
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of Gly-Gly-type (E)-methylalkene and (Z)-chloroalkene dipeptide isosteres and their application to 14-mer RGG peptidomimetics†
Hikari Okita‡
a,
Yuna Kato‡b,
Tatsuki Masuzawac,
Kosuke Araib,
Sayuri Takeoa,
Kohei Sato ab,
Nobuyuki Maseabd,
Takanori Oyoshic and
Tetsuo Narumi*abe
ab,
Nobuyuki Maseabd,
Takanori Oyoshic and
Tetsuo Narumi*abe
aDepartment of Applied Chemistry and Biochemical Engineering, Faculty of Engineering, Shizuoka University, Shizuoka, 432-8561, Japan. E-mail: narumi.tetsuo@shizuoka.ac.jp
bDepartment of Engineering, Graduate School of Integrated Science and Technology, Shizuoka University, Shizuoka, 432-8561, Japan
cDepartment of Chemistry, Graduate School of Integrated Science and Technology, Shizuoka University, Shizuoka, 422-8529, Japan
dGreen Energy Research Division, Research Institute of Green Science and Technology, Shizuoka University, Shizuoka, 432-8561, Japan
eGreen Chemistry Research Division, Research Institute of Green Science and Technology, Shizuoka University, Shizuoka, 432-8561, Japan
First published on 10th August 2020
Abstract
Stereoselective and efficient synthesis of Gly-Gly-type (E)-methylalkene and (Z)-chloroalkene dipeptide isosteres is realized by organocuprate-mediated single electron transfer reduction. The synthetic isosteres can be used in Fmoc-based solid phase peptide synthesis, resulting in the preparation of the 14-mer RGG peptidomimetics containing an (E)-methylalkene or a (Z)-chloroalkene unit.
Glycylglycine (Gly-Gly) is the smallest dipeptide and has been synthesized by many approaches in the last 100 years.1 Due to its versatile nature and ready availability, glycylglycine has been used as a chemical probe2 and buffer3 in biochemical studies and also as a reagent which can enhance the solubility of overexpressed proteins.4 In addition to the utility of Gly-Gly itself, oligoglycine (oligo-Gly) motifs are notable for being more flexible and less-functionalized than any combination of other amino acids. These features are useful in bioconjugation strategies which link multiple biomolecules without interfering with the function of each biomolecule, allowing synthesis of bioconjugated artificial molecules including dimeric, multi-domain, and fusion proteins.5 The flexibility of oligo-Gly enables the formation of unusual secondary structures of peptides and proteins.6 Thus, the oligo-Gly motif can be found in the biologically important peptides and proteins such as Met/Leu-enkephalin (
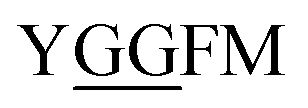 and
and 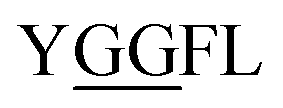 ), the C-terminus of ubiquitin
), the C-terminus of ubiquitin 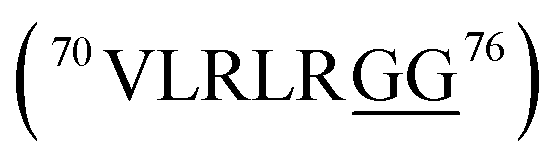 , ctenidin,7 shepherin I,8 and DNA/RNA-binding proteins with repeated
, ctenidin,7 shepherin I,8 and DNA/RNA-binding proteins with repeated 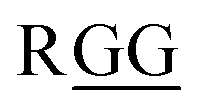 sequences related to the various physiological processes via protein–protein and protein–nucleic acids interactions.9 These proteins relate with gene expression, DNA damage signal and apoptosis, however, the detail effects of Gly-Gly with steric and electronic factors to these functions are unknown. Given the importance of oligo-Gly in various fields, non-hydrolyzable peptidomimetics of oligo-Gly could be attractive building blocks for the synthesis of novel bioconjugated molecules and complex peptidomimetics with improved chemical stability and functionality. For example, even the Gly-Gly dipeptide mimic with the tetra-substituted alkene unit replacing the Gly-Gly peptide bond has been shown to promote the β-hairpin formation, and is thus the smallest peptidomimetic that is known to control a peptide structure.10 There are two reports of the synthesis of Gly-Gly-type fluoroalkene dipeptide isosteres.11 However, the poor synthetic access to such molecules has hindered their application to the peptidomimetics. Our long-standing interest in the drug discovery with amide-to-alkene isosteric switching prompted this investigation into the stereoselective synthesis of Gly-Gly-type (E)-methylalkene and (Z)-chloroalkene dipeptide isosteres.
