
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids
Prashant S. Auti†
,
Ginson George† and
Atish T. Paul‡
 *
*
Laboratory of Natural Product Chemistry, Department of Pharmacy, Birla Institute of Technology and Science, Pilani (BITS Pilani), Pilani Campus, Pilani, 333 031, Rajasthan, India. E-mail: atish.paul@pilani.bits-pilani.ac.in
First published on 12th November 2020
Abstract
Due to the pharmacological activities of quinazoline and quinazolinone scaffolds, it has aroused great interest in medicinal chemists for the development of new drugs or drug candidates. The pharmacological activities of quinazoline and its related scaffolds include anti-cancer, anti-microbial, anti-convulsant, and antihyperlipidaemia. Recently, molecular hybridization technology is used for the development of hybrid analogues with improved potency by combining two or more pharmacophores of bioactive scaffolds. The molecular hybridization of various biologically active pharmacophores with quinazoline derivatives resulted in lead compounds with multi-faceted biological activity wherein specific as well as multiple targets were involved. The present review summarizes the advances in lead compounds of quinazoline hybrids and their related heterocycles in medicinal chemistry. Moreover, the review also helps to intensify the drug development process by providing an understanding of the potential role of these hybridized pharmacophoric features in exhibiting various pharmacological activities.
1. Introduction
Nowadays, “one drug one target one disease approach” is not appropriate in complex diseases such as cancer and infectious diseases.1,2 This conventional approach of disease treatment cannot overcome the phenomenon of drug resistance. Consequently, polypharmacy may prove beneficial by the use of two or more drugs that interact simultaneously at different targets. However, the use of multiple drug combinations is often linked with drug–drug interactions and altered pharmacokinetics, leading to severe unintended outcomes that may result in the failure of the treatment. Altered pharmacokinetics and pharmacodynamics of drugs used in combination may be due to the induction or inhibition of hepatic enzymes, which are responsible for drug metabolism.1 Hence, in order to modulate more than one disease target simultaneously, molecular hybrids were designed by combining two or more known active pharmacophores.3 A molecular hybrid is a combination of two or more independently acting pharmacophores that are covalently linked. It can be achieved by the method of linking or framework integration to form one molecule, having increased the pharmacological activity.4 The strategy of designing such hybrid molecules is widely known as molecular hybridization. The molecular hybridization strategy is emerging as a novel approach, involving the conglomeration of two or more pharmacophores in one scaffold to develop hybrid multi-functional molecules.5 The presence of two or more biologically active pharmacophores in one unit will synergize the biological activity as well as increase its ability to interact with more than one biological target.6 Due to such properties of hybrid analogues, it is possible to develop combination therapies using single multi-functional molecules.Quinazoline is a nitrogen containing fused heterocycle, having four isomeric forms, i.e., quinazoline, quinoxaline, cinnoline, and phthalazine, depending upon the position of the nitrogen atom in the heterocyclic ring system.7 Carbonyl linkage on the quinazoline ring will give rise to quinazolinone (4(3H)-quinazolinone & 2(1H)-quinazolinone).
Amongst all the isomeric forms, quinazoline and quinazolinone are medicinally important nitrogen heterocycles, displaying diverse biological activities including anti-microbial,8 anti-convulsant,9,10 anti-cancer,11 anti-malarial,12 anti-hypertensive,13 anti-inflammatory,14 anti-diabetic,15 anti-tumor,16 anti-cholinesterase,17 dihydrofolate reductase inhibition,18 cellular phosphorylation inhibition,19 and kinase inhibitory activities.20 There are many synthetic and natural product-based drugs, containing quinazoline and quinazolinone moiety, which are used clinically for treating various disease conditions (Fig. 1 and 2).21
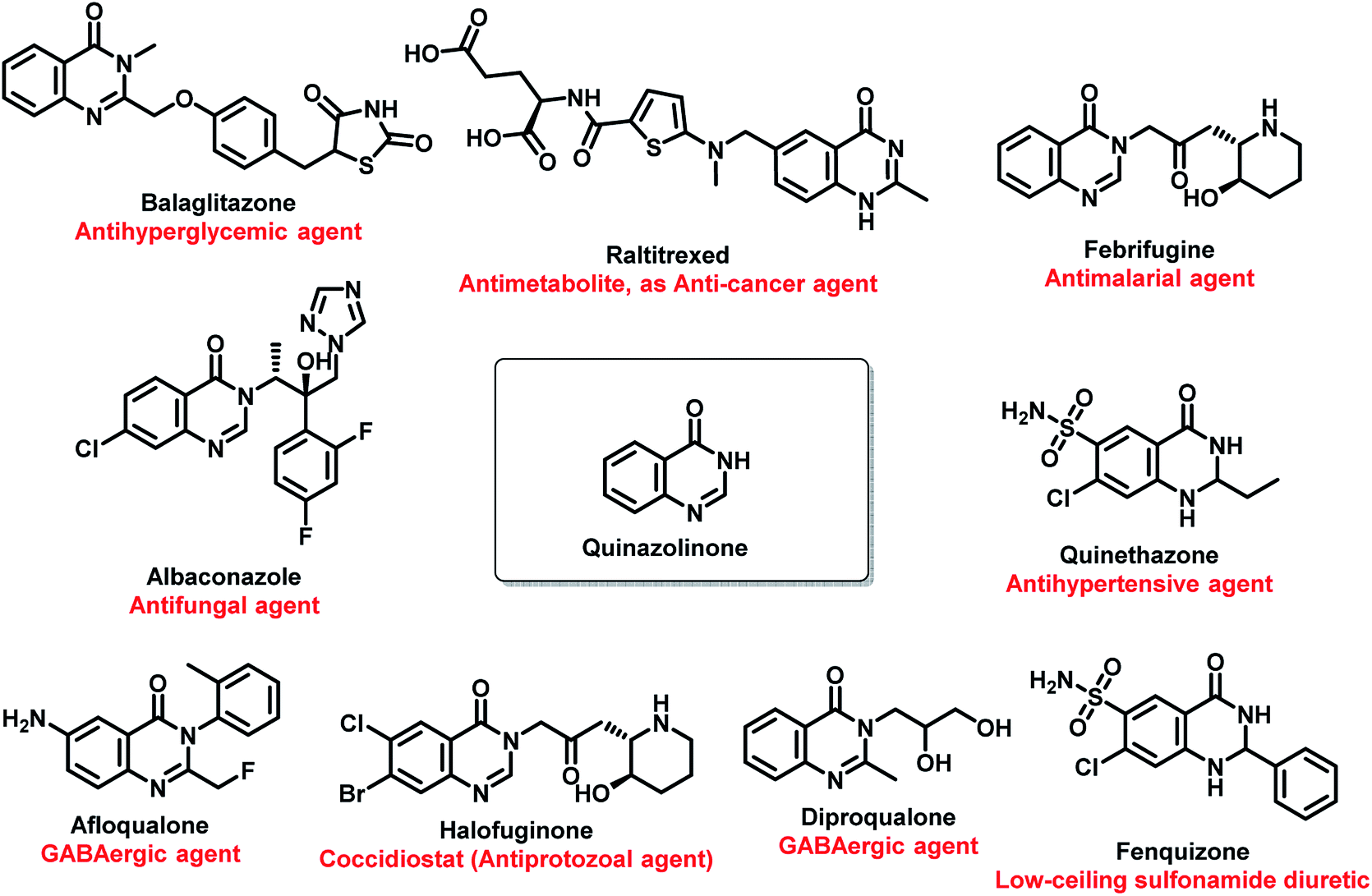 | ||
| Fig. 1 Pharmacological importance of quinazolinone-based drugs. | ||
 | ||
| Fig. 2 Pharmacological importance of quinazoline-based drugs. | ||
Hybrid analogues containing quinazoline/quinazolinone and other biologically active pharmacophores may result in drugs with increased potency and reduced drug resistance. There are many reports available on the design of various quinazoline/quinazolinone-based hybrid analogues against a variety of disease conditions.
In the current review, different strategies and approaches that were employed in the design of quinazoline/4(3H)-quinazolinone-based hybrids is discussed along with their pharmacological activities (in the areas of cancer, infectious diseases, diabetes, and CNS disorders) and the effect of various substituents on the pharmacological activity.
2. Anti-cancer hybrids
Cancer is a cell cycle disease, associated with uncontrolled cell division of abnormal cells. It continues to be one of the major health problems all over the world. There are many anti-cancer drugs in the market but still the demand is evergreen due to drug resistance. Hybrid drugs with multiple mechanism of action may be of great value for cancer treatment. Some of the hybrid analogues based on quinazoline and quinazolinone moiety are discussed below.Palem et al. synthesized novel hybrid analogues by the combination of quinazolinone and allylphenyl quinoxaline and evaluated their anti-cancer potential against 4 cancer cell lines (HeLa, MIAPACA, MDA-MB-231, and IMR32).22 Palladium (Pd)-catalyzed Heck cross coupling was utilized for the coupling of halogenated quinoxaline with substituted 3-allylquinazolinones. Two series of hybrid analogues (with alkenyl and alkyl linkages) were synthesized. These compounds of both the series have shown significant to moderate anti-cancer activity (GI50 = 0.08–0.2 μM) against all the four cancer cell lines, as compared with doxorubicin (GI50 = 0.023–0.097 μM) and paclitaxel (GI50 = 0.025–0.091 μM), which were used as standards. Compound 1 with alkenyl linker (GI50 = 0.038–1.8 μM) and compound 2 with alkyl linker (GI50 = 0.032–1.2 μM) were most potent against above cell lines (Scheme 1). The designed analogues were found to intercalate with DNA, when analyzed by docking interactions with the DNA molecule (AGACGTCT)2 (PDB code: 1N37).
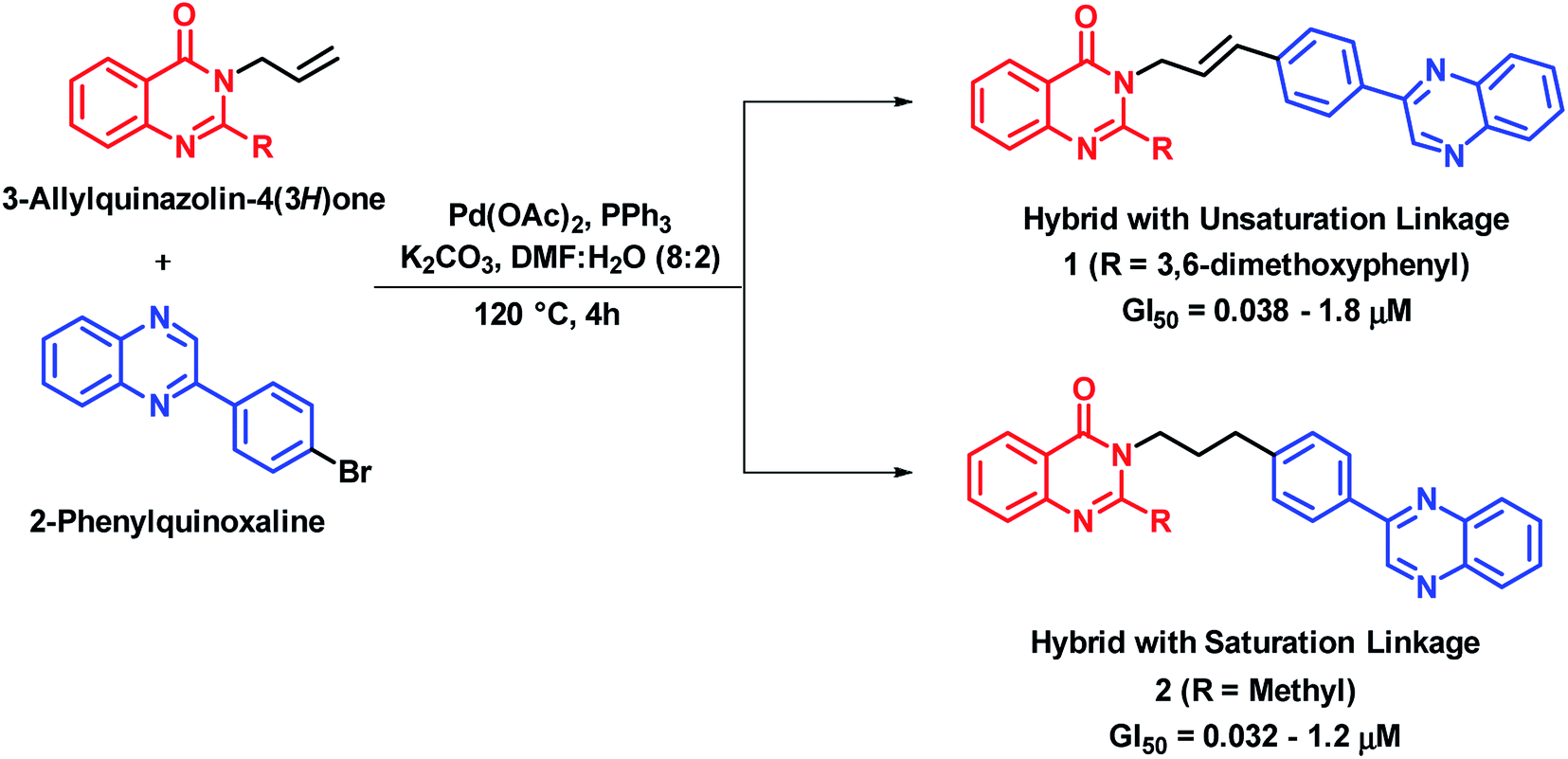 | ||
| Scheme 1 Synthetic scheme for quinazolinone-phenylquinoxaline hybrids. | ||
Venkatesh et al. have designed and synthesized sulphamide-linked dihydroquinazolinone hybrids and evaluated their anti-cancer potential on various cancer cell lines (HeLa, MDA-MB-231, PANC-1, A549 (ref. 23)). Series of compounds, with or without aliphatic linkage, have shown significant growth inhibition against all the tested cell lines with GI50 in the range of 0.045–6.94 μM. Amongst them, compound 3 was found to be potent (GI50 in the range of 0.09–0.21 μM). It was found that the alkyl linkage is also important from the activity point of view, as compound 4 with propyl linkage has shown comparable potency (GI50 in the range of 0.296–1.743 μM) as that of compound 3 (Scheme 2).
 | ||
| Scheme 2 Synthetic scheme for sulfamide-linked quinazolinone hybrids. | ||
Kamal et al.24 have designed hybrid analogues based on 2,3-dihydro-2-aryl-4-quinazolinones (DHPQZ) and diaryl-substituted isoxazoline/isoxazole, wherein both the scaffolds were reported for tubulin polymerization inhibitory activity.25–28 Four series of hybrid analogues were designed and synthesized by linking DHPQZ and isoxazoline/isoxazole with the alkyl chain linkers of varied chain length (Fig. 3) compound 8c was found to have potential anti-cancer activity, when tested against 18 human cancer cell lines. Compound 8c was found to cause significant disruption of microtubules in MCF-7 cells along with nuclear fragmentation (analyzed by DAPI staining). Further, the flow cytometric analysis of 8c suggested the G2/M phase cell cycle arrest.
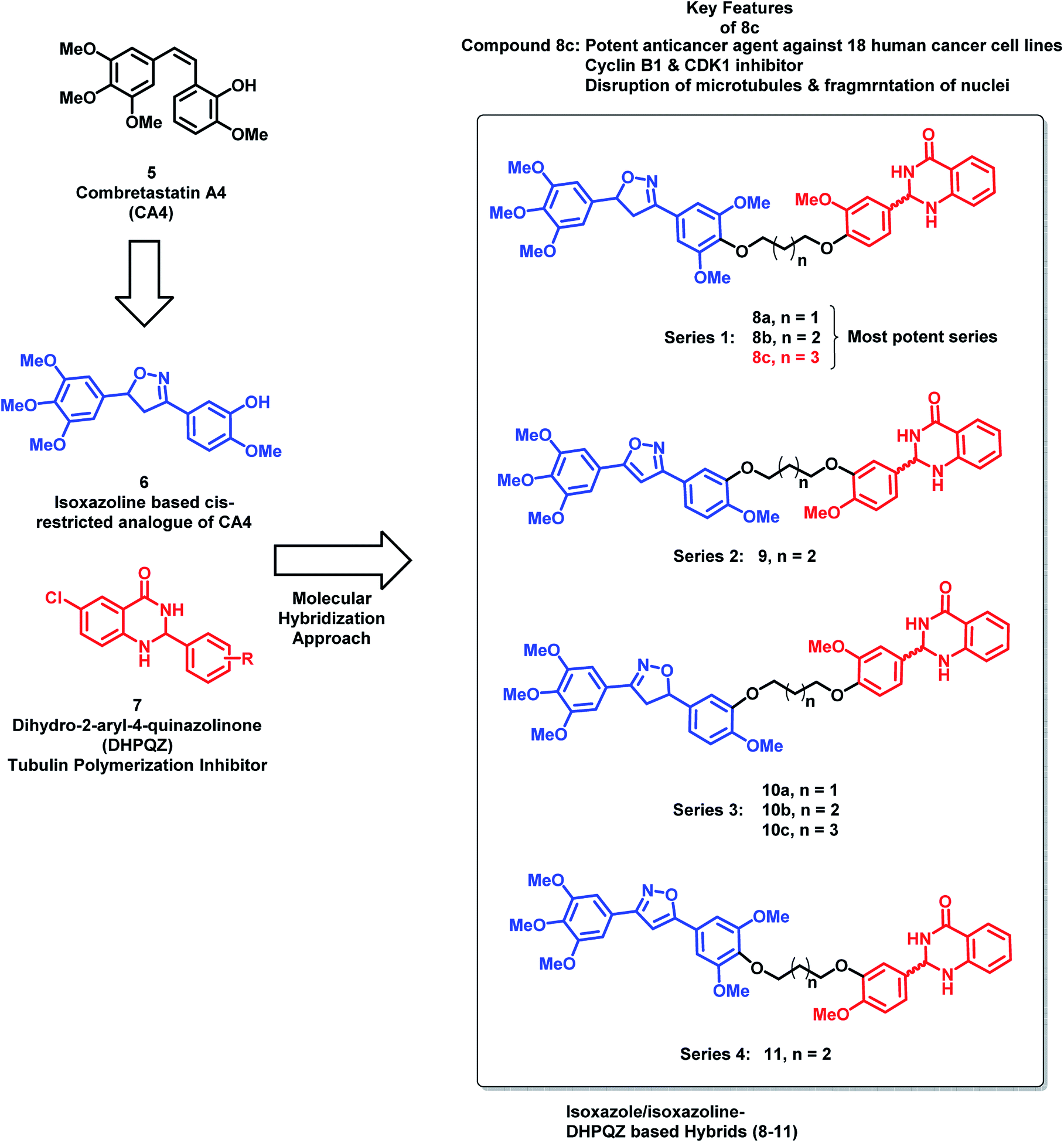 | ||
| Fig. 3 Designing strategy for oxazoline- and dihydroquinazolinone-based hybrids. | ||
In the search for potent DHFR inhibitors, Paul et al. have modified existing quinazolinone-based DHFR inhibitors (12–14)29–31 with the aim of increasing the hydrophobic π-interaction with the DHFR receptor.32 For such an attempt, the phenyl ring at the 3rd position of quinazolinone was replaced by benzimidazole, which is beneficial for exhibiting additional π-interactions due to aromatic π-system with electron rich properties (Scheme 3). Among all the synthesized analogues, hybrid analogue 15 was found to be superior in DHFR inhibition with an IC50 of 0.011 μM, as compared with methotrexate (IC50 of 0.02 μM). From the SAR study, it was found that the phenyl ring is essential at 2nd position of quinazolinone for effective DHFR inhibition. Also, the electron-donating groups at the para position of the phenyl ring lead to decreased inhibitory potential. Analogues with 5-substituted benzimidazole at 3rd position of quinazolinone have shown greater inhibition compared to that of 6-substituted ones (Scheme 3), which states the importance of orientation of benzimidazole on DHFR inhibition.
 | ||
| Scheme 3 Synthetic and design strategy for quinazolinone- and benzimidazole-based hybrids as DHFR inhibitors. | ||
By considering the various structural aspects of clinically-approved thiazole-containing anti-cancer drugs (tiazofurin and bleomycin), Hosseinzadeh et al. designed hybrid analogues containing thiazole and quinazolinone moieties.33 After anti-proliferative screening against 3 cancer cell lines (PC3, MCF-7, HT-29), compound 17 was found to be most active with IC50 values of 10, 10, and 12 μM against PC3, MCF-7, and HT-29 cells, respectively (Scheme 4).
 | ||
| Scheme 4 Synthetic scheme for quinazolinone- and thiazole-based hybrids. | ||
Hassanzadeh et al. have designed novel hybrids using 2-substituted quinazolinones and 2-phenyloxadiazole.34 As shown in Scheme 5, quinazolinone and oxadiazole derivatives show anti-proliferative activity through various mechanisms. Amongst the synthesized hybrid analogues, compound 18 exhibited potent anti-cancer activity when tested on HeLa cell lines with an IC50 of 7.52 μM. Substitution at the 2nd position of quinazolinone plays an important role in the anti-cancer activity as analogues with propyl substitution (compound 18) were found to be potent as compared with other analogues.
 | ||
| Scheme 5 Synthetic and design strategy for quinazolinone-oxadiazole hybrids. | ||
Wu et al. designed quinazolinone- and indolin-2-one-based hybrids by linking 2-methylquinazolin-4(3H)-one with indolin-2-one through an imine linkage.35 These hybrid analogues were synthesized by the condensation of 6-amino-2-methylquinazolin-4(3H)-one with indolin-2,3-dione or N-substituted indolin-2,3-dione followed by in vitro anti-cancer screening against 5 human cancer cell lines (A549, MCF-7, HeLa, HT-29, HCT-116). It was observed that N-substitution on indolin-2-one is essential for the anti-proliferative activity. Amongst both the series, compound 19 was found to be most potent with % inhibition in the range of 32–63% against above-mentioned cell lines (Scheme 6).
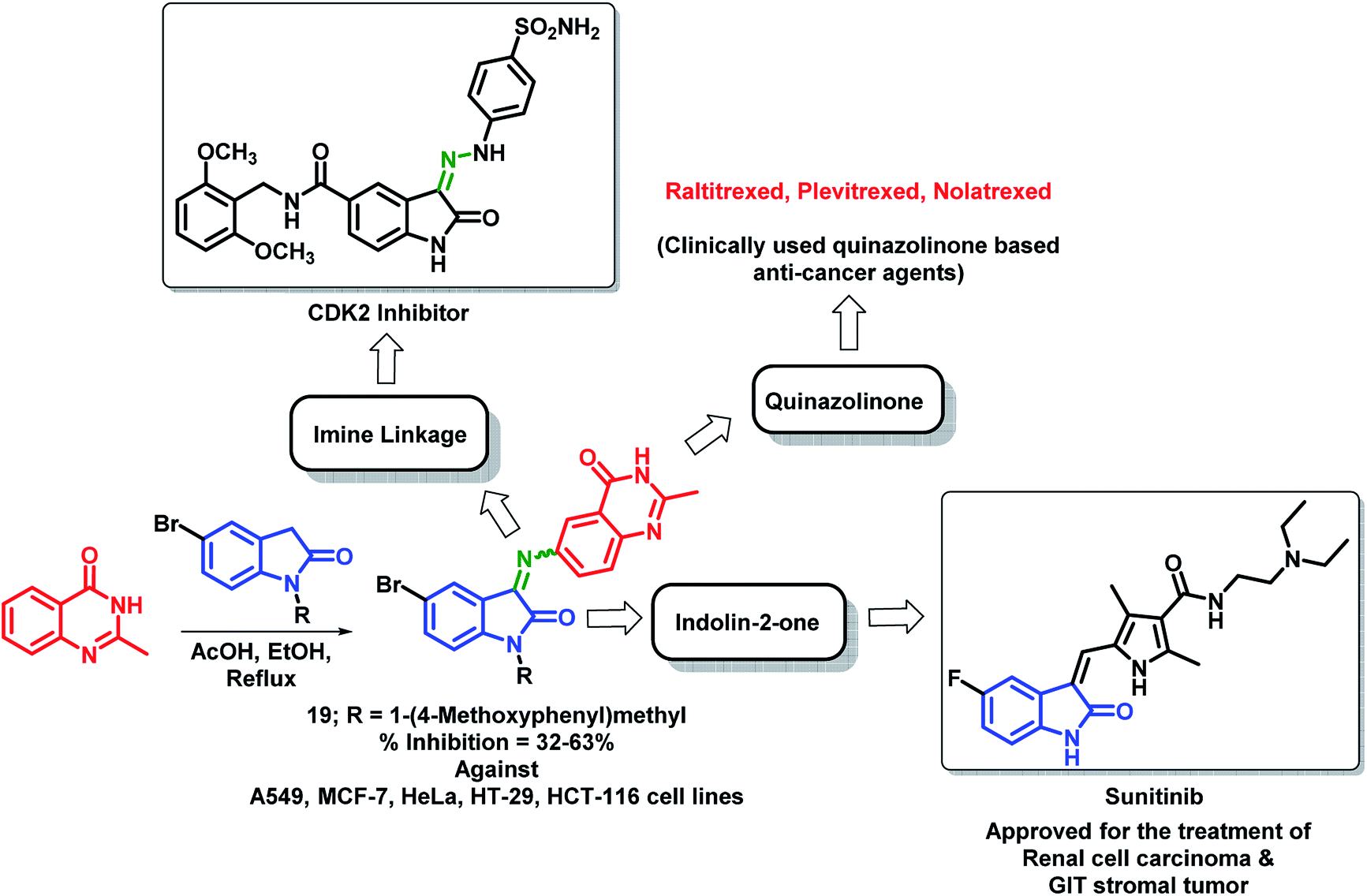 | ||
| Scheme 6 Synthetic strategy for quinazoline and indolin-2-one hybrids, showing the importance of each structural unit. | ||
Benzoxazinone, which is a widely active heterocycle,36,37 was combined with allylquinazolinone through molecular hybridization by Bollu et al.38 Pd-catalyzed Heck coupling was utilized in order to club 1,4-benzoxazinone and quinazolinone to produce novel 1,4-benzoxazinone-acetylphenylallylquinazolin-4(3H)-one hybrids. All the hybrids showed moderate to good anti-proliferative activity against 3 cancer cell lines (A549, HeLa, MDA-MB-231), with GI50 in the range of 0.32–13.4 μM. Among these, compounds 20 (GI50 = 0.58 μM; MDA-MB-231 cell line), 21 (GI50 = 0.32 μM; A549 cell line), and 22 (GI50 = 0.37 μM; HeLa cell line) were exceptionally good in the anti-proliferative activity against the particular cell lines (Scheme 7).
 | ||
| Scheme 7 Synthetic scheme for benzoxazinone and quinazolinone hybrids. | ||
Phosphodiesterase-4 (PDE-4) is known to play a vital role in the proliferation, angiogenesis, and motility of many cancerous cells;39–41 hence, PDE-4 inhibition will lead to anti-proliferative activity. N-acylhydrazones are potent inhibitors of PDE-4 (Compound 23; Scheme 8); therefore, in previous reports, quinazoline was combined with N-acylhydrazone to obtain the quinazoline-hydrazone hybrid with an IC50 of 190 nM.42,43 Further to enhance the activity of such hybrids, Rahman et al. performed lead optimization by replacing quinazoline with quinazolin-4(3H)one.44 These quinazolin-4(3H)one-hydrazone hybrids were designed, synthesized, and screened for PDE-4 inhibition. The condensation of 3-amino-2-phenylquinazolin-4(3H)one with different substituted aromatic aldehydes afforded the desired arylmethylidine-hydrazides. These synthesized Schiff base hybrids were evaluated for the in vitro inhibition of the PDE-4(B) enzyme by taking rolipram as a positive control. Many compounds were found to be potent PDE-4(B) inhibitors. Further, these active compounds were screened for the in vitro anti-cancer activity against 3 human cancer cell lines (MDA-MB-231, A549, HT-29). Amongst these compounds, compound 26 has shown more anti-proliferative activity with an IC50 of 140, 80, and 320 nm against MDA-MB-231, A549, and HT-29 cell lines, respectively (Scheme 8).
