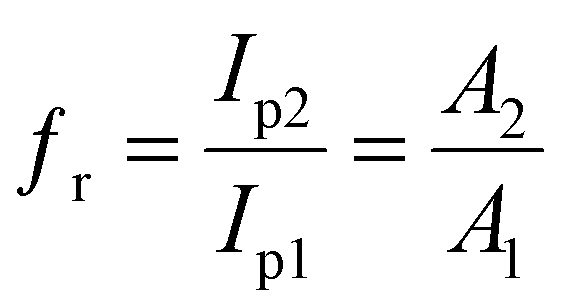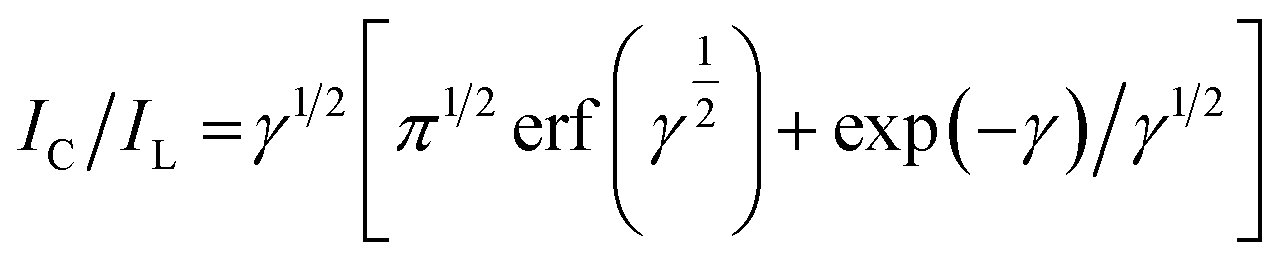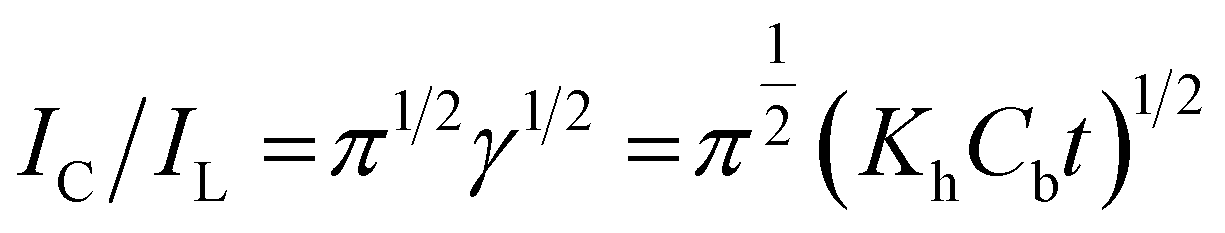DOI:
10.1039/D0RA06480G
(Paper)
RSC Adv., 2020,
10, 38532-38545
Simultaneous determination of methadone and morphine at a modified electrode with 3D β-MnO2 nanoflowers: application for pharmaceutical sample analysis
Received
26th July 2020
, Accepted 1st October 2020
First published on 19th October 2020
Abstract
The present research synthesized manganese dioxide nano-flowers (β-MnO2-NF) via a simplified technique for electro-catalytic utilization. Moreover, morphological characteristics and X-ray analyses showed Mn in the oxide form with β-type crystallographic structure. In addition, the research proposed a new efficient electro-chemical sensor to detect methadone at the modified glassy carbon electrode (β-MnO2-NF/GCE). It has been found that oxidizing methadone is irreversible and shows a diffusion controlled procedure at the β-MnO2-NF/GCE. Moreover, β-MnO2-NF/GCE was considerably enhanced in the anodic peak current of methadone related to the separation of morphine and methadone overlapping voltammetric responses with probable difference of 510 mV. In addition, a linear increase has been observed between the catalytic peak currents gained by the differential pulse voltammetry (DPV) of morphine and methadone and their concentrations in the range between 0.1–200.0 μM and 0.1–250.0 μM, respectively. Furthermore, the limits of detection (LOD) for methadone and morphine were found to be 5.6 nM and 8.3 nM, respectively. It has been found that our electrode could have a successful application for detecting methadone and morphine in the drug dose form, urine, and saliva samples. Thus, this condition demonstrated that β-MnO2-NF/GCE displays good analytical performances for the detection of methadone.
1. Introduction
It is widely accepted that morphine is one of the phenolic compounds as well as an alkaloid that is able to disrupt the central nervous system. Morphine is often utilized for alleviating serious pain in patients, in particular, following a surgery. However, it is poisonous in higher doses and thus may be deadly. For preventing over-dose, clinical medicines should necessarily detect morphine concentration sensitively in the patients' blood or urine.1 The legal blood concentration of opioid varies across European countries from 9 μg L−1 (Norway) to 20 μg L−1 (France, Poland, Netherlands) to 80 μg L−1 (England/Wales). This heterogeneity in laws within and between countries illustrates that a concentration–impairment relationship has not been established. National and international medical recommendations also reflect this uncertainty. In a UK National Health Service document, oral morphine equivalent doses above 220 mg day−1 were considered probably not safe for driving. The Royal College of Anaesthetists states that doses of about 200 mg day−1 could be as dangerous as having borderline illegal driving blood alcohol levels.2 In fact, the clear desensitization of opioid receptors may cause methadone to intervene in impacts started by morphine, and in its continual presence, may even interrupt the cascade of cellular episodes and avoid lengthy neuro-chemical modifications due to the chronic exposure to morphine.3
One of the studies reported methadone as a synthetic analgesic drug with widespread utilization for treating opioid dependence since the mid-1960s.4 According to the results, morphine and methadone have the same pharmacological features. Some studies have also shown replacement of this treatment with a short-acting opioid replacing a long-acting opioid, namely, methadone. It is known that methadone is a completely μ-opioid receptor agonist that may generate cross-tolerance with heroin or other opioids. Such a condition can itself decline the withdrawal signs in the suffering people and empower patients who have recently ceased receiving opioids for performing and maintaining normal daily performance.5 According to number of studies, methadone is one of the most frequently applied drugs in opioid substitution treatments; however, it is utilized to relieve moderate-to-severe pains.6 As the remaining pharmaceutical compounds, methadone possesses a treatment concentration range in biological fluids. Therefore, methadone is ineffective below this concentration, whereas its excess concentration can cause complications including nervousness, nausea, anxiety, sleep issues such as insomnia, dry mouth, weakness, and diarrhea.
However, researchers have taken many actions for the development of a simplified and sensitive process for accurately detecting methadone and morphine, which has been achieved by developing different methods such as liquid chromatography/mass spectrometry (LC-MS), gas chromatography, gas chromatography mass spectrometry (GC-MS), high performance liquid chromatography (HPLC), flow-injection analysis (FIA), and spectroscopy.7–12 The above methods have a significant sensitivity and high reliability; however, many of these approaches are expensive and laborious, and demand complicated sample pre-treatment phases.13,14 Thus, designing a simplified, cost-effective, sensitive, and precise analytical procedure would be favorable for the detection of methadone and morphine.
According to the studies, as electrochemical approaches are easy and robust, inexpensive preparation and miniaturization would be feasible; however, it has been demonstrated that electroanalytical procedures are beneficial for developing very selective and sensitive techniques for the detection of the organic molecules such as medicines as biological fluids and dose forms.15–17 Other studies have applied the chemically modified electrode for the improvement of selectivity, sensitivity, LOD, and other properties of the electrode. Of course, the operational system of the above chemically modified electrodes is dependent on the modifier material's features applied for improving the sensitivity towards the target samples.18–21
Consequently, one of the studies has showed the authors' interest in manganese oxide (MnOx)-based substances due to multiple benefits such as a wide range of valences (in a range between Mn0 and Mn7+), earth-abundance, non-toxicity, and more cost-effective in comparison to other metal oxides.22 Specifically, MnOx has benefits for electro-catalysis because it accounts for a large number of electro-chemically active sites that have a direct involvement with the augmented catalysis.23 Since a nano-catalyst's features largely depend on the form, dimension, structure, and morphology of the active phase and support, it would be very crucial to precisely control the above variables for further understanding their greater activity.24 However, the physico-chemical features of the MnOx-based nano-materials have a great association with their internal crystallo-graphic structure, overall compositions, form, dimension, and morphologies.25 From among MnOx, the researchers referred to MnO2 as having excellent electro-catalytic activities. Nano-flowers have a lot of useful structural properties for improving the catalysis on transition metal-based substances based on the electro-catalytic activity, affordability, and durability. In fact, other studies have showed the hierarchical architecture of the MnO2 nano-flowers as the optimized nano-structure owing to the rational open structure that could make the length of the transport path of the electron and cation shorter.26 Nonetheless, fabricating such a hierarchical architecture has been challenging as a result of the quick growing procedure.
As observed in different publications reviewed, there have been no reports for the electro-catalytic detection of methadone in the presence of morphine via the electro-chemical sensors in human saliva and urine, and pharmaceutical samples. Hence, this research aimed at investigating the electro-chemical behaviors of methadone and morphine by the means of β-MnO2-NF/GCE in the aqueous solution and developing an easy, selective, and reliable sensor for the concurrent detection of methadone and morphine without the cost and laborious pre-treatments involved.
2. Experimental details
2.1. Chemicals and reagents
Merck Co. was chosen to provide potassium permanganate (KMnO4) and manganese sulfate (MnSO4·H2O). Moreover, the American Chemical and Bio-technology Co. of Sigma-Aldrich was chosen to buy methadone and morphine. We dissolved the intended volumes of methadone and morphine in water and used a 100 mL volumetric flask to dilute the mixture to 100 mL with distilled water. Then, 0.01 M methadone and morphine solutions were obtained using this method. We used serial dilution of phosphate buffer saline (PBS) to obtain more diluted solution. Stock solution of 0.9% NaCl and 0.01 M Na2HPO4 was combined to form a 0.1 M PBS solution. In addition, HCl or NaOH was utilized for pH modification. Afterwards, the final solution was kept in the dark in a refrigerator. All other substances were of analytical reagent grade. Finally, each solution was obtained by double-distilled deionized water. Thus, we were not required to carry out any additional purification on the used chemicals. All procedures conform to the principles outlined in the declaration of Helsinki and Ethics committee of Islamic Azad University of Kerman Branch has approved the experiments. Informed consents were obtained while collecting the saliva and urine samples in this study.
2.2. Apparatus
For the illustration of the product structure, we utilized Kα radiation (α2, λ2 = 1.54439 Å) for X-ray powder diffraction (XRD), the graphite mono-chromatic Cu radiation (α1, λ1 = 1.54056 Å), as well as a Philips analytical PC-APD X-ray diffractometer. Then, EM 3200 SEM and KYKY were employed for analyzing the morphology of β-MnO2-NF. Notably, EDX spectroscopy is an example of a crucial non-destructive tool for analyzing chemical compounds. Moreover, this study utilized a SAMA 500 Electro-analyzer for electro-chemical detection. It is notable that this three electrode electro-chemical cell system included a Pt wire auxiliary electrode, a saturated calomel reference electrode (SCE), and a glassy carbon working electrode (i.e., GCE, modified or unmodified).
2.3. Hydrothermal synthesis of β-MnO2 nanoflowers
Based on the research design, 1 g of potassium permanganate (KMnO4) and 0.4 g of manganese sulfate (MnSO4·H2O) were dissolved in 30 mL of water for preparing β-MnO2 nano-flowers. Then, the mix was transferred to a 25 mL teflon-lined stainless-steel autoclave. Afterwards, an electric oven was used to heat the mix at 140 °C for one hour. Finally, ethanol and water were used to wash the precipitated precipitate, followed by drying at 80 °C for 6 hours.
2.4. Electrode modification
In this study, β-MnO2-NFs were used to modify the bare GCE. Then, we dispersed 1 mg β-MnO2-NFs with 1 hour ultra-sonication to collect the stock solution with β-MnO2-NFs in the aqueous solution (1 mL). Afterwards, we placed a 5 μL aliquot of the β-MnO2-NFs/H2O suspension solution on the carbon working electrode and thus solvent evaporation was done at room temperature.
2.5. Preparation of real samples
After the collection of the humans' blood sera and urine samples from the clinical laboratory, the samples were kept in a refrigerator. Therefore, we conducted a 2000 rpm centrifugation on 10 mL of the sample for 20 minutes and applied a 0.45 mm filter to filter the supernatant. Then, PBS with a pH equal to 7.0 was utilized for 5 times dilution of the filtered product. In the next stage, we delivered the obtained solution to the voltammetric cell for analyses without any requirement of additional purification. Finally, the standard addition procedure was used to determine the contents of methadone and morphine in the samples.
Before diluting and filtering, the serum samples were centrifuged with PBS solution with a pH of 7.0. Afterwards, different volumes of the same composite were used for adulteration.
The tablets and ampoule doses contained 40 mg methadone and 10 mg mL−1 morphine, which have been considered crucial ingredients and are electro-chemically active independent of further conventional additive medicines. We poured different contents of the diluted solution into a volumetric flask (25 mL) and used PBS with a pH of 7.0 to dilute to the mark. Then, we utilized our method to analyze methadone and morphine volumes using the standard addition procedure.
Salivette (Sarstedt, Sevelen, Iran) was used to collect saliva. Then, centrifuging of the saliva samples lasted for five minutes at 4000 rpm, followed by dilution with 25.0 mL PBS (pH = 7.0). Finally, they were employed for real samples analysis through the standard addition procedure.
3. Results and discussion
3.1. Characterization of the β-MnO2 nanoflowers
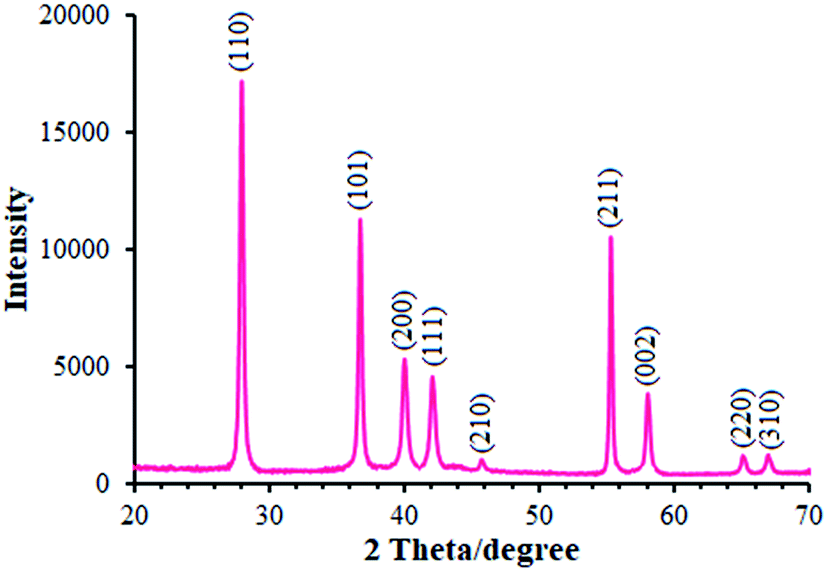
We employed XRD method for identifying the MnO2 phase form. Moreover, Fig. 1 portrays the XRD pattern of MnO2 powder. As seen in the figure, the XRD pattern showed multiple diffraction peaks that refer to MnO2 powder as a poly-crystalline structure. Therefore, each diffraction peak of the MnO2 powder was completely indexed to the tetragonal crystal system structure (JCPDS file no. 44-0141), confirming that the powder is a pure pyrolusite phase with a tetragonal MnO2 (β-MnO2).27,28 It should be noted that 9 corresponding peaks were seen at the 2θ of 28.14°, 36.91°, 40.16°, 42.13°, 45.79°, 55.46°, 58.18°, 65.14°, and 66.98° for β-MnO2 and thus were identified by the respective indicators such as (110), (101), (200), (111), (210), (211), (002), (220), and (310), respectively.
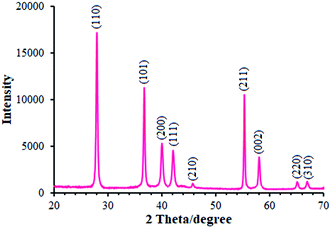 |
| | Fig. 1 XRD pattern of β-MnO2 nano-flowers. | |
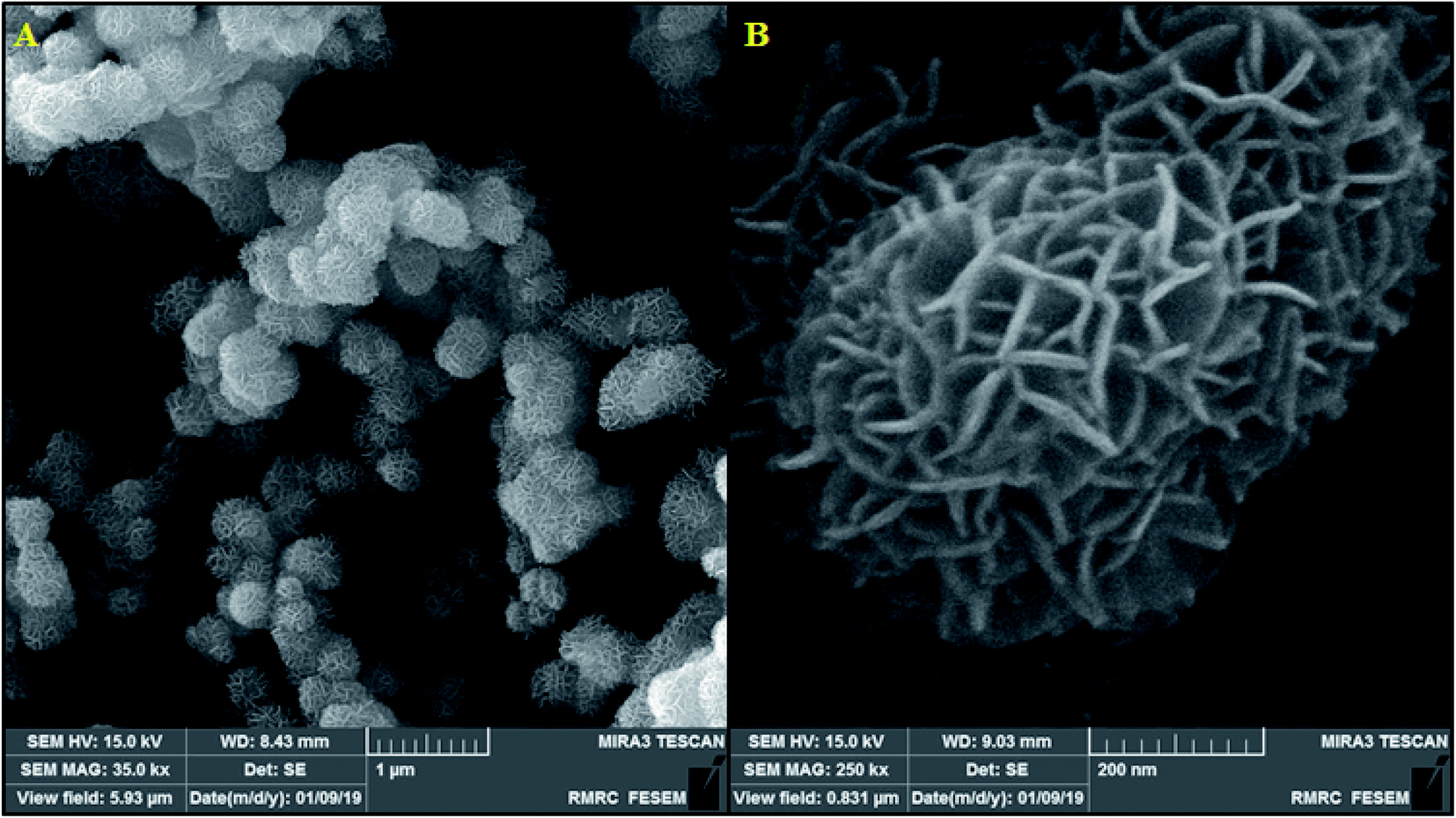
Here, it should be noted that SEM was used to examine the resulting samples' morphology. According to Fig. 2a, the as-prepared β-MnO2 samples are in a flower-shape spherical micro- or nano-structure with hundreds of nanometers of diameter. In addition, the magnified scheme (inset in Fig. 2b) showed the assembly of the flowery architecture using multiple inter-twined nano-sheets with several nanometers in thickness that are likely useful for increasing the oxidation towards morphine and methadone because of their shorter diffusion distance and greater active sites.
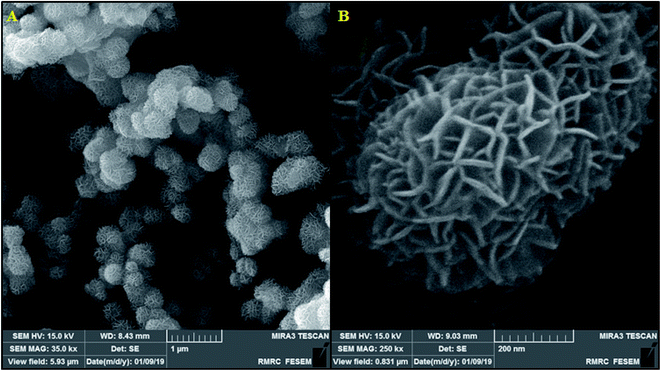 |
| | Fig. 2 (A) FESEM image of β-MnO2 nano-flowers, (B) FESEM images of a single β-MnO2 nano-flowers. | |
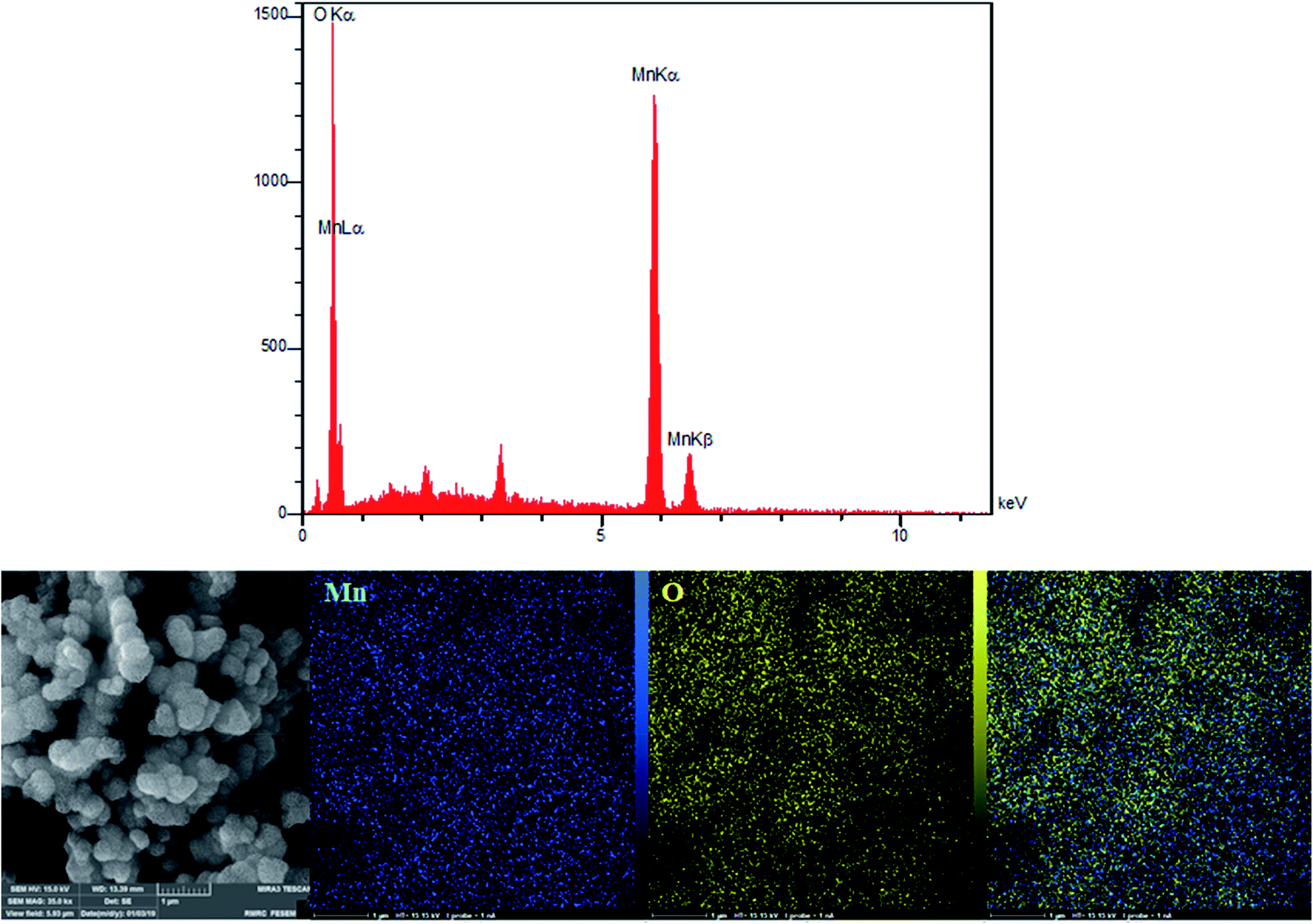
According to the research design, we utilized energy dispersive X-ray (EDX) analyses for analyzing the amounts of all the elements in the bulk materials. Fig. 3 showed the presence of oxygen and manganese. In addition, we used EDX mapping analyses for additionally examining the spatial distribution of oxygen and manganese elements. According to Fig. 3, we see the complete distribution of O (yellow zone) and Mn (blue zone) elements across the whole area of the spheres, a condition representing obvious proof for the homogeneous distribution in manganese oxide.
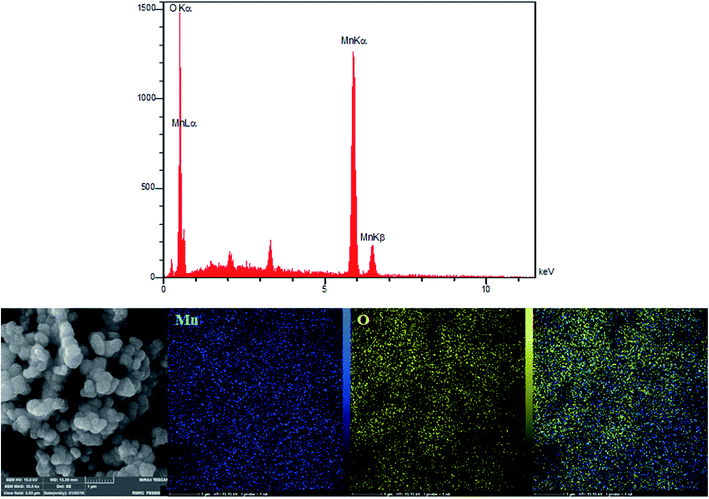 |
| | Fig. 3 EDX spectra and elemental mapping of the β-MnO2 nano-flowers. | |
3.2. Electrochemical characteristics of the β-MnO2 nanoflower-modified electrode
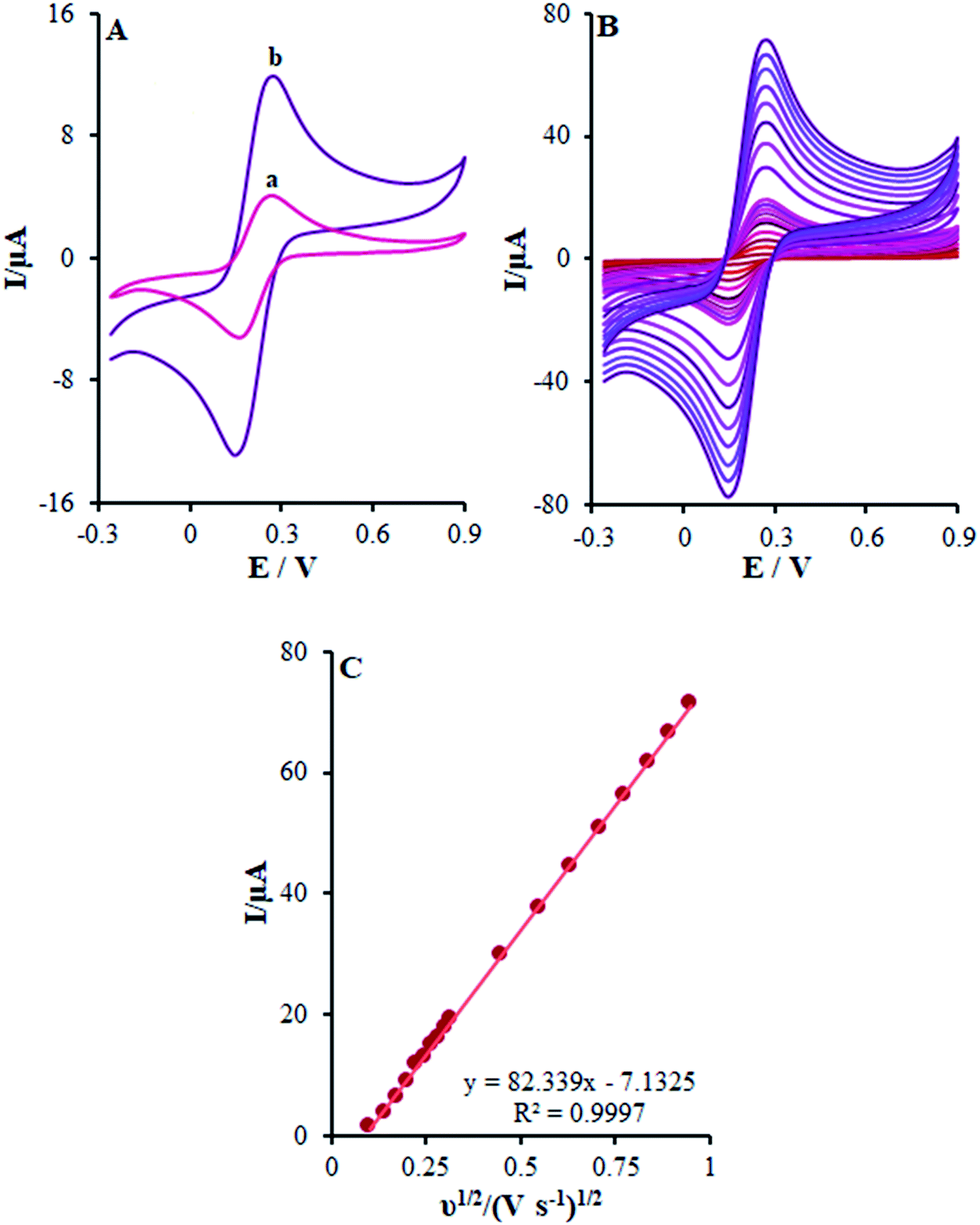
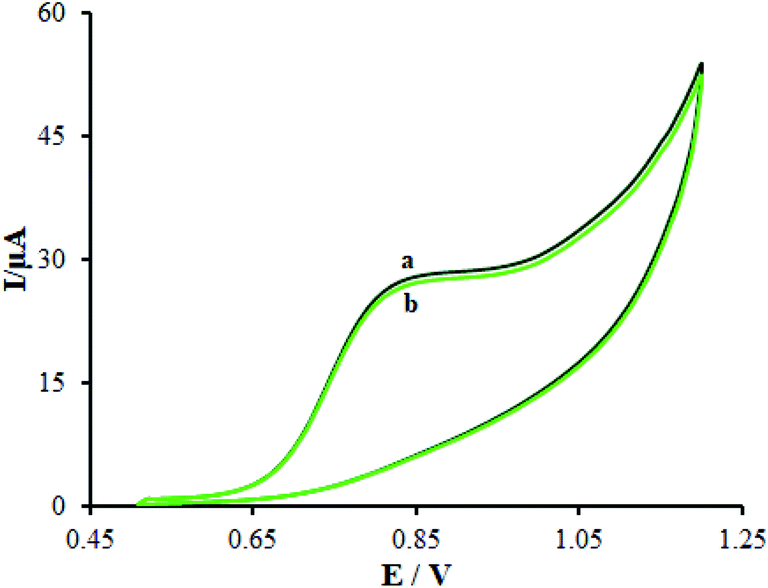
In this stage, the electro-chemical properties of β-MnO2-NF/GCE in case of the presence of the 0.5 mM [Fe(CN)6]3−/4− redox couple with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fe2+:Fe3+) with 0.1 M KCl was described by CV. The outputs observed at the β-MnO2-NF/GCE contrasted with the ones from the bare GCE (Fig. 4A).
:1 (Fe2+:Fe3+) with 0.1 M KCl was described by CV. The outputs observed at the β-MnO2-NF/GCE contrasted with the ones from the bare GCE (Fig. 4A).
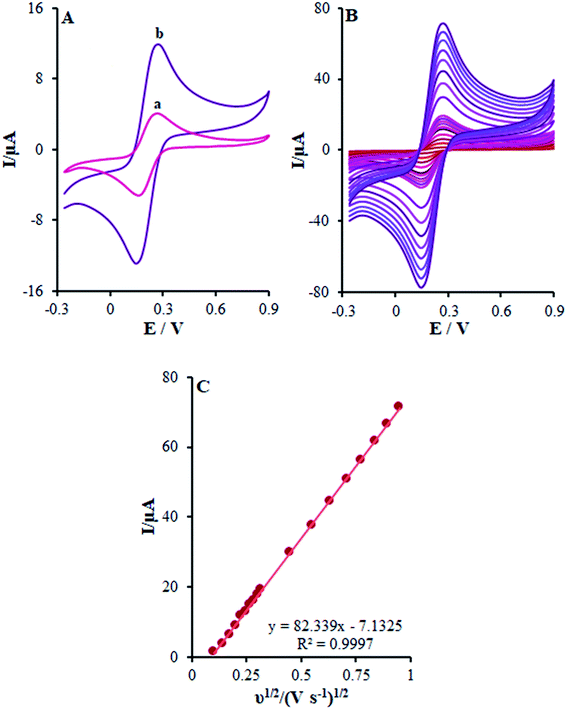 |
| | Fig. 4 (A) CV acquired at a potential scan rate 50 mV s−1 for a 0.5 mM [Fe(CN)6]3−/4− in aqueous 0.1 M KCl at the (a) bare GCE and (b) β-MnO2-NF/GCE. (B) CVs of β-MnO2-NF/GCE in the presence of 0.5 mM [Fe(CN)6]3− solution in aqueous 0.1 M KCl at various scan rates (from inner to outer curve): 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, and 900 mV s−1. (C) The plot of peak currents vs. ν1/2. | |
In other words, CV studies on the [Fe(CN)6]3−/4− solution were carried at various scan rates ranging between 10 and 900 mV s−1 on β-MnO2-NF/GCE (Fig. 4B) for the determination of the surface areas with electrochemical activity (A). After that, Randles–Sevcik equation was utilized for a reverse electrode technique.29 Here, eqn (1) characterizes the peak current (Ip):
| | |
Ip = ±(2.69 × 105)n3/2AD1/2Cv1/2
| (1) |
where
A stands for the electrode surface area (cm
2),
D refers to the diffusion coefficient of 7.6 × 10
−6 cm
2 s
−1, and
n implies number of the electron in a redox reaction, which is 1 for K
3[Fe(CN)
6]. Moreover,
C indicates the K
3[Fe(CN)
6] concentration of 0.5 mM. After that, we used the
Ipa plot slope
versus v1/2 to detect the electroactive areas for β-MnO
2-NF/GCE (
Fig. 4C). In addition, the electro-chemically active surface area of β-MnO
2-NF/GCE equaled 0.22 cm
2 and that of bare GCE equaled 0.076 cm
2. Furthermore, a roughness factor (
fr) equal to that of β-MnO
2-NF/GCE has been observed with regard to the
Ipa of the [Fe(CN)
6]
3−/4− redox couple, which corresponded to that of bare GCE. Consequently,
fr was calculated by the electrochemical method, depending on the numbers of redox cores over the electrode surface and dimension (actual surface).
30 At the end, the ratio of oxidation peak for both electrodes demonstrated changes in the real electrode surface, which equaled the electrode surface ratio (
eqn (2)):
| |
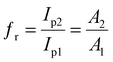 | (2) |
where
Ip1 and
Ip2 represent the peak current for bare GCE and β-MnO
2-NF/GCE. Moreover,
A1 and
A2 stand for the surface area of bare GCE and β-MnO
2-NF/GCE, respectively. Finally,
fr was estimated to be 2.89.
3.3. Electrochemical behavior of β-MnO2 nanoflower-modified and unmodified electrodes
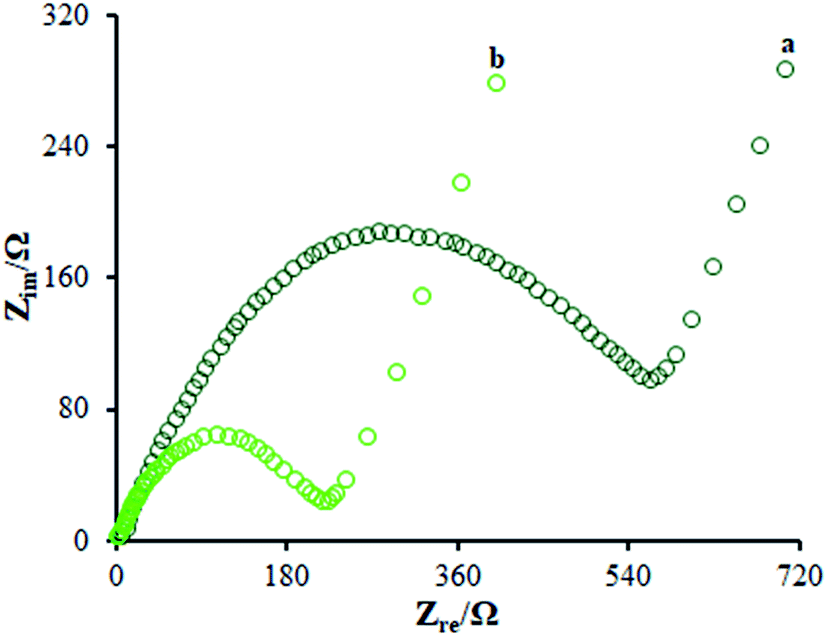
According to the studies, electro chemical impedance spectroscopy (EIS) is considered as a very good method for assessing the charge transfer technique of the electrode surfaces. Fig. 5 depicts the EIS outputs of various electrodes inserted in the same circuit. Such an equivalent circuit included the Warburg impedance (Zw), interfacial capacitance (Cdl), electron transfer resistance (Rct), and electrolyte ohmic resistance (Rs). Moreover, semicircular as well as linear sections have been detected in EIS. Notably, the contribution of Rct controlled the kinetics of electron transfer of the redox probe at the electrode interface, which has been shown by the semicircular diameter. As seen, the linear segment corresponded to the diffusion method at lower frequencies. According to Fig. 5, the bare GCE (curve a) indicated a large semi-circle with an Rct of ∼710 Ω; as seen, the Rct declined to 215 Ω in the presence of the modified electrode consisting of β-MnO2-NF (Fig. 5 (curve b)) due to insignificant conductivity, causing influential transfer of the electron between the electrode interface and the solution. Hence, the β-MnO2-NF capacity was described to enhance the electron transfer rate. Therefore, a reciprocal association has been found between the surface resistance and the electrical conductivity. Therefore, the electrical conductivity of the electrode was GCE < β-MnO2-NF/GCE, reflecting the increased electron transfer kinetics via β-MnO2-NF. Moreover, a reasonable electrical conductivity was observed for β-MnO2-NF, caused by the larger surface areas and higher electrical conductivity of β-MnO2-NF, followed by the significant enhancement in the electron transfer in the middle of the target molecule and electrode. The results demonstrate the compatibility of the outputs with the CV results.
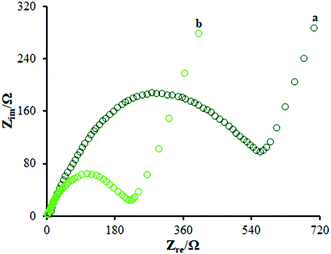 |
| | Fig. 5 EIS diagrams for 0.5 mM [Fe(CN)6]3−/4− solution at (a) unmodified GCE and (b) β-MnO2-NF/GCE in aqueous 0.1 M KCl. Frequency range from 100 kHz to 0.1 Hz. | |
3.4. Electrocatalytic-oxidation process of methadone at the β-MnO2 nanoflower-modified and unmodified electrodes
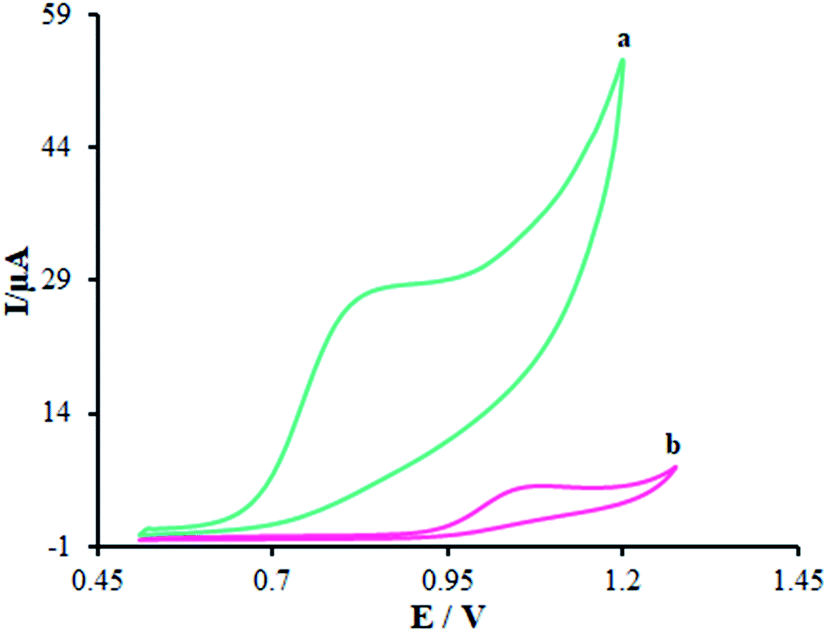
Fig. 6 displays the electro-chemical behavior of methadone at the unmodified GCE and modified GCE. Then, methadone oxidation at the surface of β-MnO2-NF/GCE (Fig. 6 (curve a)) was completely performed at the potential of 850 mV versus SCE, which was ∼320 mV more negative than the bare GCE potential (Fig. 6 (curve b)).
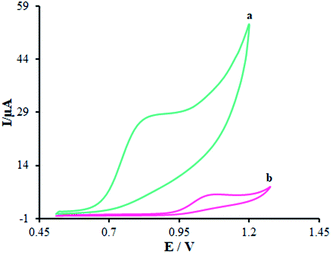 |
| | Fig. 6 CVs of (a) β-MnO2-NF/GCE and (b) unmodified GCE in the presence of methadone (100.0 μM) at a pH 7.0, respectively. In all the cases, the scan rate was 50 mV s−1. | |
Introducing β-MnO2-NF increased the electro-chemical catalytic activity of β-MnO2-NF/GCE that could be related to the greater surface areas exhibited by β-MnO2-NF and the considerable capability of electron transfer of the nanomaterials. β-MnO2-NF offered greater conductivity and quicker electron-transfer ability. Finally, electron transfer of methadone was remarkably enhanced by β-MnO2-NF/GCE.
3.5. Effect of pH and potential scan rate on the electrooxidation of methadone
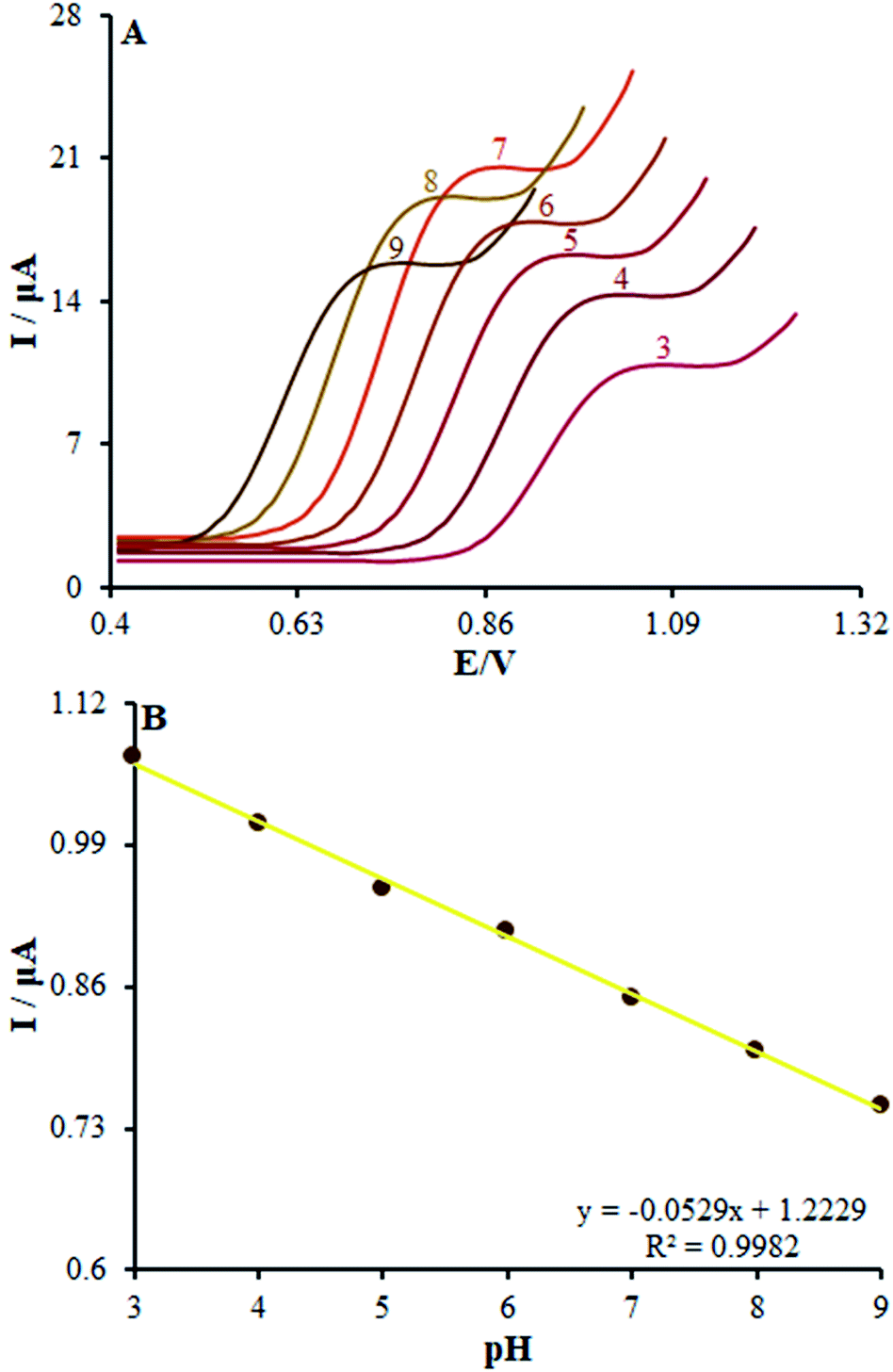
In order to acquire a favorable peak current and shape, we examined the pH impacts of the incidental electrolytes on the anodic peak currents and potential. Then, DPV on numerous buffered solutions was analyzed at pH values between 3.0 and 9.0 for methadone solutions. Then, the anodic peak potential at diverse pH-values of β-MnO2-NF/GCE was compared, showing a notable impact of the solution pH on the peak potential of methadone catalytic oxidation.
Moreover, the greatest oxidation peak current was observed at pH = 7.0. In fact, good the pH equaled 7.0. Hence, we chose 0.1 M PBS with pH of 7.0 as the suitable electrolyte for the following voltammetric analyses.
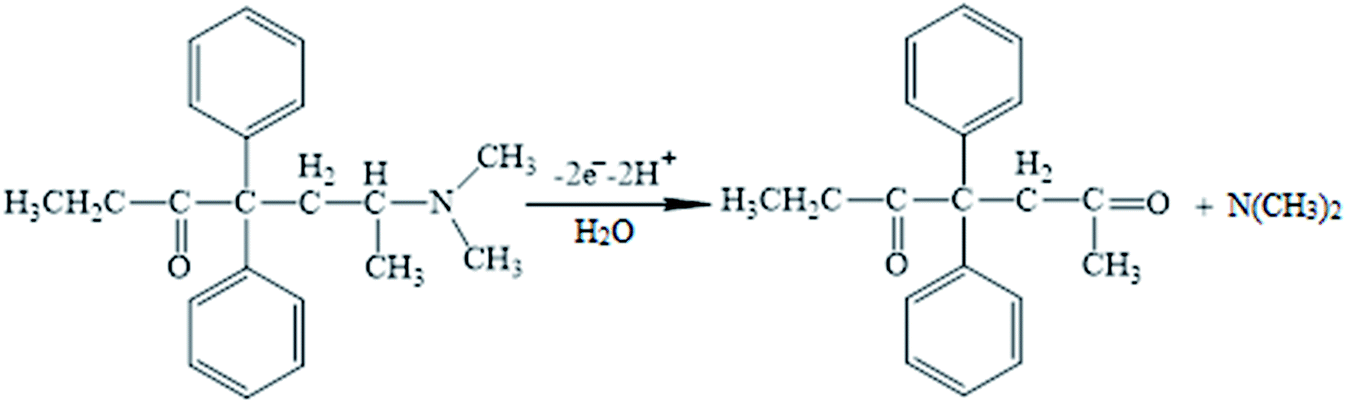
Moreover, linear correlation between the peak potential and pH may be written as Epa = 1.2229 − 0.0529 pH with R2 = 0.9982 for methadone (Fig. 7A); thus, the slopes would be approximated to the theoretical value (Nernstian value equal to 0.059 V). The above outputs reveal the same numbers of protons and electrons contributed to the electrode reactions (Scheme 1). In addition, as the pH was enhanced from 3.0 to 7.0, the redox current was enhanced to 7.0 (Fig. 7B); afterwards, it declined to a narrower range of higher pH (7.0 to 9.0). Moreover, we achieved greater oxidation peak current at a pH of 7.0 in comparison to other pH-values. As a result, pH = 7.0 has been employed for the remaining electrochemical tests.
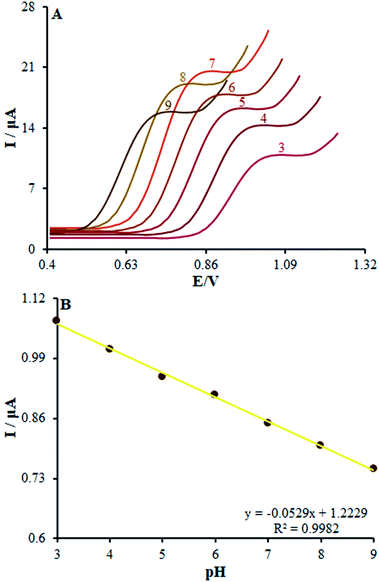 |
| | Fig. 7 (A) Effect of pH on the peak current for the oxidation of methadone (50.0 μM); pH = 3–9. In all the cases, the scan rate was 50 mV s−1. (B) The plot of peak potential vs. pH. | |
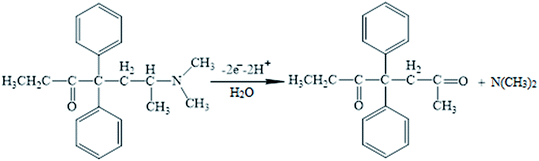 |
| | Scheme 1 The probable oxidation mechanism of methadone. | |
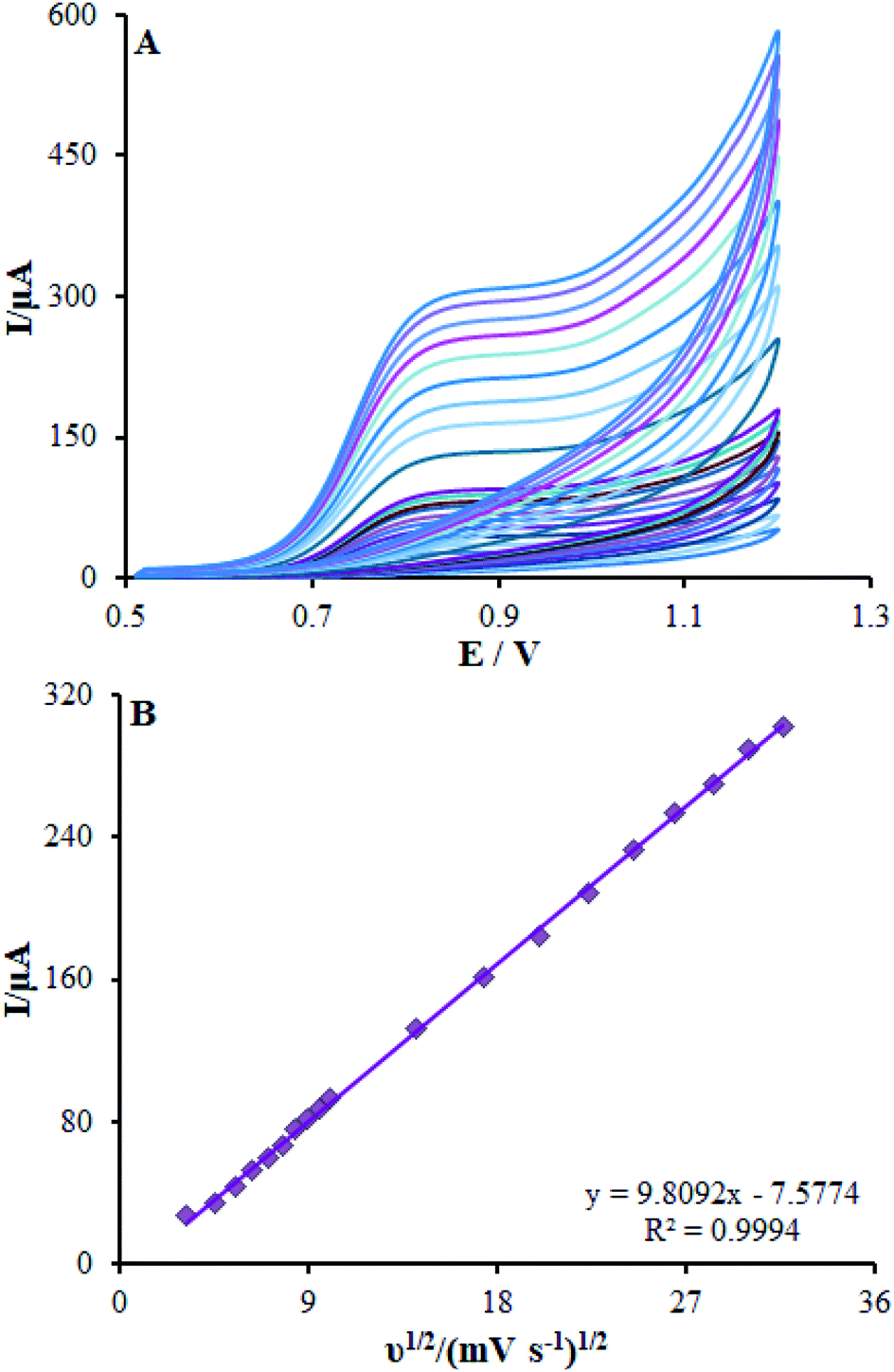
It is notable that the scan rate influences the oxidation peak current of methadone; therefore, it has been tested by CV on β-MnO2-NF/GCE. As shown in Fig. 8A, a direct proportion between the intensity of the peak current and the scan rate exists, implying the fact that enhancing the peak current intensity improved the scan rate. Moreover, a direct significant correlation has been found between the current and the square root of the scan rate in the spectra in the range of 10–1000 mV s−1 (Fig. 8B), in which the diffusion-controlled redox reactions of methadone occurred. As the charging current (ic) depends on v and the Faraday current depends on v1/2, at a higher scan rate, the charging current would be enhanced considerably. Hence, lower scan rate has been utilized for testing. Put differently, the testing would be lengthier at a very low scan rates. Finally, a scan rate equal to 50 mV s−1 was chosen as the optimum one for experimentation.
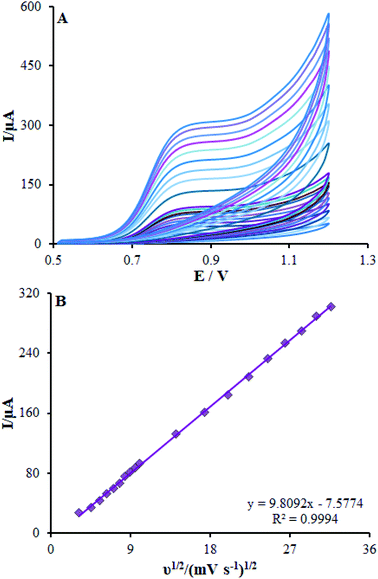 |
| | Fig. 8 (A) CVs of β-MnO2-NF/GCE in pH 7.0 in the presence of methadone (350.0 μM) at various scan rates (from inner to outer curve): 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1000 mV s−1. (B) The plots of peak currents vs. ν1/2. | |
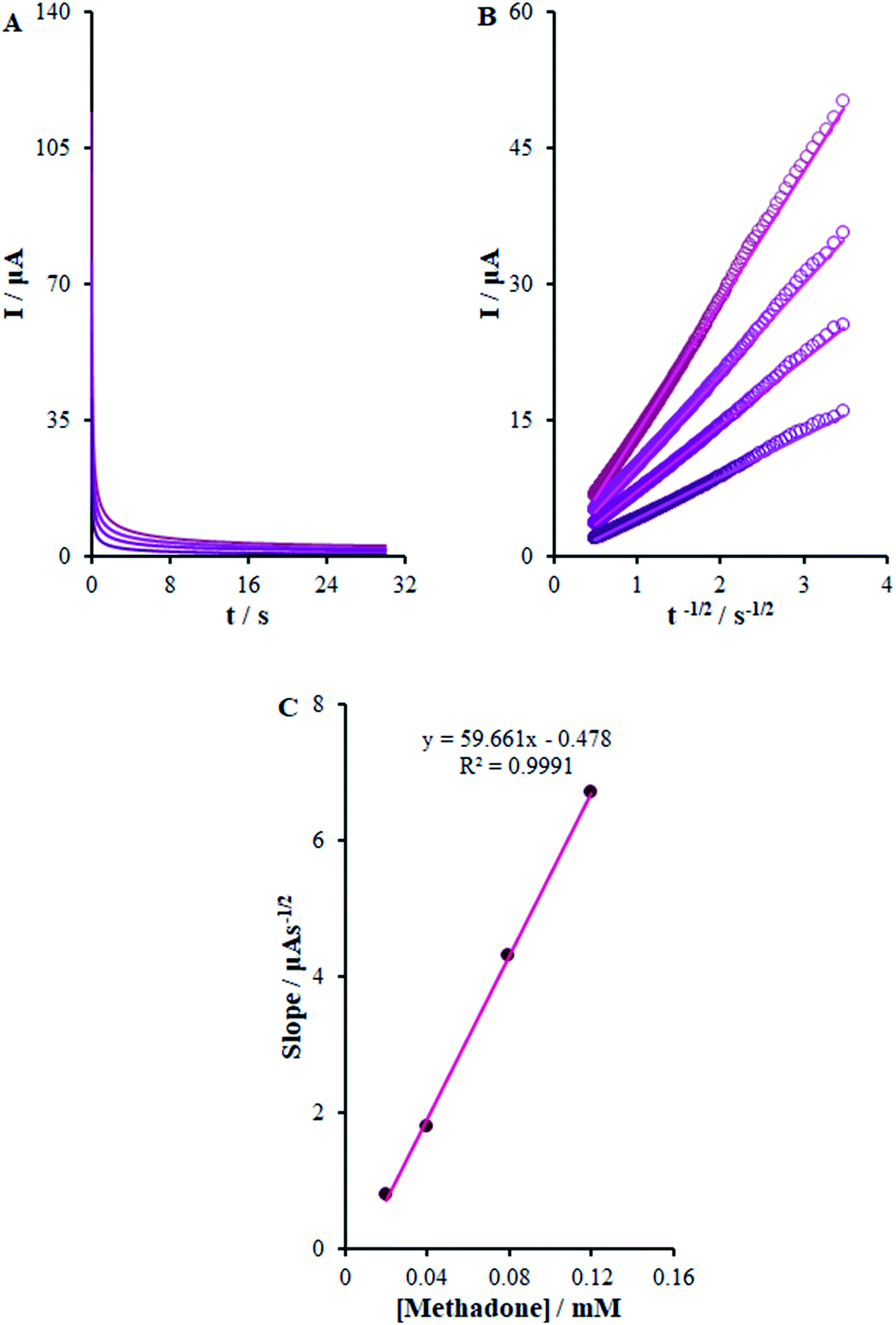
3.6. Chrono-amperometric studies
β-MnO2-NF/GCE potential was adjusted to 900 mV the and methadone concentration in PBS was altered in order to carry out chrono-amperometric evaluations (Fig. 9). Notably, Cottrell's eqn (4) was utilized to illustrate the current responses (I) for diffusion-limited electro-catalytic techniques of the electro-active materials; (for example, methadone):29| | |
I = nFAD1/2Cbπ−1/2t−1/2
| (3) |
here D (cm2 s−1) stands for the analyte diffusion coefficient and Cb implies the analyte bulk concentration (mol cm−3). Moreover, F stands for the Faraday constant of 96485 CM and A implies the electrode geometric area (0.22 cm2). In addition, n shows the electron quantity (2 for methadone) exchanged in all the reactant molecules. By drawing I against t−1/2, we observed a linear curve from the raw chrono-amperometric trace for different concentrations of methadone (Fig. 9B). Afterwards, the slope of the direct lines was plotted versus methadone concentrations (Fig. 9C). Furthermore, the estimation showed 6.2 × 10−6 cm2 s−1 diffusion coefficient for methadone.
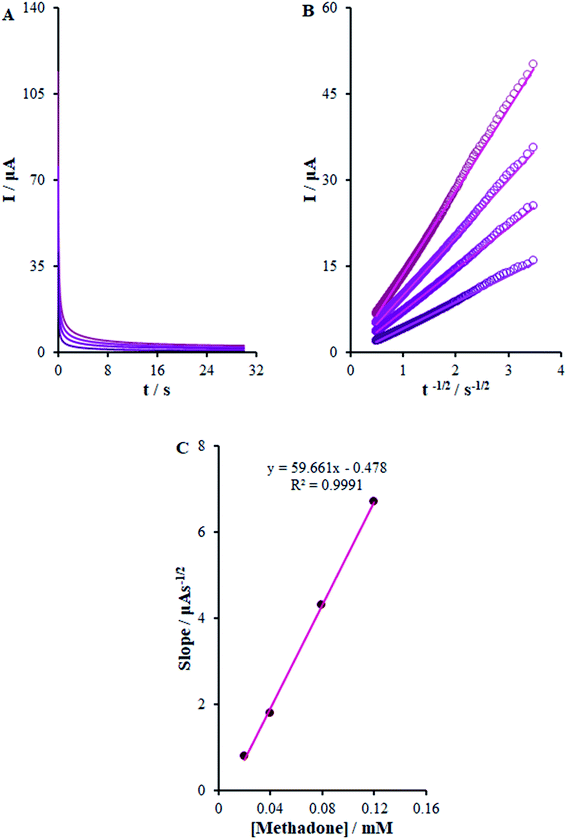 |
| | Fig. 9 (A) Chronoamperograms obtained at β-MnO2-NF/GCE in 0.1 M PBS (pH 7.0) for different concentrations of methadone (from inner to outer curve): 0.02, 0.04, 0.08, and 0.12 mM. (B) Plots of I vs. t−1/2 obtained from chronoamperograms 1–4. (C) Plot of the slope of the straight lines against methadone concentration. | |
The Galus method was utilized for the evaluation of the catalytic rate constant to the chemical reactions between methadone with β-MnO2-NF/GCE via the adjustment of the working electrode potential at 960 mV (eqn (4)).31
| |
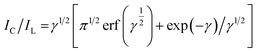 | (4) |
where
IL implies the limited current in the absence of analyte and
IC stands for the common catalytic currents of the analyte on the modified electrode surface. Moreover,
γ =
Kh Cb t indicates justifying the error function (
t,
Cb as well as
Kh stand for the time spent (s), catalytic rate constants (mol
−1 L s
−1), and analyte concentration (mol L
−1)). The error function value equaled approximately 1 as
γ exceeded 2. It has also been possible to simplify
eqn (4) here as
| |
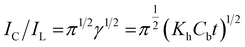 | (5) |
Eqn (5) has been used to estimate the catalytic rate constant (
Kh). It is also possible to provide a simple estimation of
Kh value
via the
IC/
IL slope
versus the
t1/2 plot for a determined substrate concentration. As seen in
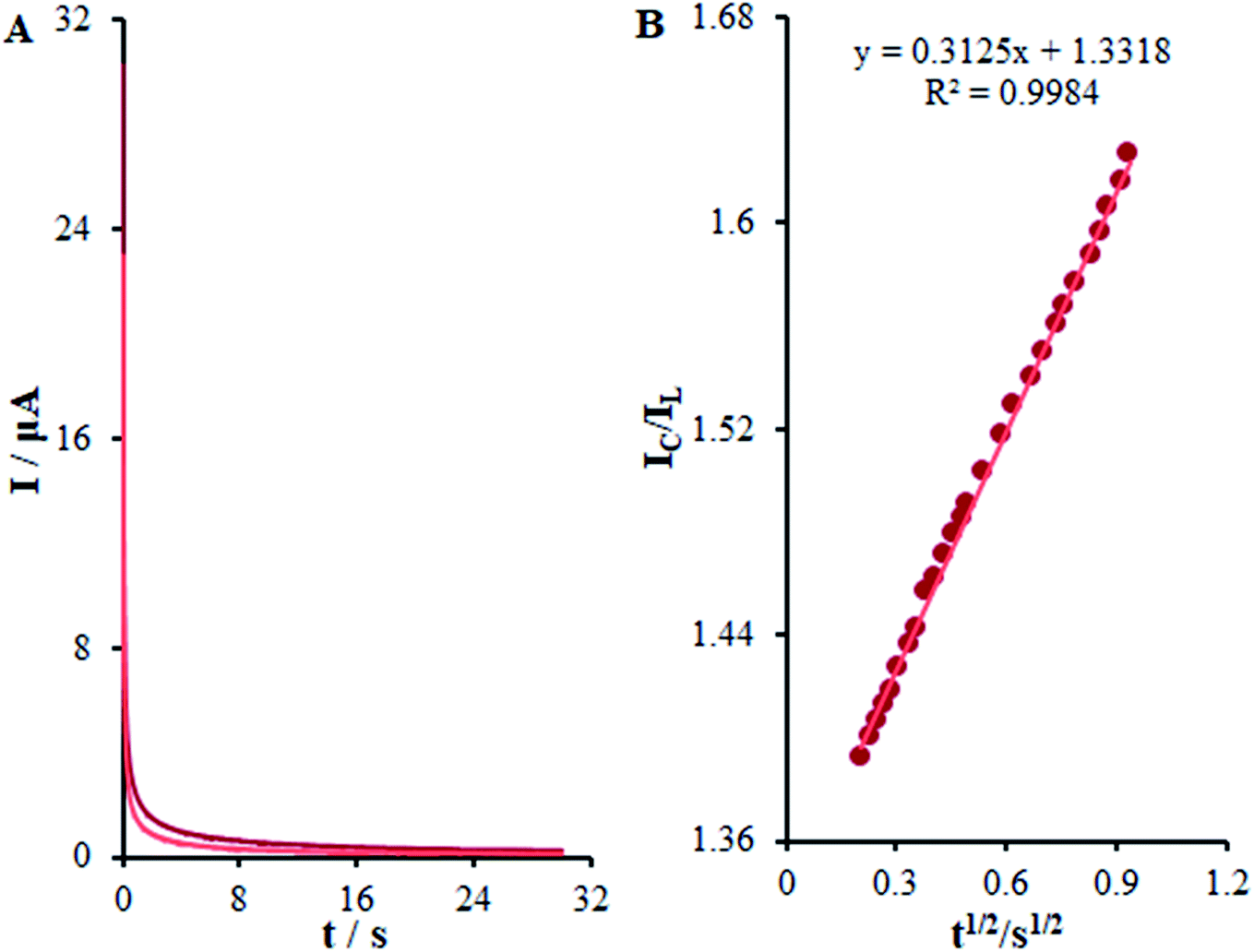
Fig. 10, the values predicted for
Kh were 3.1 × 10
3 mol
−1 L s
−1 for methadone. Consequently, we observed one of the main properties of maximum catalysis for the catalytic oxidation of methadone at the β-MnO
2-NF/GCE surface-illustrated
Kh value.
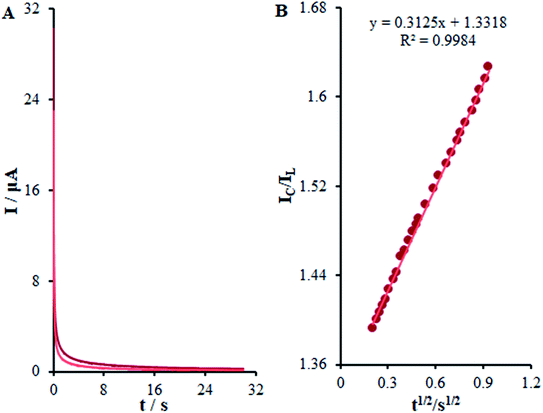 |
| | Fig. 10 (A) Chronoamperometric responses on β-MnO2-NF/GCE in the absence or presence of 0.01 mM methadone, in 0.1 M (pH 7.0); (B) Plot of IC/IL vs. t1/2. | |
3.7. The simultaneous determination of morphine and methadone at the surface of β-MnO2-NF/GCE
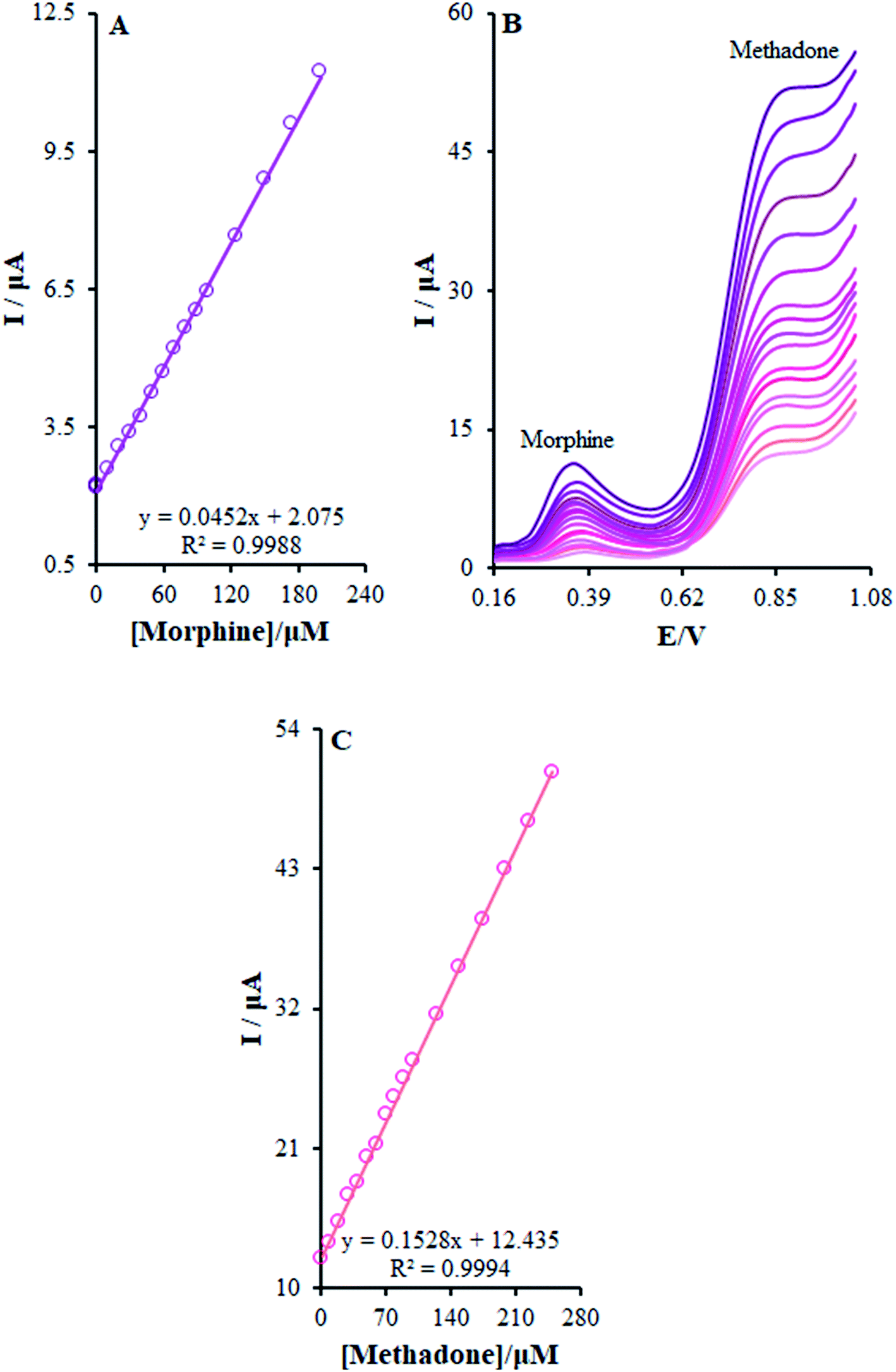
The present research utilized differential pulse voltammetry (DPV) with increased sensitivity for the possible simultaneous determination of methadone and morphine using the βH-MnO2-NF/GCE. For this reason, Fig. 11B is a schema of the DPVs for diverse methadone and morphine concentrations. As seen, a morphine slope value equal to 0.0452 μA μM−1 provided a linear range between 0.1 and 200.0 μM. Moreover, the limit of determination of morphine equaled 5.6 nM (Fig. 11A). In addition, the slope value of methadone (i.e., 0.1528 μA μM−1) provided a linear range between 0.1 and 250.0 μM. Furthermore, the limit of methadone determination equaled 8.3 nM (Fig. 11C).
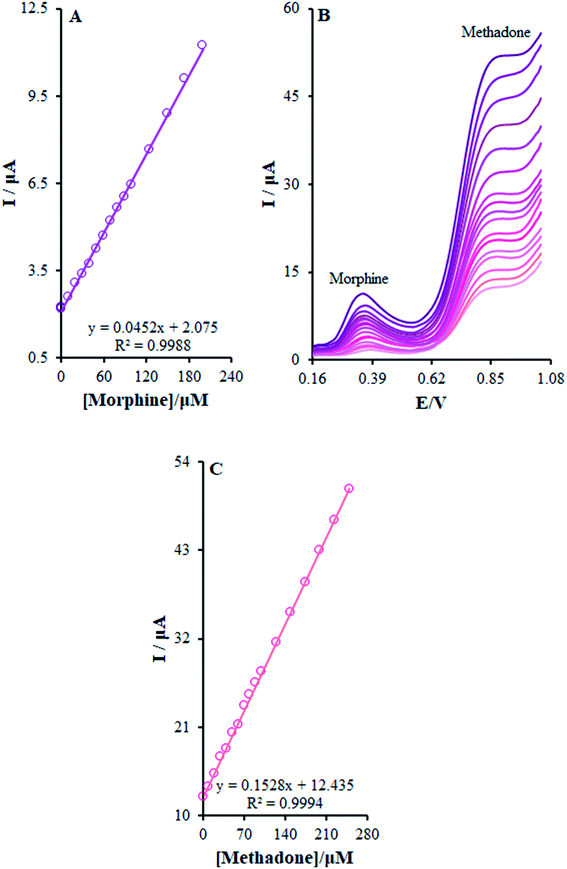 |
| | Fig. 11 (B) DPVs of β-MnO2-NF/GCE in 0.1 M (pH 7.0) containing different concentrations of morphine and methadone (from inner to outer curve): 0.1 + 1.0, 0.5 + 10.0, 1.0 + 20.0, 10.0 + 30.0, 20.0 + 40.0, 30.0 + 50.0, 40.0 + 60.0, 50.0 + 70.0, 60.0 + 80.0, 70.0 + 90.0, 80.0 + 100.0, 90.0 + 125.0, 100.0 + 150.0, 125.0 + 175.0, 150.0 + 200.0, 175.0 + 225.0, and 200.0 + 250.0 μM. (A) and (C) Plots of the electrocatalytic peak current as a function of morphine and methadone concentration in the range of 0.1 to 200.0 μM and 0.1 to 250.0, respectively. | |
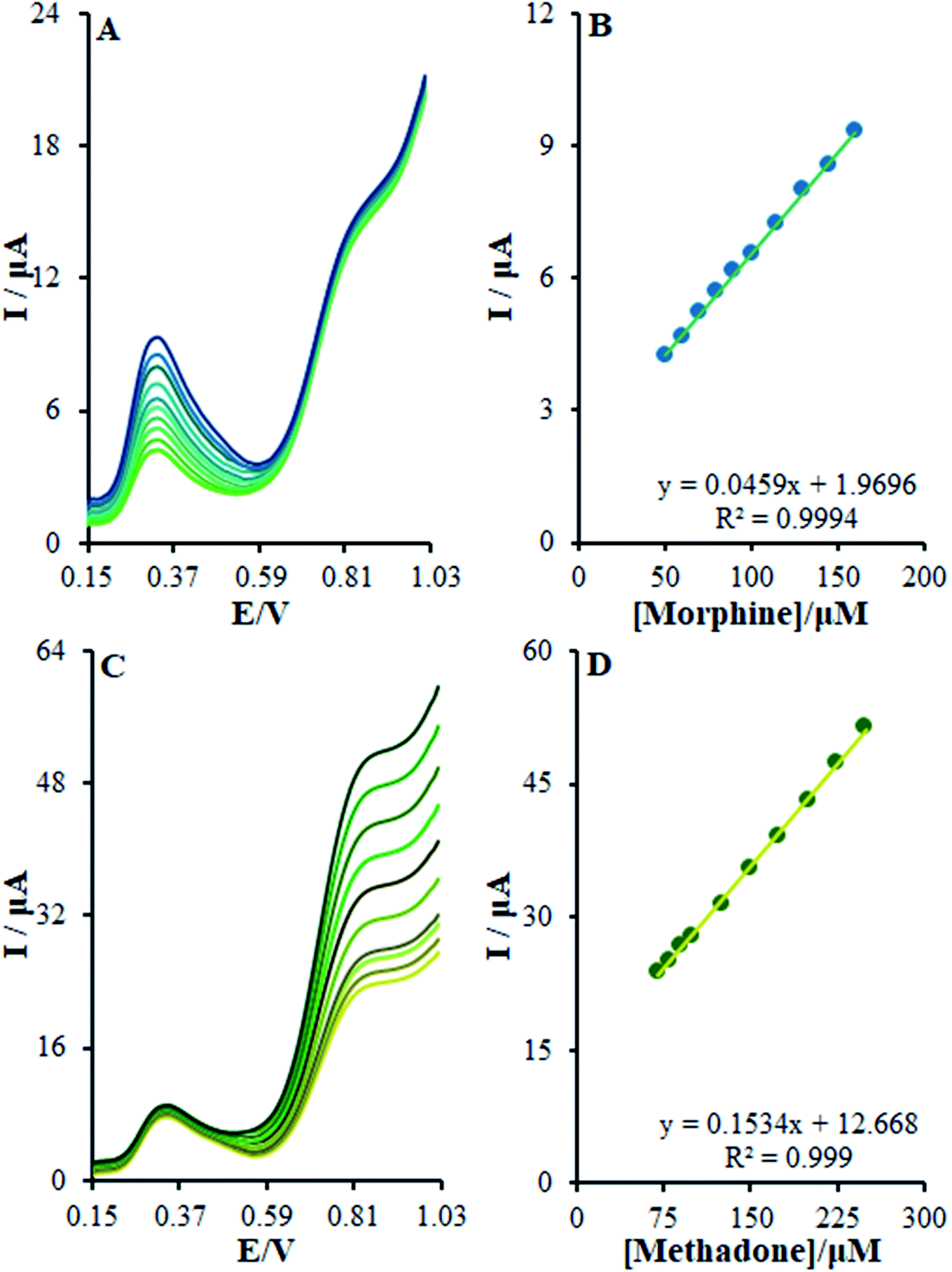
For determining the functions of βH-MnO2-NF/GCE for the simultaneous determination of methadone and morphine, the electro-oxidation of all the samples in the mixture was evaluated in the course of the changes in one sample concentration and the fixing concentration of the other species. Fig. 12A depicts the DPVs of morphine in the presence of 20.0 μM methadone. Considering the linear function Ip,morphine (μA) = 0.0459 + 1.9696 Cmorphine (μM) (R2 = 0.9994), the peak current experienced a linear enhancement from 50.0 to 160.0 μM in the concentration of morphine. Fig. 12C presents the DPV of methadone in the presence of 150.0 μM morphine. Moreover, with the linear function Ip,methadone (μA) = 0.1534 + 12.668 Cmethadone (μM) (R2 = 0.999), a linear increase from 70.0 to 250.0 μM was observed in the peak current of the concentration of methadone.
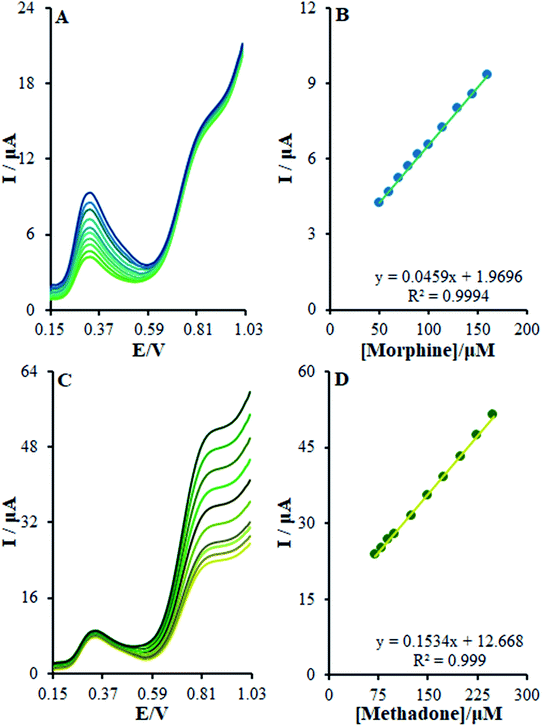 |
| | Fig. 12 (A) DPVs of β-MnO2-NF/GCE in 0.1 M (pH 7.0) containing 20.0 μM of methadone and different concentrations of morphine (from inner to outer curve): 50.0, 60.0, 70.0, 80.0, 90.0, 100.0, 115.0, 130.0, 145.0, and 160.0 μM. (B) Analytical curve from morphine, (C) 150.0 μM of AM, and different concentrations of methadone (from inner to outer curve): 70.0, 80.0, 90.0, 100.0, 125.0, 150.0, 175.0, 200.0, 225.0, and 250.0 μM, (D) analytical curve from methadone. | |
3.8. Interference, stability, and reproducibility of the β-MnO2 nanoflower-modified electrode
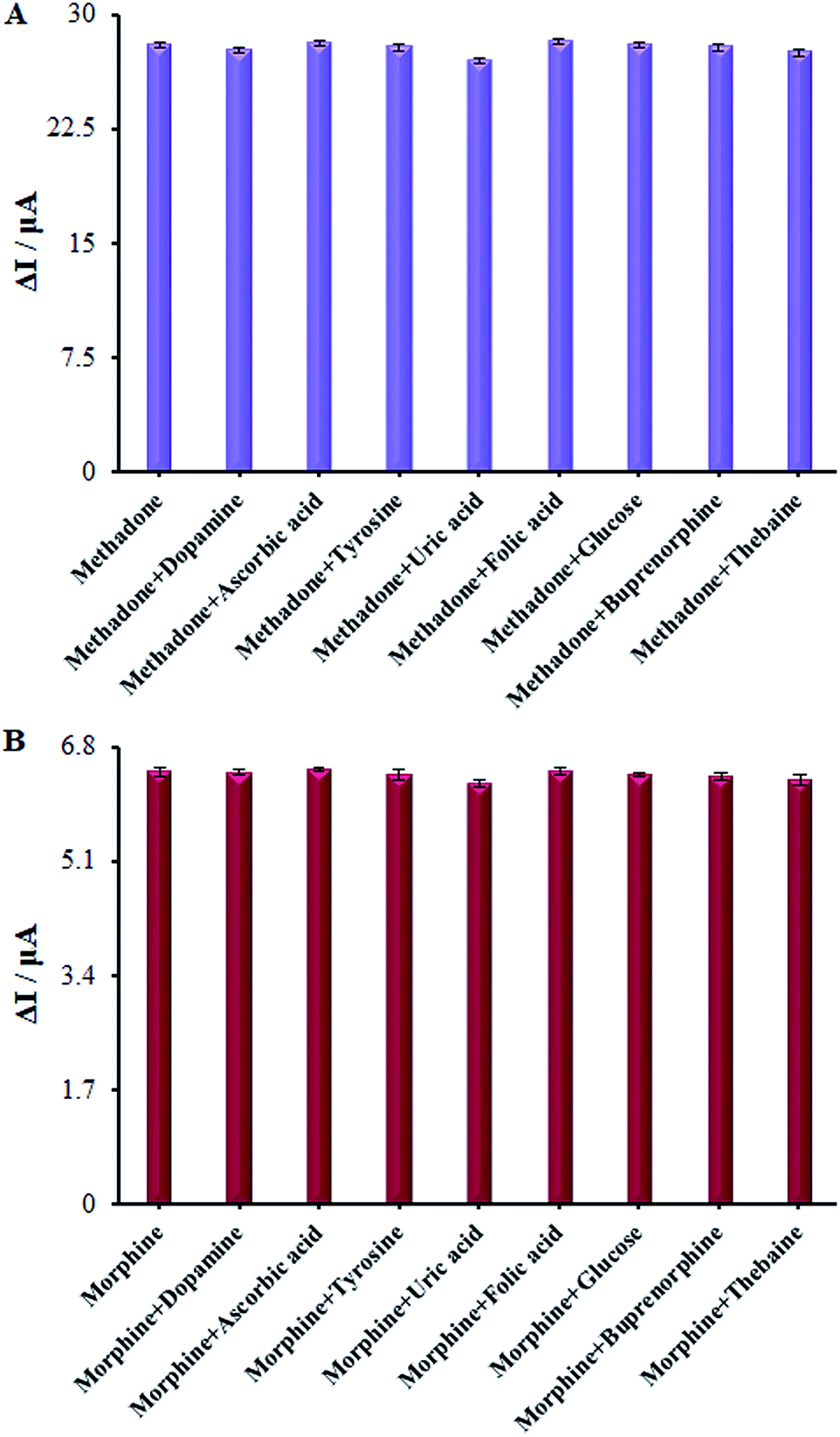
Numerous analogs such as dopamine, ascorbic acid, uric acid, folic acid, glucose, buprenorphine, and thebaine were used for checking the interference of the modified electrode, which was chosen as target molecules for investigating the influence on β-MnO2-NF/GCE (Fig. 13A and B). These changes may be illustrated by the reasonable selectivity of β-MnO2-NF/GCE for methadone and morphine.
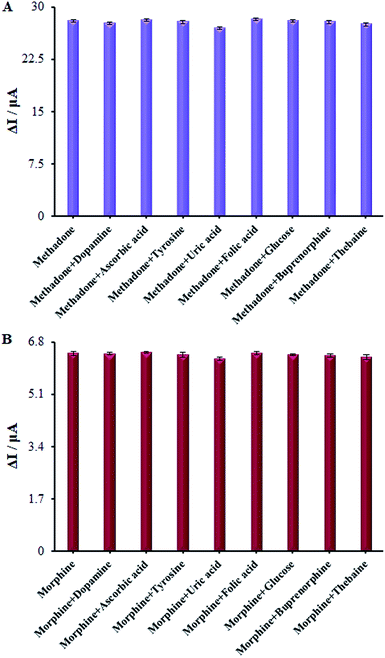 |
| | Fig. 13 (A) The columns are the current change of β-MnO2-NF/GCE in 0.1 M PBS (pH = 7.0) solution containing (a) 100.0 μM methadone, (b) a + 1.0 mM ascorbic acid, (c) a + 4.5 1.0 mM uric acid, (d) a + 4.5 1.0 mM tyrosine, (e) a + 1.0 mM naltrexone, (f) a + 1.0 mM Heroine, (g) a + 1.0 mM papaverine, (h) a + 1.0 mM buprenorphine, and (i) a + 1.0 mM thebaine. (B) The columns are the current change of β-MnO2-NF/GCE in 0.1 M PBS (pH = 7.0) solution containing (a) 100.0 μM Morphine, (b) a + 1.0 mM ascorbic acid, (c) a + 4.5 1.0 mM uric acid, (d) a + 4.5 1.0 mM tyrosine, (e) a + 1.0 mM naltrexone, (f) a + 1.0 mM heroine, (g) a + 1.0 mM papaverine, (h) a + 1.0 mM buprenorphine, and (i) a + 1.0 mM thebaine. | |
We evaluated the long-term stability of β-MnO2-NF/GCE for 30 days (Fig. 14). Prior to using the modified electrode in the course of experiment, we maintained it in the atmosphere and repeated the tests. With regard to the cyclic voltammograms, there were no changes in the oxidation peak potentials of methadone but the peak current shows a decrease of 2.94% compared to the initial response.
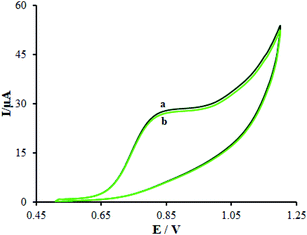 |
| | Fig. 14 (a) CVs of β-MnO2-NF/GCE in 0.1 M PBS (pH 7.0) containing 100.0 μM methadone and (b) after 30 days. | |
In addition, for the oxidation of methadone and its products, the CVs were recorded for examining the antifouling properties of the modified electrode. We registered these cyclic voltammograms around methadone following potential cycling for fifteen times at 50 mV s−1 scan rate. However, these variations were not detected at the peak potential as it decreased by <2.72%. Therefore, the sensitivity and a decrease in the analytes and the related oxidation products' fouling impacts increased using the modified β-MnO2-NF/GCE.
3.9. Application of β-MnO2-NF/GCE for the determination of morphine and methadone in pharmaceutical samples, urine, and saliva
According to the research design, we determined the utility and reliability of the suggested modified electrode in real samples with distinct matrices for confirming the β-MnO2-NF/GCE utility. Therefore, various volumes of morphine and methadone were used to spike the tablet and ampoule solutions and saliva and urine samples, as procured in Part 2.5. For this purpose, the standard addition procedure was used to carry out voltammetric measurement. Satisfactory recovery of the experimental results was found for morphine and methadone. In addition, the proposed electrodes productivity was demonstrated for the voltammetric detection of morphine and methadone in a similar solution of real samples. Table 1 presents the outputs.
Table 1 Determination of morphine and methadone in morphine ampoules, methadone tablets, saliva, and urine samples. All the concentrations are in μM (n = 5)
| Sample |
Spiked |
Found |
Recovery (%) |
| Morphine |
Methadone |
Morphine |
Methadone |
Morphine |
Methadone |
| Morphine ampoule |
5.0 |
10.0 |
5.1 ± 1.6 |
9.9 ± 2.3 |
102.0 |
99.0 |
| 10.0 |
20.0 |
9.8 ± 2.9 |
20.3 ± 1.7 |
98.0 |
101.5 |
| Methadone tablets |
15.0 |
7.5 |
15.5 ± 1.8 |
7.3 ± 3.3 |
103.3 |
97.3 |
| 25.0 |
12.5 |
25.1 ± 3.1 |
12.7 ± 2.2 |
100.4 |
101.6 |
| Saliva |
20.0 |
15.0 |
20.3 ± 3.1 |
15.1 ± 1.9 |
101.5 |
100.6 |
| 30.0 |
20.0 |
29.8 ± 2.7 |
20.4 ± 3.2 |
99.3 |
102.0 |
| Urine |
30.0 |
25.0 |
29.6 ± 2.4 |
25.2 ± 1.8 |
98.6 |
100.8 |
| 40.0 |
35.0 |
39.6 ± 1.9 |
34.6 ± 2.5 |
99.0 |
98.8 |
3.10. Comparison of the proposed method with literature methods
Table 2 presents a comparison of the analytical performance of β-MnO2-NF/GCE prepared in this research with other sensors involved in morphine analysis.1,4,5,32–40 The proposed method was inferior in terms of detection limit in comparison to the previously reported methods in the literature with the exception of two provided in ref. 1, 37–39, and 40. The advantage of the proposed method in this approach over the ref. 37 and 40 is the very sophisticated method used for the synthesis of the modifier, while this research employs a simple and one-step method for synthesizing the modifier. The strength of our research compared to ref. 1, 38 and 39 was employing non-destructive and non-toxic modifiers, and inexpensive materials (β-MnO2-NF versus carbon nanotubes, gold nanoparticles, phthalocyanines, and poly(3,4-ethylene-dioxythiophene)). However, it is noteworthy that the methods in ref. 37–40 are indirect for estimating morphine and do not utilize simultaneous determination with other compounds. The proposed method is superior as it is simple, utilizes simultaneous methadone and morphine determination, and does not need any pre-treatment processes. This proves that β-MnO2-NF/GCE possesses favorable analytical properties for methadone and morphine determination in terms of the extremely low detection limit, wide linear dynamic range, excellent repeatability and reproducibility, and high sensitivity compared to the methods mentioned in the literature.
Table 2 Comparison between various electroanalytical methods for the determination of methadone and morphine with the proposed method
| Method |
Modifier |
Detection limit |
Linear working range |
Ref. |
| Methadone |
| Voltammetry |
Functionalized multi-walled carbon nanotubes |
0.28 μM |
0.5–100.0 μM |
4 |
| Potentiometric |
MWCNT |
0.01 μM |
0.01–4600.0 μM |
5 |
| Voltammetry |
MWCNT |
87 nM |
0.01–15.0 μM |
32 |
| Voltammetry |
Cysteic acid film and gold nanoparticles |
0.14 μM |
0.024–12.67 μM |
33 |
| Voltammetry |
β-MnO2 nanoflowers |
5.6 nM |
0.1–200.0 μM |
This work |
|
| Morphine |
| Voltammetry |
MWCNT and fern-like La3+-CuO nanoleaves |
8.0 nM |
0.05–325.0 μM |
1 |
| Voltammetry |
Pretreatment of glassy carbon electrode |
0.2 μM |
4.0–100.0 μM |
34 |
| Voltammetry |
MWCNT |
0.361 μM |
2.0–300.0 μM |
35 |
| Voltammetry |
Glassy carbon electrode |
10.0 nM |
0.1–20.0 μM |
36 |
| Voltammetry |
Electrospun magnetic nanofibers |
1.9 nM |
0.0033–245 μM |
37 |
| Voltammetry |
Gold nanoparticles and different metal phthalocyanines |
5.48 nM |
0.4–900.0 μM |
38 |
| Voltammetry |
poly(3,4-ethylene-dioxythiophene)/gold-nanoparticles composite |
0.428 nM |
2.0–18.0 μM |
39 |
| Voltammetry |
Chitosan-coated magnetic nanoparticle |
3.0 nM |
0.01–720.0 μM |
40 |
| Voltammetry |
β-MnO2 nanoflowers |
8.3 nM |
0.1–250.0 μM |
This work |
4. Conclusion
This research utilized an easy hydro-thermal technique for the synthesis of β-MnO2 nano-flowers. Therefore, we examined the electro-chemical behavior of methadone and morphine at β-MnO2-NF/GCE in an aqueous buffered solution. The outputs showed a remarkable increase in the oxidation signals of methadone and a shift in their oxidation potentials to lower values because of the larger surface area, higher sorption abilities, and multiple active sites of β-MnO2-NF/GCE. According to the results, our newly-developed modified electrode could be useful for the voltammetric detection of morphine and methadone with favorable features such higher sensitivity, sub-nanomolar LOD, and decline in the overvoltage. In addition, it showed more an acceptable capability for the usual analyses of morphine and methadone in real samples.
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- M. Rajaei, M. M. Foroughi, Sh. Jahani, M. Shahidi Zandi and H. Hassani Nadiki, J. Mol. Liq., 2019, 284, 462 CrossRef CAS.
- J. W Boland, M. Johnson, D. Ferreira and D. J. Berry, J. Palliative Med., 2018, 32, 1222 CrossRef.
- J. G. Liu, X. P. Liao, Z. H. Gong and B. Y. Qin, Eur. J. Pharmacol., 1999, 373, 233 CrossRef CAS.
- M. Amiri-Aref, J. B. Raoof and R. Ojani, Colloids Surf., B, 2013, 109, 287 CrossRef CAS.
- M. Ardeshiri and F. Jalali, Mater. Sci. Eng., C, 2016, 63, 30 CrossRef CAS.
- K. A. Harris Jr, J. H. Arnsten, H. Joseph, J. Hecht, I. Marion, P. Juliana and M. N. Gourevitch, J. Subst. Abus. Treat., 2006, 31, 433 CrossRef.
- R. George, M. Lobb, A. Haywood, S. Khan, J. Hardy, P. Good, S. Hennig and R. Norris, Talanta, 2016, 149, 142 CrossRef CAS.
- P. Zahedi, S. S. H. Davarani, H. R. Moazami and S. Nojavan, J. Pharm. Biomed. Anal., 2016, 117, 485 CrossRef CAS.
- A. M. Bermejo, A. C. S. Lucas, M. J. Tabernero and P. Fernandez, Anal. Lett., 2000, 33, 739 CrossRef CAS.
- M. Jafari-Nodoushana, J. Barzinb and H. Mobedi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1011, 163 CrossRef.
- R. Montero, M. Gallego and M. Valcarcel, Anal. Chim. Acta, 1990, 234, 433 CrossRef CAS.
- M. M. Foroughi and M. Ranjbar, J. Mater. Sci., 2017, 28, 1359 CAS.
- M. Vakili Fathabadi, H. Hashemipour Rafsanjani, M. M. Foroughi, Sh. Jahani and N. Arefi Nia, J. Electrochem. Soc., 2020, 167, 027509 CrossRef.
- H. Mirzaei, A. K. Nasiri, R. Mohamadee, H. Yaghoobi, M. Khatami, O. Azizi, M. A. Zaimy and H. Hakim Azizi, Microchem. J., 2018, 142, 343 CrossRef CAS.
- N. Sheibani, M. Kazemipour, Sh. Jahani and M. M. Foroughi, Microchem. J., 2019, 149, 103980 CrossRef CAS.
- N. Farvardin, Sh. Jahani, M. Kazemipour and M. M. Foroughi, Anal. Methods, 2020, 12, 1767 RSC.
- S. R. Yan, M. M. Foroughi, M. Safaei, Sh. Jahani, N. Ebrahimpour, F. Borhani, N. Rezaei Zade Baravati, Z. Aramesh-Boroujeni and L. K. Foog, RSC Adv., 2020, 155, 184 CAS.
- A. Mahanta, K. Barman and S. k. Jasimuddin, RSC Adv., 2019, 9, 38713 RSC.
- N. Jandaghi, Sh. Jahani, M. M. Foroughi, M. Kazemipour and M. Ansari, Microchim. Acta, 2020, 187, 24 CrossRef CAS.
- P. Viswanathan, S. Manivannan and R. Ramaraj, RSC Adv., 2015, 5, 54735 RSC.
- N. Arefi Nia, M. M. Foroughi, Sh. Jahani, M. Shahidi Zandi and N. Rastakhiz, J. Electrochem. Soc., 2019, 166, B489 CrossRef.
- M. S. Ahmed, D. Park and S. Jeon, J. Power Sources, 2016, 308, 180 CrossRef CAS.
- R. W. Chen, J. Yan, Y. Liu and J. H. Li, J. Phys. Chem. C, 2015, 119, 8032 CrossRef CAS.
- M. S. Ahmed and Y. B. Kim, Sci. Rep., 2017, 7, 43279 CrossRef.
- H. Begum, M. S. Ahmed and S. Jeon, J. Nanosci. Nanotechnol., 2017, 17, 4961 CrossRef CAS.
- H. Begum, M. Shamsuddin Ahmed and S. Jeon, Electrochim. Acta, 2019, 296, 235 CrossRef CAS.
- A. Bahloul, B. Nessark, F. Habelhames and C. M. Julien, Ionics, 2011, 17, 239 CrossRef CAS.
- X. Wang and Y. Li, J. Am. Chem. Soc., 2002, 124, 2880 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical methods fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- M. M. Foroughi, Sh. Jahani and M. Rajaei, J. Electrochem. Soc., 2019, 166, B1300 CrossRef CAS.
- Z. Fathi, Sh. Jahani, M. Shahidi Zandi and M. M. Foroughi, Anal. Bioanal. Chem., 2020, 412, 1011 CrossRef CAS.
- E. Alipour, M. R. Majidi and O. Hoseindokht, J. Chin. Chem. Soc., 2015, 62, 461 CrossRef CAS.
- G. Karim-Nezhad and Z. Khorablou, Anal. Bioanal. Electrochem., 2017, 9, 698 Search PubMed.
- F. Li, J. Song, D. Gao, Q. Zhang, D. Han and L. Niu, Talanta, 2009, 79, 845 CrossRef CAS.
- J. M. P. J. Garrido, C. Delerue-Matos, F. Borges, T. R. A. Macedo and A. M. Oliveira-Brett, Electroanalysis, 2004, 16, 1419 CrossRef CAS.
- F. Li, J. Song, C. Shan, D. Gao, X. Xu and L. Niu, Biosens. Bioelectron., 2010, 125, 1408 CrossRef.
- G. Bahrami, H. Ehzari, S. Mirzabeigy, B. Mohammadi and E. Arkan, Mater. Sci. Eng., C, 2020, 106, 110183 CrossRef CAS.
- N. F. Atta, A. Galal, F. M. Abdel-Gawad and E. F. Mohamed, Electroanalysis, 2014, 26, 1 CrossRef.
- N. F. Atta, A. Galal and E. H. El-Ads, Int. J. Electrochem. Sci., 2014, 9, 2113 Search PubMed.
- S. Dehdashtian, M. B. Gholivand, M. Shamsipur and S. kariminia, Mater. Sci. Eng., C, 2016, 58, 53 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Mohammad Mehdi Foroughi
bc,
Mohammad Mehdi Foroughi