sequences related to the various physiological processes via protein–protein and protein–nucleic acids interactions.9 These proteins relate with gene expression, DNA damage signal and apoptosis, however, the detail effects of Gly-Gly with steric and electronic factors to these functions are unknown. Given the importance of oligo-Gly in various fields, non-hydrolyzable peptidomimetics of oligo-Gly could be attractive building blocks for the synthesis of novel bioconjugated molecules and complex peptidomimetics with improved chemical stability and functionality. For example, even the Gly-Gly dipeptide mimic with the tetra-substituted alkene unit replacing the Gly-Gly peptide bond has been shown to promote the β-hairpin formation, and is thus the smallest peptidomimetic that is known to control a peptide structure.10 There are two reports of the synthesis of Gly-Gly-type fluoroalkene dipeptide isosteres.11 However, the poor synthetic access to such molecules has hindered their application to the peptidomimetics. Our long-standing interest in the drug discovery with amide-to-alkene isosteric switching prompted this investigation into the stereoselective synthesis of Gly-Gly-type (E)-methylalkene and (Z)-chloroalkene dipeptide isosteres.
In this report, we describe the beginning of our oligo-Gly-based peptidomimetic study with the stereoselective synthesis of Gly-Gly-type (E)-methylalkene and (Z)-chloroalkene dipeptide isosteres and their use in Fmoc-based solid phase peptide synthesis (SPPS). In the first application of isosteres of this type to access complex peptidomimetics, we synthesize 14-mer RGG peptidomimetics containing (E)-methylalkene or (Z)-chloroalkene unit as surrogates for a Gly-Gly peptide bond. These two isosteres were selected because the potentials of both isosteres have not been fully uncovered, though there are several promising examples.12,13 Since the carbonyl oxygen equivalents of those isosteres are similar in their size14 but differ in their electronic properties,15 comparative studies of these isosteres would have the advantage of exploring the role of peptide bonds in terms of their steric and electronic natures. This work also uncovered the unique ability of the Gly-Gly-type (Z)-chloroalkene isostere to induce β-turn structures in the almost unfoled peptides (Fig. 1).