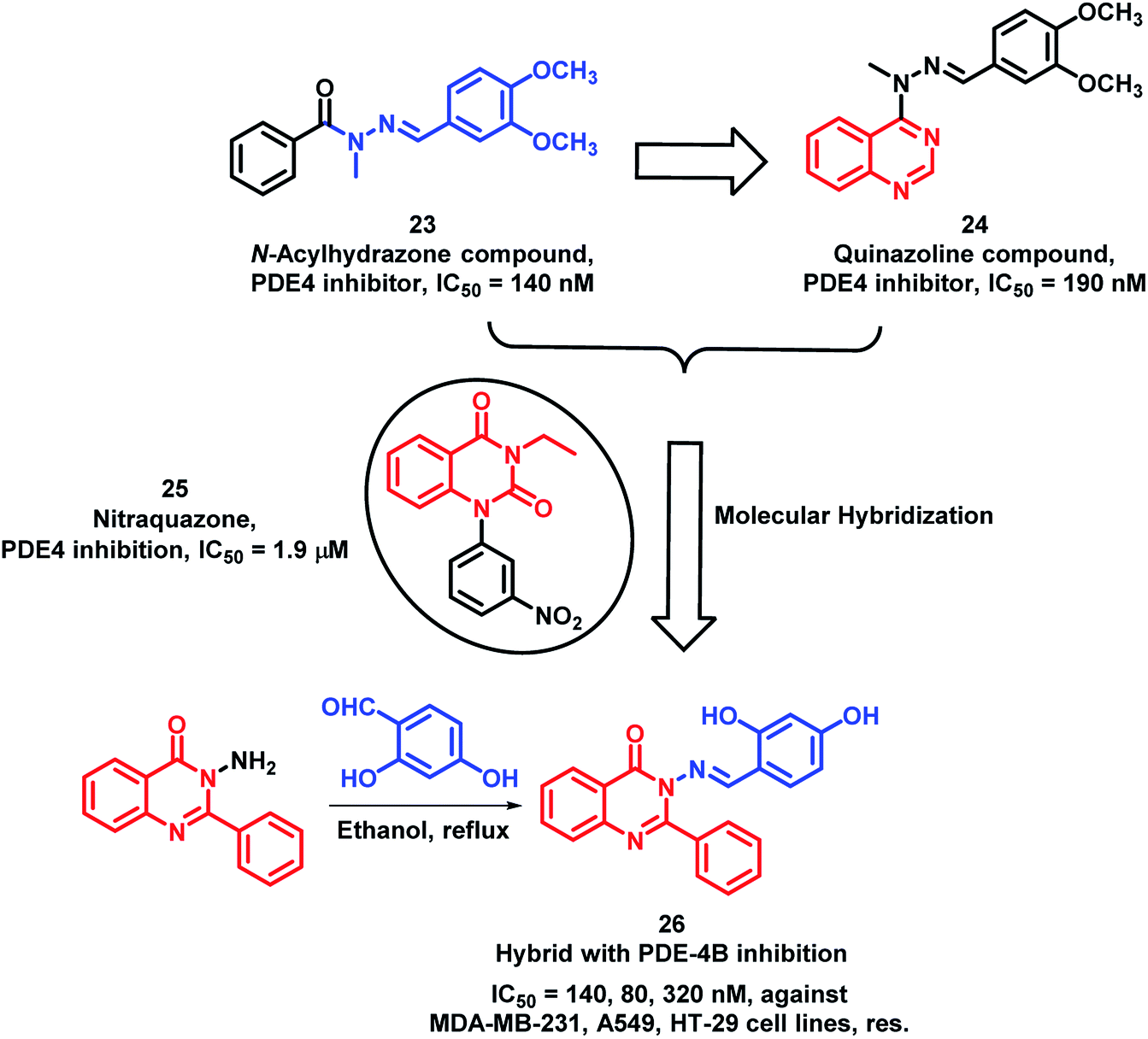 | ||
| Scheme 8 Synthetic and design strategy for the Schiff base hybrids of quinazolinone as PDE-4 inhibitors. | ||
Thiazolidin-4-one, a saturated 4-oxo form of thiazole, occupies an important place in medicinal chemistry. Due to its diverse biological activities, Thakral et al. combined thiazolidin-4-one with quinazolinone scaffold via molecular hybridization approach.45 For the synthesis of the desired hybrids, 3-(4-aminophenyl)-2-phenyl-quinazolin-4(3H)-one was treated with aromatic aldehydes to produce Schiff bases of quinazolin-4(3H)one. Further, cyclocondensation of the obtained products (Schiffs base) with thioglycolic acid in DMF resulted in desired hybrid derivatives. Hybrid compounds 27 (IC50 = 1.79 μM) and 28 (IC50 = 1.94 μM) showed good anti-cancer activity against Hep-G2 and MCF-7 cell lines, respectively (Scheme 9), as compared with doxorubicin (IC50 = 0.09 μM against Hep-G2 and MCF-7 cell lines), used as a standard. Moreover, the results of the molecular docking study of the designed hybrids with topoisomerase-II enzyme (PDB code: 1ZXM) have shown positive correlation of topoisomerase-II inhibition with the anti-proliferative activity.
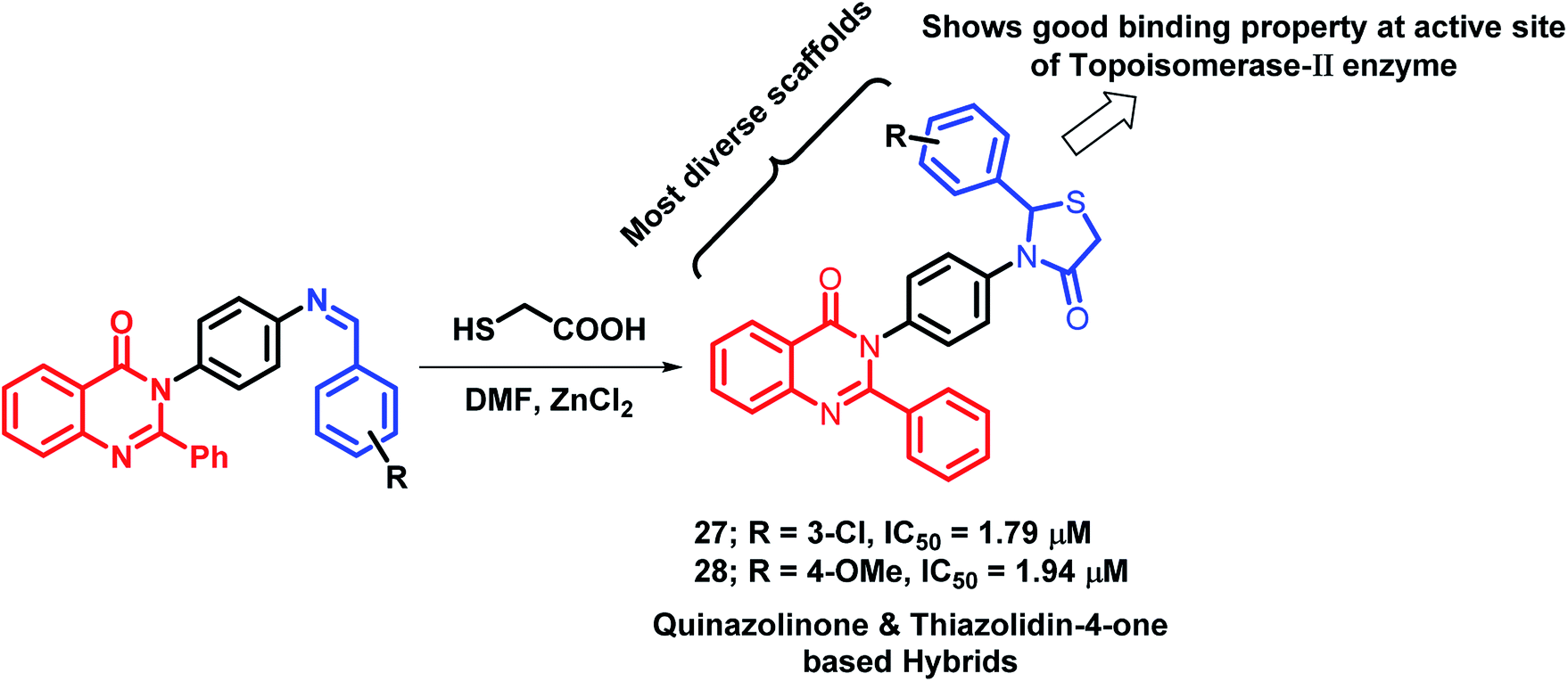 | ||
| Scheme 9 Synthetic scheme for quinazolinone- and thiazolidinone-based hybrids. | ||
Sulphonamide derivatives have shown anti-cancer activity through various mechanisms including the disruption of microtubule assembly, carbonic anhydrase (CA) inhibition, and cell cycle perturbation.46–48 By considering such points, Zayed et al. optimized previously designed potent quinazoline-based analogues (29)49 by attaching the benzenesulfonamido moiety on quinazolinone backbone50 (Scheme 10). The target compounds were synthesized by the simple fusion of various N-substituted 4-Aminobenzene sulphonamides with benzoxazin-4-one in pyridine. Amongst the synthesized analogues, compound 30 was found to be potent as a cytotoxic agent with an IC50 of 2.51 μM against NCI cell line.
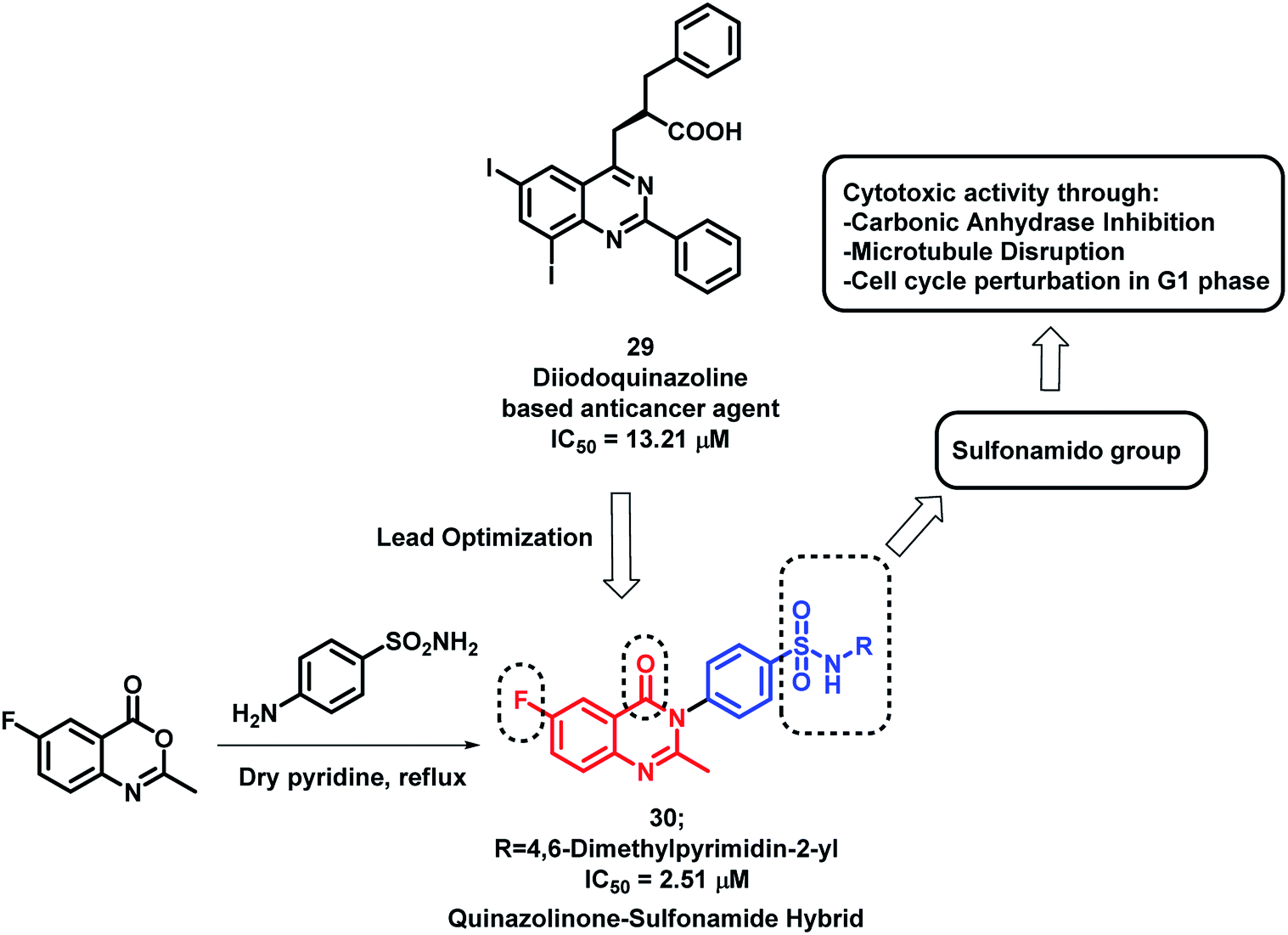 | ||
| Scheme 10 Synthetic and design strategy for quinazolinone-sulfonamide hybrids. | ||
As triazole was found to increase the existing activity of many bioactive scaffolds,4 Hassanzadeh et al. have designed hybrids with the structural features of triazole and quinazolinone.51 These triazole and quinazolinone scaffolds were linked through a 2-thioacetamido linkage and synthesized via the substitution reaction of 2-chloro-N-(4-oxo-2-quinazolin3(3H)-yl)acetamide derivatives with 4-methyl-4-H-1,2,4-triazole-3-thiol. These synthesized hybrids were tested against MCF-7 and HeLa cell lines and MCF-7 cell line was found to be susceptible to compound 31 with an IC50 of 38.38 μM (Scheme 11).
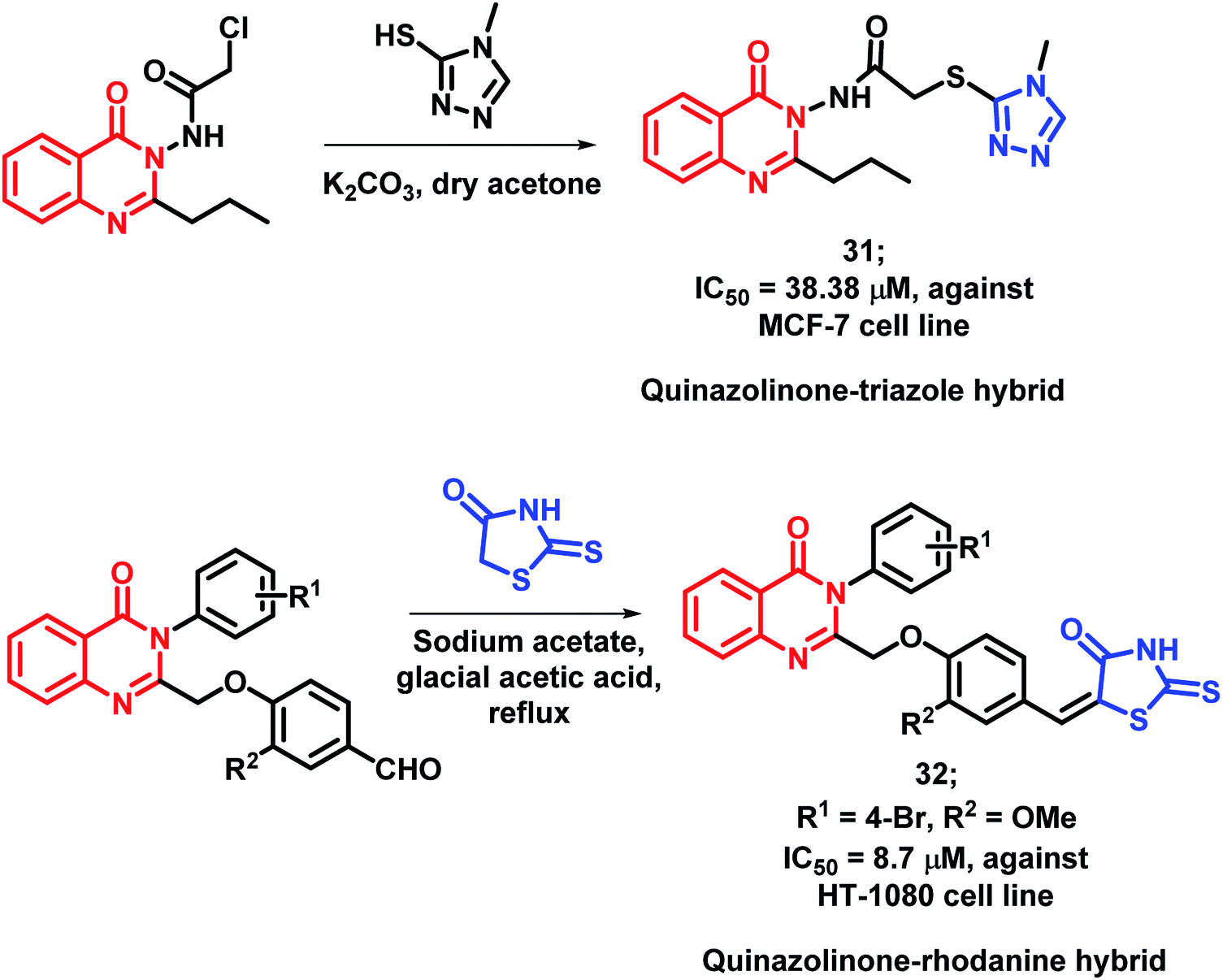 | ||
| Scheme 11 Synthetic schemes for quinazolinone-based rhodanine (31) and triazole (32) hybrids. | ||
Rhodanine derivatives were found to induce apoptosis through the modulation of the Bcl-2 family of proteins. Hence, Sayed et al. linked rhodanines with quinazolinones in order to design rhodanine-based quinazolinone hybrids.52 All the synthesized compounds were found to be active with an IC50 value in the range of 10–60 μM, when screened against the HT-1080 cell line. The SAR study revealed that bulky, hydrophobic, and electron-withdrawing substituents at the para position of the phenyl ring linked at the 3rd position of quinazolinone moiety is essential for anti-proliferative activity. Compound 32 (IC50 = 38.38 μM) was the most potent analogue, having desired substituents (Scheme 11).
Gokhale et al. have designed indole and quinazolinone-based amides for anti-cancer activity53 by considering the apoptosis inducing property of various indole-based amides.54 Also, the introduction of amide at the 2nd position of indole leads to cytotoxicity through histone deacetylase (HDAC) inhibition. Based on such observations, quinazolinone-based indolyl amides were designed (Scheme 12) and synthesized, followed by the screening of their anti-cancer activity. It was observed that the substituent at the phenyl ring attached to the amide nitrogen is essential for the anti-proliferative activity. Various substituents such as cyclopropyl, cyclohexylmethyl, fluoro, electron releasing groups were found to be active against the cancer cell lines (HepG2 and MCF-7). Compounds 33 and 34 had potential cytotoxicity (IC50 of 15.8 and 17.8 μM, respectively) against the HepG2 cell line as compared with doxorubicin and 5-fluorouracil as positive controls (IC50 of 6.5 and 18.8 μM, respectively). All the synthesized compounds were less toxic against the non-cancerous Vero cell line, which confirms the selectivity of the compounds towards the cancerous cells.
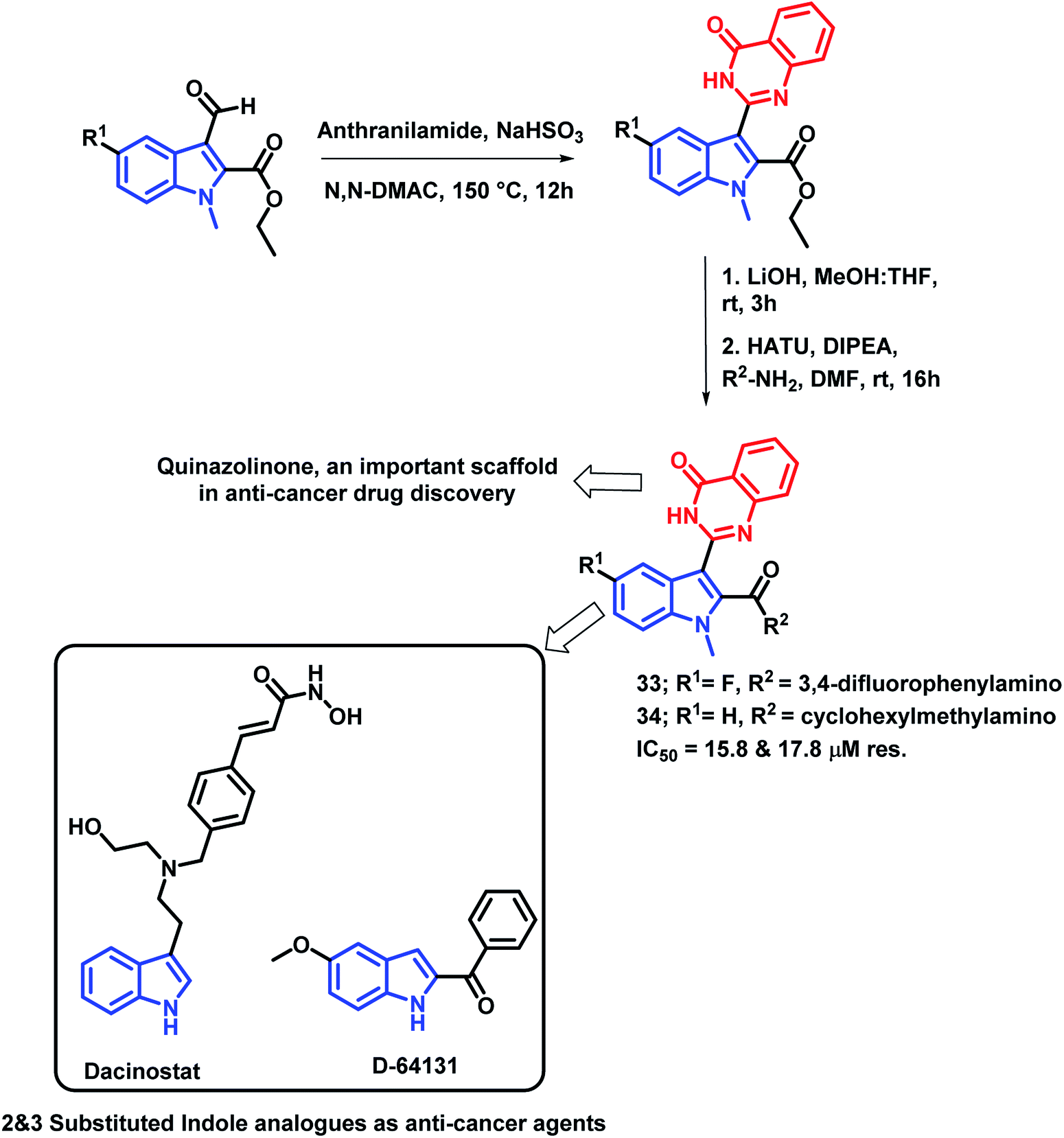 | ||
| Scheme 12 Synthetic and design strategy for indole- and quinazolinone-based hybrids for anti-cancer potential. | ||
Luotonin A (pyrroloquinazolinoquinoline alkaloid) possesses anti-cancer activity through the inhibition of human topoisomerase-I. By taking such facts into consideration, Venkatesh et al. attempted to develop luotonin A- and triazole-based hybrid analogues by targeting HDAC and enhancer of zeste homolog 2 (EZH2) proteins.55 The designed quinazolinone hybrid analogues were synthesized by two methods from 3-(2-(hydroxymethyl)phenyl)-2-methylquinazolin-4(3H)-ones. In the first method, click chemistry approach was applied by using various acetylenes. The second method was applied by the direct attachment of substituted triazoles in the presence of para-toluenesulfonic acid (pTSA). Amongst all, compound 35 was found to be active with IC50 values of 8.0, 5.94, 20.16, and 256 μM against the MCF-7, HeLa, K562, and HEK cell lines, respectively (Scheme 13). Further, the effect of compound 35 on the cell cycle was identified by flow cytometry, showing G1 phase arrest on the HeLa cell line.
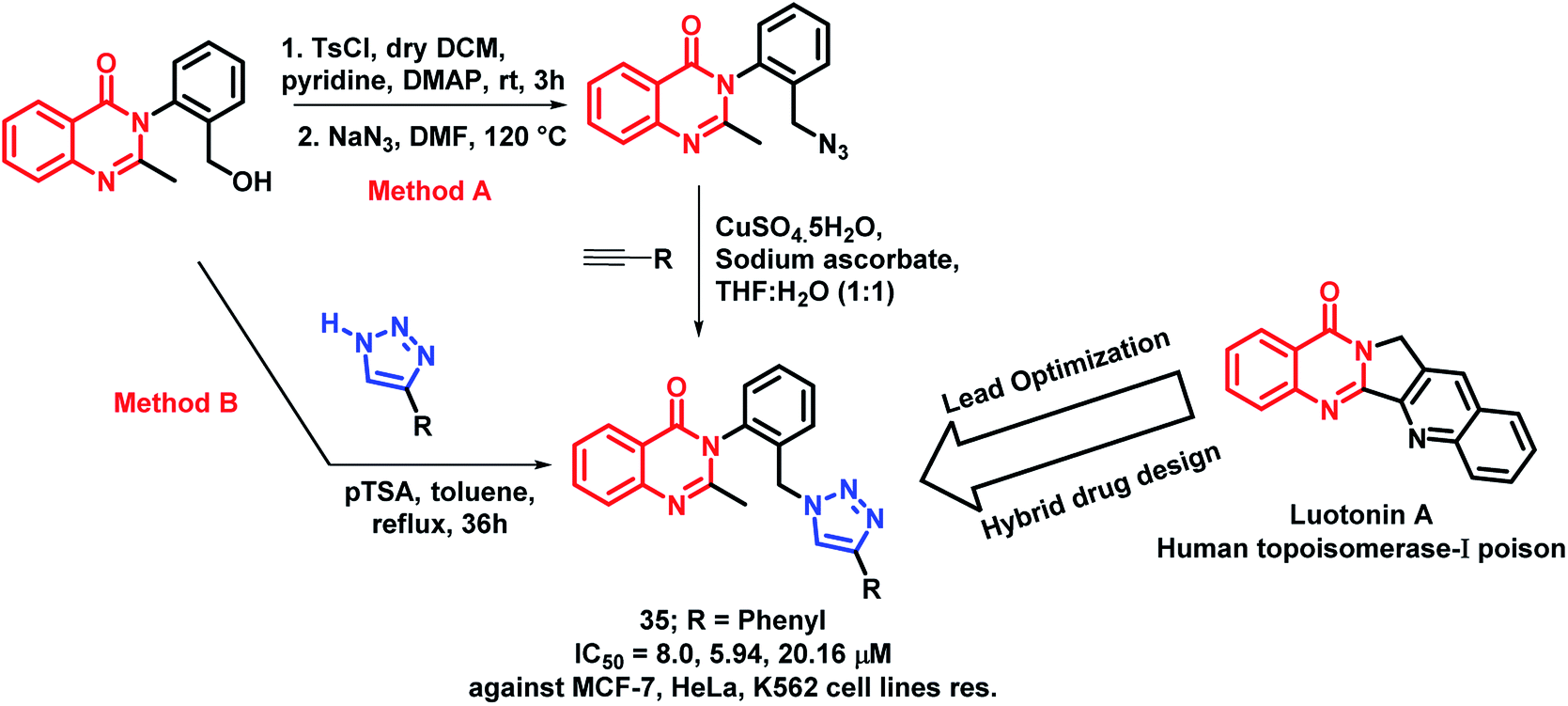 | ||
| Scheme 13 Synthetic scheme for luotonin A-inspired quinazolinone-triazole hybrids. | ||
Zayed et al. modified the quinazolinone scaffold by attaching desired fragments that may be helpful in EGFR binding.56 In this way, various amino acid-linked quinazolinone hybrids were synthesized by using various amino acids (Scheme 14). The amino acids were supposed to provide additional H-bonding interactions at the target site. Amongst the synthesized hybrids, phenylalanine- and glutamine-based hybrid analogues were potent against the MCF-7 cell line (IC50 of 0.44 and 0.43 μM, respectively) compared to that of the reference drug erlotinib (IC50 of 1.14 μM). The docking study was also performed by taking the EGFR enzyme (PDB; 1M17) and tubulin (PDB; 1SA0), followed by EGFR inhibition and tubulin polymerization inhibition assays. The results of both the docking and enzyme inhibition studies were correlated with the results of the cell-based cytotoxicity study. Hence, the authors concluded that the anti-cancer activity of these hybrid analogues was possibly due to EGFR and tubulin polymerization inhibition.
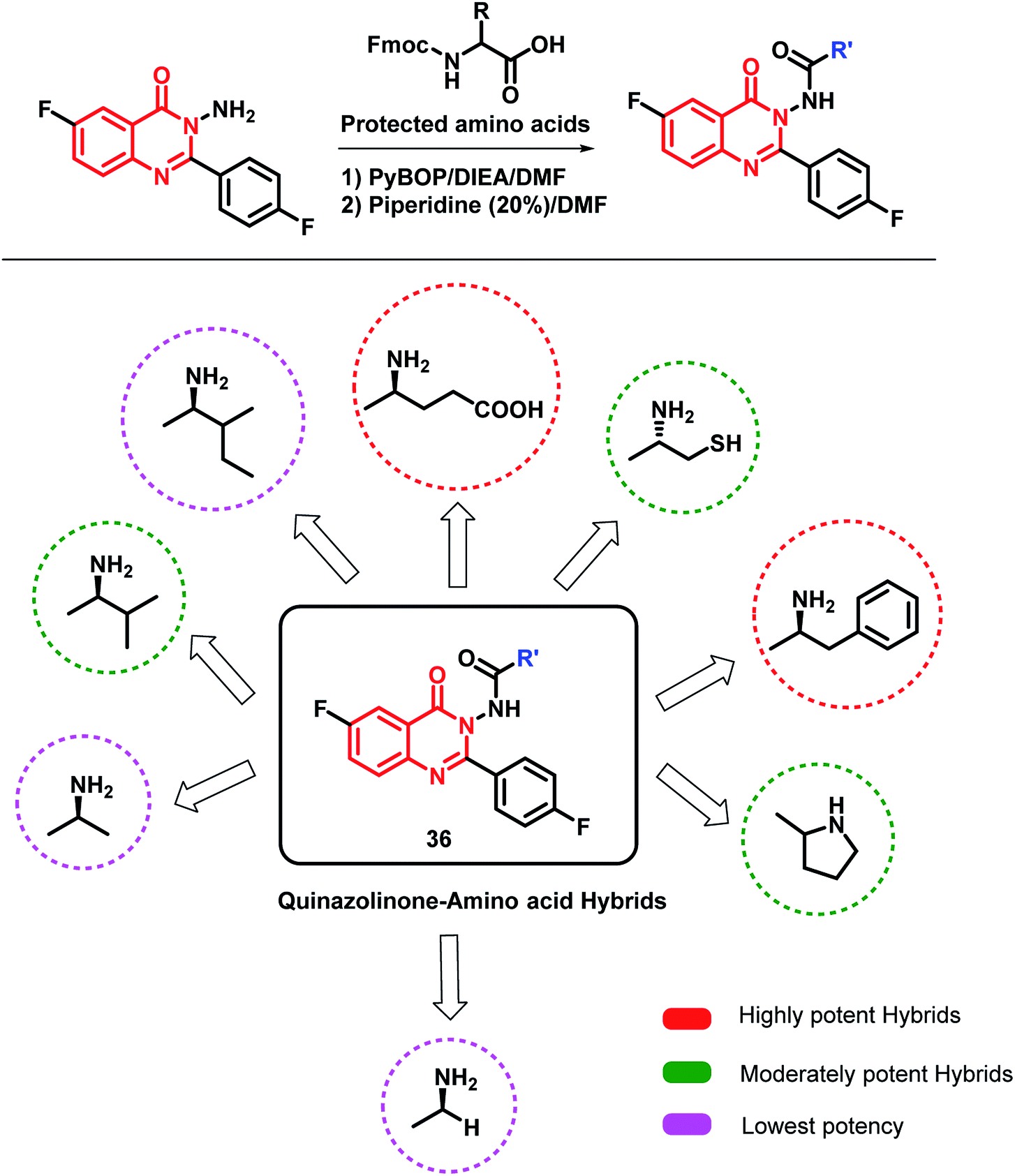 | ||
| Scheme 14 Synthetic scheme for amino acid-linked quinazolinone hybrids. | ||
The inhibition of tumor angiogenesis is one of the targets for cancer treatment and the VEGFR-2 receptor is one of the regulators of tumoral angiogenesis.57,58 Vandetanib is one of the drugs used as an anti-cancer agent, which acts through VEGFR-2 inhibition. Peng et al. designed molecular hybrids having desired features for VEGFR-2 and HDAC inhibition.59 The hybrid analogues were designed by considering the structural features of vandetanib (VEGFR-2 inhibitor) and vorinostat (HDAC inhibitor). As shown in Scheme 15, 4-aminoquinazoline (pharmacophore of vandetanib) was linked with hydroxamic acid (for HDAC inhibition) via a long alkyl chain. Those designed analogues have shown proper binding at the active site of VEGFR-2 kinase (PDB: 2QU5) and Histone Deacetylase-Like Protein (HDLP) (PDB: 1C3S). These synthesized compounds were tested for HDAC and VEGFR-2 inhibition, followed by anti-cancer screening against the MCF-7 cell line. Compound 37 was found to have potent inhibition of both VEGFR-2 and HDAC, with IC50 values of 74 and 2.2 nM, respectively. Also, it was found to be cytotoxic against the MCF-7 cell line with an IC50 of 0.85 μM.
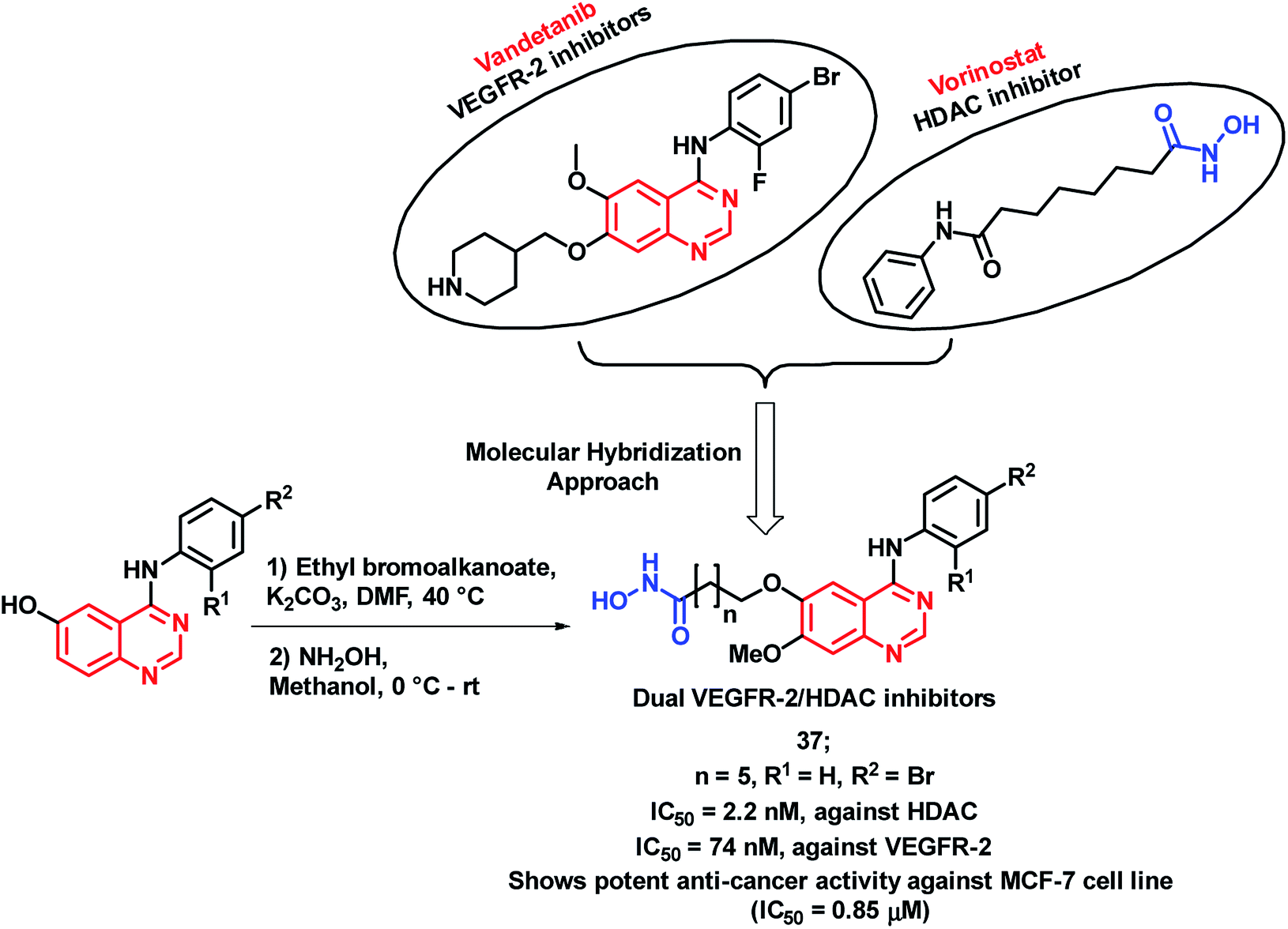 | ||
| Scheme 15 Synthetic and design strategy for quinazoline-hydroxamic acid hybrids. | ||
1,2,3-Triazole is one of the important fragments in medicinal chemistry and can be used for the synthesis of a variety of bioactive heterocycles.60–64 Also, various triazole-containing hybrids were synthesized recently with optimum activities. By considering such factors, Le-Nhat-Thuy et al. designed triazole linked 4-aminoquinazoline-based hybrids (Scheme 16), followed by synthesis and in vitro anti-cancer screening.65 Two structural requirements were found to be essential for cytotoxic activity: (a) oxygenated substituents on quinazoline, as derivatives with fused deoxygenated ring were effective as anti-cancer agents, (b) substituents on the phenyl ring, attached to the triazole moiety. With such structural features, compound 38 was found to be most potent with IC50 of 0.04, 0.14, and 1.03 μM against KB, HepG2, and Lu cell lines, respectively.
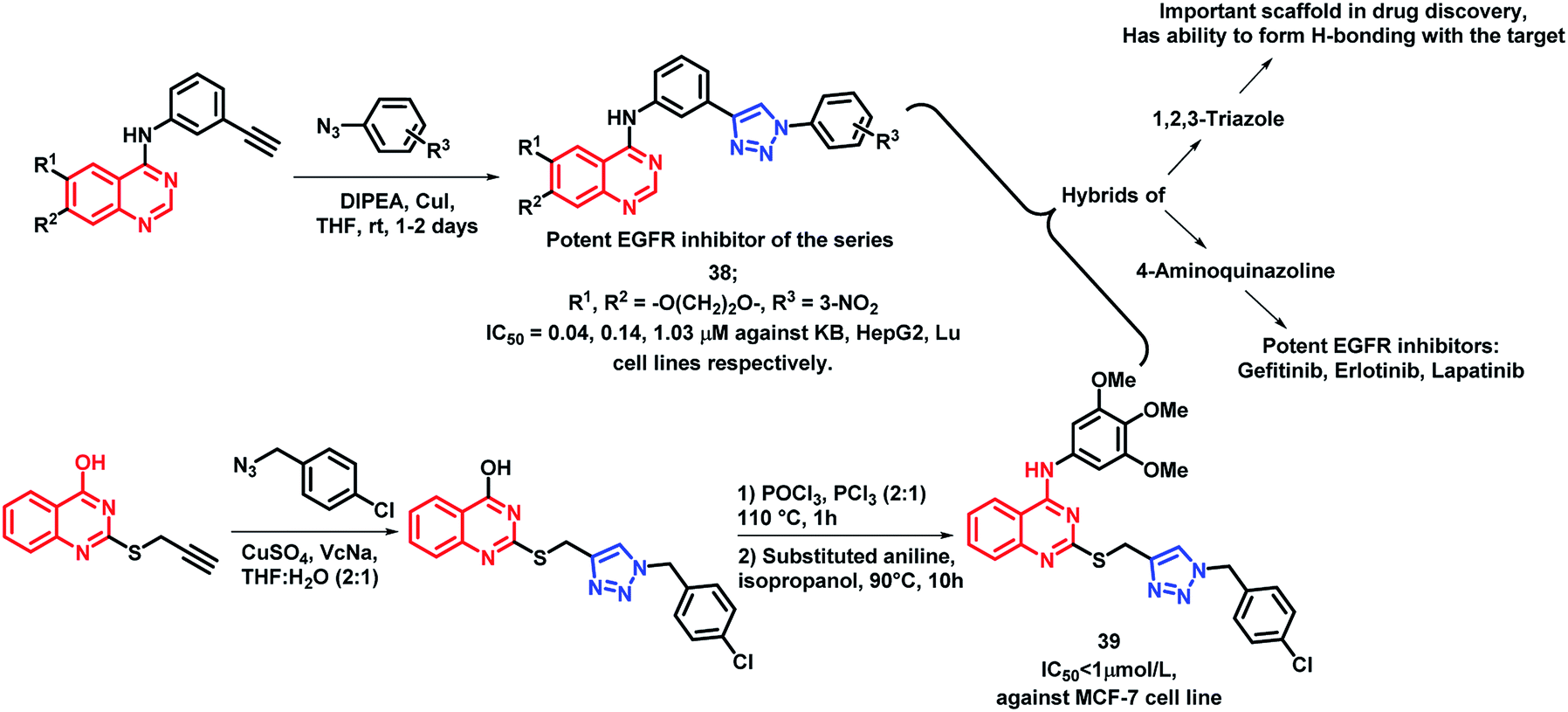 | ||
| Scheme 16 Synthetic scheme for 4-aminoquinazoline and triazole hybrids. | ||
Quinazoline and triazole were linked through thioether linkage via the click chemistry approach by Song et al. and the synthesized analogues were screened for in vitro anti-cancer activity.66 Compound 39 was found to be highly active as compared with other analogues, having an IC50 value less than 1 μmol L−1 (Scheme 16).
The indole nucleus was found to be prevalent in many bioactive heterocycles, with anti-microbial, anti-oxidant, and anti-cancer activities.67–70 Due to such diverse properties of the indole nucleus, Mphahlele et al. designed hybrids of indole and 4-aminoquinazoline (Scheme 17) for increased anti-cancer potential.71 Those hybrids were found to bind at the ATP binding region of EGFR (PDB code: 1M17), with similar interactions as that of erlotinib. Compound 40 has shown potential EGFR inhibition with an IC50 of 40.7 nM, as compared with the standard drug gefitinib (IC50 = 38.9 nM). Many compounds of the series were found to be active in the in vitro cytotoxicity screening against various cancer cell lines (A549, Caco-2, C3A, MCF7, and HeLa).
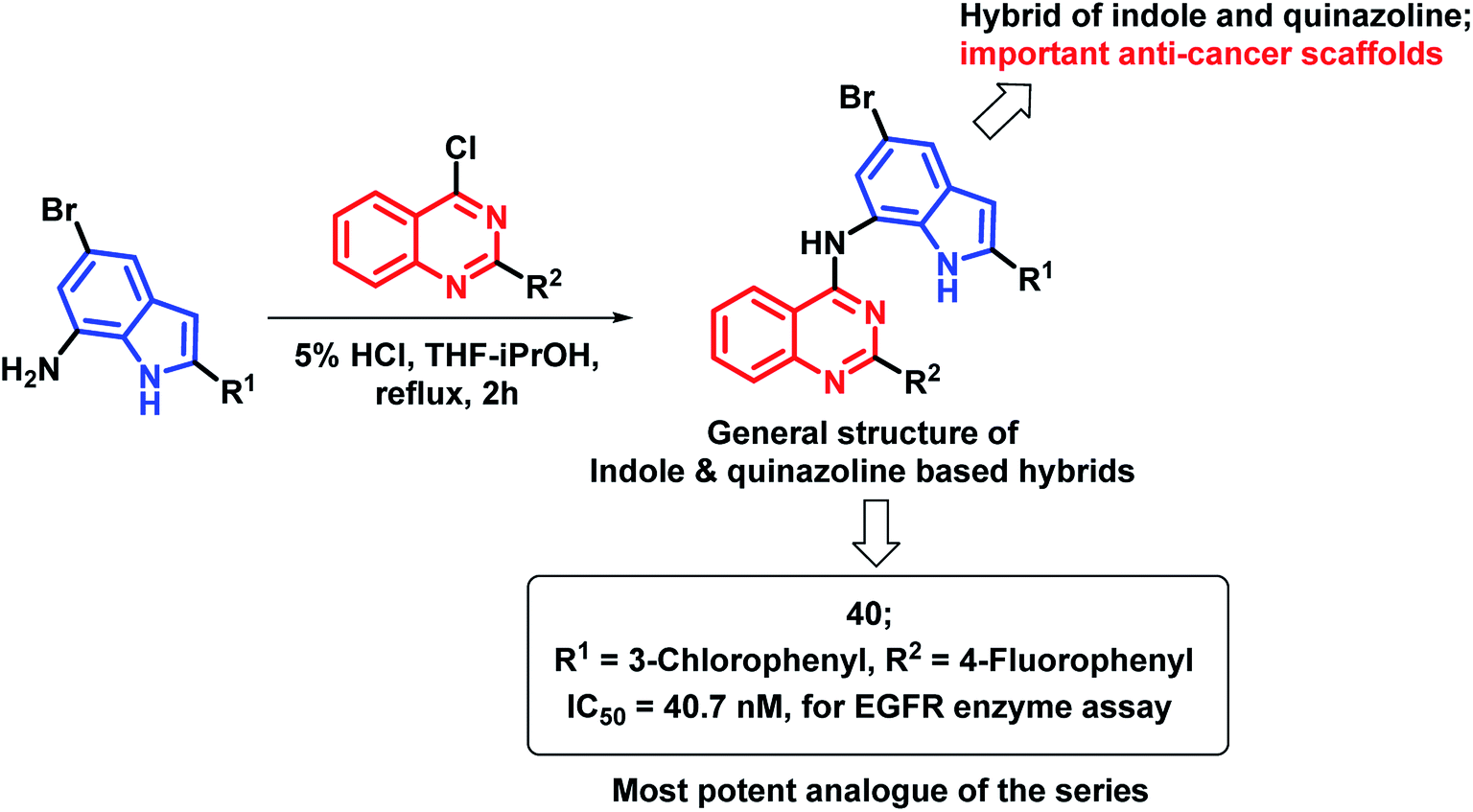 | ||
| Scheme 17 Synthetic scheme for indole and 4-aminoquinazoline hybrids. | ||
Another indole derivative, i.e., 1H-indole-2,3-dione (isatin), was linked with 4-aminoquinazoline scaffold via the molecular hybridization approach (Scheme 18) by Fares et al.72 Imine linkage was used as it is present in many of the isatin-containing EGFR inhibitors such as sunitinib. Compounds 41, 42, and 43 were the most potent anti-cancer agents in the series, with IC50 values in the range of 1–2.4 μM against the HepG2 cell line. These active compounds were found to induce the activation of caspase-3 along with a reduction in the Bcl-2/Bax ratio in the HepG2 cells. These results revealed that the anti-cancer activity of these hybrid analogues is due to the activation of the mitochondrial apoptotic pathway.
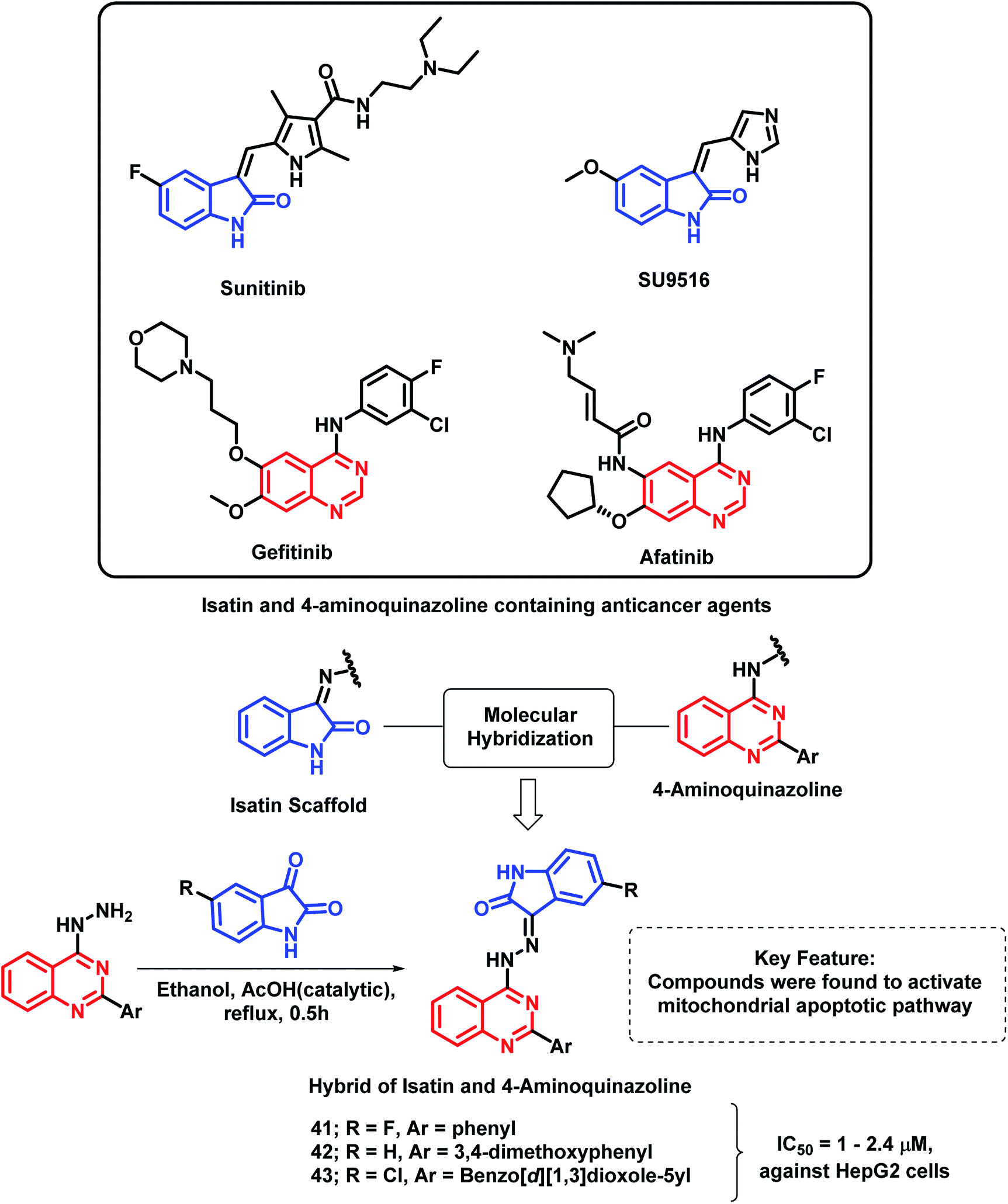 | ||
| Scheme 18 Synthetic and design strategy for isatin and 4-aminoquinazoline hybrids. | ||
Aurora kinase is one of the enzymes responsible for the proliferation of cancer cells. Hence, by targeting such enzymes, cancerous cells can be inhibited from proliferation. Considering the biological importance of the benzimidazole heterocycle, Luxami et al. have designed hybrid analogues having the structural features of benzimidazole and 4-aminoquinazoline (important scaffold with anti-cancer potential).73 Amongst the synthesized analogues, compounds 44 and 45 have shown arora kinase inhibition with IC50 values of 7.50 and 0.53 μM, respectively (Scheme 19). Along with aurora kinase inhibition, these hybrids were also found to be useful as ratiometric chemosensors for the estimation of lead and cyanide concentration in the given sample. Heavy metals are found to be toxic to the environment as well as the human health; hence, chemosensors will be effective for the detection of such ions. Pb2+ and CN− binding property was investigated via UV-Vis spectroscopy, fluorescence spectroscopy, 1H NMR titration experiment, and DFT calculations. Compound 44 was used to estimate CN− (2–500 μM) and compound 45 was used to estimate the Pb2+ ion (0.05–1500 μM) ratiometrically.
 | ||
| Scheme 19 Synthetic and design strategy for benzimidazole and 4-aminoquinazoline hybrids. | ||
EGFR inhibitors containing 4-aminoquinazoline such as gefitinib and erlotinib are beneficial for anti-cancer activity but they lack clinical efficiency due to the point mutation of the EGFR enzyme. To address such problems, Shao et al. prepared hybrid analogues consisting of oxooxazolidine and 4-aminoquinazoline.74 As shown in Scheme 20, these hybrids were designed as per the structural requirements for EGFR inhibition. Many of the synthesized hybrids exhibited potent EGFR kinase inhibitory activity against the mutant forms of EGFR (EGFRT790M and EGFRL858R) but amongst these hybrid analogues, compound 46 was highly potent. Through Western blotting analysis, it was found that compound 46 reversibly inhibited EGF-mediated phosphorylation of EGFR in A431 cells.
 | ||
| Scheme 20 Synthesis of oxazolidinone and 4-aminoquinazolinone hybrids. | ||
Sharma et al. synthesized novel regioisomeric quinazoline/benzimidazole hybrids and evaluated the in vitro anti-tumor effects in 60 cell lines.75 The hybrid analogues were designed for increased lipophilicity (via the substitution of alkyl/allyl groups in benzimidazole and aryl groups in the quinazoline moiety) and for electron donor and acceptor effects (by substitution of secondary amines). The thrust effort is focused to evaluate the effects of substitution at the 2nd position of quinazoline for potential anti-tumor effect with desired selectivity. The anti-tumor activity of the synthesized compounds was evaluated against nine tumor cell subpanels such as leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancer cells. As shown in Fig. 4, compound 47 exhibited selectivity towards leukaemia cancer cells, colon cancer cells, melanoma cancer cells, and breast cancer cell lines (GI value 45.0–98%, 76.6–94.3%, 97.5%, and 58%, respectively).
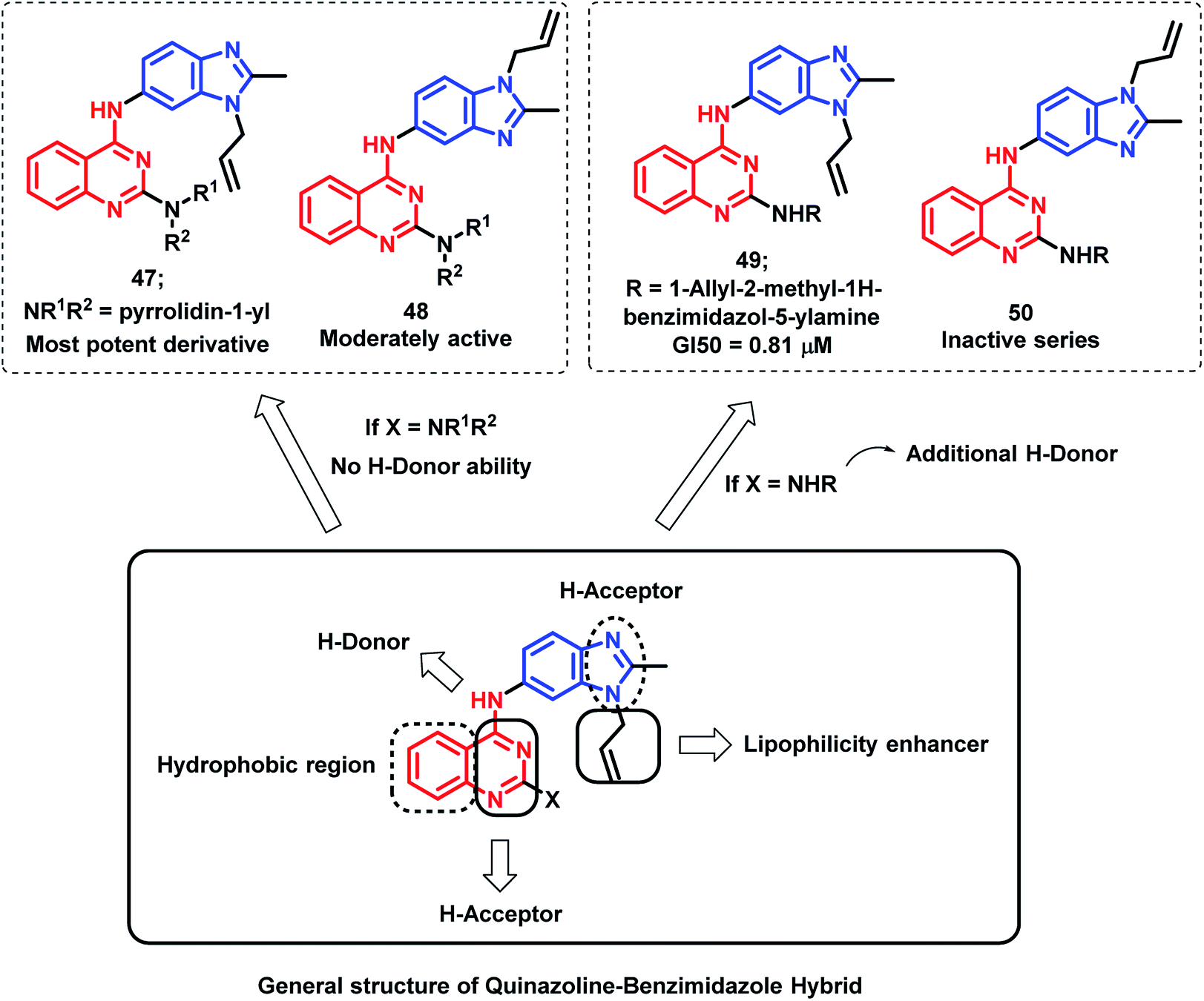 | ||
| Fig. 4 Designing strategy for benzimidazole- and 4-aminoquinazoline-based hybrids. | ||
Banerji et al. synthesized a series of triazole-substituted quinazoline hybrid analogues and evaluated their anti-cancer effect against HCT116 (human colorectal cancer cell line), HEPG2 (human liver cancer cell line), MCF-7 (human breast cancer cell line), and PC-3 (human prostate cancer cell line).76 The preliminary SAR of erlotinib and lapatinib (EGFR inhibitors) revealed that the presence of 5-aminoquinazoline moiety was helpful for EGFR inhibition. The triazole scaffold has also been previously reported due to its anti-cancer activity via various mechanisms. Hence, an attempt was made for a triazole-substituted quinazoline hybrid analogues as anti-cancer agents (Scheme 21). The designed molecules were expected to exhibit anti-cancer activity by inhibiting EGFR, induction of oxidative stress, and thereby, excessive ROS generation by mitochondrial dysfunction. In anti-cancer evaluation, erlotinib was used as the positive control, which exhibited an activity of 11.57 to 26.87 μM. From the results, it can be concluded that MCF-7 is more susceptible to the synthesized compounds than the remaining cell lines. Compound 53 exhibited the highest cytotoxicity (20.71 μM). Further, the activity of the compounds highlighted that the substitution of –Cl at the 2nd position and the –OCH3 groups at the 6 and 7 positions of quinazoline are essential for the cytotoxicity activity. The effects of compound 53 on the EGFR pathway in the MCF-7 cell lines were identified by using Western blot analysis. The levels of EGFR and p-EGFR were decreased by the treatment of various concentrations of compound 53. Hence, it is concluded that the cytotoxicity effects are mainly attributed to the decrease in EGFR and phosphorylation of EGFR and its downstream process. The effects of ROS-mediated pathway on apoptosis are confirmed by using flow cytometric analysis. The study highlighted that the mean intensity of fluorescein isothiocyanate (FITC) was lesser in the control cells than the compound 53-treated cells. These results also suggested the ROS-mediated pathways in the apoptosis of the MCF-7 cell lines. Fluorescence-activated cell sorting (FACS) was performed on the MCF-7 cell lines treated with compound 5b at different time intervals for identifying the apoptosis effects of compound 53. At the initial stage of apoptosis, phosphatidyl serine was exposed from inside the cell membrane to the outside, which can bind with annexin V. It was observed that early apoptosis rates increased from 5.9 to 24.6% and the late apoptosis rates increased from 0.7 to 14.2%. Time-dependent cellular apoptosis was shown by compound 53. These results indicate that compound 53 might induce apoptosis by the generation of ROS via the mitochondrial pathway as it leads to the lowering of the mitochondrial membrane potential.
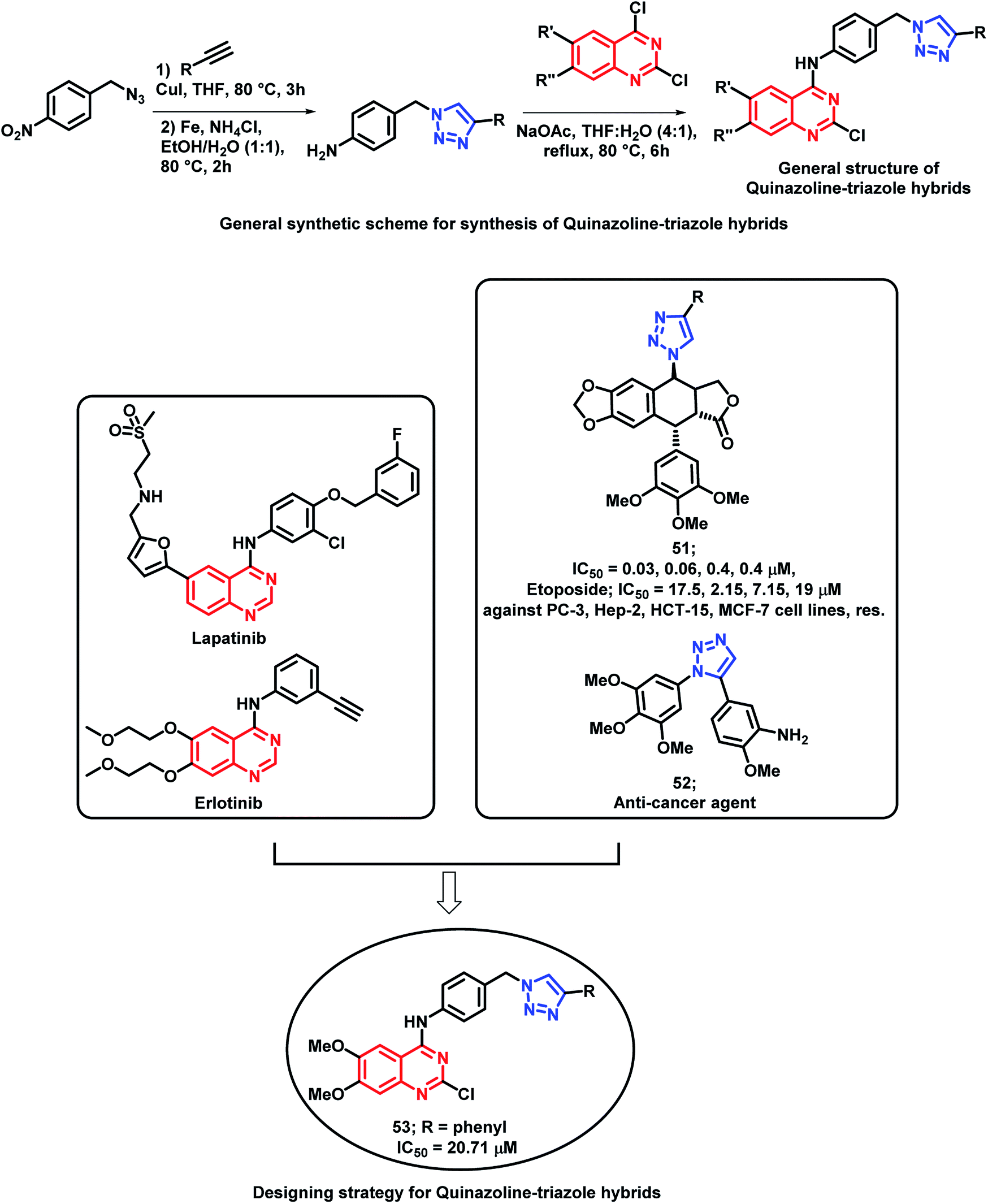 | ||
| Scheme 21 Synthetic and design strategy for triazole-linked 4-aminoquinazoline hybrids. | ||
Peng et al. synthesized a series of 4-anilinoquinazoline and hydroxamic acid hybrids as dual inhibitors for vascular endothelial growth factor receptor-2 (VEGFR-2) and histone deacetylase (HDAC).77 HDAC inhibitors are claimed to possess minimal toxicities toward the normal cells; hence, their combinations with other anti-cancer agents could be proved to be a successful application. As shown in Scheme 22, the synthesized analogues exhibited moderate VEGFR-2 inhibitory activities, wherein compound 54 exhibited a comparable activity (84 nM) to that of the positive drug, VEGFR-2 TKI vandetanib (IC50 = 62 nM). Further, an increased activity was observed with the increment in the length of the hydroxamic acid side chain. The most active VEGFR-2 in the series, i.e., compound 54 exhibited maximum HDAC inhibition (IC50 = 2.8 nM) with 5 times higher potency than the reference compound vorinostat (IC50 = 12 nM). The isoform selectivity was evaluated by conducting the HDAC inhibition assay in HDAC1, HDAC2, HDAC6, and HDAC8 (IC50 = 3.5 nM, 6.8 nM, 25.5 nM, and 5.2 nM, respectively). Further, the anti-proliferative activity against four types of human cancer cell lines, including a human breast cancer cell line (MCF-7), a human liver cancer cell line (HepG2), a human lung cancer cell line (A549), and a human colon cancer cell line (HCT-116) revealed a potent effect (IC50 ranges in 1.2 to 5.4 μM).
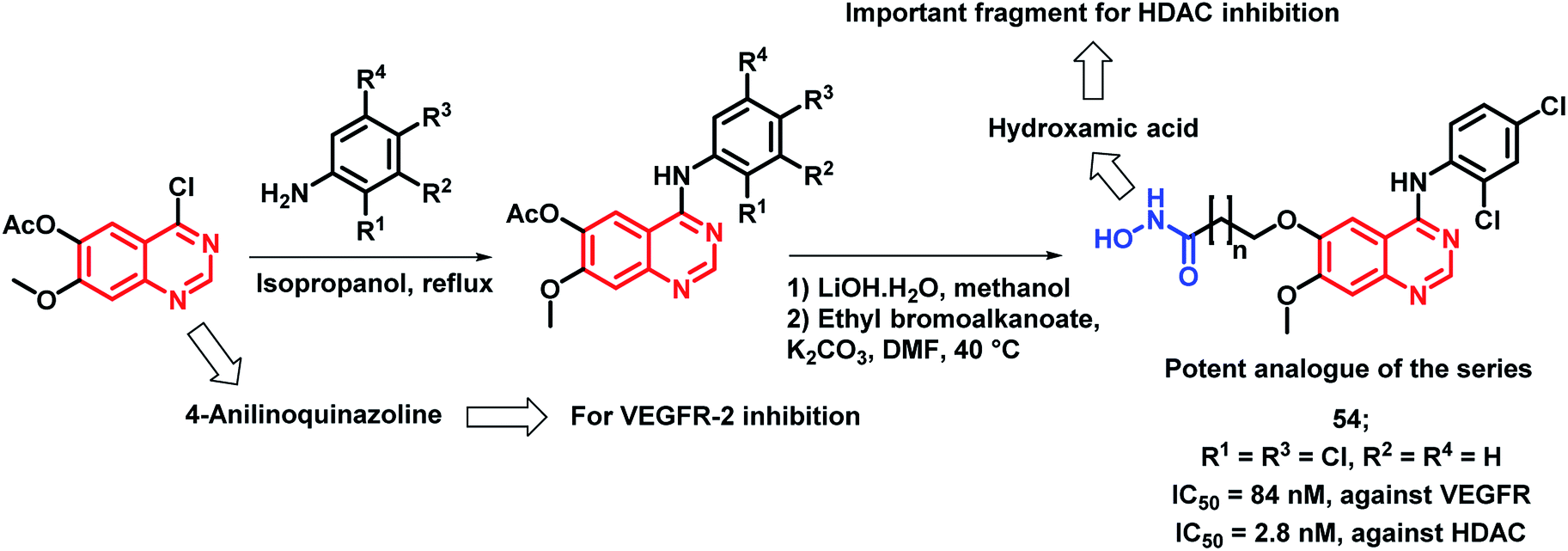 | ||
| Scheme 22 Synthetic scheme for hydroxamic acid and 4-aminoquinazoline-based hybrids for HDAC and VEGFR inhibition. | ||
Sulfonamide-containing drugs were found to be biologically active, with various activities such as carbonic anhydrase (CA) inhibition, anti-malarial, and anti-microbial activities.78–80 Hence, Ghorab et al. designed sulfonamido-linked 4-aminoquinazoline hybrids as anti-cancer agents.81 The screening of the synthesized analogues was done against human cancer cell lines (A549, HeLa, LoVo, MDA-MB-231). Most of the compounds were found to be inactive against the HeLa cell line. Compounds 55 and 56 were found to be cytotoxic against the above-mentioned cell lines (Scheme 23).
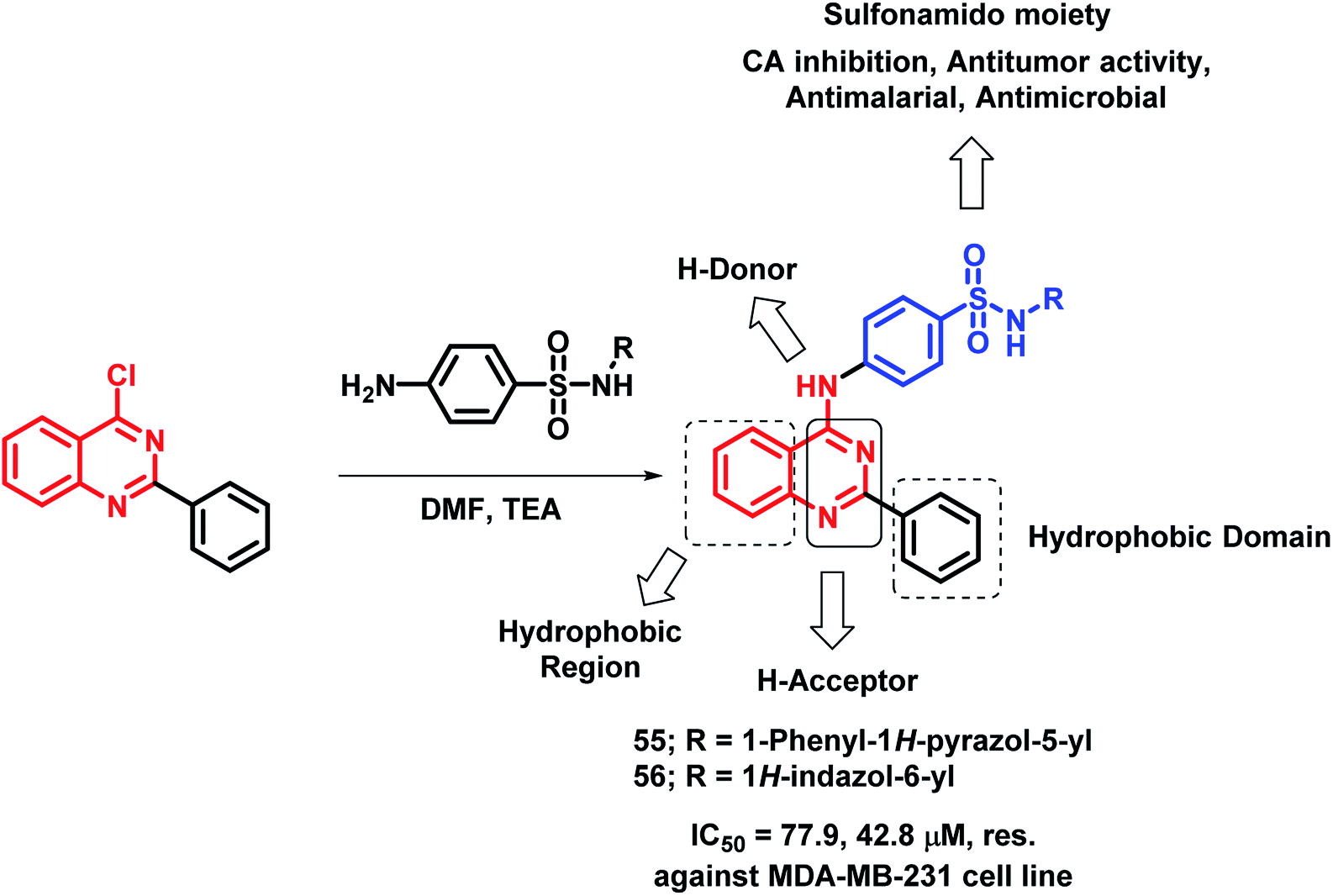 | ||
| Scheme 23 Synthetic scheme for sulfonamide linked 4-aminoquinazoline hybrids. | ||
3. Anti-microbial hybrids
By considering the anti-microbial potential of both triazine and quinazolinone moieties, Dinari et al. designed a novel chemical framework by combining both the heterocycles to get potential anti-microbial agents.82 The synthesized hybrid analogues were screened for anti-bacterial and anti-fungal potential by using microplate alamar blue assay (MABA) against Gram-negative (Salmonella enterica, Pseudomonas aeruginosa, Escherichia coli), Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus, Listeria monocitogenes), and Candida albicans, a yeast-like fungi, by taking ciprofloxacin and ketoconazole as the positive controls. Compound 57 was found to have strong anti-bacterial property against four bacterial strains with low minimum inhibitory concentration (MICs) in the range of 16–32 μg mL−1 (Scheme 24). The docking study of all the synthesized hybrids shows better binding in the pocket of S. aureus dihydrofolate reductase (DHFR) enzyme (PDB; 2W9S). The docking study revealed that the most potent analogue 57 showed the best in vitro anti-bacterial activity due to the presence of the sulfonamido group, which provides additional H-bonding interactions with Ser 49, Ileu 50, Gln 19, and Phe 92 at the binding site.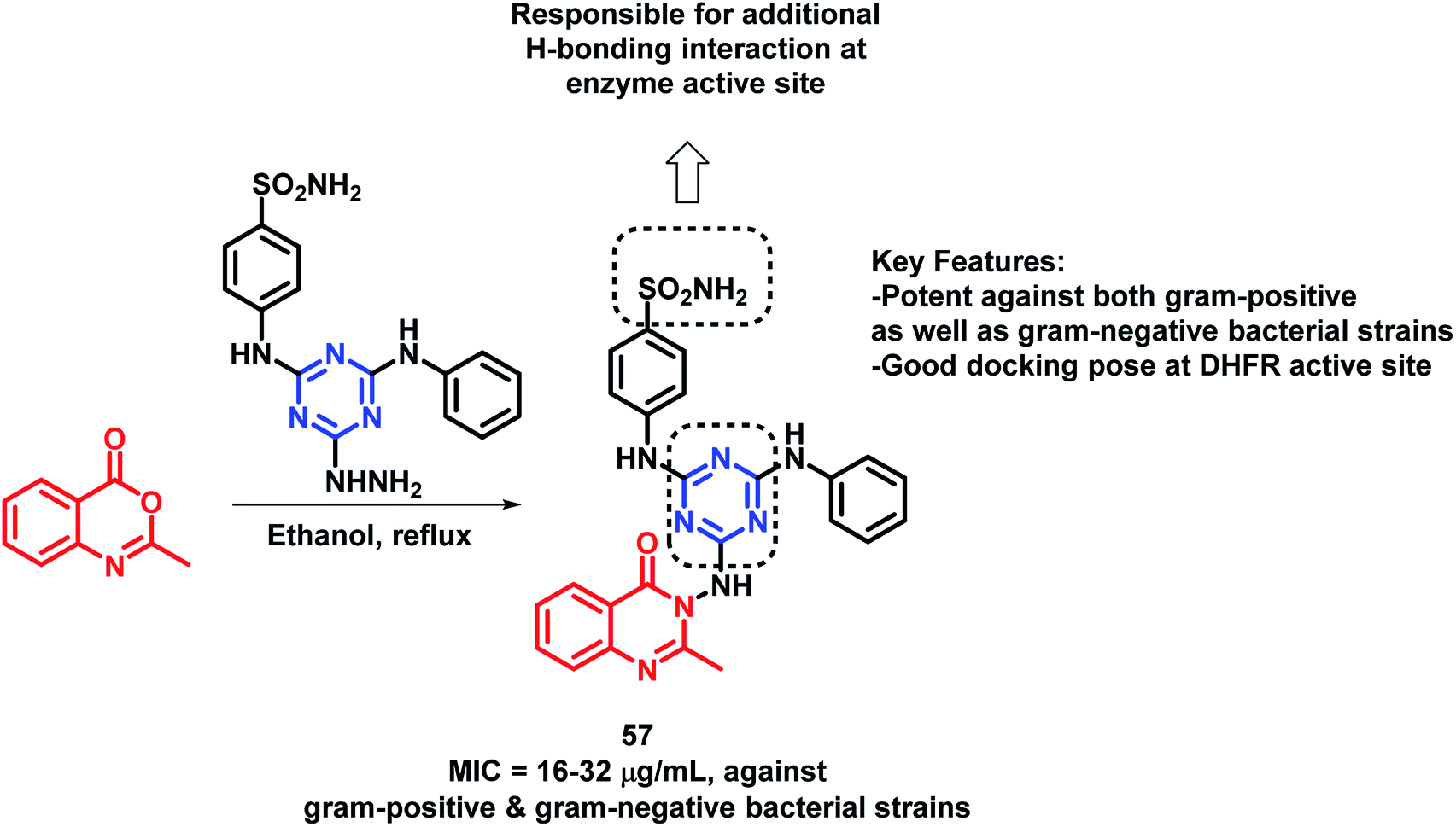 | ||
| Scheme 24 Synthetic scheme for quinazolinone- and triazole-based hybrids, showing its key features. | ||
Several triazole containing molecules are reported to have many biological activities; also, β-lactam antibiotic cefatrizine and the β-lactamase inhibitor tazobactam contain 1,2,3-triazole as a structural feature.83 Based on such observations, Gatadi et al. synthesized hybrids by a combination of structural features of triazole and quinazolinone moieties.84 Triazole was attached to the quinazolinone scaffold via the click chemistry approach. After the anti-bacterial evaluation of the synthesized compounds against the ESKAP panel of bacteria, many of the synthesized compounds were found to be active as anti-bacterial agents against Staphylococcus aureus. Compounds 58 and 59 were highly potent as anti-bacterial agents against S. aureus, with an of MIC 0.5 μg mL−1 (Scheme 25). These compounds were inactive against E. coli (Gram-negative bacteria) and were also found to be non-toxic against the Vero cell line. Compounds 58 and 59 also displayed inhibitory activity against 8 MRSA strains (IC50 in the range of 0.5–2 μg mL−1) & 3 VRSA strains (IC50 in the range of 0.5–32 μg mL−1), indicating the ability of these compounds to overcome the drug resistance mechanism.
 | ||
| Scheme 25 Synthetic scheme for quinazolinone- and triazole-based hybrids. | ||
In another report, Veeramreddy et al. synthesized triazolyl-quinazolinone hybrids via the click chemistry approach.85 The paper disc method was utilized for the anti-bacterial screening of these analogues against Escherichia coli (MTCC 443) and Staphylococcus aureus (MTCC 096). All the compounds have shown moderate to good anti-bacterial activity with MIC in the range of 7–30 μg mL−1, as compared with Ciprofloxacin (MIC of 20 and 21 μg mL−1 against E. coli and S. aureus, respectively), used as the positive control. Compound 60 was the most potent analogue (Scheme 26).
 | ||
| Scheme 26 Synthetic strategy for quinazolinone-thiazole hybrids. | ||
Azolyl ethanol fragment is widely present in various biologically-active compounds.86 Many azolyl ethanols have been successfully developed and are used to treat clinical infections (fluconazole and voriconazole, which are triazole type of azolyl ethanol, are used as first line drugs to treat fungal infections).87 Various azolyl ethanol fragments such as imidazolyl, triazolyl, tetrazolyl, benzoimidazolyl, and benzotriazolyl were incorporated at the 3rd position of quinazolin-4(3H)one by Peng et al. to get a series of hybrid analogues as potent anti-microbial agents.88 These synthesized analogues were screened for in vitro anti-bacterial activity against Gram-positive (Micrococcus luteus ATCC4698, MRSA, Staphylococcus aureus ATCC25923, and Bacillus subtilis ATCC6633) and Gram-negative strains of bacteria (Pseudomonas aeruginosa ATCC27853, Escherichia coli DH52, Bacillus proteus ATCC13315, and Eberthella typhosa ATCC14028). Amongst the synthesized hybrid analogues, triazolyl ethanol hybrids were found to effectively inhibit all the bacterial strains, except for P. aeruginosa, and their activities were comparable or even superior than that of norfloxacin (MIC of 8 μg mL−1) and chloromycin (MIC of 16 μg mL−1) against MRSA. Analogue 61 (MIC of 8 μg mL−1) was found to be potent in the series. Further, the docking study of analogue 61 has shown good interaction with MRSA DNA (PDB; 2XCS), which further supports the in vitro results. Imidazolyl ethanol hybrids has shown potential inhibitory activity against E. coli DH52 (MIC of 16 μg mL−1), while nitroimidazolyl analogues are potent against M. luteus and P. aeruginosa with MIC values between 16 and 32 μg mL−1. The most potent analogues of each series (61, 62, 63, and 64), along with their MIC values, are depicted in Scheme 27.
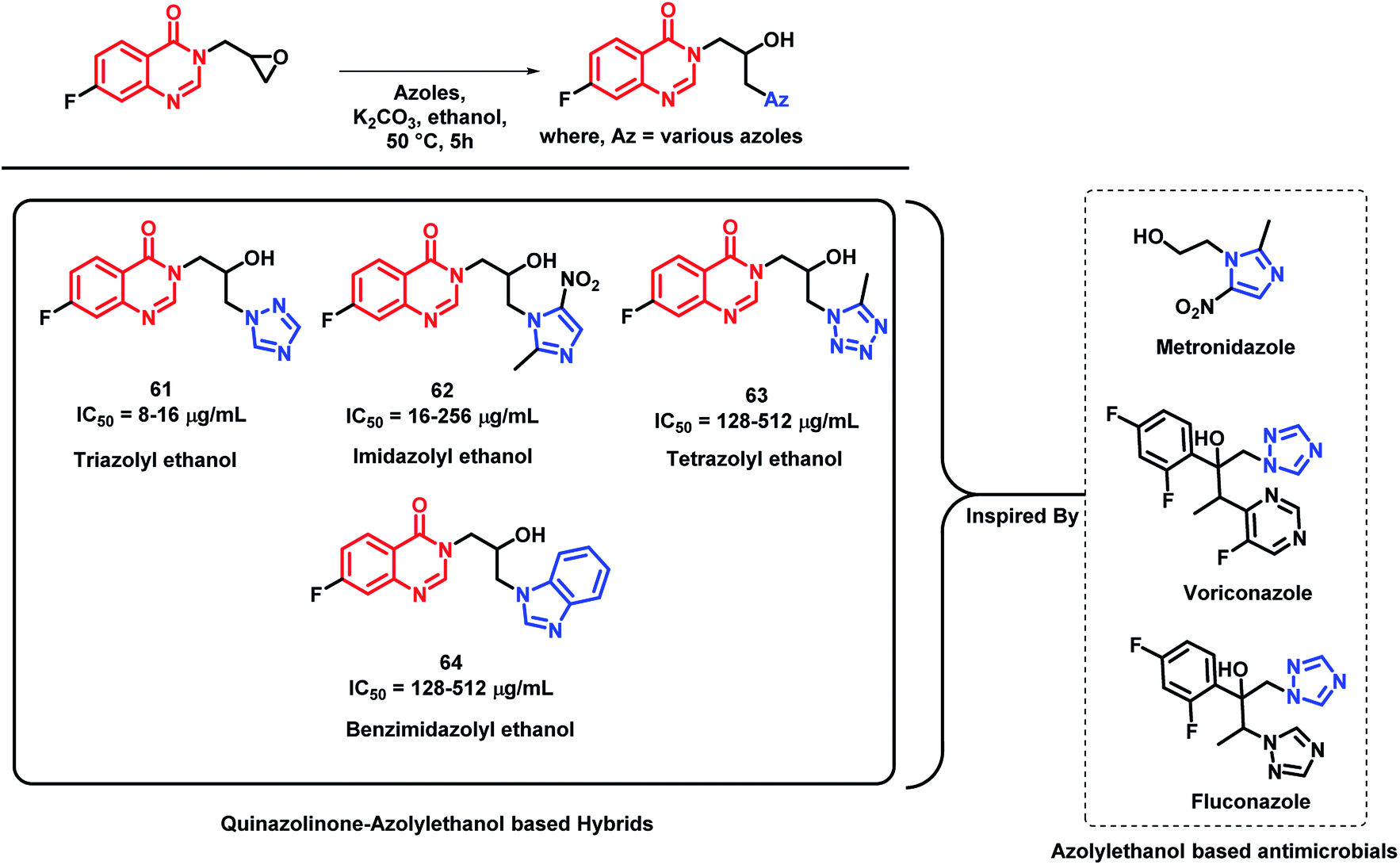 | ||
| Scheme 27 Synthetic scheme for various azolyl ethanol-based quinazolinone hybrids. | ||
Desai et al. have synthesized novel 5-arylidine derivatives of thiazolidin-4-one and quinazolin-4(3H)one.89 No derivative was found to be exceptionally potent in anti-microbial screening. Amongst them, compound 65a was found to be potent, with the MIC value of 25 μg mL−1 against E. coli.
In another study, in order to improve the anti-bacterial activity of previously synthesized quinazolinone and thiazolidin-4-one hybrids, the hydroxyl group at the para position of the phenyl ring directly attached to thiazolidinone was replaced by fluorine by Desai et al.90 The concept of increasing the hydrophobicity and the permeability of the compounds was effective, which resulted in potent fluorinated analogues. As compared with hybrid analogue 65a (MIC = 25, 100 μg mL−1 against E. coli and P. aeruginosa, respectively), analogue 66 was most potent with an MIC of 12.5 μg mL−1 against both E. coli and P. aeruginosa. The presence of electron withdrawing groups such as chloro, bromo, fluoro, and nitro at the arylidine ring are effective for anti-bacterial activity. The comparison of compound 65b and 66 is shown in Scheme 28.
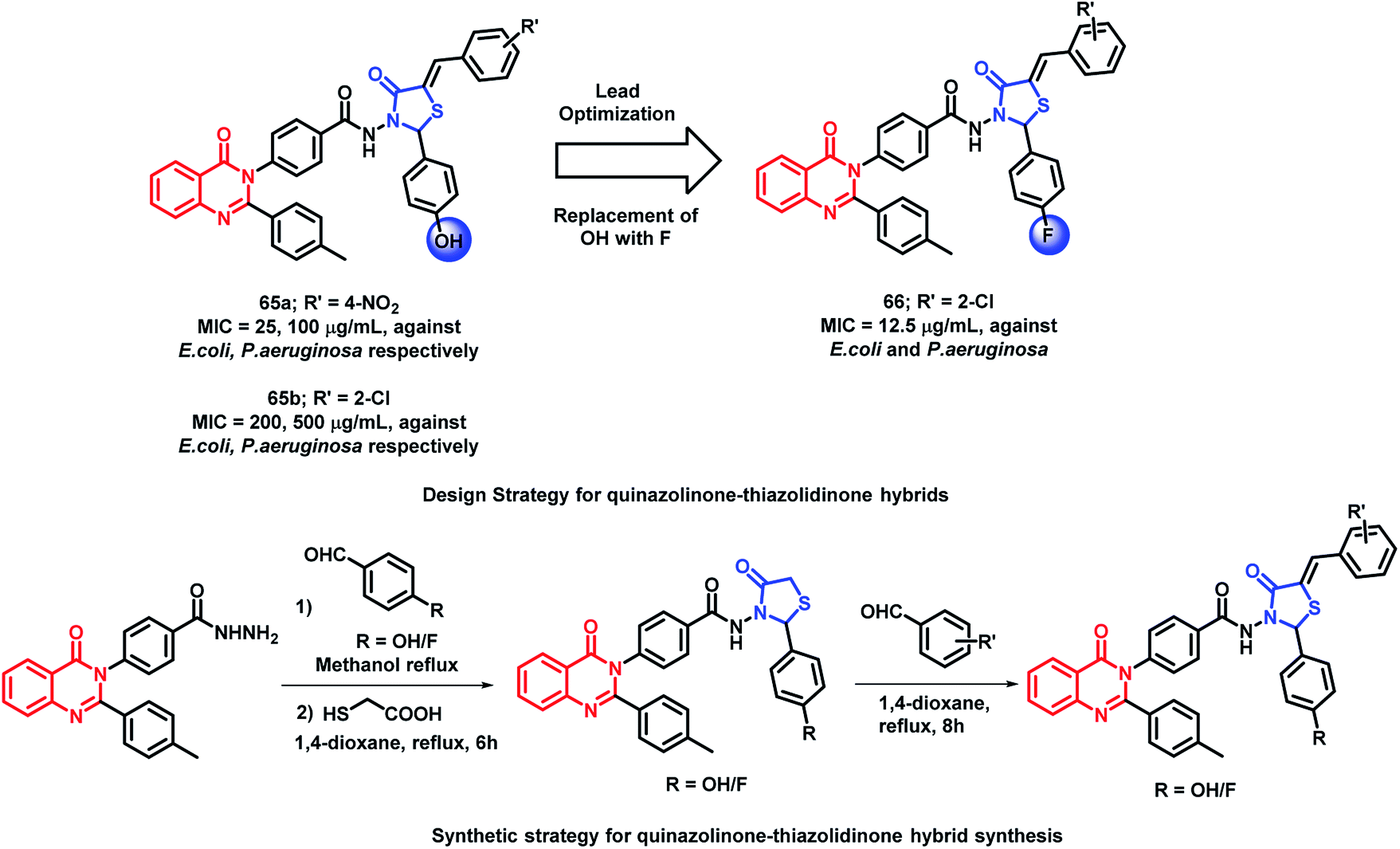 | ||
| Scheme 28 Synthesis and design strategy for quinazolinone-thiazolidinone hybrids, showing the importance of fluorine in the anti-microbial potential. | ||
By considering various thiadiazole-containing anti-microbial agents (67, 68, 69), Patel et al. synthesized novel quinazolino-thiadiazole hybrids and evaluated their anti-bacterial and anti-fungal potential.91 The MIC values of all the synthesized compounds were found in the range of 62.5–250 μg mL−1 against all the evaluated strains. From the SAR study of the synthesized analogues, it was found that the electron withdrawing groups at the phenyl ring are important for the anti-microbial activity. Compounds 70 and 71 with Cl and NO2 groups at the para position of the phenyl ring attached at C-2 of thiadiazole were most active with an MIC value of 62.5 μg mL−1 against Gram-negative bacteria (S. aureus, B. subtilis, E. coli, P. vulgaris) and fungal strains (C. albicans, A. niger) (Scheme 29).
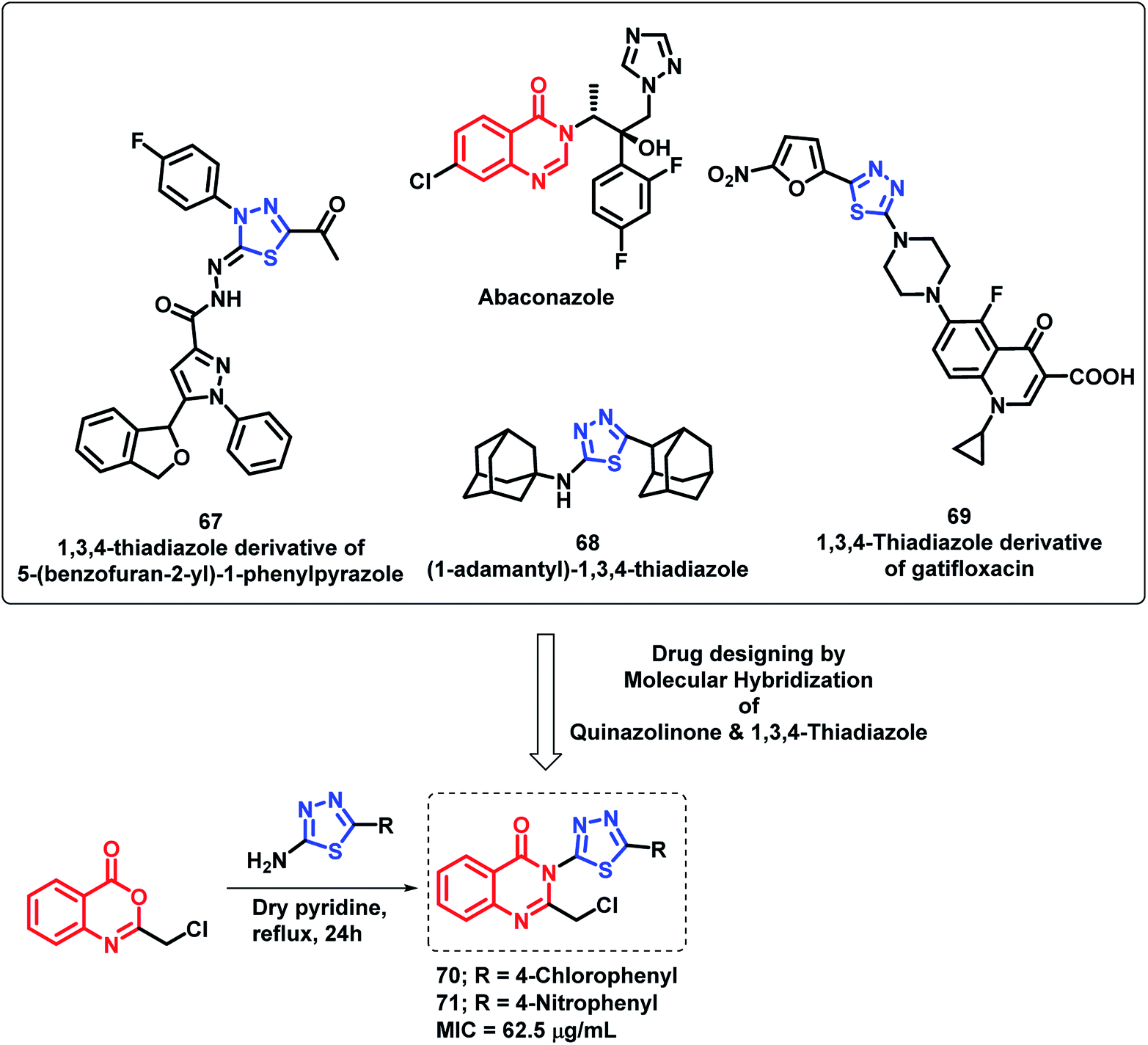 | ||
| Scheme 29 Synthesis and design strategy for quinazolinone-thiadiazole hybrids. | ||
4. Anti-tubercular hybrids
Pandit et al. designed hybrid analogues based on pyrazole and quinazolinone for targeting M. tuberculosis.92 Radiometric BACTEC and broth dilution assay methods were utilized for the in vitro screening of the synthesized analogues against M. tuberculosis H37Rv. Isoniazid was taken as a standard with % inhibition of 99% at an MIC < 6.5 μg mL−1. Some of the compounds (72, 73, 74, 75, 76) exhibited excellent anti-tubercular activity with % inhibition of 96, 90, 94, 93, and 92, respectively, at an MIC of less than 6.25 μg mL−1 (Scheme 30). According to the SAR study, the electron donating groups (Cl, CH3, OCH3) at the ortho position (compounds 72, 74, 75) and the electron withdrawing groups at the meta position (compound 76) are responsible for the increased anti-tubercular activity.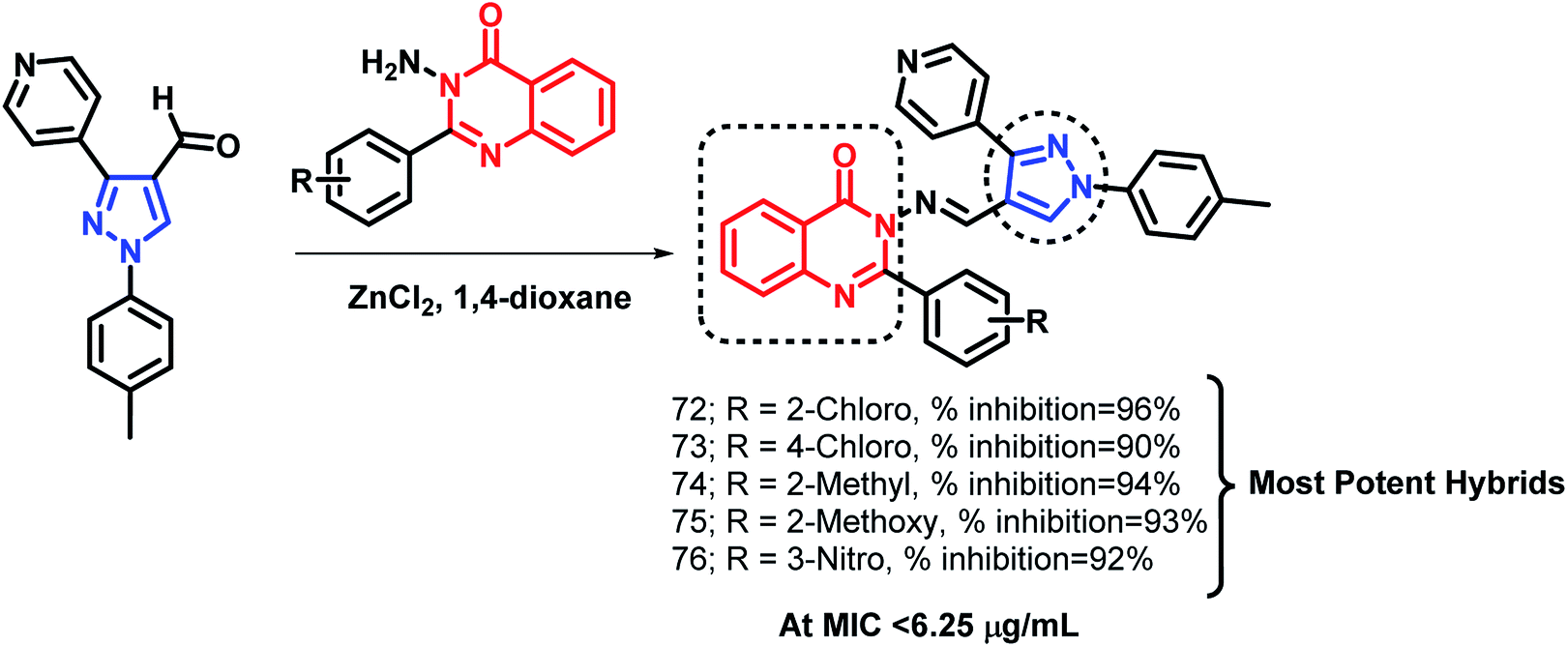 | ||
| Scheme 30 Synthetic scheme for pyrazole-linked quinazolinone hybrids. | ||
In the study by Maddali et al., triazole- and quinazolinone-linked hybrid analogues were designed through the molecular modelling study, targeting pantothenate synthase protein (PDB code: 3IVX).93 The effects of substitutions on the anti-tubercular activity was studied, with respect to the quinazolinone core, 1,2,3-triazole moiety, and phenyl ring attached to triazole. Through the SAR study, it was found that electron donating groups on the phenyl ring, electron withdrawing groups on triazole, and electron donating groups on the quinazolinone ring enhanced the anti-tubercular activity. Compound 77 was the most potent hybrid analogue with an MIC of 7 μg mL−1 against M. tuberculosis H37Rv (Scheme 31), using Rifampicin as a standard drug.
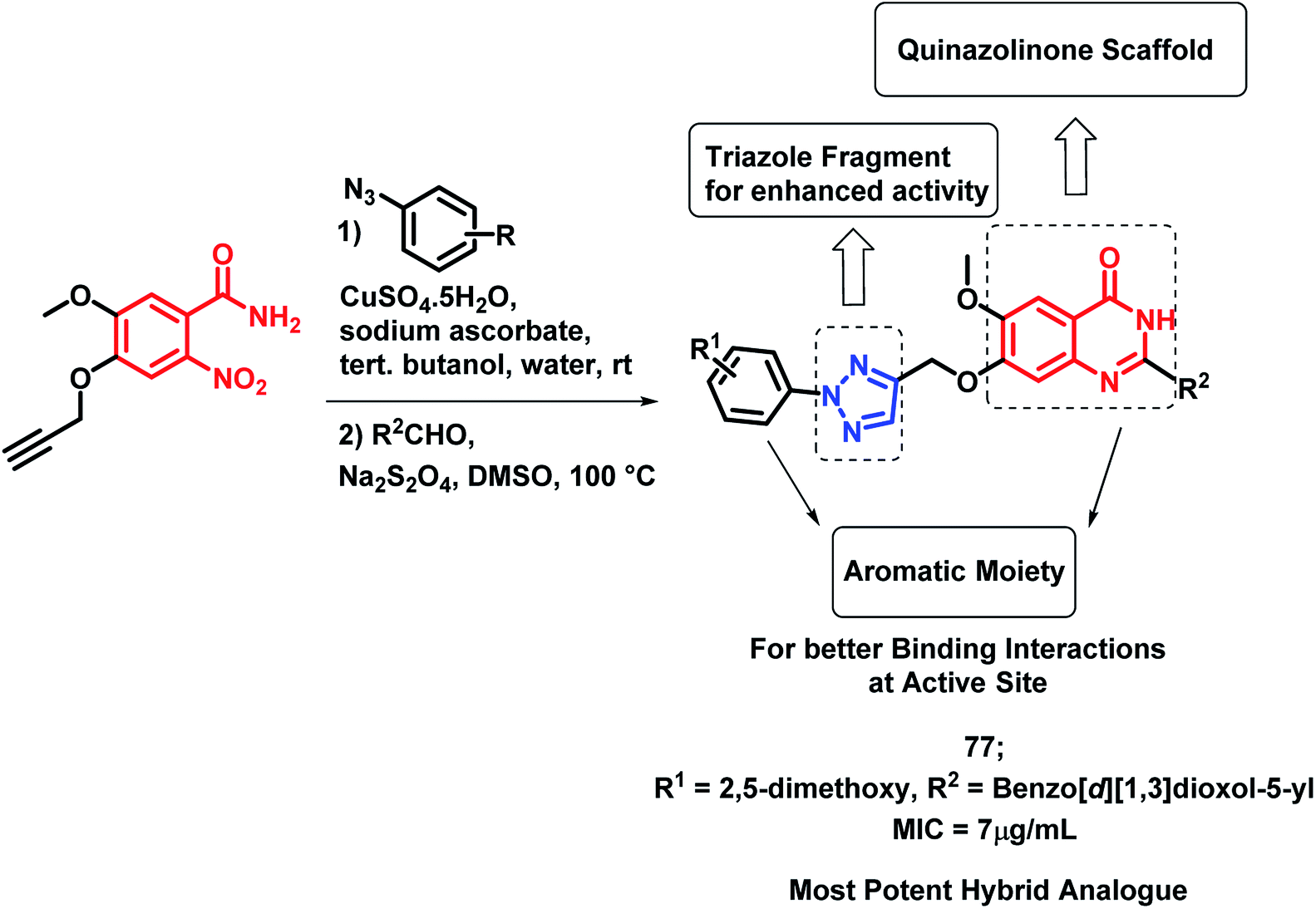 | ||
| Scheme 31 Synthetic scheme for quinazolinone-triazole hybrids, showing the importance of each structural unit. | ||
Hybrids of quinazolinones and thiazolidinones were designed by Shah et al. and evaluated for their anti-tubercular as well as the anti-microbial potential against M. tuberculosis H37Rv, fungal strains, and Gram-positive and Gram-negative bacteria.94 Compounds with Cl and NO2 group at the 2nd position (compounds 78 and 83, respectively) were found to be active (MIC of 6.25 and 3.12 μg mL−1, respectively) against Gram-positive S. aureus. Compounds 81, 82, and 83 showed good activity against Gram-negative E. coli (MIC of 3.12 μg mL−1). The S. typhi strain was found to be inhibited by 80 and 81 (MIC of 3.12 μg mL−1); 81 also showed excellent inhibition of K. Pneumonia (MIC of 6.25 μg mL−1). Ciprofloxacin was used as a standard for the in vitro anti-bacterial screening. As shown in Scheme 32, compounds 80 and 81, with NO2 and CH3 groups attached, were found to be active against M. tuberculosis H37Rv, with % inhibition of 99% at the MIC of 6.25 μg mL−1. The fungal strain C. albicans was inhibited by compound 81 with an MIC of 3.12 μg mL−1 as compared with the standard drug ketoconazole (MIC = 1.56 μg mL−1). All the anti-tubercular results were compared with isoniazid, rifampicin, ethambutol, and pyrazinamide standards (MIC = 0.20, 0.25, 3.12, and 6.25 μg mL−1, respectively).
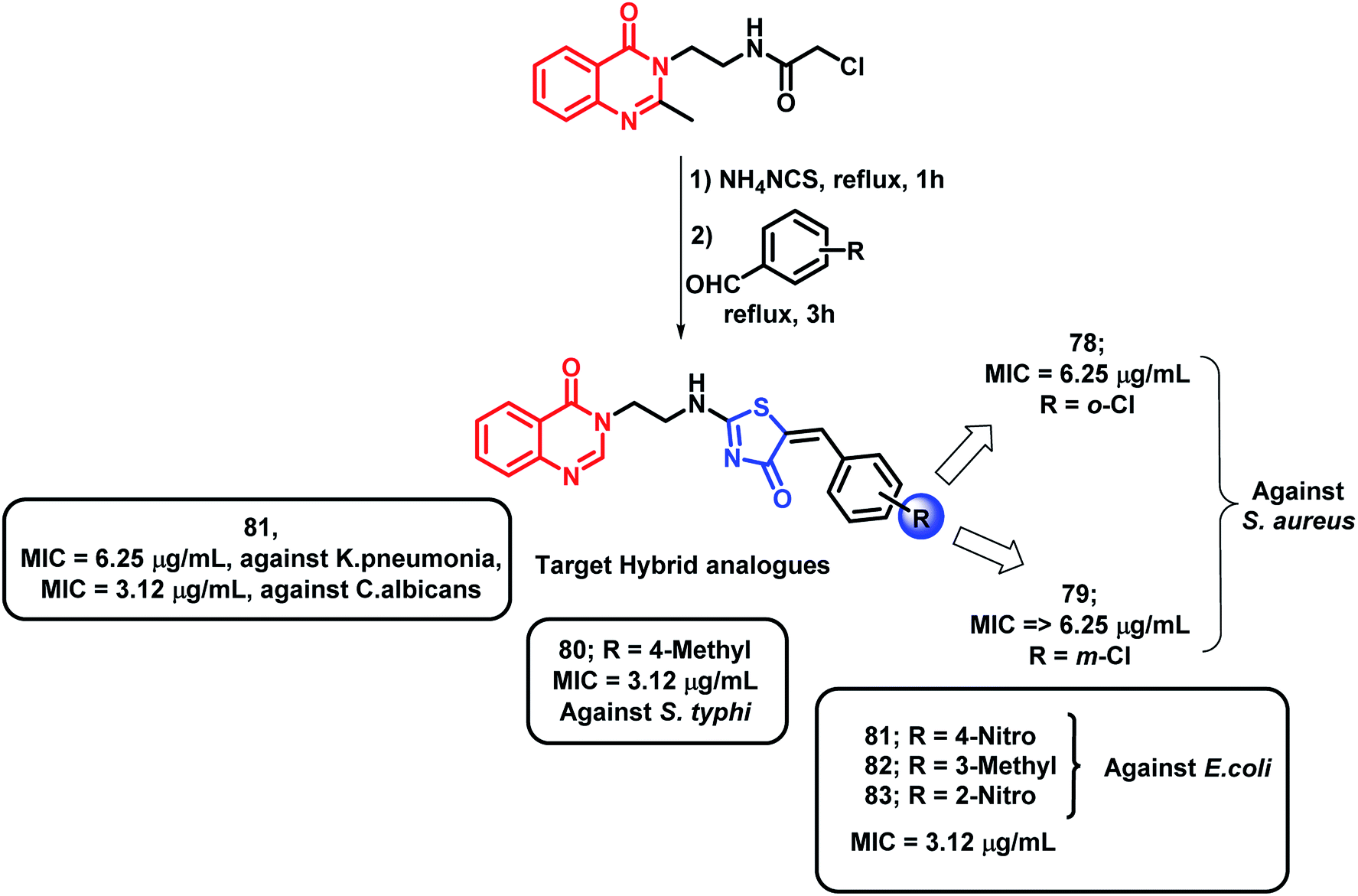 | ||
| Scheme 32 Synthetic scheme for quinazolinone- and thiazolidinone-based hybrids. | ||
Plant pathogenic bacteria are widely studied because of their threats towards agricultural production. Molecular hybridization technology can be used in order to inhibit such plant pathogens in the era of drug resistance. Quinazoline is a biologically-diverse scaffold, including some clinically approved pesticides, for e.g., fluquinconazole (fungicide), fenazaquin (acaricide), and pyrifluquinazon (insecticide). Also, by taking into consideration the various furan-based pesticides (cyclafuramid and furamizole), Long et al. designed quinazoline-furan-based hybrids.95 The designed furan-functionalized quinazolin-4-amines were synthesized and tested for their anti-bacterial activity. Compounds 84 and 85 were found to be effective in inhibiting the growth of Xanthomonas oryzae pv oryzae and X. axonopodis pv. citri, with EC50 values of 7.13 and 10.3 mg L−1, respectively (Scheme 33).
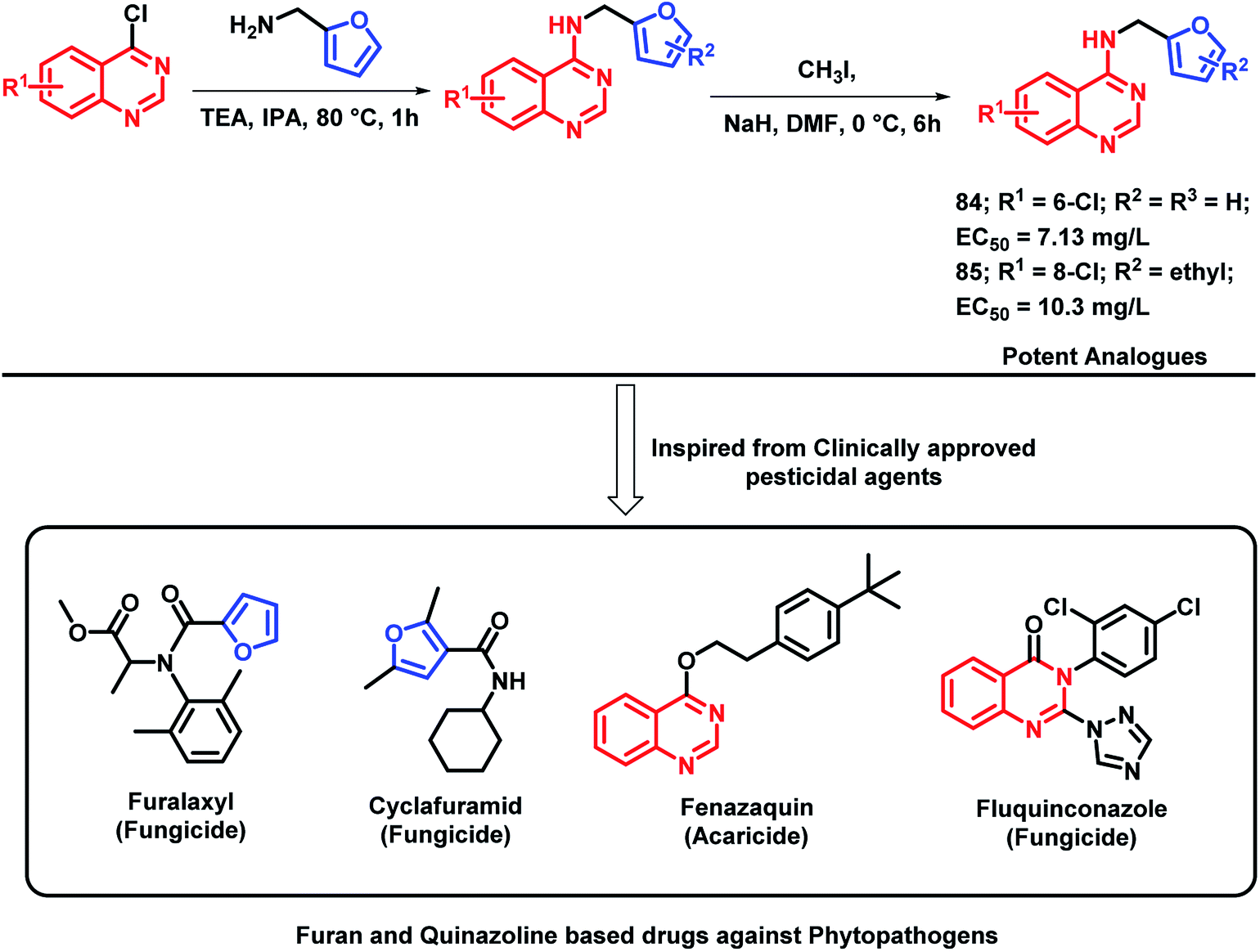 | ||
| Scheme 33 Synthesis and design strategy for furan-linked quinazoline hybrids. | ||
In continuation of the development of anti-microbial agents against phytopathogens, the hybrid drug approach was utilized by Fan et al.96 In an attempt to design such hybrids, quinazoline and 1,2,4-triazolo[4,3-a]pyridine (privileged scaffold for the discovery of drug/pesticide leads) were linked through a thioether linkage (Scheme 34). Thus, all the hybrids were designed, synthesized, and evaluated as anti-microbial agents. Amongst all the analogues, 86 and 87 were found to be potent with EC50 of 10.0 and 24.7 μg mL−1, respectively. The SAR study revealed some pharmacophoric features: (a) carboxylic acid ester at the 2nd position of quinazoline is essential for the anti-bacterial activity; (b) amongst the halogenated compounds, fluorine-containing molecules were found to have greater potential, probably due to the improved lipophilicity, bioavailability, and metabolic stability imparted by fluorine.
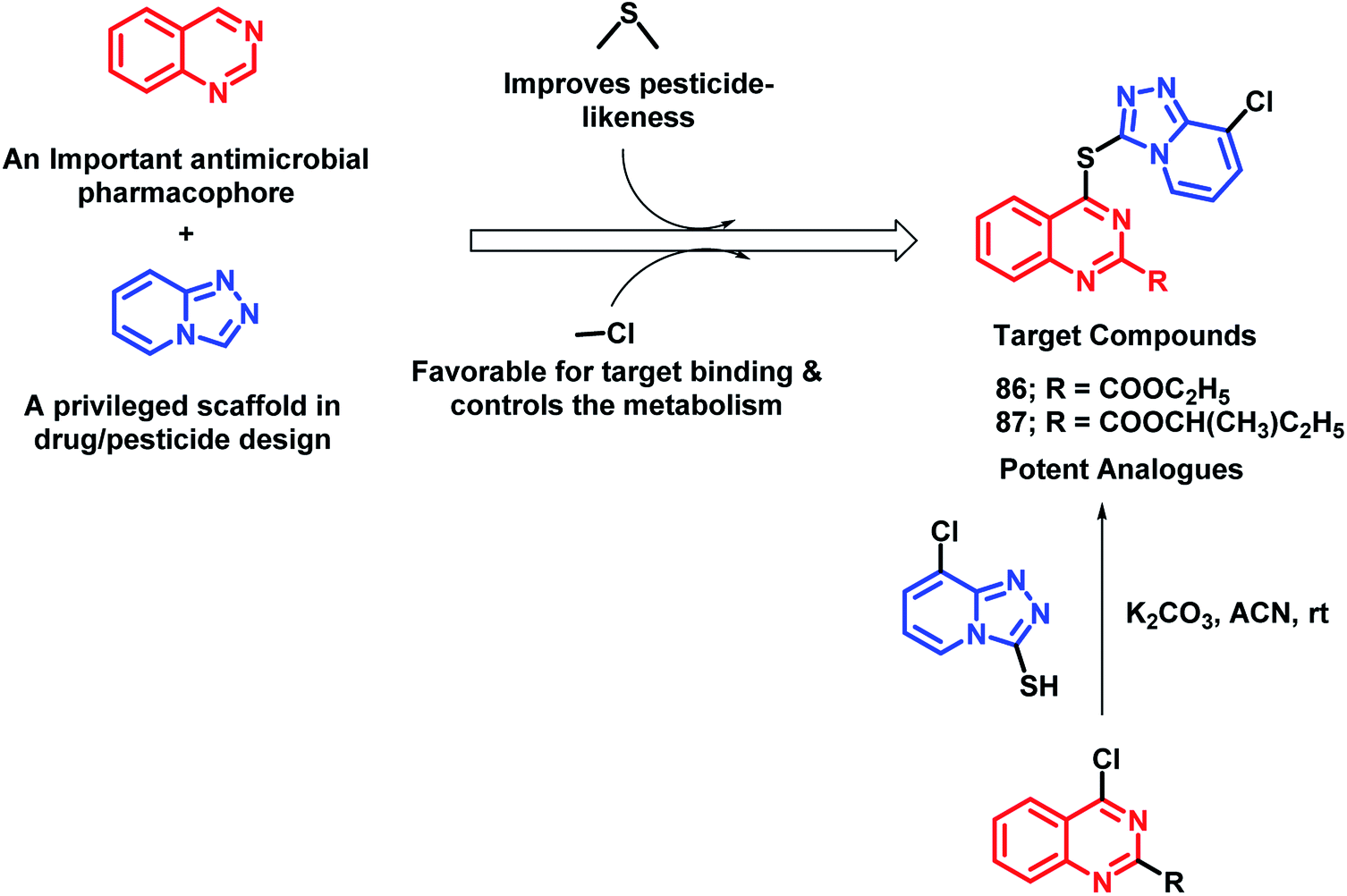 | ||
| Scheme 34 Synthesis and design strategy for quinazoline-linked 1,2,4-triazolopyridine hybrids. | ||
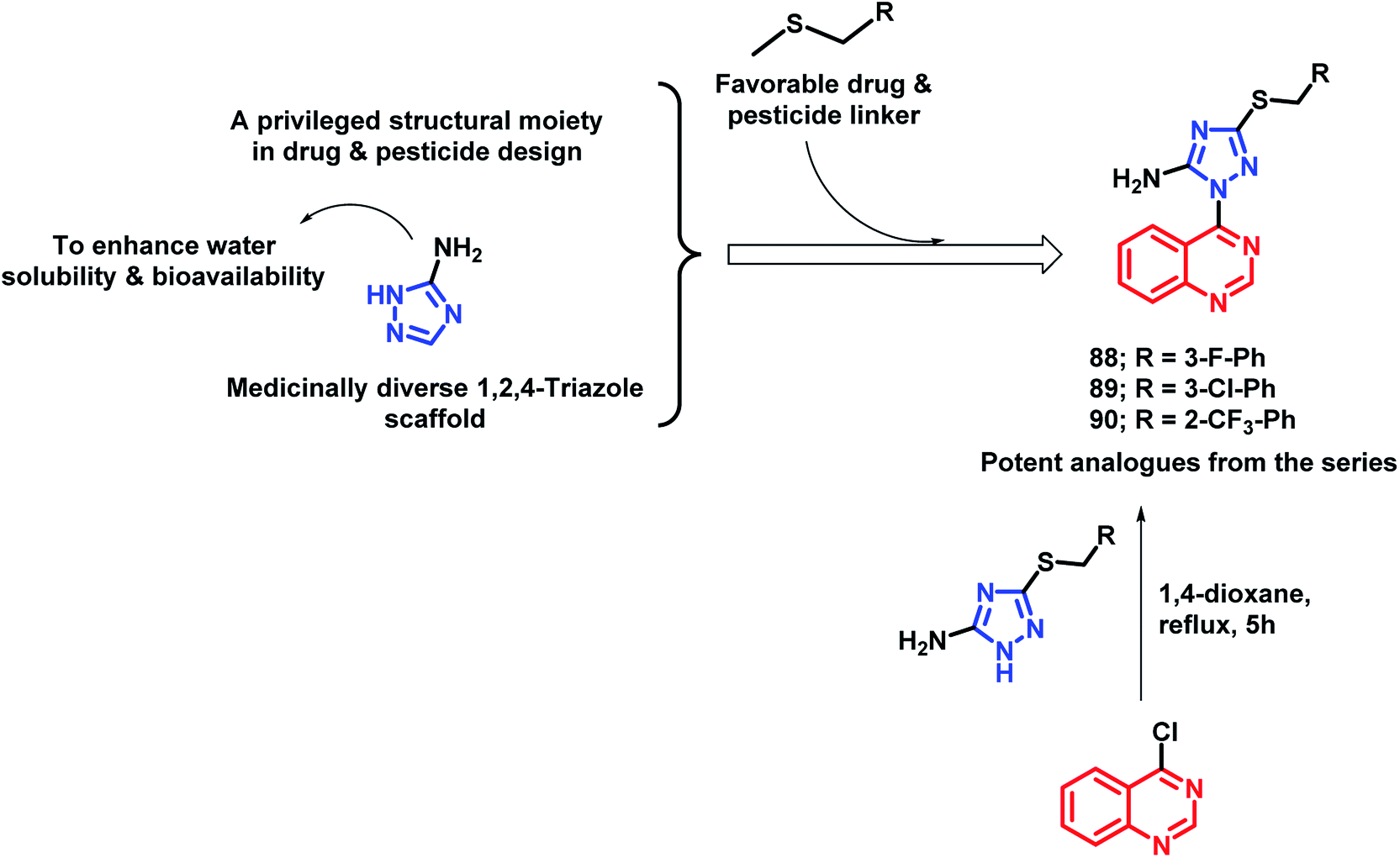
In another study by Fan et al., quinazoline was linked with 5-amino-1,2,4-triazole by the pharmacophore hybrid approach to obtain anti-microbial agents against plant pathogens.97 Along with quinazoline and triazole that have pesticidal effects, two additional features were incorporated into the designed molecules: (a) an amino group was placed on triazole in order to increase the water solubility and bioavailability, (b) thioether linkage for H-bond interaction and decreasing lipophilicity. Anti-bacterial activities of all the tested analogues were tested against 3 pathogenic bacteria, namely, Xanthomonas axonopodis pv. citri (Xac), Ralstonia solanacearum (Rs), and Xanthomonas oryzae pv. oryzae (Xoo), by taking Bismerthiazol (BMT) and Thiodiazole-copper (TDC) as the control agents. Compounds 88, 89, and 90 were found to be potent with EC50 of 46.9, 47.8, and 43.2 μg mL−1, respectively, against Xac (Scheme 35). These analogues were found to be more potent as compared with the commercial agrobactericide bismerthiazol (56.9 μg mL−1).
 | ||
| Scheme 35 Synthesis and design strategy for quinazoline and 5-aminotriazole hybrids. | ||
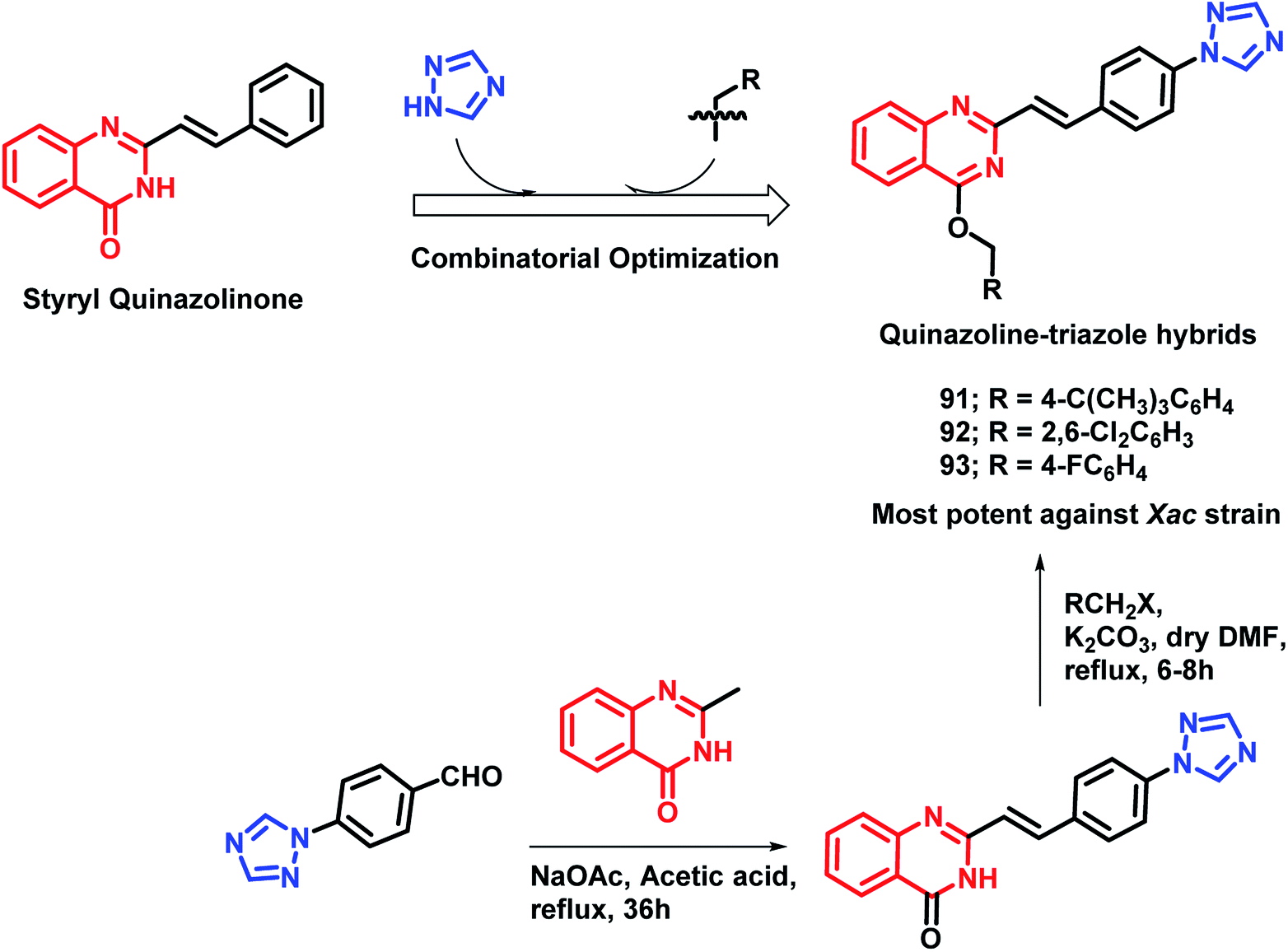
As quinazoline and triazole are well-explored as anti-microbial agents,98–105 Yang et al. designed triazolyl-linked quinazoline hybrids.106 As many styryl quinazolines have been reported to have anti-microbial and anti-mycobacterial activities,107,108 hence, styryl quinazolines were considered over simple ones. Also, various alkyl (aryl) methyleneoxy units were incorporated on the quinazoline backbone in order to study its effect on the anti-microbial activity. Turbidometric assay was used for the anti-bacterial screening of the synthesized analogues against phytopathogenic bacteria (Xoo, Xac, Rs). Bismerthiazol (BMT), which is a commercial bactericide, was used as a positive control. After the bioassay, compounds 91, 92, and 93 (% inhibition of 57.3, 66.6, and 57.8, respectively; at a concentration of 100 μg mL−1) were found to be potent (Scheme 36), comparable or even better than that of BMT (% inhibition of 58.0 at a concentration of 100 μg mL−1 against the Xac strain).
 | ||
| Scheme 36 Synthesis and design strategy for triazole- and styryl quinazoline-based hybrids. | ||
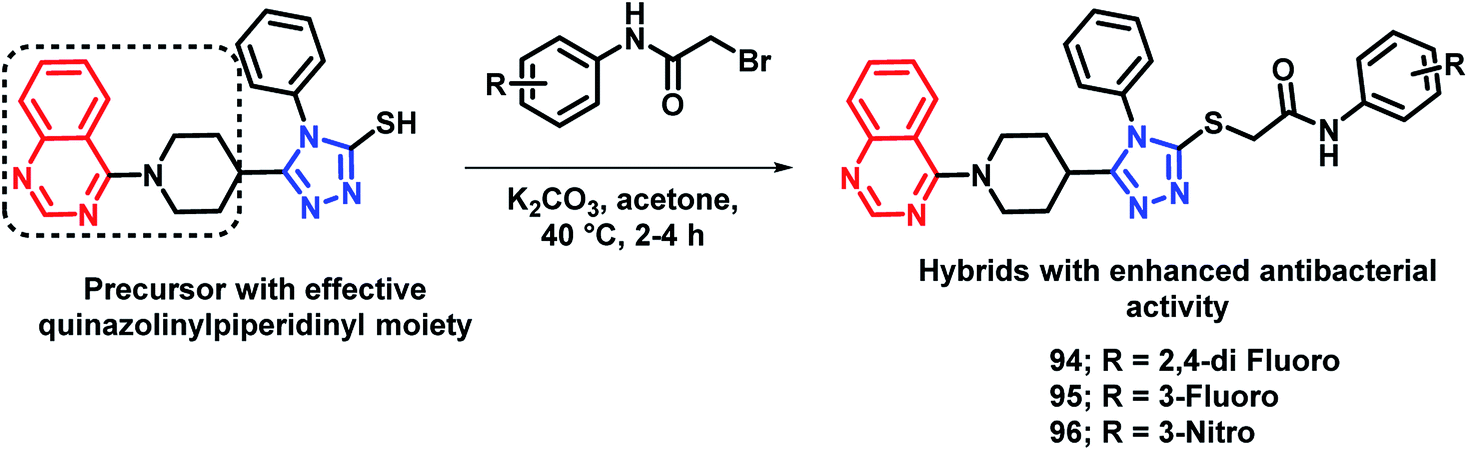
By considering the diverse range of activities of quinazoline and triazole scaffolds, Yang et al. designed hybrid analogues by linking these motifs together.109 Molecules were designed with the following rationale: (a) 1,2,4-triazole-based analogues are well reported with anti-bacterial activities,105,110,111 with Triadimefon, Triadimenol, Diniconazole, Flusilazole, and Difenoconazole, which are clinically used agrofungicides. (b) The quinazoline moiety is found in many pesticidal drugs such as Fluquinconazole and Pyrifluquinazon. (c) Acetamide is an important linkage as it is a part of a fungicide, Oxathiapiprolin. (d) Quinazoline with the piperidinyl linker is important to achieve effective anti-bacterial properties. By taking the above points in consideration, various N-(substituted phenyl)acetamide groups were linked with the quinazolinylpiperidinyl-modified 1,2,4-triazole ring (Scheme 37). These hybrid molecules were synthesized and screened for anti-bacterial activities. Compounds 94, 95, and 96 were found to be more potent, with EC50 in the range of 34.5–39.0 μg mL−1 as compared with that of the standard BMT (EC50 of 85.6 μg mL−1) against the Xoo pathogenic strain of bacteria.
 | ||
| Scheme 37 Synthesis of quinazoline- and triazole-linked hybrids. | ||
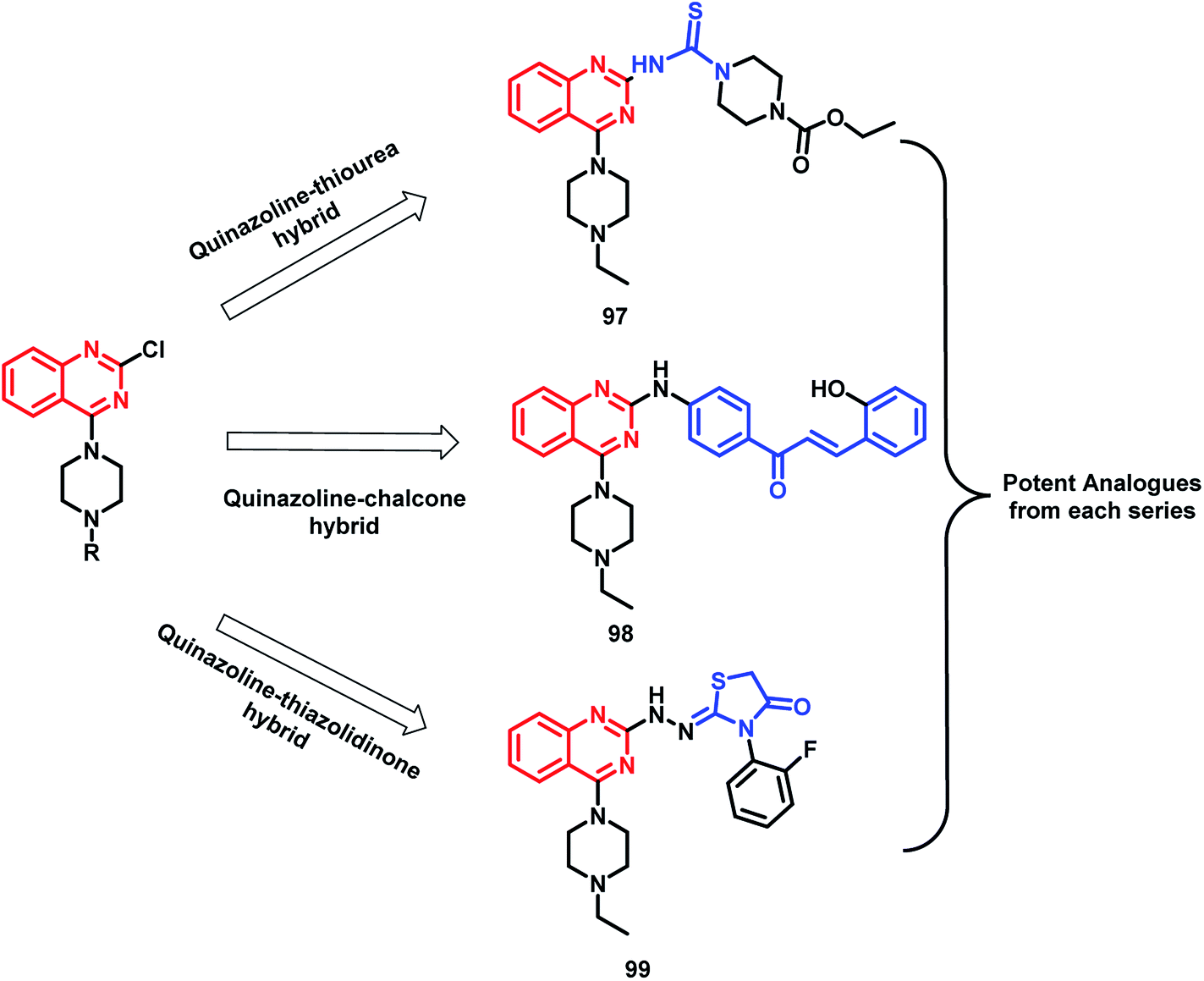
In the study by Shah et al., various hybrids were synthesized by fixing the quinazoline moiety and linking various other pharmacophores on it in order to study their effect on the activity.112 Quinazoline-thiourea, quinazoline-chalcone, and quinazoline-thiazolidinone hybrids were designed (Fig. 5, Scheme 38), followed by in vitro anti-bacterial and anti-fungal screening. Compound 97 was potent from the thiourea series. Compound 98, a chalcone hybrid, was found to be the most potent amongst all the series of compounds. From the last thiazolidinone series, compound 99 was found to be potent as both an anti-bacterial and anti-fungal agent.
 | ||
| Fig. 5 Design strategy for quinazoline-linked thiourea (97), chalcone (98), and thiazolidinone (99) hybrids. | ||
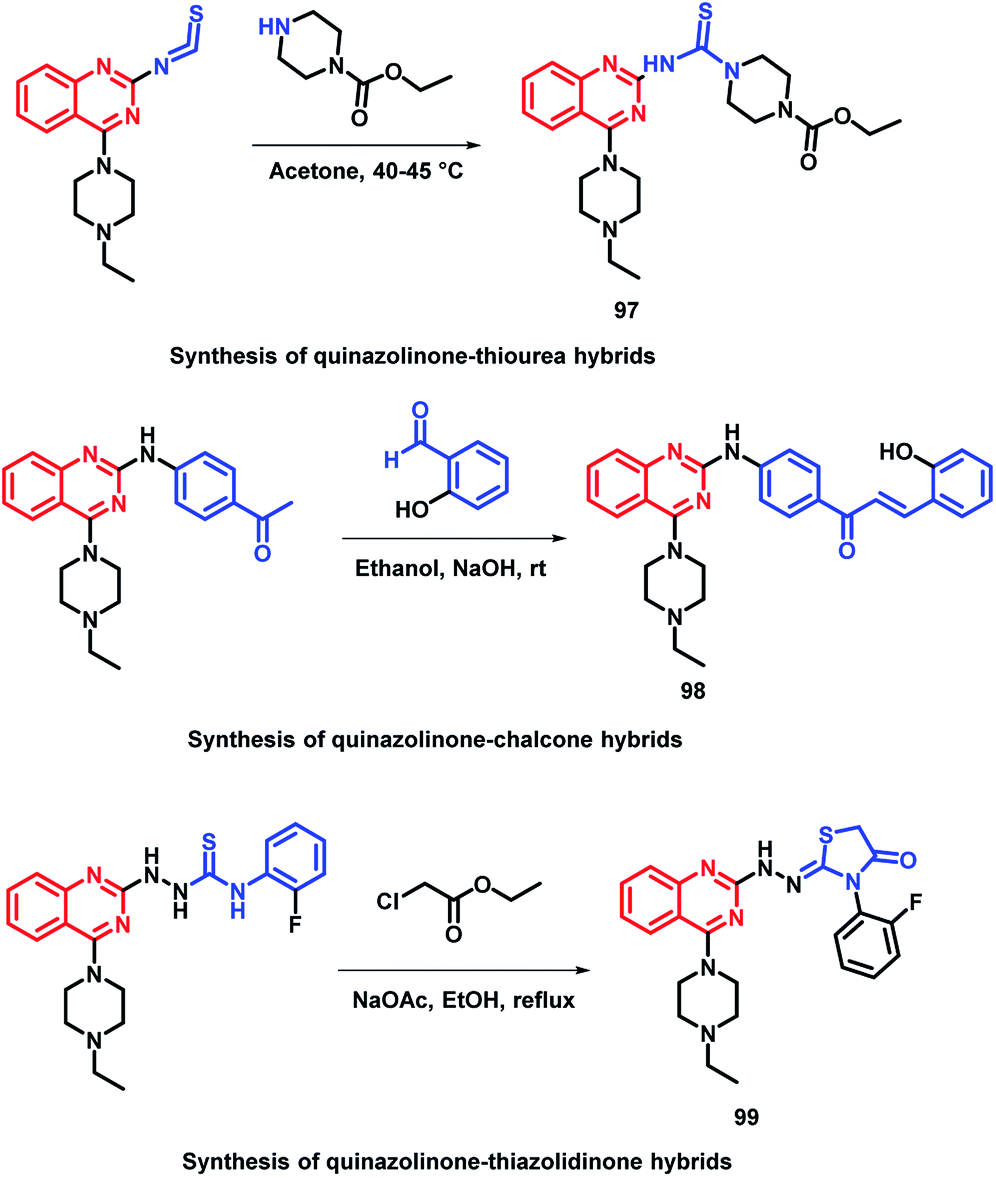 | ||
| Scheme 38 Synthesis of quinazoline-linked thiourea (97), chalcone (98), and thiazolidinone (99) hybrids. | ||
5. Anti-leishmanial hybrids
Destruction of the metabolic pathways that are essential for the survival of the parasite in the host is found to be a major target for the development of anti-leishmanial agents. Trypanothione reductase (TR) is one of the essential enzymes responsible for the anti-oxidant defence of the parasite. Previously, various anti-leishmanial agents, (with β-carboline, dihydro-β-carboline, and quinazolinone) were synthesized with potent activity towards each series. By taking such factors into consideration, Chauhan et al. synthesized quinazolinone and β-carboline-based hybrids, followed by their in vitro screening for anti-leishmanial activity.113 Many of the compounds were active in inhibiting the LdTR enzyme after in vitro enzyme inhibition performed against the LdTR enzyme. Compounds with aromatic groups on quinazolinone were found to be as active as the aliphatic substituents. Aromatic groups with electron withdrawing substituents (compounds 100, 101, and 102) show better inhibition as compared with the unsubstituted phenyl or phenyl ring with electron donating substituents (Scheme 39). All the enzyme inhibitors have shown competitive inhibition. Anti-leishmanial activity was also performed and the results of the enzyme assay were correlated with the anti-leishmanial activity. Compounds 100, 101, and 102 were found to be the most potent in the series with IC50 of 4.4, 6.0, and 4.3 μg mL−1, respectively, against intracellular amastigotes of L. donovani by taking Miltefosine (IC50 of 8.1 μg mL−1) and SSG (IC50 of 54.4 μg mL−1) as the standards.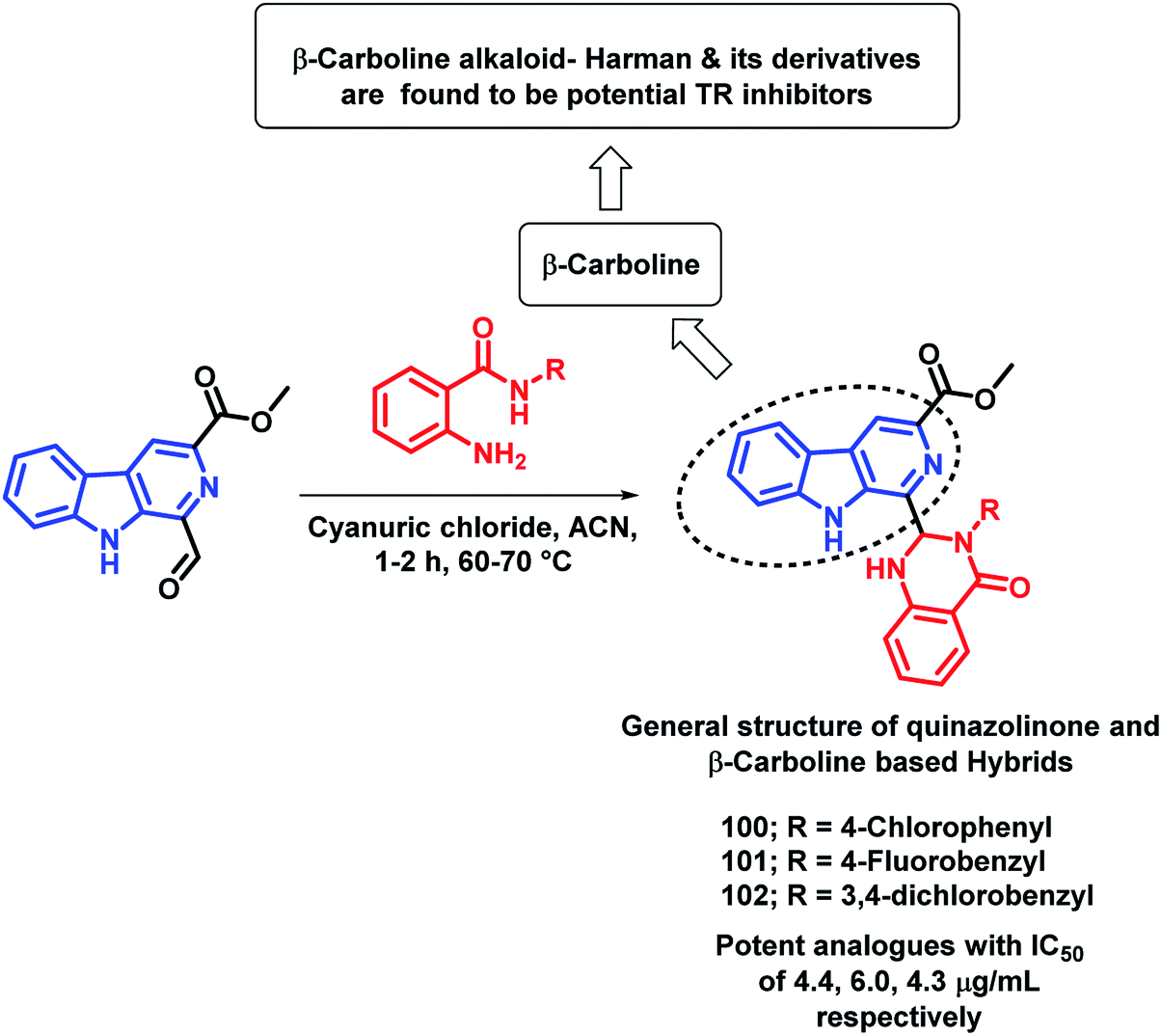 | ||
| Scheme 39 Synthetic scheme for β-carboline-quinazolinone hybrids. | ||
Sharma et al. designed 4 series of quinazolinone-based hybrids, namely, (a) quinazolinone-pyrimidine hybrids, (b) quinazolinone-triazine hybrids, (c) quinazolinone-peptide hybrids, and (d) quinazolinone-tetrazole hybrids (Fig. 6 and Scheme 40).114 After the screening of anti-leishmanial activity, all the series of compounds were found to be active except for the tetrazole series.
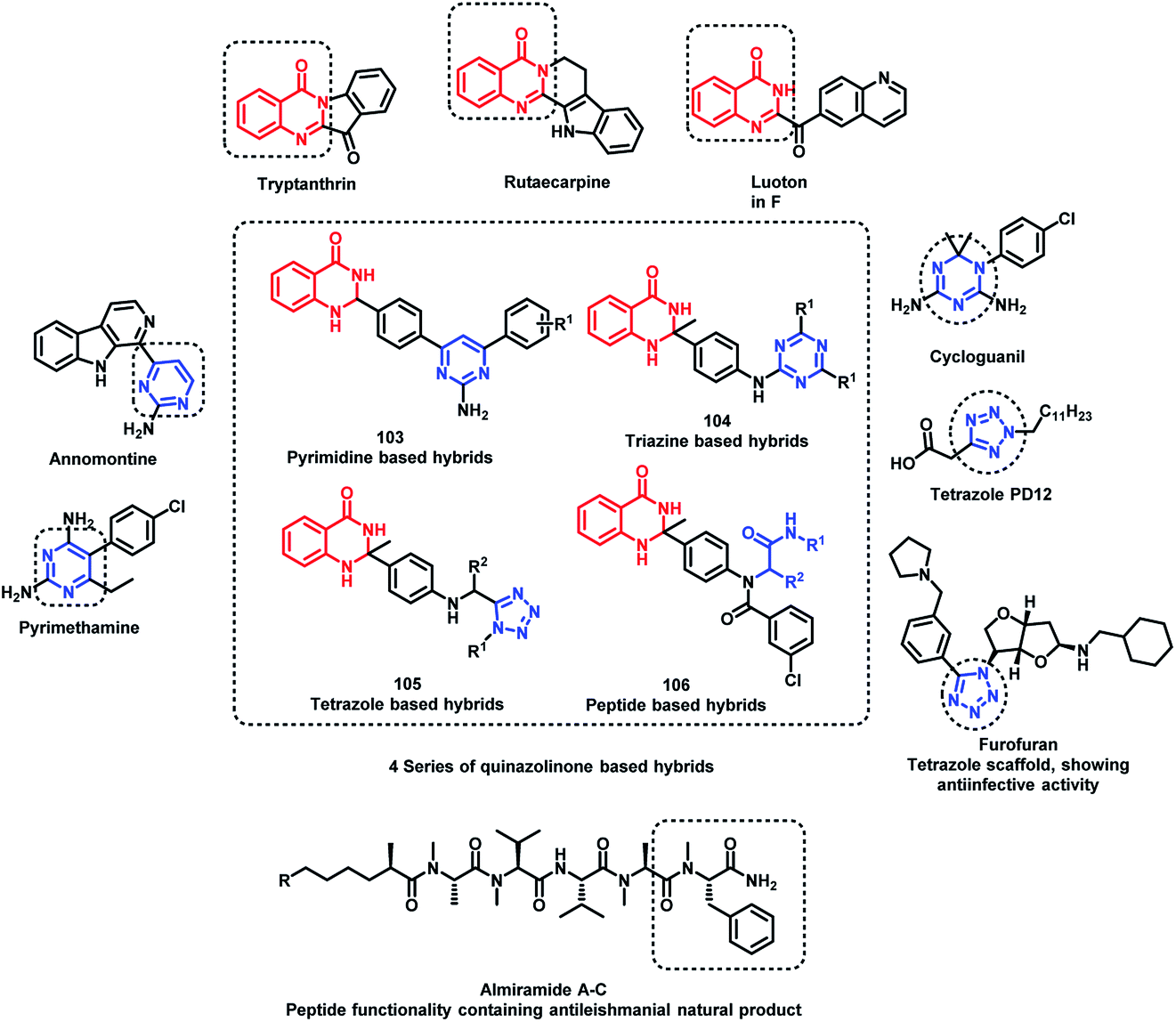 | ||
| Fig. 6 Design strategy for quinazoline hybrids with pyrimidine, triazine, tetrazole, and peptide. | ||
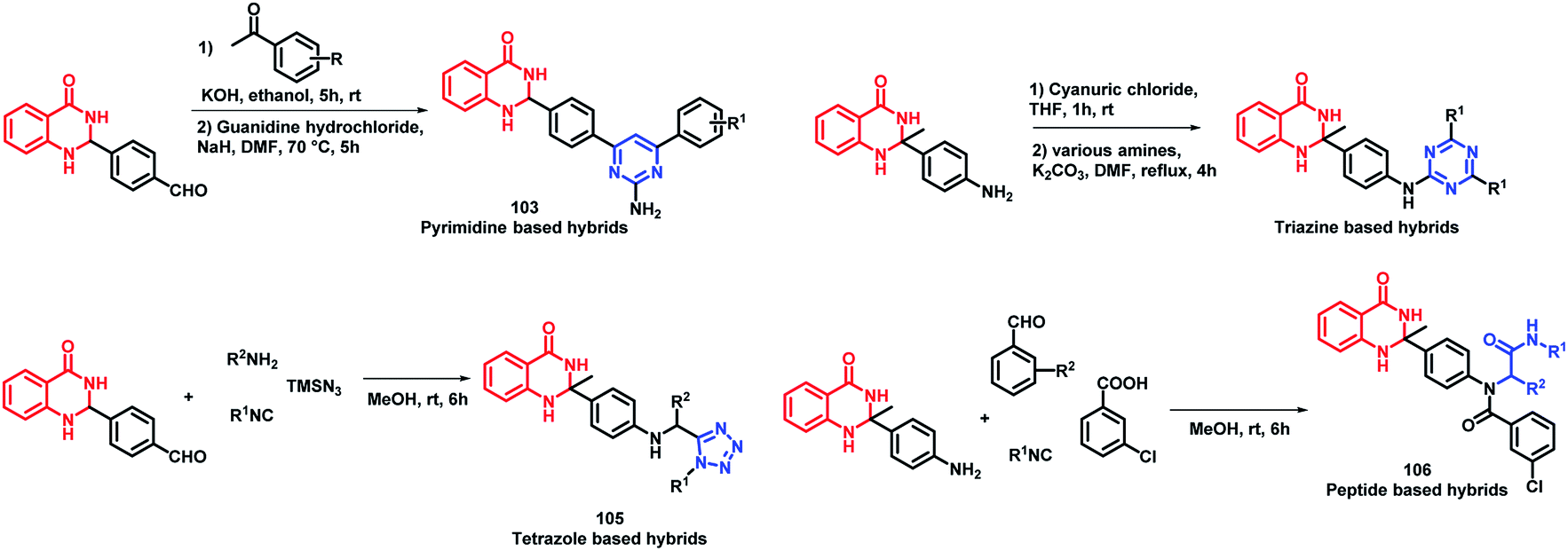 | ||
| Scheme 40 Synthetic scheme for quinazoline hybrids with pyrimidine, triazine, tetrazole, and peptide. | ||
6. Anti-diabetic hybrids
α-Glucosidase is an important enzyme responsible for the digestion of carbohydrates, which leads to the absorption of glucose and other monosaccharides. Inhibition of this enzyme reduces the postprandial blood glucose level, which results in decreased risk of postprandial hyperglycemia. Previous reports have depict the importance of quinazolinone115–117 as well as triazole118–120 in α-glucosidase inhibition. Taking such points into consideration, Saeedi et al. designed quinazolinone- and triazole-based hybrids and evaluated their α-glucosidase inhibition.121 All the synthesized hybrid analogues were found to inhibit the α-glucosidase enzyme through competitive inhibition with IC50 value in the range of 181–474.5 μM, using acarbose (IC50 of 750 μM) as the standard. Compounds 111 and 112 were the most potent with an IC50 of 181.0 and 192.3 μM, respectively (Scheme 41).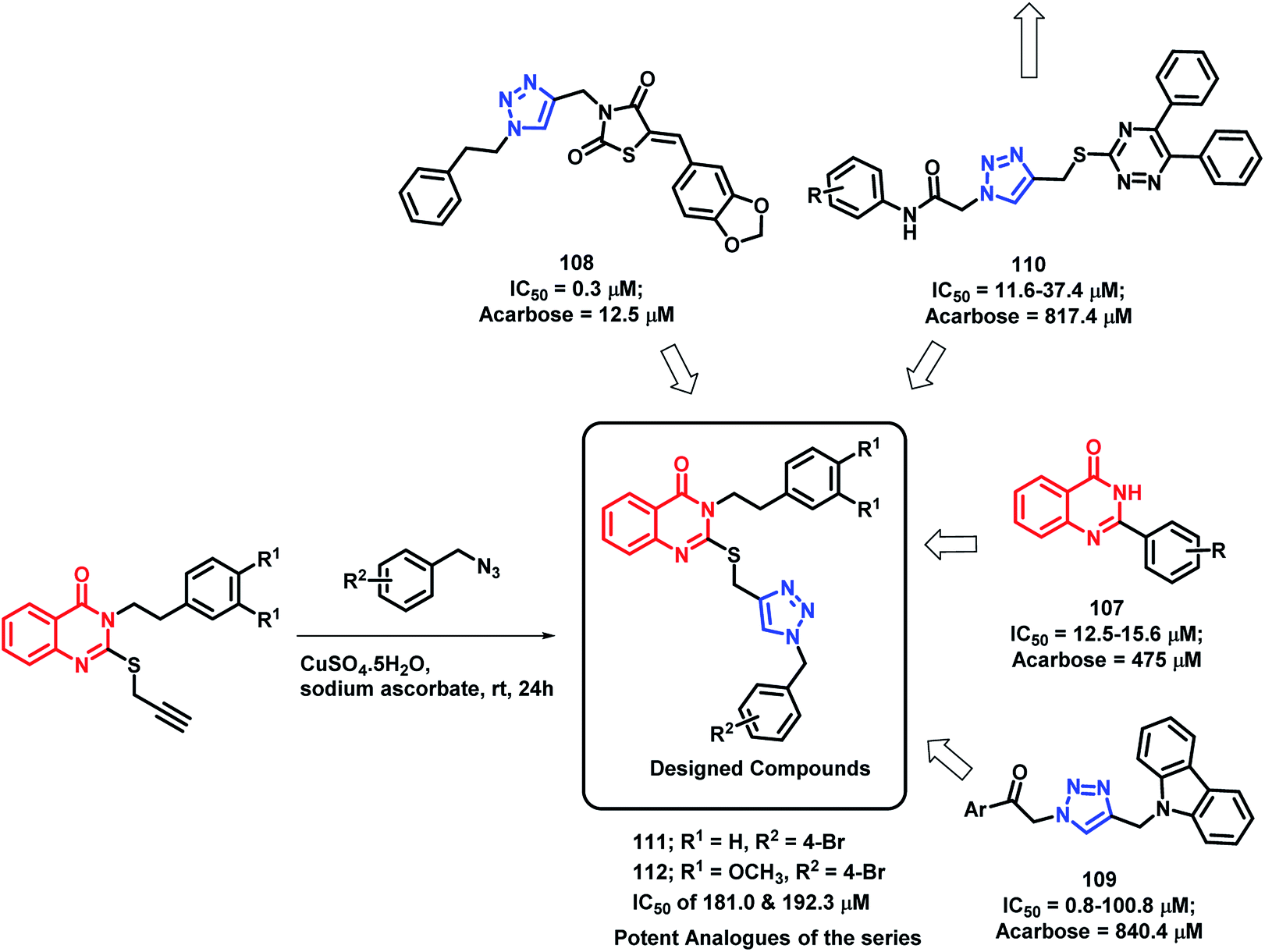 | ||
| Scheme 41 Synthesis and design strategy for triazole-linked quinazolinone hybrids, targeting α-glucosidase. | ||
By considering the importance of thiazolidinediones as PPAR-γ agonists (rosiglitazone, pioglitazone, troglitazone), it was hybridized with the medicinally important quinazolinone scaffold by Jangam et al.122 The docking study of all the designed compounds showed good binding at the active site of the PPAR-γ receptor (PDB: 4PRG). The anti-diabetic activity of the synthesized compounds was tested against Streptozotocin-induced diabetic rats and compounds 113, 114, 115, 116 showed significant anti-diabetic potential (Scheme 42). All the tested compounds were tested for various biochemical parameters such as alkaline phosphatise (ALP), alanine transaminase (ALT), aspartate transaminase (AST), insulin, glycated haemoglobin (HbA1C), and lipid profile such as high density lipoprotein (HDL), low density lipoprotein (LDL), cholesterol, triglycerides, as well as total protein level, and all were found to be significant.
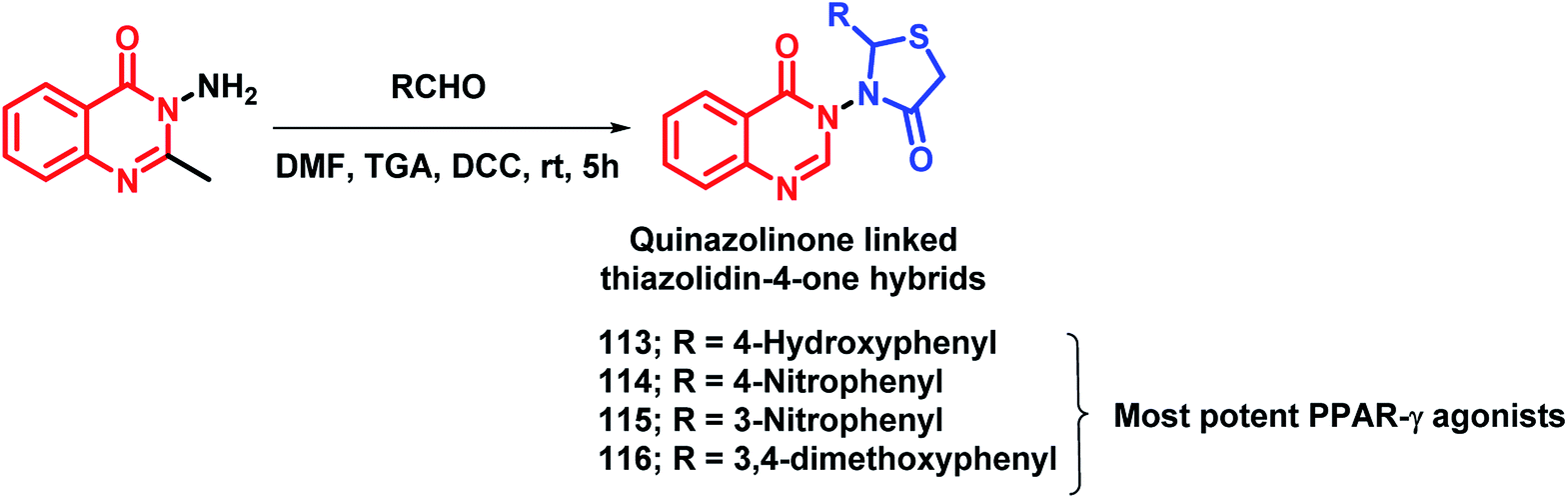 | ||
| Scheme 42 Synthesis of quinazolinone-thiazolidinone hybrids. | ||
7. Anti-malarial hybrids
Patel et al. performed the N-heterocyclization of leucine-linked sulphonamides in order to synthesize quinazolinone- and sulphonamide-based hybrids via modified Grimmel's method (Scheme 43).123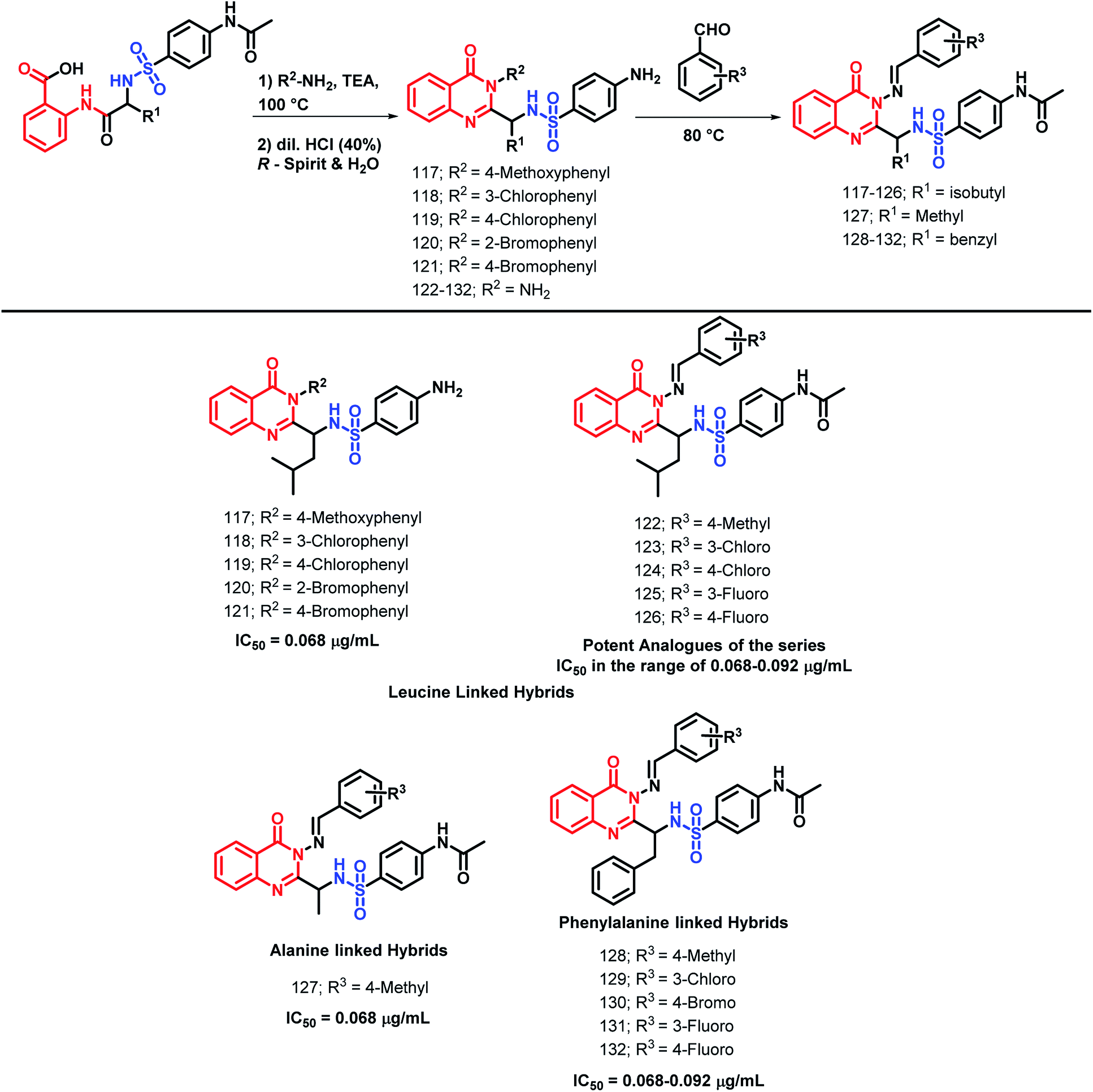 | ||
| Scheme 43 Synthetic scheme for various amino acid-linked quinazolinone hybrids. | ||
4-Amino-N-[(4-oxo-3-substituted aryl-3,4-dihydroquinazolin-2-yl)alkyl]benzenesulfonamide derivatives were synthesized and tested for anti-malarial activity against the P. falciparum strain. Compounds 117, 118, 119, 120, and 121 have shown comparable anti-malarial activity with an IC50 of 0.068 μg mL−1 as compared with that of Chloroquine, Pyrimethamine, and Trimethoprime standards, with IC50 of 0.020, 0.25, and 0.38 μg mL−1, respectively.
The further optimization of these compounds (4-amino-N-[(4-oxo-3-substituted aryl-3,4-dihydroquinazolin-2-yl)alkyl]benzenesulfonamide derivatives) was performed by the replacement of the phenyl ring at the 3rd position of quinazolinone with the benzylideneamino group.124 The activity of such novel lead optimized analogues was tested and amongst them, compounds 122, 123, 124, 125, and 126 were found to be active as anti-malarial agents when screened against P. falciparum.
Patel et al. synthesized alanine-linked quinazolinone-sulfonamide hybrids by N-heterocyclization of the alanine-linked sulphonamides.125 Compound 127 was found to be potent against anti-malarial screening (IC50 = 0.068 μg mL−1) as well as the DHFR inhibition assay (IC50 = 0.068 μg mL−1).
Also, the utility of phenylalanine was checked by the synthesis and anti-malarial screening of phenylalanine-linked quinazolinone-sulfonamide hybrids by Patel et al.126 Compounds 128, 129, 130, 131, and 132 were found to be potent for anti-malarial efficacy against the P. falciparum strain with IC50 in the range of 0.068–0.092 μg mL−1.
8. Anti-convulsant hybrids
Epilepsy is a group of disorders, characterized by neuronal hyper excitability with episodes of sensory, motor, or autonomic phenomenon with or without the loss of consciousness. Quinazolinone derivatives are found to be potential in the treatment of epilepsy, with many clinically approved drugs such as Methaqualone, Afloqualone, and Mecloqualone. Derivatives of benzothiazole are also gaining attention for their anti-convulsant effects, mainly due to AMPA antagonism (e.g., Riluzole). Hence, Malik et al. designed some hybrid analogues by combining GABA agonistic and AMPA antagonistic features of quinazolinones and benzothiazoles, respectively, in one hybrid scaffold.127 The pharmacophoric features, which are listed below, were incorporated into the molecule for enhancing the anti-convulsant potential of the designed analogues. Such features are (a) hydrophobic domain, (b) hydrogen bonding domain, (c) electron donor moiety, and (d) distal hydrophobic domain. In this way, quinazolinone- and benzothiazole-based hybrids were designed (Scheme 44) and synthesized. Maximal electroshock (MES) and subcutaneous pentylenetetrazole (scPTZ) seizure models were developed in mice for the anti-convulsant screening of the synthesized hybrids. Compound 133 with ED50 values of 82.5 μmol kg−1 (MES) and 510.5 μmol kg−1 (scPTZ) was found to be more potent than phenytoin (ED50 = 92 μmol kg−1 (MES) and > 3540 μmol kg−1 (scPTZ)) and ethosuximide (ED50 ≥ 3540 μmol kg−1 (MES) and 1182 μmol kg−1 (scPTZ)), which were used as the positive control.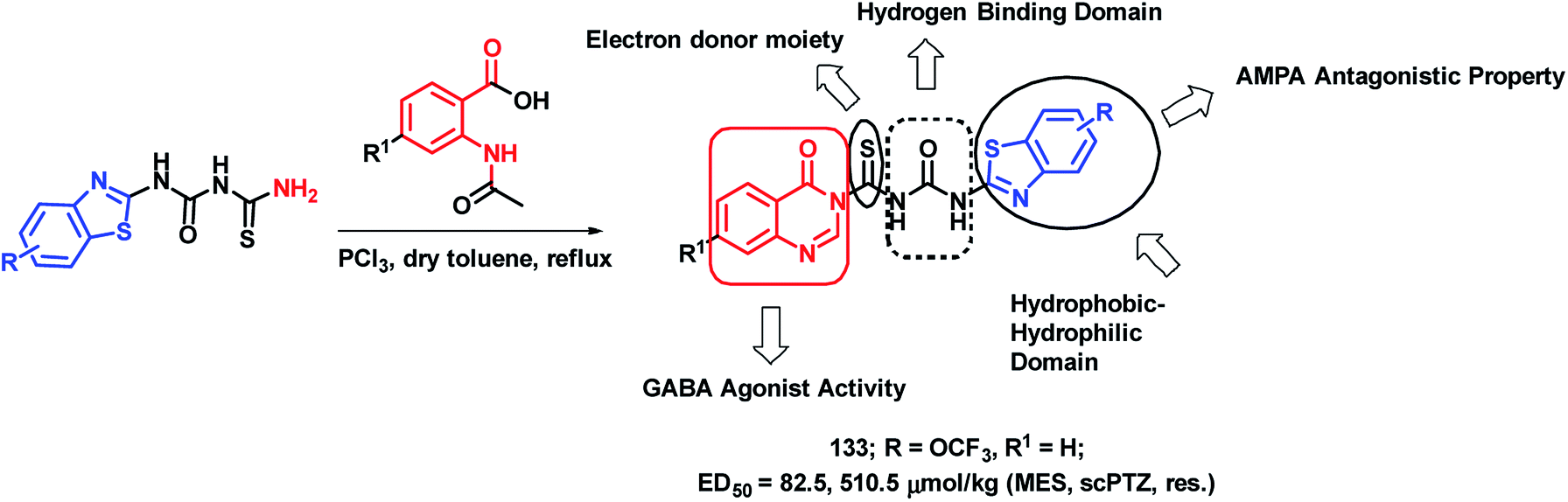 | ||
| Scheme 44 Synthetic strategy for quinazolinone-benzothiazole hybrids, showing the structural importance. | ||
Based on quinazolinone-containing anti-convulsant analogues (methaqualone, ethaqualone, mecloqualone), it can be concluded that the aromatic ring substitution at the 3rd position of quinazolinone is essential for CNS depressant and anti-convulsant activities. Many researchers have tried to substitute several aromatic ring substituents to obtain potent CNS acting drugs. By taking such facts into consideration, Jangam et al. designed quinazolinone-based hybrids by linking 4-oxothiazolidine at the 3rd position of the quinazolinone backbone (Scheme 45).128 Many of the synthesized compounds have shown moderate to good anti-convulsant potential when screened using MES-induced mice model. The SAR study showed the importance of electron withdrawing groups at the ortho, meta, and para position of the phenyl ring attached at the 2nd position of thiazolidine for anti-convulsant activity. These designed analogues were docked at the active site of the sodium channel IIA inactivation gate (PDB ID: 1BYY) and their binding energy was in the range from −5.15 to −6.13 kcal mol−1, which shows proper binding at the receptor active site.
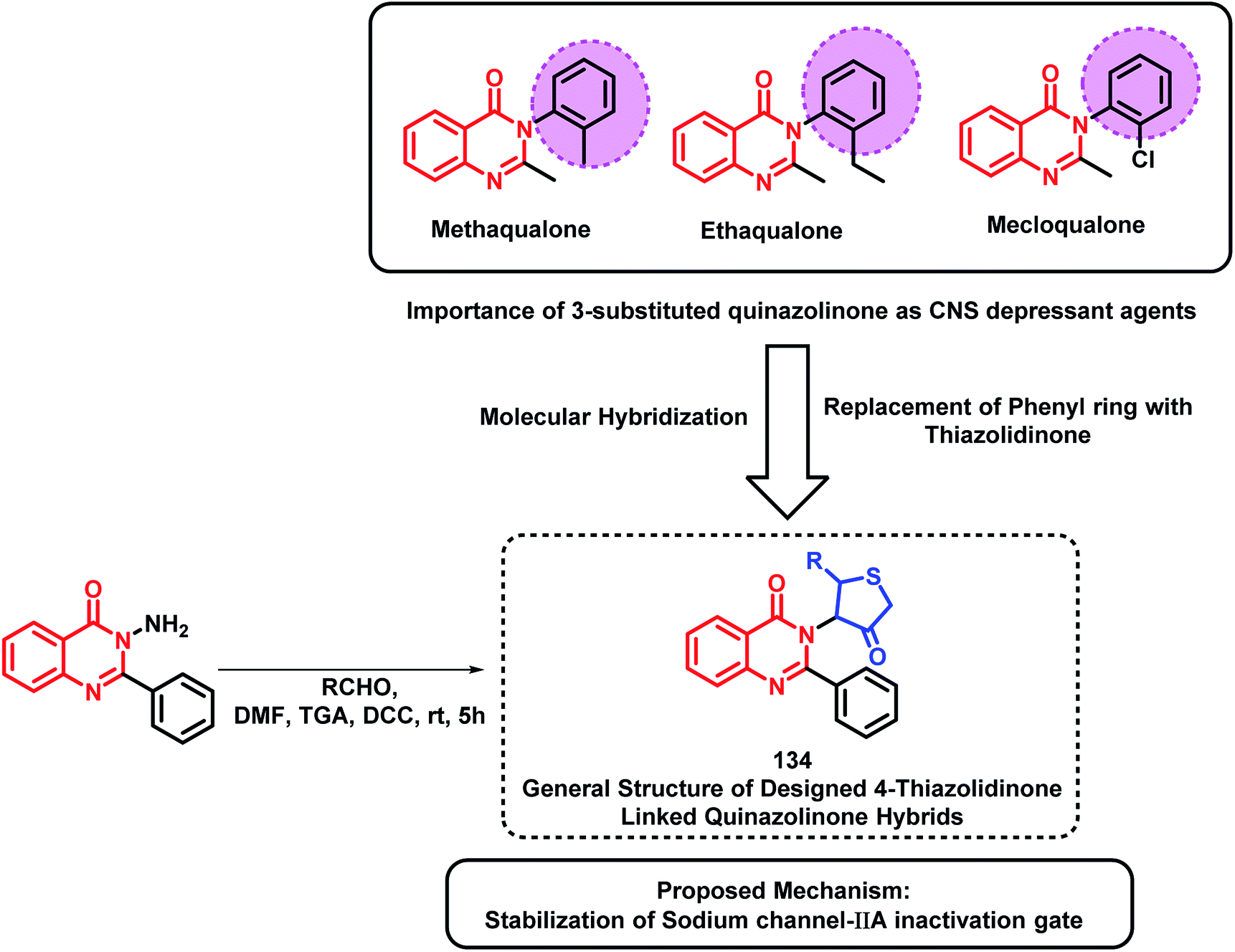 | ||
| Scheme 45 Synthesis and design strategy for 4-oxothiazolidine and quinazolinone hybrids. | ||
9. Hybrids with dual inhibition
Quinazoline derivatives are found to have diverse pharmacological properties including anti-malarial property (febrifugine, a well known anti-malarial drug). Hence, the quinazoline scaffold was combined with artemisinin, a known anti-malarial agent, by Frohlich et al. to form a hybrid pharmacophore.129 Five novel quinazoline-artemisinin hybrids were designed, synthesized, and screened for their inhibitory potency against Plasmodium falciparum 3D7 strain, leukemia cell lines CCRF-CEM and CEM/ADR5000, and human cytomegalovirus (HCMV). For anti-malarial activity against P. falciparum, all the hybrids were found to be active with EC50 in the range of 1.4–39.9 nM, comparable to that of artesunic acid (EC50 = 9.7 nM). Against HCMV replication in primary cell cultures, compounds 135 and 136 were found to be the most potent inhibitors, surpassing the anti-viral activity of ganciclovir by about 12–17 times. Compound 135 also showed anti-leukemia effect, with an IC50 of 0.6 μM against the multidrug-resistant CEM/ADR5000 cells (Scheme 46).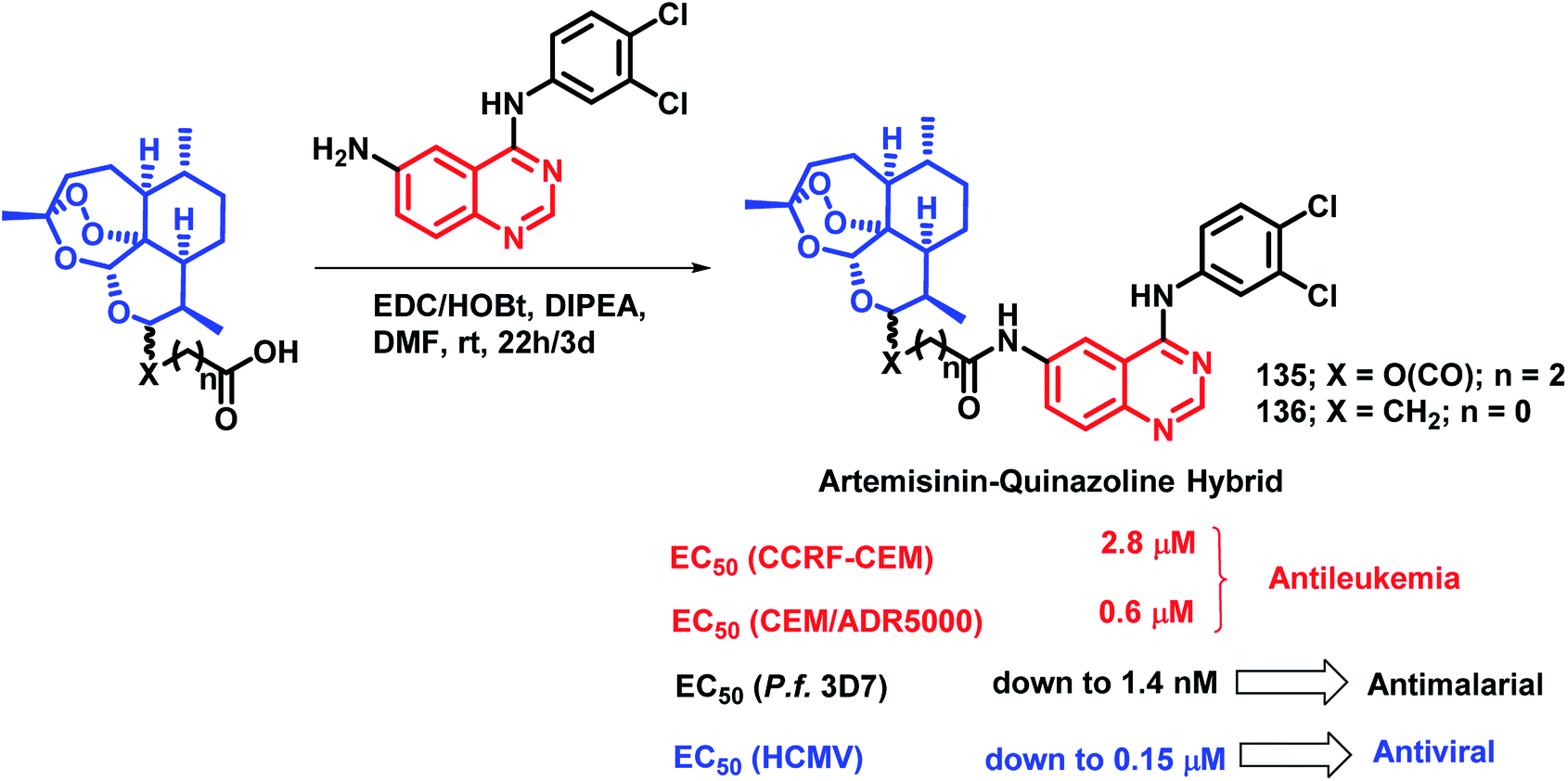 | ||
| Scheme 46 Synthetic scheme for artemisinin and 4-aminoquinazoline hybrids. | ||
Cancer and diabetes are common diseases with tremendous impact on the health of human beings. It is observed that people with diabetes are at a greater risk of developing various forms of cancer.130,131 1-Deoxynojirimycin (1-DNJ) is a naturally occurring aza sugar, having anti-cancer as well as α-glucosidase inhibitory potential. In the way of making drugs for the treatment of diabetes as well as cancer, Zhang et al. linked quinazoline (having anti-cancer potential) with an anti-diabetic, 1-DNJ, with α-glucosidase inhibitory potential (Scheme 47).132 In such a way, dual inhibitors, targeting EGFR and α-glucosidase, were designed and synthesized, followed by their in vitro evaluation. EGFR inhibition was evaluated by the ELISA method against recombinant EGFRwt tyrosine kinase, with gefitinib and lapatinib as the reference compounds. For the α-glucosidase inhibition assay, 1-DNJ and miglitol were selected as the standards. Many compounds were active against individual enzymes but compound 137 was found to be a dual inhibitor with high inhibition against EGFRwt tyrosine kinase (IC50 = 1.79 nM) and α-glucosidase (IC50 = 0.39 μM).
 | ||
| Scheme 47 Synthesis and design strategy for 1-deoxynojirimycin and 4-aminoquinazoline hybrids as dual inhibitors of EGFR and α-glucosidase enzymes. | ||
Modh et al. designed some analogues as dual inhibitors for HIV and microbial infections by combining triazine, quinazoline, and piperazine moieties in one hybrid structure.133 These hybrids were designed by joining various fragments found in clinically approved drugs, such as triazine in R106168, quinazoline in DPC 961, and piperazine type linkage in Abacavir. With this rationale, they synthesized novel ethylenediamine-linked quinazoline-triazine derivatives (Scheme 48), followed by their anti-microbial and anti-HIV screening. All the compounds were moderately active against HIV infection but were highly potent as anti-bacterial agents. Hence, it was found that quinazoline with ethylenediamine linkage is important for anti-microbial activity. Amongst all, compound 138 was found to be the most potent from the series, with MIC in the range of 6.25–50 μg mL−1 against S. aureus, B. cereus, P. aeruginosa, K. pneumonia, A. clavatus, and C. albicans.
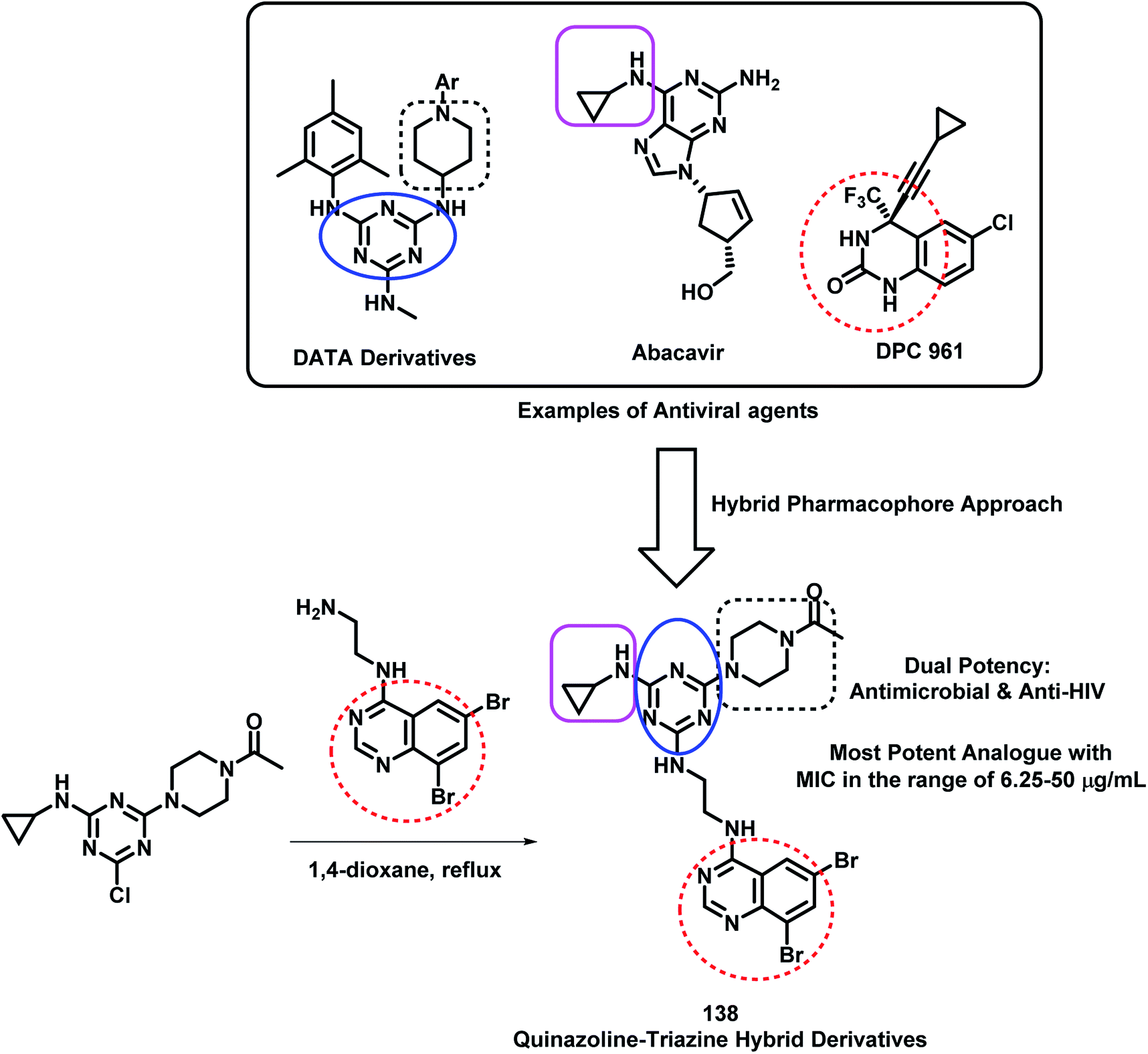 | ||
| Scheme 48 Synthesis and design strategy for quinazoline-triazine hybrids as anti-viral and anti-microbial agents. | ||
10. Current status, challenges, and future directions
Research interest in the synthesis and pharmacological evaluation of quinazoline/quinazolinone hybrids has increased over the years due to their diversified biological activities. Though hybrid analogues have shown to possess enhanced efficacy compared to the parent scaffold or their combinations, to the best of our knowledge, none of the hybrid analogues in the present review have reached the clinical phase. However, a detailed literature search (https://clinicaltrials.gov/) revealed the following quinazoline scaffold-containing analogues in clinical trials (Table 1).| Name | Status | Structure | Indication |
|---|---|---|---|
| Afatinib dimaleate | Phase II | 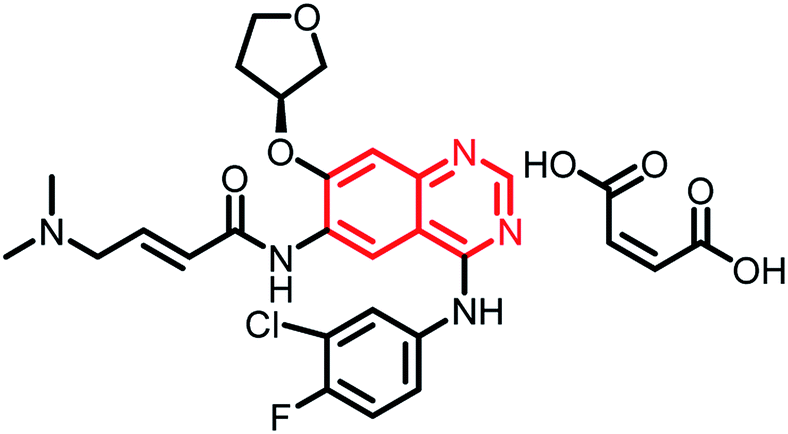 |
Targeted treatment in cancers with HER2 genetic changes134 |
| RXDX-105 | Phase I/Ib | 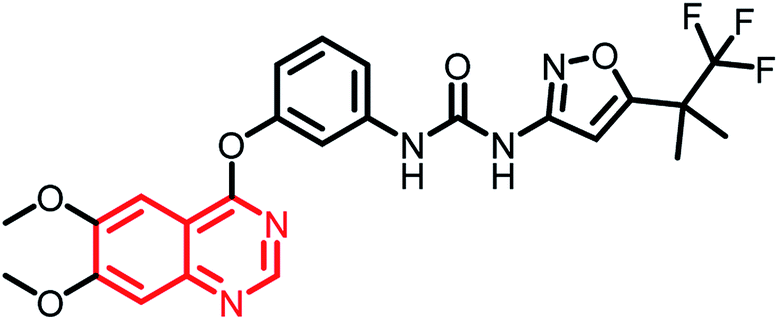 |
Non-small cell lung cancer (NSCLC)135 |
| Icotinib | Phase II | 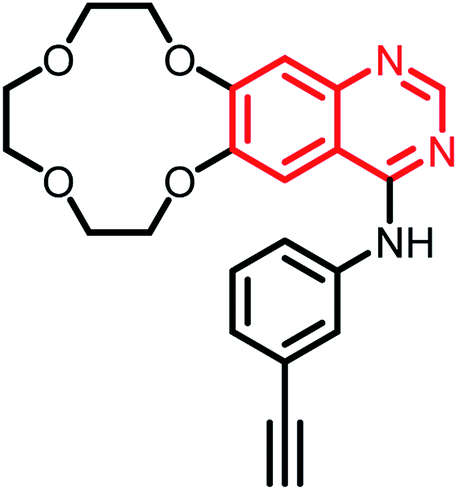 |
Neurofibromatosis type 2 (NF2)136 |
| Dacomitinib | Phase II |  |
Advanced non-small cell lung cancer137 |
| Doxazosin mesylate | Phase II |  |
Nightmares, sleep disturbance, and non-nightmare clinical symptoms in PTSD138 |
| Letermovir (MK-8228) | Phase II | 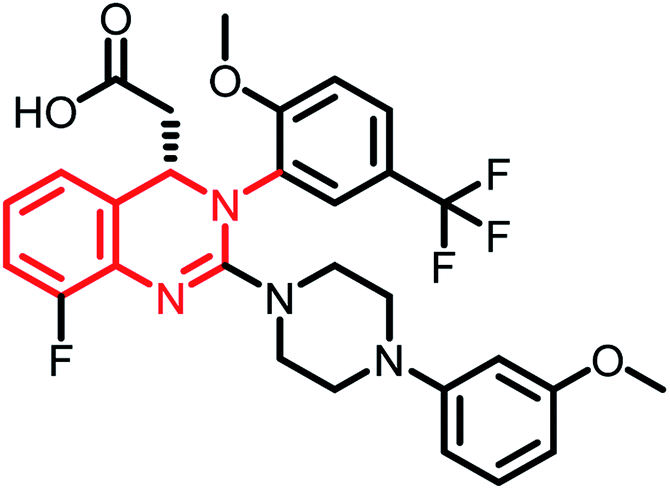 |
Prevention of cytomegalovirus reactivation in patients with haematological malignancies treated with alemtuzumab139 |
The present review summarizes the various synthetic strategies of quinazoline/quinazolinone hybrids. However, in many reports, it was observed that the hybrid analogues were designed without proper support of in silico studies. Most of the research in such cases has relied on the mere combination of the active functionalities from the potential agents. Nevertheless, these hybrid analogues possess good activity and proper substitution/structural modification strategy using in silico approaches will further help to achieve a potent activity profile.
Numerous quinazoline/quinazolinone hybrid analogues discussed above have exhibited similar or better pharmacological activity compared to that of the standard drugs. For instance, Peng et al. designed a hybrid of quinazoline-hydroxamic acid as a dual inhibitor for VEGFR and HDAC enzymes.59 The study resulted in compound 37, which exhibited a potential HDAC inhibition (IC50 = 2.2 nM) and VEGFR 2 inhibition (IC50 = 74 nM) as compared to the standard drugs [vorinostat (IC50 = 15 nM) and vandetanib (IC50 = 54 nM) respectively]. Apart from that, Saeedi et al. synthesized novel quinazolinone-1,2,3-triazole hybrids as novel anti-diabetic agents (acts via the inhibition of α-glucosidase).121 All the synthesized derivatives exhibited potential inhibition (IC50 = 181 to 474 μM) compared to acarbose (standard drug) with an IC50 of 750 μM. However, these studies are mainly focused on the evaluation of their in vitro potential towards the corresponding targets. As no further pre-clinical studies have been reported for a majority of such analogues, their clinical evaluation data could not be found in the literature.
A few studies have been reported the in vivo pharmacological efficacy of the quinazoline/quinazolinone hybrid. For instance, Jangam et al. evaluated the anti-diabetic potential of 4-thiazolidinone and 4(3H)-quinazolinone hybrids against streptozotocin-induced diabetic rats.122 The synthesized compounds exhibited a comparable anti-diabetic activity to that of pioglitazone. Also, Malik et al. synthesized benzotriazole-quinazolinone hybrids and evaluated the anti-convulsant activities in Wistar rats.127 Compound 133 exhibited potential activity in maximal electroshock (MES) induced convulsion model and subcutaneous pentylenetetrazole seizure test (ED50 = 82.5 and 510.5 μmol kg−1, respectively). Phenytoin exhibited an ED50 of 92.0 and >3450 μmol kg−1, respectively, for the above tests. However, no reports are available for the clinical studies of these molecules and no further information is available for their current status.
In addition to the above challenges, there are certain other points to be addressed, including the reliance on the synthesis of such hybrid analogues on traditional methodologies. Efforts are required for the synthesis of these analogues in a greener, cost-effective, eco-friendly manner along with the utility of multicomponent reactions. In addition, there is a need to explore the dual inhibition ability of these molecules as they contain two scaffolds. Although some dual inhibitory activities of the hybrids are reported, such analogues exhibited non-selectivity towards the targets. Further, there are no reports on the pharmacokinetic properties of the active analogues. Studies are required in these directions for solving the same.
However, so far, the reported quinazoline/quinazolinone hybrids are still limited and their structural diversity needs further refinement for finding potential drug candidates. In the near future, continuous efforts will provide further vital discoveries as clinical drugs with quinazoline/quinazolinone hybrid scaffolds.
11. Conclusion
The present review is comprised of the rationale and various techniques involved in the design of hybrids of quinazoline/quinazolinone with other bioactive scaffolds, targeting individual or multiple targets. Major diseases, such as cancer, microbial infections, tuberculosis, leishmania, diabeties, and epilepsy, are covered in this review. Quinazoline and quinazolinone scaffolds are diverse as far as their various biological activities are concerned. Hence, many research groups have carried out extensive research for enhancing the activity of such scaffolds by hybridization with another bioactive moiety. Hybrid drug design approach has been found to be very attractive and efficient, especially in the research area of cancer and infectious diseases. The triazole moiety was present in most of the quinazoline/quinazolinone hybrids against a myriad of disease conditions. In the overall study, we found that the linking approach was used most prominently instead of framework integration for designing molecular hybrids. Overall, there is much scope of molecular hybridization approach in order to make various hybrids for better selectivity, mixed mechanism of action, overcoming the drug resistance, and so on.Conflicts of interest
There is no conflict of interest among the authors.Acknowledgements
The authors acknowledge Birla Institute of Technology and Science, Pilani (BITS Pilani), Pilani Campus, Rajasthan (India) for providing necessary infrastructural support. The authors acknowledge the financial support received from DST-SERB (Grant No. CRG/2018/002608). Mr Ginson George thankfully acknowledges the Council for Scientific and Industrial Research (CSIR), New Delhi, India, for providing a fellowship (SRF-file no: 09/719 (0100)/2019-EMR-I).References
- F. Prati, E. Uliassi and M. Bolognesi, RSC Med. Chem., 2014, 5, 853–861 RSC.
- M. L. Bolognesi and A. Cavalli, ChemMedChem, 2016, 11, 1190–1192 CrossRef CAS.
- V. Ivasiv, C. Albertini, A. E. Gonçalves, M. Rossi and M. L. Bolognesi, Curr. Top. Med. Chem., 2019, 19, 1694–1711 CrossRef CAS.
- V. Abbot, P. Sharma, S. Dhiman, M. N. Noolvi, H. M. Patel and V. Bhardwaj, RSC Adv., 2017, 7, 28313–28349 RSC.
- S. Sandhu, Y. Bansal, O. Silakari and G. Bansal, Bioorg. Med. Chem., 2014, 22, 3806–3814 CrossRef CAS.
- N. Kerru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 142, 179–212 CrossRef CAS.
- A. Hameed, M. Al-Rashida, M. Uroos, S. A. Ali, Arshia, M. Ishtiaq and K. M. Khan, Expert Opin. Ther. Pat., 2018, 28, 281–297 CrossRef CAS.
- L. F. Kuyper, D. P. Baccanari, M. L. Jones, R. N. Hunter, R. L. Tansik, S. S. Joyner, C. M. Boytos, S. K. Rudolph, V. Knick and H. R. Wilson, J. Med. Chem., 1996, 39, 892–903 CrossRef CAS.
- H. Georgey, N. Abdel-Gawad and S. Abbas, Molecules, 2008, 13, 2557–2569 CrossRef CAS.
- V. Srivastava and A. Kumar, Eur. J. Med. Chem., 2002, 37, 873–882 CrossRef.
- P. M. Chandrika, T. Yakaiah, A. R. R. Rao, B. Narsaiah, N. C. Reddy, V. Sridhar and J. V. Rao, Eur. J. Med. Chem., 2008, 43, 846–852 CrossRef CAS.
- P. Verhaeghe, N. Azas, M. Gasquet, S. Hutter, C. Ducros, M. Laget, S. Rault, P. Rathelot and P. Vanelle, Bioorg. Med. Chem. Lett, 2008, 18, 396–401 CrossRef CAS.
- M. A. Ismail, S. Barker, D. A. Abou El Ella, K. A. Abouzid, R. A. Toubar and M. H. Todd, J. Med. Chem., 2006, 49, 1526–1535 CrossRef CAS.
- V. Alagarsamy, S. Meena, K. Ramaseshu, V. R. Solomon, T. D. A. Kumar and K. Thirumurugan, Chem. Biol. Drug Des., 2007, 70, 254–260 CrossRef CAS.
- M. S. Malamas and J. Millen, J. Med. Chem., 1991, 34, 1492–1503 CrossRef CAS.
- N. M. A. Gawad, H. H. Georgey, R. M. Youssef and N. A. El-Sayed, Eur. J. Med. Chem., 2010, 45, 6058–6067 CrossRef.
- M. Decker, Eur. J. Med. Chem., 2005, 40, 305–313 CrossRef CAS.
- A. Rosowsky, J. E. Wright, C. M. Vaidya and R. A. Forsch, Pharmacol. Therapeut., 2000, 85, 191–205 CrossRef CAS.
- D. W. Fry, A. J. Kraker, A. Mcmichael, L. A. Ambroso, J. M. Nelson, W. R. Leopold, R. W. Connors and A. J. Bridges, Science, 1994, 265, 1093–1095 CrossRef CAS.
- A. Garofalo, L. Goossens, A. Lemoine, S. Ravez, P. Six, M. Howsam, A. Farce and P. Depreux, RSC Med. Chem., 2011, 2, 65–72 RSC.
- I. Khan, S. Zaib, S. Batool, N. Abbas, Z. Ashraf, J. Iqbal and A. Saeed, Bioorg. Med. Chem., 2016, 24, 2361–2381 CrossRef CAS.
- J. D. Palem, G. R. Alugubelli, R. Bantu, L. Nagarapu, S. Polepalli, S. N. Jain, R. Bathini and V. Manga, Bioorg. Med. Chem. Lett, 2016, 26, 3014–3018 CrossRef CAS.
- R. Venkatesh, S. Kasaboina, N. Jain, S. Janardhan, U. D. Holagunda and L. Nagarapu, J. Mol. Struct., 2019, 1181, 403–411 CrossRef CAS.
- A. Kamal, E. V. Bharathi, J. S. Reddy, M. J. Ramaiah, D. Dastagiri, M. K. Reddy, A. Viswanath, T. L. Reddy, T. B. Shaik and S. Pushpavalli, Eur. J. Med. Chem., 2011, 46, 691–703 CrossRef CAS.
- E. Hamel, C. M. Lin, J. Plowman, H.-K. Wang, K.-H. Lee and K. D. Paull, Biochem. Pharmacol., 1996, 51, 53–59 CrossRef CAS.
- H. L. Yale and M. Kalkstein, J. Med. Chem., 1967, 10, 334–336 CrossRef CAS.
- D. Simoni, G. Grisolia, G. Giannini, M. Roberti, R. Rondanin, L. Piccagli, R. Baruchello, M. Rossi, R. Romagnoli and F. P. Invidiata, J. Med. Chem., 2005, 48, 723–736 CrossRef CAS.
- J. Kaffy, R. Pontikis, D. Carrez, A. Croisy, C. Monneret and J.-C. Florent, Bioorg. Med. Chem., 2006, 14, 4067–4077 CrossRef CAS.
- S. T. Al-Rashood, G. S. Hassan, S. M. El-Messery, M. N. Nagi, E.-S. E. Habib, F. A. Al-Omary and H. I. El-Subbagh, Bioorg. Med. Chem. Lett, 2014, 24, 4557–4567 CrossRef CAS.
- S. T. Al-Rashood, I. A. Aboldahab, M. N. Nagi, L. A. Abouzeid, A. A. Abdel-Aziz, S. G. Abdel-Hamide, K. M. Youssef, A. M. Al-Obaid and H. I. El-Subbagh, Bioorg. Med. Chem., 2006, 14, 8608–8621 CrossRef CAS.
- F. A. Al-Omary, G. S. Hassan, S. M. El-Messery, M. N. Nagi, E.-S. E. Habib and H. I. El-Subbagh, Eur. J. Med. Chem., 2013, 63, 33–45 CrossRef CAS.
- P. Singla, V. Luxami and K. Paul, J. Photochem. Photobiol., B, 2017, 168, 156–164 CrossRef CAS.
- L. Hosseinzadeh, A. Aliabadi, M. Rahnama, H. M. M. Sadeghi and M. R. Khajouei, Res. Pharm. Sci., 2017, 12, 290 CrossRef.
- F. Hassanzadeh, H. Sadeghi-Aliabadi, E. Jafari, A. Sharifzadeh and N. Dana, Res. Pharm. Sci., 2019, 14, 408 CrossRef.
- W.-Y. Wu, S.-L. Cao, B.-B. Mao, J. Liao, Z.-F. Li, H.-B. Song and X. Xu, Lett. Drug Des. Discovery, 2013, 10, 61–66 CrossRef CAS.
- B. Parrino, C. Ciancimino, A. Carbone, V. Spano, A. Montalbano, P. Barraja, G. Cirrincione and P. Diana, Tetrahedron, 2015, 71, 7332–7338 CrossRef CAS.
- J.-H. Wu, F.-R. Chang, K.-I. Hayashi, H. Shiraki, C.-C. Liaw, Y. Nakanishi, K. F. Bastow, D. Yu, I.-S. Chen and K.-H. Lee, Bioorg. Med. Chem. Lett, 2003, 13, 2223–2225 CrossRef CAS.
- R. Bollu, S. Banu, S. Kasaboina, R. Bantu, L. Nagarapu, S. Polepalli and N. Jain, Bioorg. Med. Chem. Lett, 2017, 27, 5481–5484 CrossRef CAS.
- S. Pullamsetti, G. Banat, A. Schmall, M. Szibor, D. Pomagruk, J. Hänze, E. Kolosionek, J. Wilhelm, T. Braun and F. Grimminger, Oncogene, 2013, 32, 1121–1134 CrossRef CAS.
- M. Narita, T. Murata, K. Shimizu, T. Nakagawa, T. Sugiyama, M. Inui, K. Hiramoto and T. Tagawa, Oncol. Rep., 2007, 17, 1133–1139 CAS.
- K. Murata, T. Sudo, M. Kameyama, H. Fukuoka, M. Mukai, Y. Doki, Y. Sasaki, O. Ishikawa, Y. Kimura and S. Imaoka, Clin. Exp. Metastasis, 2000, 18, 599–604 CrossRef CAS.
- A. E. KüMmerle, M. Schmitt, S. V. Cardozo, C. Lugnier, P. Villa, A. B. Lopes, N. C. Romeiro, H. L. N. Justiniano, M. A. Martins and C. A. Fraga, J. Med. Chem., 2012, 55, 7525–7545 CrossRef.
- A. E. Kümmerle, M. M. Vieira, M. Schmitt, A. L. Miranda, C. A. Fraga, J.-J. Bourguignon and E. J. Barreiro, Bioorg. Med. Chem. Lett, 2009, 19, 4963–4966 CrossRef.
- H. M. Abdel-Rahman, M. Abdel-Aziz, J. C. Canzoneri, B. D. Gary and G. A. Piazza, Arch. Pharmazie, 2014, 347, 650–657 CrossRef CAS.
- S. Thakral, D. Saini, A. Kumar, N. Jain and S. Jain, Med. Chem. Res., 2017, 26, 1595–1604 CrossRef CAS.
- J. Sławiński, K. Szafrański, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2013, 69, 701–710 CrossRef.
- F. Xu, H. Xu, X. Wang, L. Zhang, Q. Wen, Y. Zhang and W. Xu, Bioorg. Med. Chem., 2014, 22, 1487–1495 CrossRef CAS.
- C. T. Supuran, F. Briganti, S. Tilli, W. R. Chegwidden and A. Scozzafava, Bioorg. Med. Chem., 2001, 9, 703–714 CrossRef CAS.
- M. Zayed and M. Hassan, Drug Res., 2013, 63, 210–215 CrossRef CAS.
- M. F. Zayed, H. E. Ahmed, S. Ihmaid, A.-S. M. Omar and A. S. Abdelrahim, J. Taibah Univ. Medical Sci., 2015, 10, 333–339 Search PubMed.
- F. Hassanzadeh, H. Sadeghi-Aliabadi, S. Nikooei, E. Jafari and G. Vaseghi, Res. Pharm. Sci., 2019, 14, 130 CrossRef.
- S. El-Sayed, K. Metwally, A. A. El-Shanawani, L. M. Abdel-Aziz, H. Pratsinis and D. Kletsas, Chem. Cent. J., 2017, 11, 1–10 CrossRef.
- N. Gokhale, N. