting the possible oxygen–carbon (C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO) and SiO2 contributions determined from the C 1s and Si 2p fitted peaks, respectively, from the total oxygen O 1s signal. The absence of carbon throughout the in-depth profiles of the films allows us to define the (O–Ti) amount in the volume by subtracting the contribution corresponding to SiO2 only from the total oxygen O 1s signal.
O and O–CO) and SiO2 contributions determined from the C 1s and Si 2p fitted peaks, respectively, from the total oxygen O 1s signal. The absence of carbon throughout the in-depth profiles of the films allows us to define the (O–Ti) amount in the volume by subtracting the contribution corresponding to SiO2 only from the total oxygen O 1s signal.
2.3. Optical characterisations
Photoluminescence (PL) measurements were carried out on a Renishaw inVia confocal micro-Raman spectrometer. The PL spectra were acquired using a near UV laser excitation at 325 nm (7.8 mW) at room temperature.2.4. Photocatalytic tests
The validation of the photocatalytic activity of mesoporous samples was rapidly estimated by the photocatalytic degradation tests on organic dye molecules. The photocatalytic degradation tests were carried out using weakly powered lamps (UV lamp at 365 nm (8 W) and visible irradiation range lamp at 400–700 nm (8 W)) on methylene blue (MB), rhodamine B (RhB) and salicylic acid (SA) with an initial concentration of 5 mg L−1. Standard quartz 10 mm light path Hellma® fluorescence cuvettes were used as photocatalytic reactors. Samples were pre-cut in order to fit the cuvette side of ∼9 mm, placed along one side of the cuvette. Cuvettes were filled with 4 mL of pollutant solution and closed with an appropriated lid. One cuvette from the set underwent a blank test – control of the pollutant photodegradation without a photocatalyst. The negative time scale (−60 min up to 0 min) corresponds to measurements in the dark without irradiation to evaluate the pollutant adsorption–desorption phenomenon on the sample surface. The photocatalytic degradation corresponds to the positive time scale where the light was switched on. The control of the photocatalytic degradation kinetic was performed every 30–60 min by the UV-vis spectra acquisition and the intensity of the pollutant absorption peak. UV-vis measurements were performed on an UV-visible Infinite M1000 Pro spectrometer from TECAN. The principal absorption peak for methylene blue (MB) is at 666 nm, for rhodamine B (RhB) at 554 nm and for salicylic acid (SA) at 298 nm. The intensity of the absorbance spectra was measured twice on two points along the photocatalytic reactor and the average value was used to evaluate the pollutant concentration and the experimental error.To ensure a homogenous degradation, the cuvettes were placed on the mini-shaker 3D plate. One cuvette filled with MB and a mesoporous film sample was stored in the dark for 18 h and did not show any remarkable MB concentration modification. Therefore, the detected change in MB concentration was related to the photocatalytic degradation of the molecule. In the case of the visible range measurements, a thin polystyrene plate was used to filter eventual UV presence. The photocatalytic activity of the samples was compared with a 90 nm reference sample of mixed anatase and rutile phases, deposited by ALD using TiCl4 and H2O at 350 °C.28
3. Results and discussion
The non-conventional ALD process was performed at RT using two growth conditions with different purge times, 5 and 30 s. Usually, established mass evolution during the standard TiCl4 and water surface reaction at 300–350 °C goes as follows: a mass increment for the TiCl4 pulse and then achieving a stable value during the purge time (Fig. S1, ESI†).28,29 A further water supply releases HCl molecules that lower the recorded mass signal, which is subsequently stabilised during the second purge time. Contrary to the conventional ALD process, QCM mass curves recorded for the room temperature deposition presented an anomalous behaviour while the water pulse was supplied. Instead of the mass decrease due to the HCl release, we observed a mass increase during the pulse time, followed by a continuous mass decrease when the purge time did not attain a constant value, even after an exceedingly long purge time of 1800 s (Fig. 1b).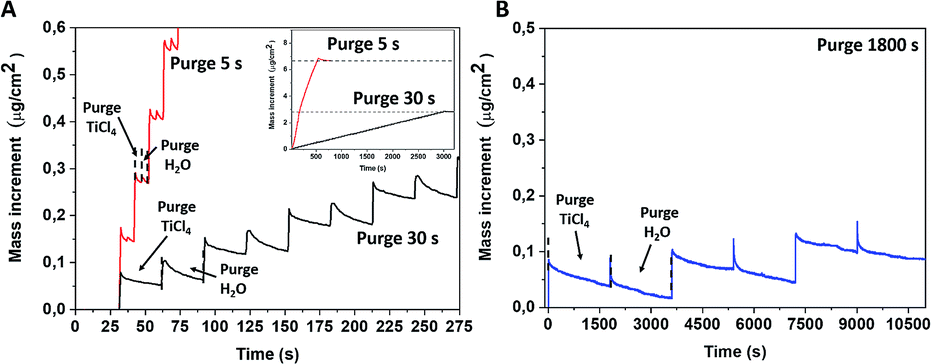 | ||
| Fig. 1 In situ QCM monitoring on the ALD mass increment recorded for depositions at RT for both precursors with purge time of 5 s and 30 s (A) (inset: full time range of the same experiments) and 1800 s (B). | ||
The average mass increments per cycle are strongly correlated with the purge time and were determined to be ∼0.14, 0.06 and 0.02 μg cm−2 for a purge time of 5 s, 30 s and 1800 s, respectively. The mass increment after 50 cycles was twice as large in the case of a 5 s purge time regime as for a 30 s purge time (inset of Fig. 1a). This led to different thicknesses of deposited amorphous films of 220 and 100 nm when 5 s and 30 s purge times were, respectively, used for an identical number of ALD cycles. This observation suggests that the unreacted or partly reacted precursors and by-products are likely accumulated within the film grown at room temperature and this may also correspond to the “condensation” regime from ALD fundamentals. Several theoretical studies have reported that the first half-reaction of TiCl4 and H2O is endothermic, meaning that the hydrolysis reaction at room temperature is not completed and thus the desorption of HCl is hampered.30–32 According to Gu and Tripp,33 the first half reaction of TiCl4 on isolated Si–OH groups at room temperature can be completed within a 10 s contact time, while the addition of water vapour led to the formation of the “titania-like” Ti(OH)xCl4−x compounds. Further ALD cycles led to TiCl4 polymerisation with adsorbed water.
This accumulated acidic medium in amorphous titania-like films can bring certain advantages to a post-deposition thermal treatment. The annealing process will induce the crystallisation of amorphous films, but it will also complete the hydrolysis reaction for non- or partly-reacted precursors accumulated in the films and release an acidic medium within the films. This allows an in situ acidic treatment to be performed at a high temperature, which is expected to improve titania photocatalytic properties by introducing structural defects. According to the results reported by Park et al.,16 Ti3+ defects could be introduced in TiO2 with a thermal treatment in an acidic medium (HCl, H2SO4). Therefore, we purposely performed the deposition in such a non-conventional regime followed by the post-deposition annealing at 600 °C and at atmospheric pressure to preserve the accumulated volatile by-products within the films prior to the thermal recrystallisation.
After the post-deposition annealing, the film thickness was reduced by 10–25%, down to 170 and 90 nm for the purge time regimes of 5 and 30 s, respectively. This observation is mainly due to the thermal shrinkage of the film, but could also be related to the desorption of volatile compounds. The previously studied annealing of amorphous films deposited at a higher temperature (100 °C) did not show such a significant reduction in thickness after the post-deposition annealing.28 The film crystallinity, characterised by XRD, evidenced the anatase phase formation in both annealed samples (Fig. S2, ESI†). The morphology of the samples, characterised by SEM and AFM, underwent a significant modification after annealing (Fig. 2, S3 and S4, ESI†). The thermal treatment at 600 °C induced the formation of a mesoporous film structure. This kind of structure is likely to be formed by degassing some volatile compounds from the amorphous films and/or thermal shrinking during annealing. From the SEM and AFM images, we notice that a sample grown with the longer purge time (30 s) presents lower porosity and roughness than films grown with shorter purge times.
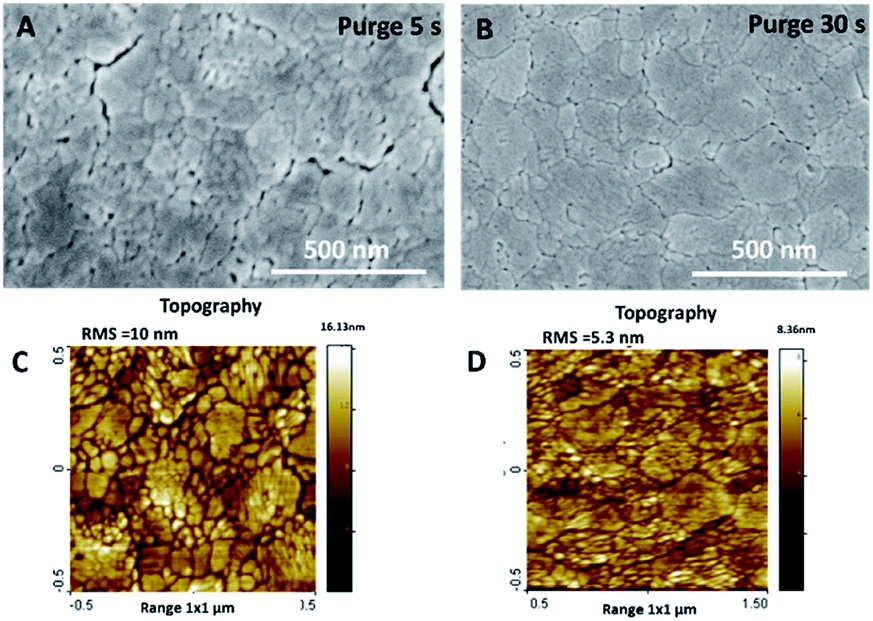 | ||
| Fig. 2 SEM and AFM topography images of annealed TiO2 films deposited with purge time 5 s (A and C) and 30 s (B and D). | ||
The chemical characterisation of the films was achieved by performing an XPS analysis on the amorphous and annealed films (Fig. 3). On the spectra acquired at the surface, the O 1s peaks are asymmetric and their initial position was shifted by 0.3 eV after annealing (Fig. 3a). The Ti 2p3/2 peak shape and position indicate that titanium is in its Ti4+ oxidation state in all samples, without any significant contribution of Ti3+, even when chlorine is detected before annealing (Fig. 3b). It should be noted that chlorine is no longer detected at the surface of the samples after the annealing process (Fig. 3c).
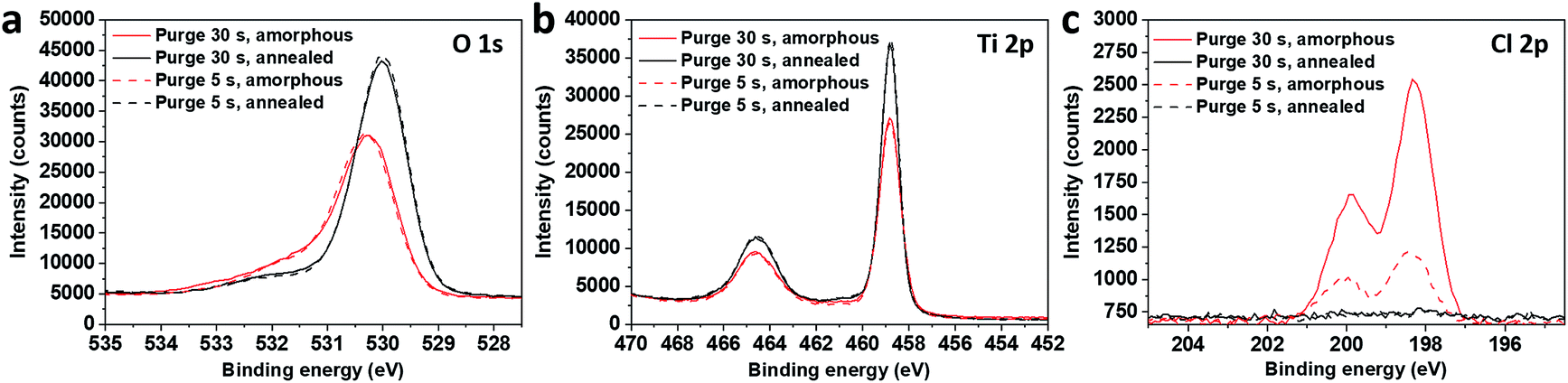 | ||
| Fig. 3 XPS O 1s (a), Ti 2p (b) and Cl 2p (c) spectra on surface of amorphous and annealed samples, grown in short (5 s) and long (30 s) purge time regimes. | ||
The binding energy gap ΔE (Ti 2p3/2, O 1s) was found to be 71.5 eV and 71.2 eV on amorphous and annealed films, respectively. According to Park and Shin,16 the binding energy difference (ΔE) between Ti 2p3/2 and O 1s for bare TiO2 and HCl-treated TiO2 was in the same range at 71.25 eV, that is, in agreement with our results. The authors performed their spectra calibration on the C 1s contribution and found an important shift of 0.75 eV for both the O 1s and Ti 2p3/2 peaks after the acidic treatments (HCl and H2SO4) of their TiO2 samples, which was attributed to the TiO2 protonation. In our case, the calibration was performed on Ti 2p3/2 peaks and the higher value of O 1s binding energy after the annealing process is clearly not related to an additional differential charging effect between the carbonaceous species and the underlying TiOx matrix. The phenomenon is due to changes in the chemical environment of Ti and O, which potentially underwent protonation and chlorine removal from the matrix.
The chemical composition was further investigated in the films volume by sputtering the samples with a monoatomic argon beam sputtering. The relative elemental ratio [(O–Ti)/Ti] on the surface and in the volume was found to be between 1.8 and 2, respectively (Table 1). The presence of chlorine was detected only in the amorphous samples fabricated by both deposition processes, and it was completely removed by annealing (Fig. 4c, d, g and h), quantified by the (Cl/Ti) ratio in Table 1. The depth profiles also confirm the difference in the film thickness: the film grown in the long purge time regime (30 s) was sputtered away faster than the film grown with the short purge time regime.
| Sample | (O–Ti)/Ti | Cl/Ti | (O–Ti) | OH | ||
|---|---|---|---|---|---|---|
| Purge time 30 s | Surface | Amorphous | 2 | 0.2 | 42 | 12 |
| Annealed | 1.9 | 0 | 51 | 8 | ||
| Volume | Amorphous | 1.8 | 0.1 | 63 | 15 | |
| Annealed | 1.8 | 0 | 65 | 17 | ||
| Purge time 5 s | Surface | Amorphous | 1.9 | 0 | 43 | 13 |
| Annealed | 2 | 0 | 52 | 8 | ||
| Volume | Amorphous | 1.9 | 0.2 | 63 | 14 | |
| Annealed | 1.8 | 0 | 65 | 17 | ||
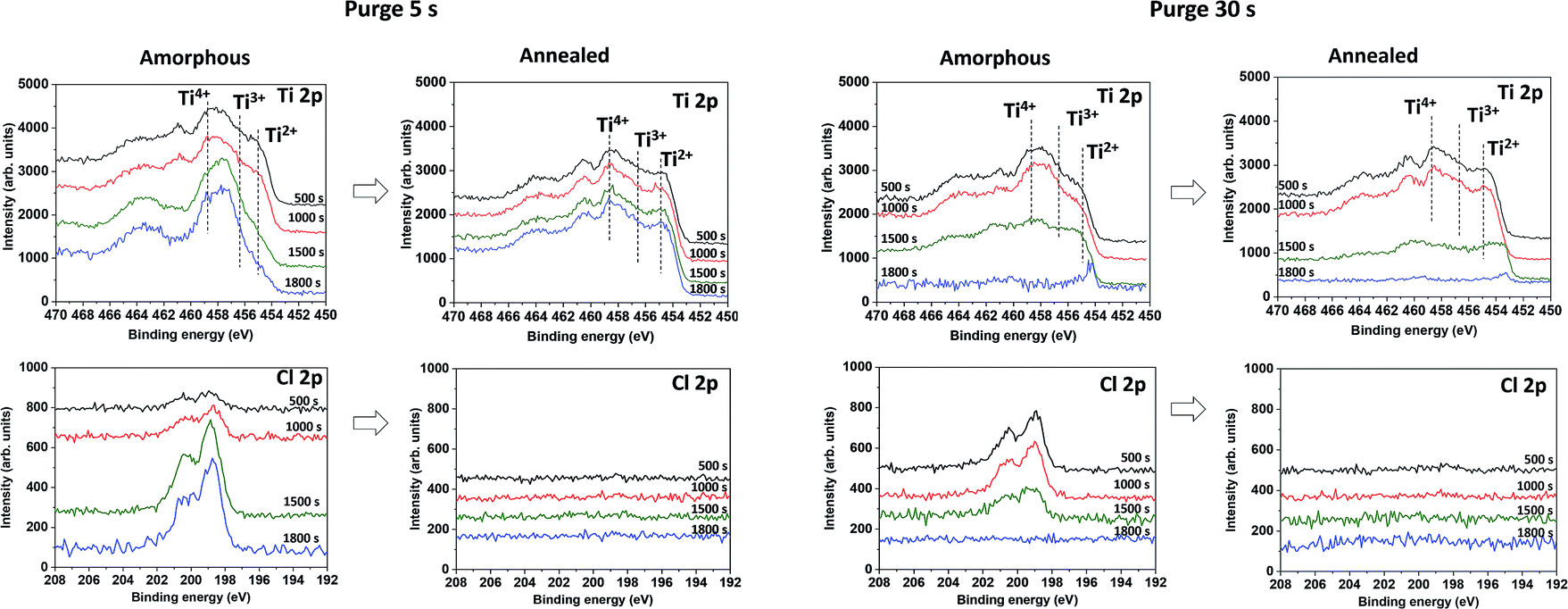 | ||
| Fig. 4 In depth XPS Ti 2p and Cl 2p spectra on amorphous and annealed samples, grown in short (5 s) and long (30 s) purge time regimes at RT after different sputtering times. | ||
The XPS spectra of sputtered amorphous samples grown with the long purge time (30 s) show a homogeneous decrease of the chlorine contribution in the volume (Fig. 4g). For samples grown with a short purge time, the presence of chlorine was lower on the surface, but increased with depth from 2 to 6% (Fig. 4c). The Ti 2p peaks in the depth profile of oxides are known to be modified by Ar+ beam sputtering.34 In TiO2, the Ti 2p peak shape changes due to the partial reduction of Ti4+ under the Ar beam into Ti3+ and Ti2+. Moreover, the typical relative atomic ratio [(O–Ti)/Ti] in a reduced TiO2 standard reference is in the range of 1.3–1.6 due to the preferential sputtering of oxygen atoms.34–37 Interestingly, in our case, the samples studied demonstrate an almost constant atomic ratio [(O–Ti)/Ti] close to 2, making it clear that the films are not constituted by a pure TiO2 matrix. Furthermore, within the depth profiles of the amorphous samples grown in the short purge-time (5 s) process, an evolution of the Ti 2p peak shape was observed for the 1500 s and 1800 s sputtering times. From a broad peak due to the Ti4+, Ti3+, Ti2+ contributions, the Ti 2p spectra shape is modified to a sharper peak, mainly due to the decrease of the Ti2+ component and a predominant contribution at 457.8 eV (Fig. 4a). The noticeable increase of chlorine and non-reduced Ti4+ after sputtering indicates the presence of another additional compound. Therefore, in-depth profiles of amorphous films correspond to the mixture of TiOx and another titanium-based compound, where Ti is not in a pure oxide phase, likely TiCl4−x(OH)x. Moreover, the amount of the last compound is significantly predominant in the volume of the films grown in the short purge time regime. Scheme 1 summarises the XPS results on the chemical composition difference in films grown in two regimes, before and after the annealing process.
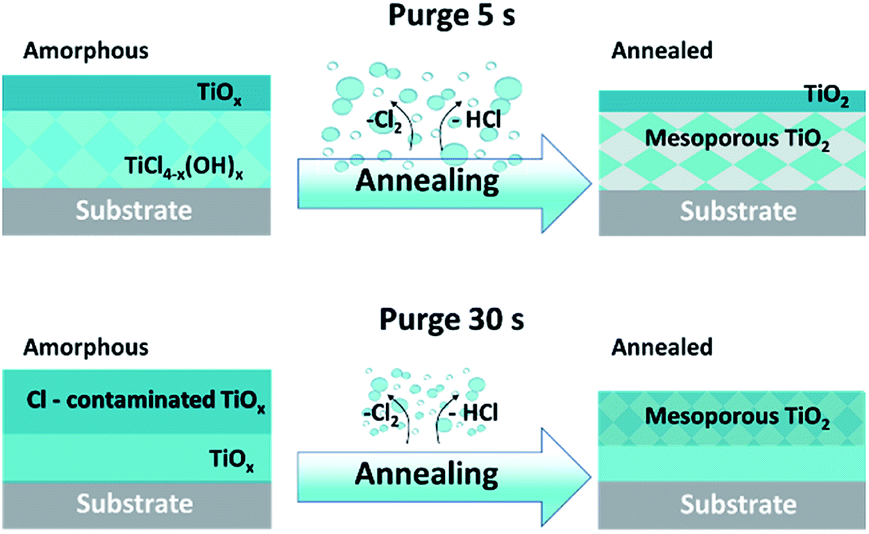 | ||
| Scheme 1 Schematic illustration of the evolution of films composition grown in two purge time regimes, amorphous and after annealing. | ||
The fine deconvolution of the O 1s peaks on the surface (Fig. S5, ESI†) was performed by considering three components, corresponding to (i) the titanium oxide (O–Ti), (ii) hydroxides and (OC) carbonaceous species and (iii) adsorbed water and (O–C) carbonaceous species. The amounts of (O–C) and (OC) carbonaceous species were estimated from the C 1s peak fit (Fig. S6, ESI†). The corresponding percentages of the presence of the two main oxygen components, O–Ti and OH, are included in Table 1. Thus, the quantity of hydroxyl groups present at the surface of the amorphous samples significantly decreases after annealing. The deconvolution of the O 1s spectra collected from the surface and in the film volume clearly shows that annealed films are more hydroxylated in the volume than on the surface (Table 1). Furthermore, the amount of hydroxides is similar in the bulk of amorphous and annealed films. However, the quantity of hydroxides found in the bulk can be controlled by the in situ adsorption of –OH groups to stabilise the dandling bonds created during the sputtering. The quantitative data of the chemical composition on the surface and in the volume of amorphous and annealed samples are summarised in Tables 1 and S1, ESI.† Whereas no change was observed in the Ti 2p spectra, the O 1s peak was systematically shifted towards higher binding energy (+0.3 eV) after annealing, and the shoulder close to 532 eV, attributed to the hydroxylation of TiO2 (Ti–OH), was decreased.13,38,39 Similar results were reported by Chen and co-workers for highly hydrogenated black TiO2 and by Wang and co-workers for hydrogen-treated rutile nanowires.13,39
For black TiO2, Chen et al.13 noted two contributions in the O 1s peak at 530 to 530.9 eV without any modification of the Ti 2p spectra, and Wang et al.39 reported these peak positions at 530.4 and 532 eV, respectively. The authors claimed that the hydrogenation of TiO2 by H2 treatment induces a significant disorder of the crystalline structure with, for instance, the creation of oxygen vacancies or the hydroxylation of the dangling bonds.13 These structural defects (mainly VO) have a significant impact on the electronic properties via the creation of inter-band gap energy levels. Theoretical predictions performed by Chen et al. confirm that disordered hydrogenated-TiO2 crystals exhibit mid-gap electronic states at 1.8 eV and high-energy states at 3 eV, leading to a band gap narrowing. Such a band gap modification allows the hydrogenated TiO2 to be photoactive in both UV and visible ranges. Thus, hydrogenated TiO2 was intensively investigated and demonstrated a significant enhancement of its photocatalytic activity.13,40,41 Such a significant improvement is still not fully understood. However, all reports agree that hydrogenation leads to a formation of a surface-disordered “shell” layer on TiO2 nanograins, which creates additional inter-band gap energy levels that enhance the absorption in the visible light range.42 Lu and co-workers43 found that a similar layer was formed on classical TiO2 nanoparticles during the water splitting process under UV irradiation. The thickness of this layer depends on the duration of the UV exposure and clearly plays a role in H2 production. The authors followed the formation of the self-hydrogenated surface layer by in situ TEM measurements, while ex situ measurements on the same dried sample show the reconstruction of the crystalline surface layer, but with a lower oxygen content (Ti2O3). The analogy between the self-hydrogenated surface layer and the surface-disordered layer due to hydrogenation confirms their significant role in the photocatalytic properties.
In the present films grown in non-conventional ALD regime, the XPS analysis before and after thermal annealing did not detect any significant presence of Ti3+ defects on the films' surface. However, the volume characterisation of annealed films is puzzled by the preferential oxygen sputtering. With respect to the volume characterisation of the amorphous films, the important amount of chlorine detected in the film grown in the short purge time regime could indirectly confirm the introduction of the structural defects in the annealed TiO2. Moreover, the similarity of our results with data on the hydrogenated TiO2 reported in the literature suggests that our non-conventional ALD process could lead to the fabrication of mesoporous thin layers with introduced lattice defects and oxygen vacancies in the film volume. For a better insight of inter-band gap states in mesoporous TiO2 films, we performed photoluminescence (PL) measurements. The recorded PL spectra of mesoporous TiO2 films at room temperature are presented in Fig. 5. The PL peak intensity of TiO2 films deposited on Si varies with the film thickness, and the peak wavelength is blue-shifted for the thickest film deposited with a 5 s purge time. The samples have a broad PL response with specific contributions (Fig. 5B) at ∼530 nm (2.34 eV, green), ∼585 nm (2.11 eV, yellow) and ∼680 nm (1.82 eV, red). For samples grown with a 5 s purge time, we note the large green contribution to the spectra compared to the lower energy ones and against the PL-response from the 30 s purge time film.
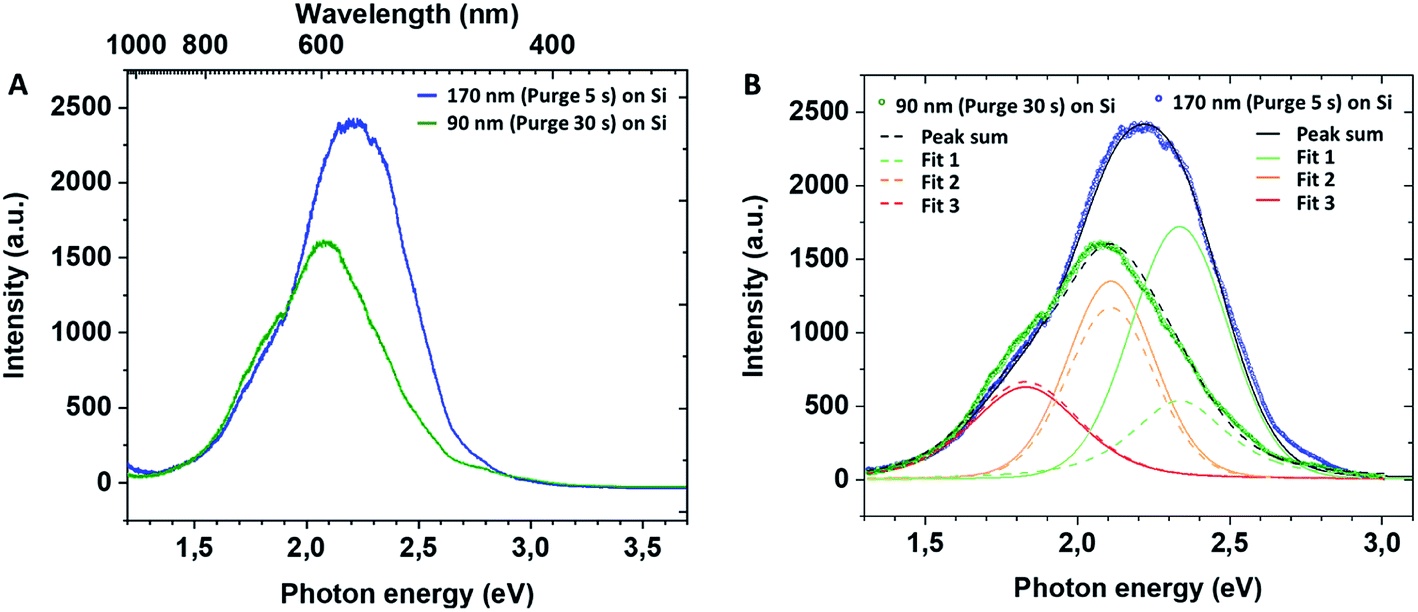 | ||
| Fig. 5 PL spectra of mesoporous TiO2 films (A) and their deconvolution (B). | ||
As a signature of the photo-generation of carriers and their radiative recombination, PL profiles provide specific information related to the localised photo-active electronic states associated with defects by the emission of light at sub-band gap levels.44 McHale and Knorr demonstrated that the anatase TiO2 PL spectra include the superposition of two types of radiative recombinations: one of the mobile electrons from the conduction band and shallow traps (type 1 green PL), another one from trapped electrons on defects to valence band holes (type 2 red PL):
| Type 1 (green PL): eCB− + htrap+ → hνgreen |
| Type 2 (red PL): hVB+ + etrap− → hνred |
Later, Jin et al.45 evidenced a correlation between the presence of red (600 nm) and green (515 nm) PL bands with specific defects. The green band (515 nm) is related to oxygen vacancies, while the red band (600 nm) indicates under-coordinated Ti3+ ions. These reported results (Fig. 6) concur with our PL profile features, highlighting that similar point defects and chemical doping of mesoporous films could be at the origin of the PL spectra observed.
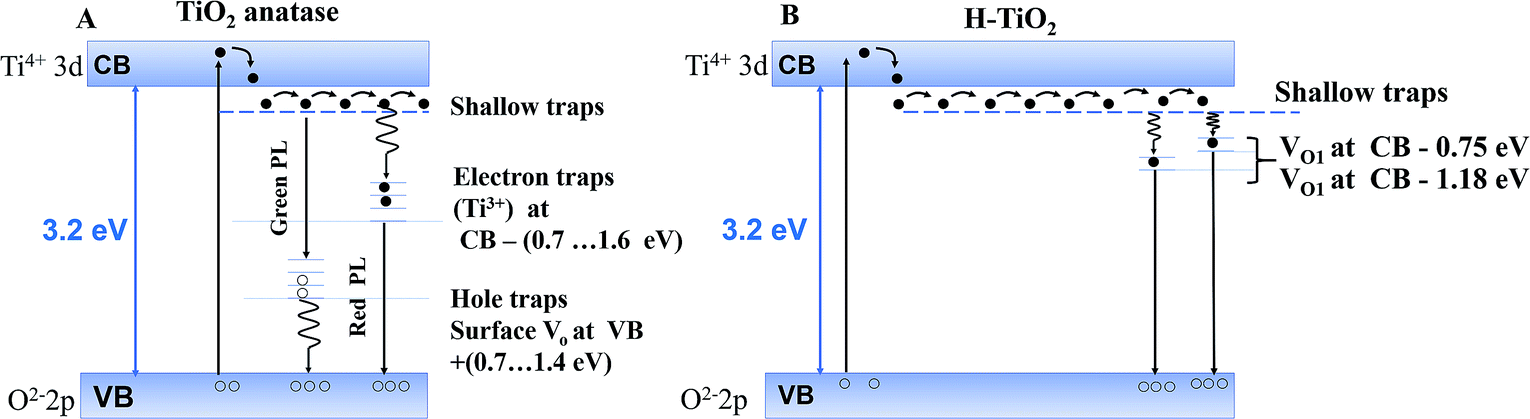 | ||
| Fig. 6 Models for photoluminescence from the electronic transition of trap states (A) for anatase reproduced from Jin et al.45 and (B) for hydrogenated TiO2 reproduced from Wang et al.39 | ||
In our case, PL profiles (Fig. 5) evidence defects contributing to the radiative recombination of carriers at ∼530 nm (2.34 eV), ∼585 nm (2.11 eV) and ∼680 nm (1.82 eV). Therefore, we propose the following interpretation for our PL signals: (i) inter-band gap recombination states situated at 0.86 eV below the CB correspond to oxygen vacancies and are found to be in-between values reported by Wang at al.39 (Fig. 6B), (ii) the signal at 1.09 eV below the conduction band may be attributed either to oxygen vacancies39 or Ti3+ states45 and (iii) the signal at 1.38 eV below the conduction band may be specifically attributed to the H–TiO2 mid-gap states.
3.1. Photocatalytic properties of mesoporous TiO2 films
The investigation of the photocatalytic activity of mesoporous TiO2 anatase films was performed on 170 and 90 nm thick mesoporous samples deposited on silicon substrates. The photocatalytic degradations of MB under UV irradiation by the mesoporous TiO2 films deposited on Si substrates were compared with a 90 nm thick reference TiO2 sample grown in the conventional ALD regime at 350 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 (Fig. 7). TiO2 films deposited at room temperature demonstrate significantly higher photocatalytic activity compared to the reference film, with a photocatalytic degradation constant of up to 10 times higher (Fig. 7 and Table 2). The films grown with a purge time of 5 s are more photocatalytically active than films grown with a 30 s purge time. In a first approximation, this improvement of the degradation rate for the short purge time can be related to the higher film thickness (XRR data presented on Fig. S7 and Table S2, ESI†). However, the normalisation of the photocatalytic degradation constant per mass unit confirms higher photocatalytic activity of films grown with short purge time regimes. These results are in agreement with the PL measurements discussed above and suggest that the concentration of structural defects is more important on samples deposited in a short purge time regime and could be correlated with chlorine-concentration detected in amorphous films.
28 (Fig. 7). TiO2 films deposited at room temperature demonstrate significantly higher photocatalytic activity compared to the reference film, with a photocatalytic degradation constant of up to 10 times higher (Fig. 7 and Table 2). The films grown with a purge time of 5 s are more photocatalytically active than films grown with a 30 s purge time. In a first approximation, this improvement of the degradation rate for the short purge time can be related to the higher film thickness (XRR data presented on Fig. S7 and Table S2, ESI†). However, the normalisation of the photocatalytic degradation constant per mass unit confirms higher photocatalytic activity of films grown with short purge time regimes. These results are in agreement with the PL measurements discussed above and suggest that the concentration of structural defects is more important on samples deposited in a short purge time regime and could be correlated with chlorine-concentration detected in amorphous films.
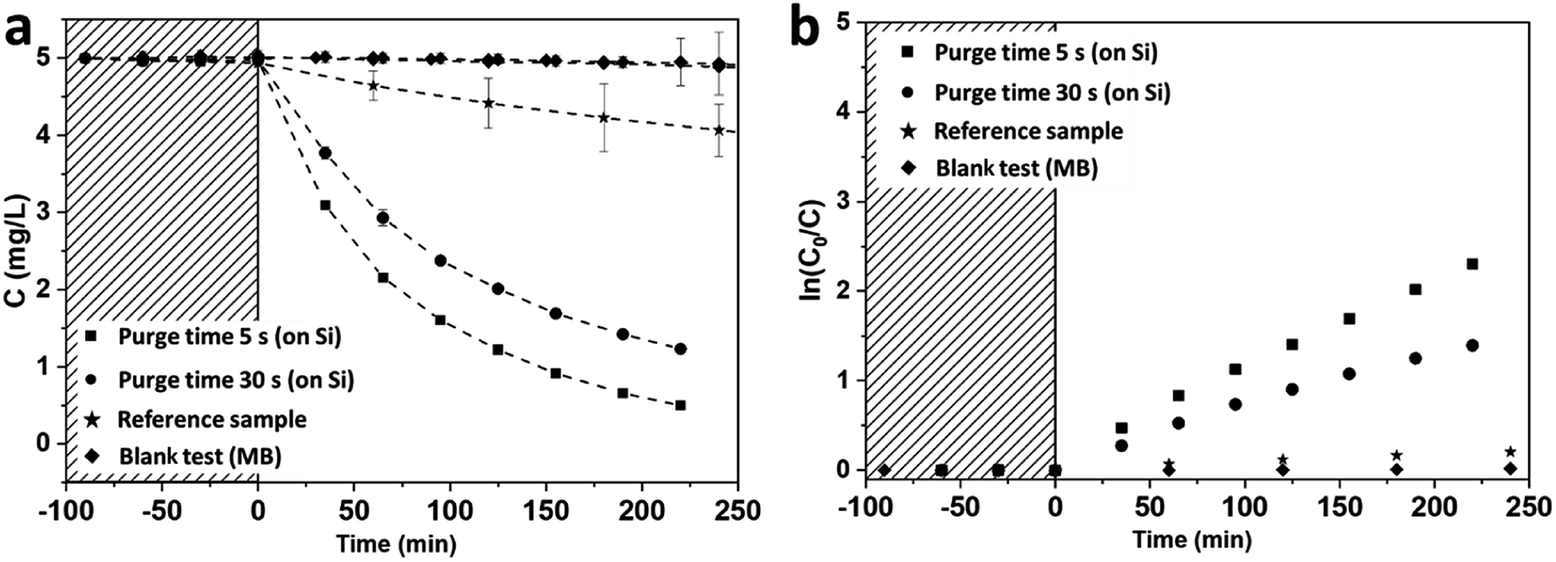 | ||
| Fig. 7 Photocatalytic degradation of MB in UV range on TiO2 films grown on Si(100) with native silicon oxide (a); associated kinetics of MB degradation (b). | ||
| kr, ×10−3 min−1 | kr, normalised by surface unit, ×10−4 min−1 cm−2 | kr, normalised by mass unit, min−1 g−1 | |
|---|---|---|---|
| Purge 5 s | 10.5 | 50.9 | 92.6 |
| Purge 30 s | 4.7 | 15.7 | 53.4 |
| Reference sample | 0.8 | 2.8 | 9.7 |
The degradation constants normalised per surface unit are significantly higher for our mesoporous hydrogenated films than values reported in the literature for pure TiO2 mesoporous films. For instance, the degradation constant obtained using similar photocatalytic test conditions on hierarchically ordered macro–mesoporous TiO2 films is 20.5 × 10−4 min−1 cm−2 as demonstrated by Du and co-workers46 while our degradation constant (50.9 × 10−4 min−1 cm−2) is more than twice their reported value.
In the visible range, both types of mesoporous TiO2 samples show that the photocatalytic degradation of BM is higher than its photodegradation of the blank sample. Samples grown with a purge time of 5 s demonstrate an improvement in the photocatalytic degradation rate compared to samples grown with a 30 s purge time (Fig. 8).
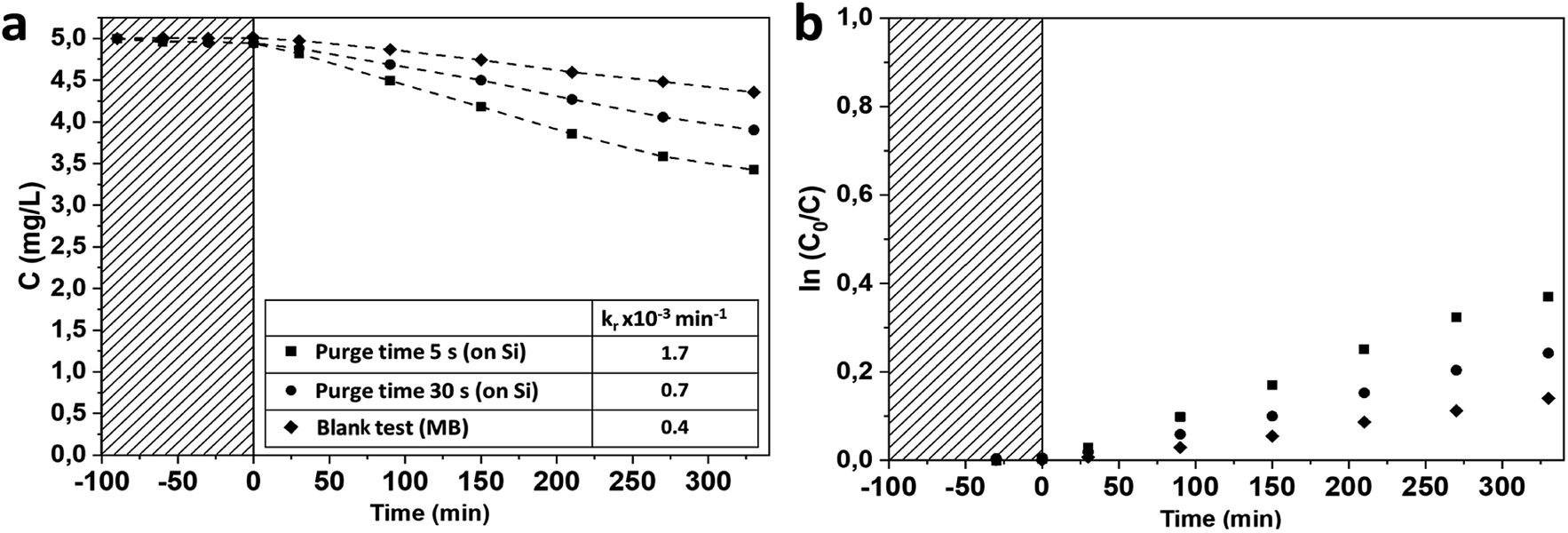 | ||
| Fig. 8 (a) Photocatalytic degradation rate of MB in visible range (400–700 nm) on TiO2 films grown on Si(100) native silicon oxide and blank test; inset table of the photocatalytic degradation constant; (b) associated kinetics of MB degradation. | ||
These results of photocatalytic activity corroborate the physico-chemical characterisations reported above and suggest that our mesoporous TiO2 films feature specific oxygen vacancies and Ti3+ coordination that likely improve the activity in both the UV and visible ranges. Due to the low temperature deposition in the non-conventional ALD process, the incorporation of chlorine species such as TiCl4−x(OH)x into amorphous films is promoted. The HCl degassing and potentially its thermal decomposition at 600 °C are likely to generate the formation of numerous structural defects.
The mesoporous TiO2 films were first grown on Si(100) substrates, however SiO2 is known to be a better substrate for the carriers' separation, which potentially increases the photocatalytic efficiently.47,48 Therefore, we also performed the TiO2 deposition on 40 nm thermally oxidised SiO2 (Si/SiO2) substrates, using only the short purge time regime (5 s). The structural and morphological characterisations of these samples were similar to the ones deposited on Si substrates with a native SiO2 layer (Fig. S8, ESI†). The photocatalytic activity measured in both ranges (UV and visible) on samples with a 40 nm buffer SiO2 layer was unexpectedly higher than that presented above for Si substrates (Fig. S9 and Table S3, ESI†). The reason for such an exaltation of photocatalytic properties is still unclear and requires further additional investigations. However, PL spectra measured on samples deposited in a 5 s purge time regime on Si/SiO2 substrates reveal the remarkable increase of the PL intensity for the identical thickness of TiO2 as for samples deposited on the Si substrate. The deconvolution of PL spectra (Fig. S10, ESI†) show the significant contribution at 2.11 eV that could be attributed to both oxygen vacancies and Ti3+ defects. Therefore, the role of the substrate reveals its importance. In our case, it could be related to film hydrogenation upon thermal annealing. The high quality thermal silica layer can prevent hydrogen diffusion from the film to the Si substrate49,50 and thus increase its impact in terms of structural defect introduction in the TiO2 film. This suggestion requires further investigations that lie outside the scope of the present study. The surface saturation (or surface deactivation) of the mesoporous sample deposited on the Si/SiO2 substrate was controlled by the cycling of the MB photocatalytic degradation under UV irradiation. After the fourth run of complete degradation, the MB degradation rate was identical (Fig. S11, ESI†). A known particular affinity of the MB to the TiO2 surface facilitates its degradation51 while an efficient photocatalytic system degrades multiple molecule chemistries. Therefore, we also exposed mesoporous TiO2 samples (series 2) deposited on Si/SiO2 substrates to other relevant molecules, such as rhodamine B (RhB) and salicylic acid (SA), at the same time as MB. The degradation rate in the UV range was found to be almost identical for the three selected molecules. In our case, the photocatalytic degradation under UV light was not dependent on the molecular affinity towards our photocatalyst surface (Fig. 9, Table 3). This feature is of high interest for depolluting water typically characterised by a mixture of polluting molecules having a significantly different chemical nature, steric volume, polarity, and chemical terminations. For instance, when considering emerging photocatalysts such as Bi2WO6 nanosheet with a positively charged surface, the MB and RhB were found to have higher adsorption, while neutrally-charged molecules like SA are hardly adsorbed (and thus less degraded) on such a surface.52 In our case, a slightly higher degradation constant for SA was measured compared to the two other molecules.
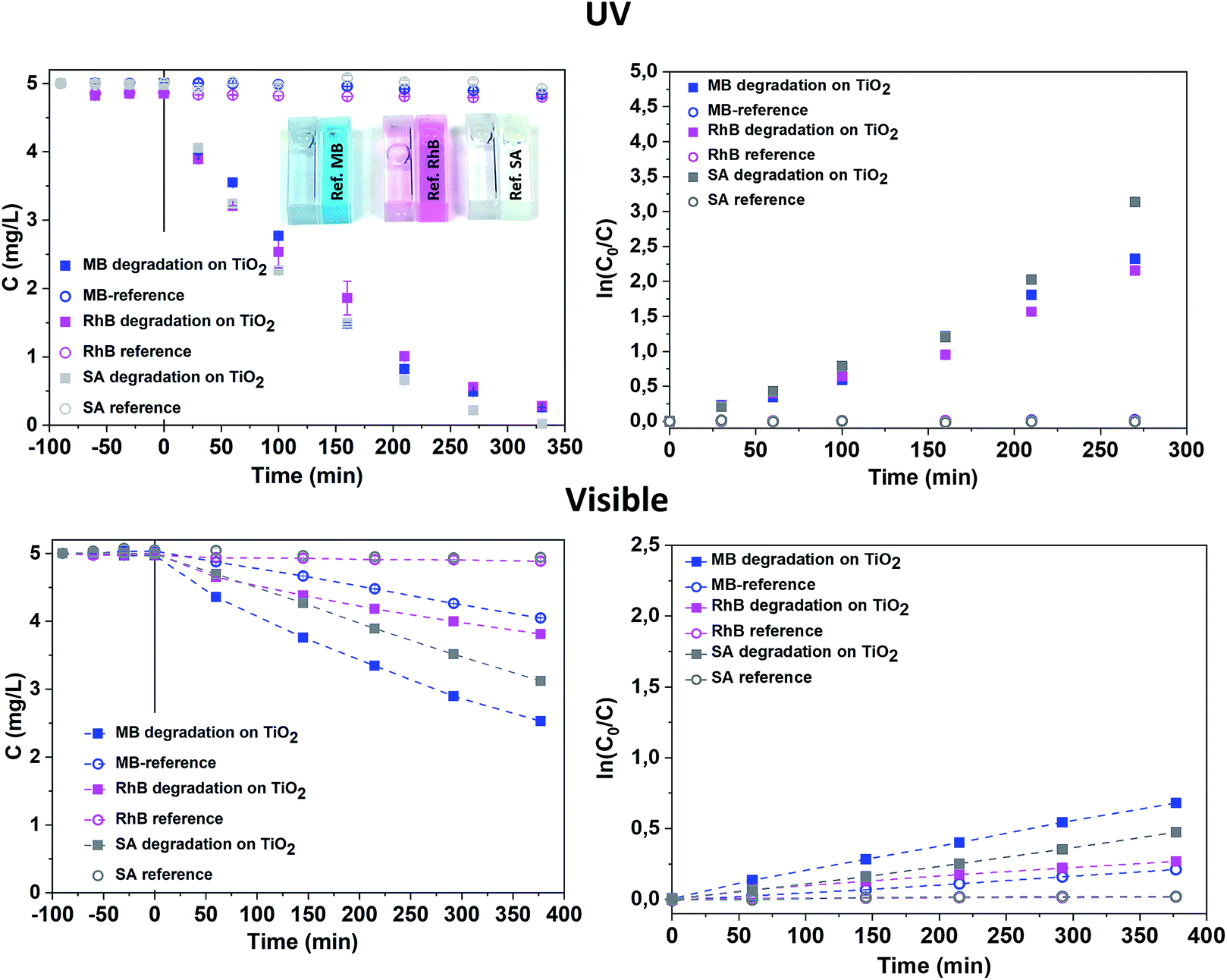 | ||
| Fig. 9 Photocatalytic degradation of MB, RhB and SA on RT TiO2 films deposited on Si/SiO2 substrate in the UV and visible ranges. | ||
| Target molecule | kr, normalised by the sample surface area, min−1 cm−2 | |||
|---|---|---|---|---|
| UV range | Visible range | |||
| On TiO2 | Blank test | On TiO2 | Blank test | |
| BM | 3.8 × 10−3 | 9 × 10−5 | 1.7 × 10−3 | 5.8 × 10−4 |
| RhB | 3.1 × 10−3 | 4 × 10−5 | 0.6 × 10−3 | 4.3 × 10−5 |
| SA | 5.0 × 10−3 | 4 × 10−5 | 1.2 × 10−3 | 5.0 × 10−5 |
Under visible light irradiation, the degradation constant of MB was the highest, but the photodegradation of MB under visible light must be considered, as evidenced by the significant photodegradation of the reference solution. The SA and RhB molecules are more stable under visible light, as proven by the concentration plateau of the reference solution. The photocatalytic degradation under visible light is even more efficient for neutral molecules such as SA compared to BM and RhB, showing that our mesoporous TiO2 material is particularly efficient. The photocatalytic degradation rate for different molecules was formerly explained by a better adsorption due to the charge difference between the surface and the molecule.52 However, our results in the UV range contradict this assumption, showing an identical degradation rate for all molecules. The lower activity in the visible range may be due to the lower carrier density and thus lower radical concentration. Therefore, degradation in the visible range occurs according to the stability of the molecules.
The reproducibility of the proposed system was controlled under UV light exposure for series 1 and 2 deposited on Si/SiO2 substrates. The comparison between these two series confirmed the efficiency of the developed photocatalytic systems while the noted difference of the degradation rate is related to the technical limitations of the deposition setup and requires further detailed investigation (Fig. S12, ESI†).
4. Conclusion
The deposition of mesoporous anatase TiO2 films was carried out using a non-conventional room temperature ALD process, using TiCl4 and water as precursors. This work demonstrates an important interplay between the deposition conditions (purge time) and the physico-chemical properties of TiO2 films. The process developed led to an accumulative growth of amorphous titania-like films, composed of by-products of a partial hydrolysis. The growth in the short purge time regime enhances this by-product accumulation into the films and results in a higher growth rate and consequently higher content of TiCl4−x(OH)x. The post-deposition annealing at 600 °C completes the partial hydrolysis into the films and leads to the degassing of the volatile acidic by-products. Such a non-conventional approach leads to the formation of a mesoporous anatase structure, while the presence of an acidic medium leads to the generation of structural defects in the TiO2. The evaluation of the photocatalytic performances of mesoporous hydrogenated anatase films confirms the photocatalytic activity enhancement of the films, both under UV and visible irradiation. A particular improvement of the photocatalytic activity was noted for the mesoporous films deposited with a short purge time, which is likely related to the initial amount of chlorine ions in the amorphous films that led to a larger amount of structural defects through annealing, determined from the physico-chemical studies and enhancement of green photoluminescence in these films. Unexpectedly, high photocatalytic activity in both UV and visible light ranges was found for films deposited on thermally oxidised silicon substrates. The reason for this improvement remains unclear and will require further investigations. Moreover, we observed that the high degradation rate constants in the UV range were almost identical for our three tested pollutant molecules (BM, RhB, SA), which indicates a broader applicability of our films for depollution purposes. The significant improvement of photocatalytic activity in both the UV and visible ranges of mesoporous TiO2 ALD films, without the need for additional doping or a plasmonic approach, can be considered an important step forward. Moreover, the affordability of the precursors used and the scalability of the ALD process developed here are favourable for an industrial implementation of water purification applications or for the developments of new concept of electrodes of Na-ion batteries.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from the Fonds National de la Recherche (FNR) Luxembourg via the INTER/NSF/MAT/11/01 VISICAT project. The authors are also thankful to Dr Yves Fleming for the XPS, XRD and XRR measurements, as well as to P. Grysan and Dr S. Girod for the AFM measurements. Dr O. Ishchenko expresses her acknowledgement to Dr G. Garry (TE-OX) for the fruitful discussions.Notes and references
- P. J. J. Alvarez, C. K. Chan, M. Elimelech, N. J. Halas and D. Villagrán, Nat. Nanotechnol., 2018, 13, 634–641 CrossRef CAS.
- K. Honda and A. Fujishima, Nature, 1972, 238, 37–38 CrossRef.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemannt, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. N. Banerjee, Nanotechnol., Sci. Appl., 2011, 4, 35–65 CrossRef CAS.
- O. M. Ishchenko, V. Rogé, G. Lamblin and D. Lenoble, in Semiconductor Photocatalysis - Materials, Mechanisms and Applications, ed. W. Cao, IntechOpen, 2016, pp. 3–30 Search PubMed.
- M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS.
- W. Fang, M. Xing and J. Zhang, Appl. Catal., B, 2014, 160–161, 240–246 CrossRef CAS.
- X. Liu, H. Xu, L. R. Grabstanowicz, S. Gao, Z. Lou, W. Wang, B. Huang, Y. Dai and T. Xu, Catal. Today, 2014, 225, 80–89 CrossRef CAS.
- F. Zuo, L. Wang and P. Feng, Int. J. Hydrogen Energy, 2014, 39, 711–717 CrossRef CAS.
- J. Cai, Z. Huang, K. Lv, J. Sun and K. Deng, RSC Adv., 2014, 4, 19588 RSC.
- A. Sasinska, T. Singh, S. Wang, S. Mathur and R. Kraehnert, J. Vac. Sci. Technol., A, 2015, 33, 1–6 CrossRef.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- R. Ren, Z. Wen, S. Cui, Y. Hou, X. Guo and J. Chen, Sci. Rep., 2015, 5, 10714 CrossRef CAS.
- A. Naldoni, M. Altomare, G. Zoppellaro and N. Liu, ACS Catal., 2019, 9, 345–364 CrossRef CAS.
- S. K. Park and H. Shin, J. Nanosci. Nanotechnol., 2014, 14, 8122–8128 CrossRef CAS.
- R. Yuan, T. Chen, E. Fei, J. Lin, Z. Ding, J. Long, Z. Zhang, X. Fu, P. Liu, L. Wu and X. Wang, ACS Catal., 2011, 1, 200–206 CrossRef CAS.
- Y. Xu, S. Wu, P. Wan, J. Sun and Z. D. Hood, RSC Adv., 2017, 7, 32461–32467 RSC.
- H. Li, S. Wu, Z. D. Hood, J. Sun, B. Hu, C. Liang, S. Yang, Y. Xu and B. Jiang, Appl. Surf. Sci., 2020, 513, 145723 CrossRef CAS.
- D. Pugliese, A. Lamberti, F. Bella, A. Sacco, S. Bianco and E. Tresso, Org. Electron., 2014, 15, 3715–3722 CrossRef CAS.
- F. Bella, A. Lamberti, A. Sacco, S. Bianco, A. Chiodoni and R. Bongiovanni, J. Membr. Sci., 2014, 470, 125–131 CrossRef CAS.
- F. Bella, S. Galliano, G. Piana, G. Giacona, G. Viscardi, M. Grätzel, C. Barolo and C. Gerbaldi, Electrochim. Acta, 2019, 302, 31–37 CrossRef CAS.
- T. Xia, W. Zhang, J. B. Murowchick, G. Liu and X. Chen, Adv. Energy Mater., 2013, 3, 1516–1523 CrossRef CAS.
- M. Serrapede, U. Savino, M. Castellino, J. Amici, S. Bodoardo, E. Tresso and A. Chiodoni, Materials, 2020, 13, 21 CrossRef CAS.
- Y. Liu and Y. Yang, J. Nanomater., 2016, 1–15 Search PubMed.
- R. M. Tamgadge and A. Shukla, Electrochim. Acta, 2018, 289, 342–353 CrossRef CAS.
- A. Massaro, A. B. Muñoz-García, P. Maddalena, F. Bella, G. Meligrana, C. Gerbaldi and M. Pavone, Nanoscale Adv., 2020, 2, 2745–2751 RSC.
- O. M. Ishchenko, G. Lamblin, D. Arl, N. Adjeroud, J. Guillot, P. Grysan, P. Nukala, J. Guyon, I. Fechete, F. Garin, P. Turek and D. Lenoble, Cryst. Growth Des., 2018, 18(9), 4929–4936 CrossRef CAS.
- J. Aarik, A. Aidla, H. Mandar and T. Uustare, Appl. Surf. Sci., 2001, 172, 148–158 CrossRef CAS.
- Z. Hu and C. H. Turner, J. Phys. Chem. B, 2006, 110, 8337–8347 CrossRef CAS.
- J. Leem, I. Park, Y. Li, W. Zhou, Z. Jin, S. Shin and Y. Min, Bull. Korean Chem. Soc., 2014, 35, 1195–1201 CrossRef CAS.
- T.-H. Wang, A. M. Navarrete-López, S. Li, D. a. Dixon and J. L. Gole, J. Phys. Chem. A, 2010, 114, 7561–7570 CrossRef CAS.
- W. Gu and C. P. Tripp, Langmuir, 2005, 21, 211–216 CrossRef CAS.
- S. Pétigny, H. Mostéfa-Sba, B. Domenichini, E. Lesniewska, A. Steinbrunn and S. Bourgeois, Surf. Sci., 1998, 410, 250–257 CrossRef.
- M. Iwaki, Y. Okabe and K. Yabe, Nucl. Instrum. Methods Phys. Res., Sect. B, 1990, 45, 212–215 CrossRef.
- V. S. Lusvardi, M. A. Barteau, J. G. Chen, J. Eng, B. Frühberger and A. Teplyakov, Surf. Sci., 1998, 397, 237–250 CrossRef CAS.
- S. Hashimoto and A. Tanaka, Surf. Interface Anal., 2002, 34, 262–265 CrossRef CAS.
- E. McCafferty and J. P. Wightman, Surf. Interface Anal., 1998, 26, 549–564 CrossRef CAS.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS.
- Y. Liu, H. Feng, X. Yan, J. Wang, H. Yang, Y. Du and W. Hao, Dalton Trans., 2017, 46, 10694–10699 RSC.
- J. Tian, Y. Leng, H. Cui and H. Liu, J. Hazard. Mater., 2015, 299, 165–173 CrossRef CAS.
- T. Xia, C. Zhang, N. A. Oyler and X. Chen, Adv. Mater., 2013, 25, 6905–6910 CrossRef CAS.
- Y. Lu, W. J. Yin, K. L. Peng, K. Wang, Q. Hu, A. Selloni, F. R. Chen, L. M. Liu and M. L. Sui, Nat. Commun., 2018, 9, 1–9 CrossRef.
- J. L. McHale and F. J. Knorr, Handbook of Luminescent Semiconductor Materials, Taylor & Francis Group, LLC, 2012 Search PubMed.
- C. Jin, B. Liu, Z. Lei and J. Sun, Nanoscale Res. Lett., 2015, 10(95), 1–9 CAS.
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590–596 CrossRef CAS.
- S. Hu, F. Li and Z. Fan, Bull. Korean Chem. Soc., 2012, 33, 1895–1899 CrossRef CAS.
- R. Sun, Z. Chen, Y. Yang, J. Peng and T. Zheng, Mater. Res. Express, 2019, 6, 046409 CrossRef.
- B. Sopori, M. I. Symko, R. Reedy, K. Jones and R. Matson, Conf. Rec. IEEE Photovoltaic Spec. Conf., 1997, 25–30 CAS.
- B. Sopori, Y. Zhang and N. M. Ravindra, J. Electron. Mater., 2001, 30, 1616–1627 CrossRef CAS.
- T. S. Natarajan, H. C. Bajaj and R. J. Tayade, J. Colloid Interface Sci., 2014, 433, 104–114 CrossRef CAS.
- Y. Zhou, Y. Zhang, M. Lin, J. Long, Z. Zhang, H. Lin, J. C. S. Wu and X. Wang, Nat. Commun., 2015, 6, 1–8 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06455f |
| This journal is © The Royal Society of Chemistry 2020 |