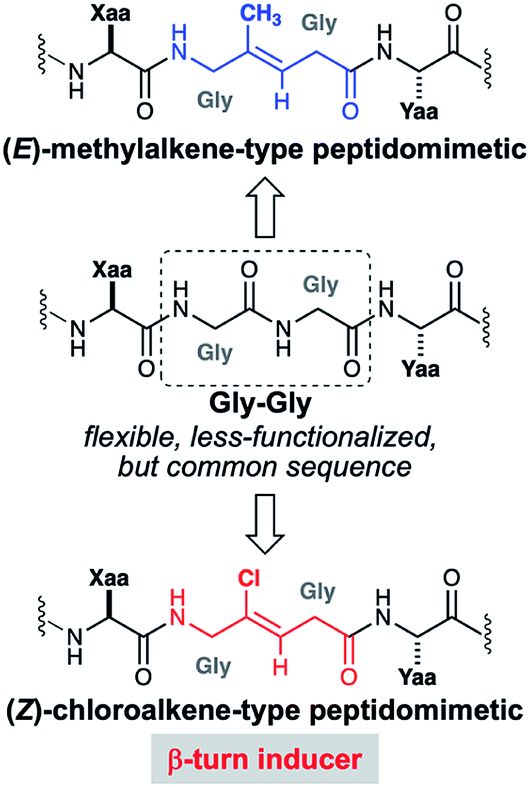 | ||
| Fig. 1 Gly-Gly peptide and its alkene-type peptidomimetics. | ||
The main challenges in this study include the stereoselective formation of the (E)-methylalkene and (Z)-chloroalkene moieties as the surrogates of the Gly-Gly trans-peptide bond, together with the control of the olefine isomerization under the condition of the isostere synthesis and Fmoc-based SPPS. There are several synthetic approaches toward tri-substituted alkene-type isosteres, including the Overman rearrangement,13 the SN2′-type opening of alkenylaziridines,12a Cu-mediated CF3-coupling of vinyl iodide,16 and cross-couplings of vinyl stannane.17 Organocuprate-mediated reactions of α,β-unsaturated carbonyl compounds with γ-leaving group(s) are also powerful methods to produce tri-substituted alkene-type isosteres, and are particularly suitable for the stereoselective synthesis of (Z)-fluoroalkene18 and (Z)-chloroalkene isosteres.19
The preparation of Gly-Gly-type alkene dipeptide isosteres was facilitated by the organocuprate-mediated SET reduction that was used in the stereoselective formation of the (E)-methylalkene and (Z)-chloroalkene moieties. Scheme 1 shows the synthesis of (E)-methylalkene isosteres (5). A Witting reaction of methacrolein (1) with ethyl (triphenylphosphoranylidene)acetate followed by epoxidation with m-CPBA afforded the alkenyl oxirane (2) which, treated with Gillman reagent (n-Bu2CuLi), produced the allylic alcohol (3) with high E-selectivity. A Mitsunobu reaction of 3 with Ns(Boc)NH and deprotection provided the Gly-Gly-type (E)-methylalkene dipeptide isostere 5 that can be applied to the Fmoc-based SPPS. All reactions leading to the synthesis of 5 were performed on a gram scale.
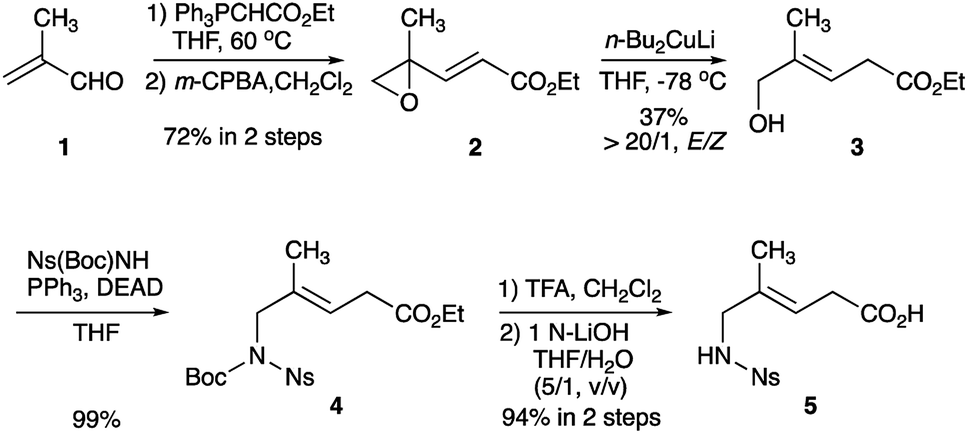 | ||
| Scheme 1 Synthesis of Gly-Gly-type (E)-methylalkene dipeptide isosteres (5). | ||
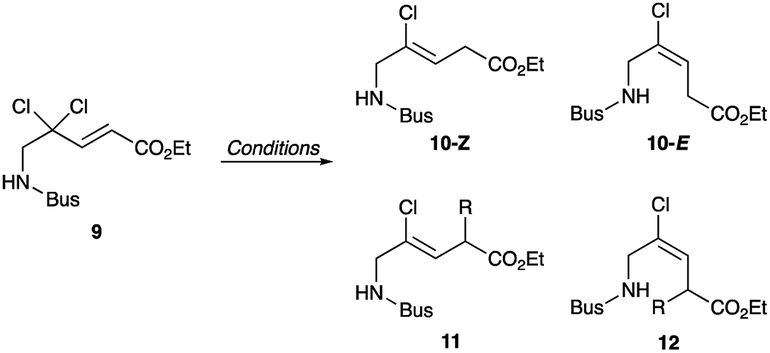
Synthesis of chloroalkene isostere (14) is shown in Scheme 2. Based on previous results for the successful, efficient synthesis of Val-Xaa-type chloroalkene isosteres,19c we assumed that a similar protocol would allow for the synthesis of 14. However, our attempts revealed that the Gly substrate used has different reactivity and selectivity compared to the other substrates, particularly in the SET reduction step. Consequently, Gly-specific reaction conditions are necessary. The nucleophilic addition of the lithium enolate of methyl dichloroacetate to the N-sulfinylaldimine (7), prepared from (±)-tert-butylsulfinamide (6) and paraformaldehyde and m-CPBA oxidation of 6 gave the N-tert-butylsulfonyl (Bus)-protected α,α-dichloro-β-amino ester (8). Precise control of the amount of DIBAL-H at low temperatures enables the partial reduction of 8, and this is followed by a Horner–Wadsworth–Emmons reaction to produce the corresponding (E)-enoate (9). Initial efforts to apply our established conditions for SET reduction with Me2CuLi identified the poor Z-selectivity of the reaction and also its propensity to form the α-methylated side products 11 and 12 (Table 1, entry 1). Changing the methyl groups on the copper to n-butyl groups decreased the formation of (E)-chloroalkene compounds and allowed the easy separation of 10-Z from side products (entry 2). Addition of Lewis bases such as HMPA, NMP, or DMSO proved to be ineffective for the reductive dechlorination step (entries 3–5). Bulky alkyl groups on copper can improve the product selectivity and favor the desired (Z)-chloroalkene product (10-Z) (entries 6–8). Although the organocuprates derived from tert-BuLi and tert-BuMgCl gave slightly better yields and selectivity (entries 7 and 8), we favoured the organocuprate derived from sec-BuLi due to its safety and reproducibility; treatment of the enoate (9) with sec-Bu2CuLi gave the (Z)-chloroalkene product (10-Z) in 67% yield (entry 6). Manipulations of the amine protecting group followed by acidic hydrolysis produced the Gly-Gly-type (Z)-chloroalkene dipeptide isostere (14). During the conversion of both esters 4 and 13 to the isosteres 5 and 14, suitable for Fmoc-based SPPS, we observed no olefin isomerization products.
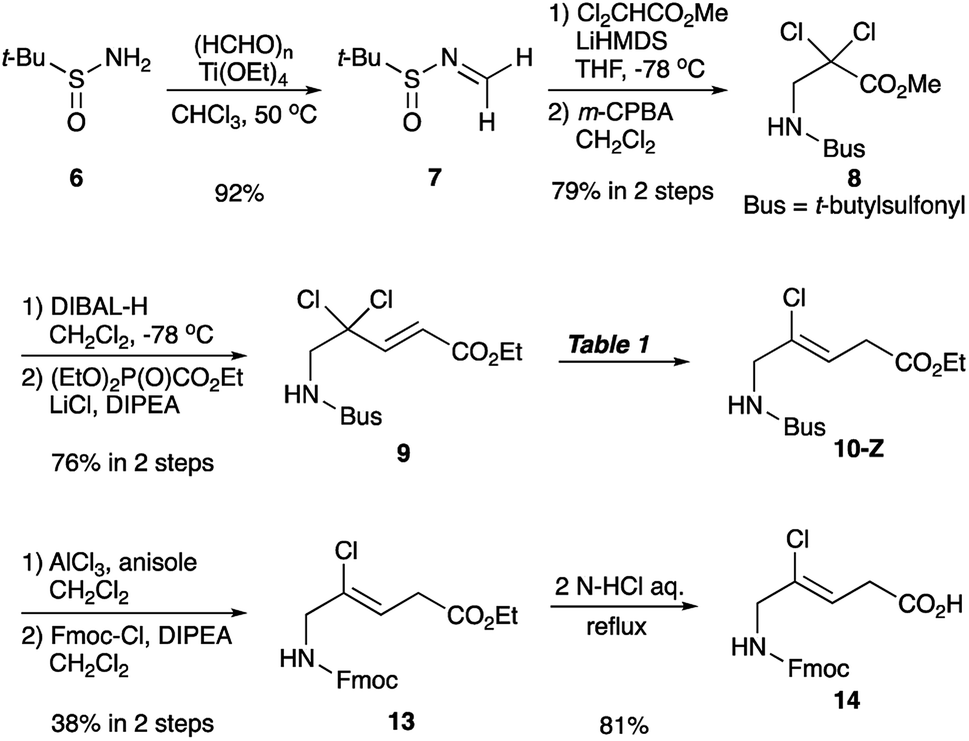 | ||
| Scheme 2 Synthesis of Gly-Gly-type (Z)-chloroalkene dipeptide isosteres (14). | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Yieldb (%) | |||
| 10-Z | 10-E | 11 | 12 | ||
| a All reactions were carried out at −78 °C for 30 min on a 0.25 mmol scale with 4 equiv. of organocuprates in the presence of metal salts.b Yield is determined by 1H NMR analysis of the crude mixture utilizing mesitylene as an internal standard.c R = Me.d R = n-Bu.e R = sec-Bu.f R = tert-Bu. | |||||
| 1 | Me2CuLi | 48 | 19 | 15c | 1c |
| 2 | n-Bu2CuLi | 55 | 0 | 25d | 20d |
| 3 | n-Bu2CuLi, HMPA | 0 | 0 | 46d | 13d |
| 4 | n-Bu2CuLi, NMP | 44 | 0 | 29d | 6d |
| 5 | n-Bu2CuLi, DMSO | 46 | 7 | 19d | 2d |
| 6 | sec-Bu2CuLi | 67 | 6 | 27e | 0 |
| 7 | tert-Bu2CuLi | 74 | 4 | 2f | 0 |
| 8 | tert-Bu2CuMgCl | 63 | 0 | 0 | 0 |
With these isosteres in hand, we explored their use in Fmoc-based SPPS for the preparation of peptidomimetics of the 14-mer RGG peptide derived from translocation in lipo-sarcoma/fused in sarcoma (TLS/FUS) related to the RNA processing (Schemes 3 and 4).20 Starting from the Rink Amide ChemMatrix resin, standard Fmoc-based SPPS with DIC/Oxyma for peptide couplings and 20% (v/v) piperidine/DMF for Fmoc removals were performed for the construction of the peptide resin (15). The synthesized isosteres were incorporated into the peptide-chain by HATU/DIPEA in DMF affording the peptide resins 16 and 18. For the synthesis of (E)-methylalkene-type peptidomimetic, deprotection of Ns group with thiophenol/K2CO3 in DMF and chain elongation followed by global deprotection with TFA/m-cresol/thioanisole/H2O (87.5/5/5/2.5, v/v/v/v) provided the desired (E)-methylalkene-type peptidomimetic (17). Synthesis of the (Z)-chloroalkene-type peptidomimetic (19) was achieved using standard conditions. NMR analysis of the purified peptidomimetics revealed that although (Z)-chloroalkene-type peptidomimetic (19) can be purified solely, a trace amount of olefin isomerized compounds of 17, possibly generated under the coupling, is observed as a side product and was difficult to remove from the desired product.21 Since we used a single coupling protocol with HATU for this study, optimization for the coupling without olefin isomerization is likely to be possible.
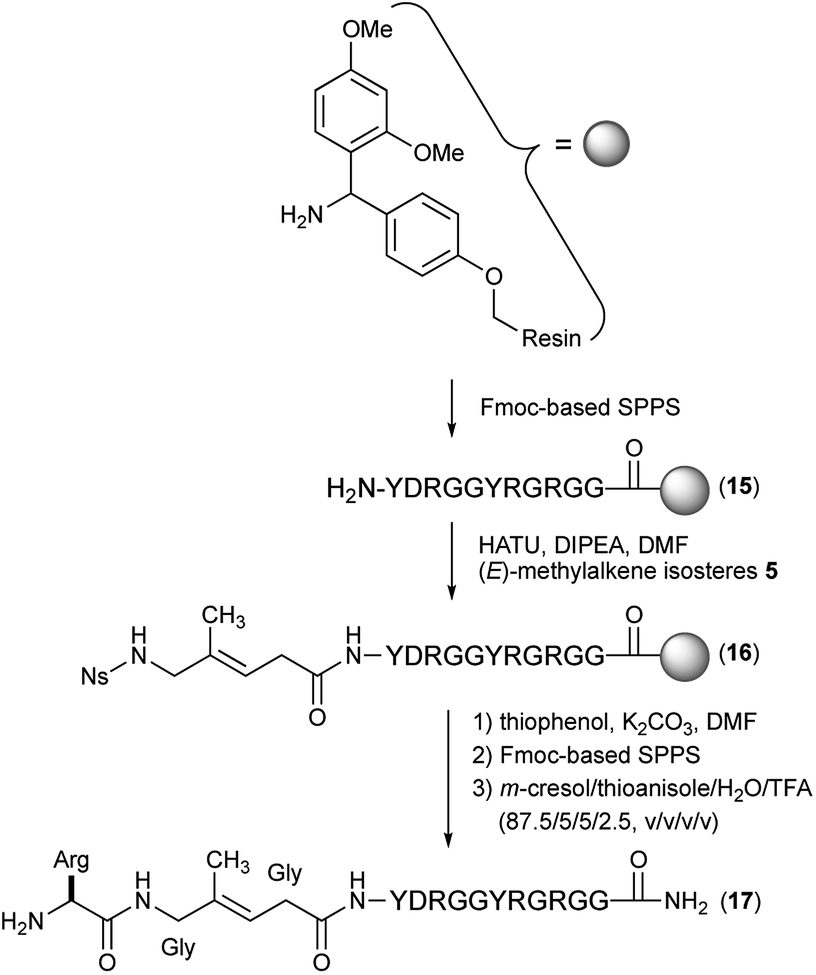 | ||
| Scheme 3 Synthesis of Gly-Gly-type (E)-methylalkene-type peptidomimetic (17). | ||
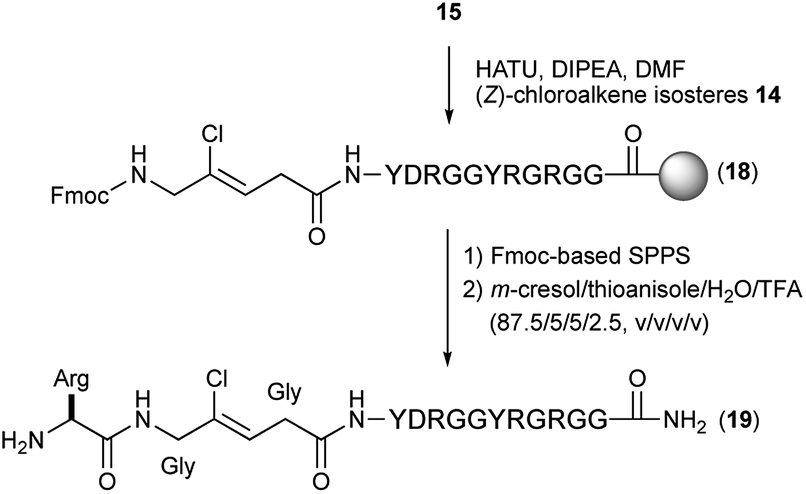 | ||
| Scheme 4 Synthesis of (Z)-chloroalkene-type peptidomimetic (19). | ||
It has been demonstrated that D-Ala-L-Ala-type (E)-methylalkene isostere sequence shows a higher preference for a type-II′ β-turn than the corresponding (E)-alkene isostere.12a To determine whether amide-to-alkene isosteric switching in Gly-Gly peptide bonds affects the ability of a peptide to form a β-turn structure, CD spectra were obtained for peptidomimetics (17 and 19) in 50 mM Tris–HCl (pH 7.5) with 100 mM KCl (Fig. 2). The native peptide (20) was included as the control. The CD spectra analysis of turn structures has been discussed in the literature, albeit with lower accuracies.22 Although (E)-methylalkene-type peptidomimetic (17) appears to be random coil, the spectra of 19 exhibited a minimal absorbance peak at 202 nm, which is a typical characteristic of a β-turn conformation.20 On the other hand, the peptide 20 appears to form a β-turn conformation slightly. These results indicated that isosteric switching of Gly-Gly peptide bond with a (Z)-chloroalkene unit can induce a β-turn conformation in the secondary structure of peptides and also that the β-turn inducing ability of (Z)-chloroalkene isosteres is superior to that of (E)-methylalkene isosteres. To the best of our knowledge, this is the first example of such drastic structural control effects of (Z)-chloroalkene isosteres on peptides. We speculated that the electronic effects of the chlorine substituent are responsible for the superior β-turn inducing ability. Efforts to determine their biological activity, including DNA/RNA-binding affinity, are currently in progress.
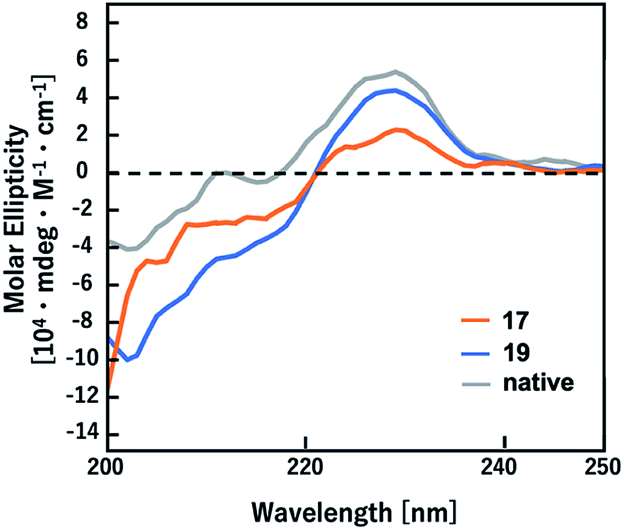 | ||
| Fig. 2 CD spectra of peptidomimetics (17 and 19) and the corresponding native peptide (20). | ||
Conclusions
In conclusion, we have achieved the synthesis of amide-to-alkene 14-mer peptidomimetics containing (E)-methylalkene or (Z)-chloroalkene dipeptide isosteres employing a combination of a key organocuprate-mediated SET reduction and Fmoc-based SPPS. Organocuprate-mediated reduction enables the rapid construction of two-types of isostere skeletons in good yields with high trans-selectivity. The use of organocuprates with bulky alkyl groups was found to improve the chemical yields and the product selectivity of the reductive dechlorination for the (Z)-chloroalkene isostere. The esters obtained in this ways can be converted to the Ns/Fmoc-protected acids without isomerization of the olefin and can be coupled with the N-terminus of peptides on a resin, affording the 14-mer RGG pepidomimetics containing an (E)-methylalkene or a (Z)-chloroalkene unit. Also, we have identified the β-turn inducing ability of the Gly-Gly-type (Z)-chloroalkene isostere. Such oligo-Gly motifs are common in peptides and proteins and show unique characteristics. Thus, the synthesized Gly-Gly-type isosteres can offer new opportunities for development of enzymatically stable and structurally defined bioconjugated molecules and peptidomimetics. Continuing studies on the applications of both isosteres to the synthesis of mimics of peptides and proteins are ongoing.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. S. Dunn, A. W. Butler and T. Deakers, J. Biol. Chem., 1932, 99, 217 CAS.
- For examples: (a) H. Arakawa, S. Saito, M. Kanagawa, H. Kamioka, K. Yano, K. Morimoto and T. Ogihara, Drug Metab. Pharmacokinet., 2014, 29, 470 CrossRef PubMed; (b) D. A. Buckingham, C. E. Davis, D. M. Foster and A. M. Sargeson, J. Am. Chem. Soc., 1970, 92, 5571 CrossRef CAS PubMed; (c) E. Nakashima and A. Tsuji, J. Pharmacobio-Dyn., 1985, 8, 623 CrossRef CAS PubMed.
- M. E. Smith and L. B. Smith, Biol. Bull., 1949, 96, 233 CrossRef CAS PubMed.
- S. Ghosh, S. Rasheedi, S. S. Rahim, S. Banerjee, R. K. Choudhary, P. Chakhaiyar, N. Z. Ehtesham, S. Mukhopadhyay and S. E. Hasnain, BioTechniques, 2004, 37, 418 CrossRef CAS PubMed.
- V. P. R. Chichili, V. Kumar and J. Sivaraman, Protein Sci., 2013, 22, 153 CrossRef PubMed.
- K. Mohle, M. Gußmann and H. J. Hofmann, J. Comput. Chem., 1997, 18, 11141 CrossRef.
- T. Baumann, U. Kämpfer, S. Schürch, J. Schaller, C. Largiadèr, W. Nentwig and L. Kuhn-Nentwig, Cell. Mol. Life Sci., 2010, 67, 2787 CrossRef CAS PubMed.
- C. Remuzgo, T. S. Oewel, S. Daffre, T. R. S. Lopes, F. H. Dyszy, S. Schreier, G. M. Machado-Santelli and M. T. Machini, Amino Acids, 2014, 46, 2573 CrossRef CAS PubMed.
- (a) P. Thandapani, T. R. O'Connor, T. L. Bailey and S. Richard, Mol. Cell, 2013, 50, 613 CrossRef CAS PubMed; (b) P. A. Chong, R. M. Vernon and J. D. F. Kay, J. Mol. Biol., 2018, 430, 4650 CrossRef CAS PubMed.
- R. R. Gardner, G. B. Liang and S. H. Gellman, J. Am. Chem. Soc., 1995, 117, 3280 CrossRef CAS.
- (a) S. Sano, Y. Kuroda, K. Saito, Y. Ose and Y. Nagao, Tetrahedron, 2006, 62, 11881 CrossRef CAS; (b) C. Calata, E. Pfund and T. Lequeux, Tetrahedron, 2011, 67, 1398 CrossRef CAS.
- (E)-Methylalkene-type: (a) P. Wipf, T. C. Henninger and S. J. Geib, J. Org. Chem., 1998, 63, 6088 CrossRef CAS PubMed; (b) S. Oishi, K. Miyamoto, A. Niida, M. Yamamoto and K. Ajito, Tetrahedron, 2006, 62, 1416 CrossRef CAS.
- (Z)-Chloroalkene-type: (a) R. Waelchli, R. Gamse, W. Bauer, H. Meigel, E. Lier and J. H. M. Feyen, Bioorg. Med. Chem. Lett., 1996, 6, 1151 CrossRef CAS; (b) T. Kobayakawa, Y. Matsuzaki, K. Hozumi, W. Nomura, M. Nomizu and H. Tamamura, ACS Med. Chem. Lett., 2018, 9, 6 CrossRef CAS PubMed.
- (a) R. W. Taft Jr, J. Am. Chem. Soc., 1952, 74, 2729 CrossRef; (b) R. W. Taft Jr, J. Am. Chem. Soc., 1953, 75, 4538 CrossRef; (c) R. W. Taft Jr, Steric Eff. Org. Chem., 1956, 556 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. Xiao, B. Weisblum and P. Wipf, J. Am. Chem. Soc., 2005, 127, 5742 CrossRef CAS PubMed.
- P. Wipf, J. Xiao and C. R. J. Stephenson, Chimia, 2009, 63, 764 CrossRef CAS PubMed.
- (a) A. Otaka, H. Watanabe, E. Mitsuyama, A. Yukimasa, H. Tamamura and N. Fujii, Tetrahedron Lett., 2001, 42, 285 CrossRef CAS; (b) A. Otaka, H. Watanabe, A. Yukimasa, S. Oishi, H. Tamamura and N. Fujii, Tetrahedron Lett., 2001, 42, 5443 CrossRef CAS; (c) Y. Nakamura, M. Okada, A. Sato, H. Horikawa, M. Koura, A. Saito and T. Taguchi, Tetrahedron, 2005, 61, 5741 CrossRef CAS; (d) T. Narumi, A. Niida, K. Tomita, S. Oishi, A. Otaka, H. Ohnoa and N. Fujii, Chem. Commun., 2006, 4720 RSC.
- (a) T. Narumi, T. Kobayakawa, H. Aikawa, S. Seike and H. Tamamura, Org. Lett., 2012, 14, 4490 CrossRef CAS PubMed; (b) T. Kobayakawa, T. Narumi and H. Tamamura, Org. Lett., 2015, 17, 2302 CrossRef CAS PubMed; (c) T. Kobayakawa and H. Tamamura, Tetrahedron, 2016, 72, 4968 CrossRef CAS; (d) T. Kobayakawa and H. Tamamura, Tetrahedron, 2017, 73, 4464 CrossRef CAS.
- (a) R. Yagi, T. Miyazaki and T. Oyoshi, Nucleic Acids Res., 2018, 46, 5894 CrossRef CAS; (b) N. Hamad, T. Mashima, Y. Yamaoki, K. Kondo, R. Yoneda, T. Oyoshi, R. Kurokawa, T. Nagata and M. Katahira, Sci. Rep., 2020, 10, 2629 CrossRef CAS PubMed; (c) K. Takahama and T. Oyoshi, J. Am. Chem. Soc., 2013, 135, 18016 CrossRef CAS PubMed.
- See ESI† for the details.
- P. Manavalan and W. C. Johnson Jr, Anal. Biochem., 1987, 167, 76 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06554d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |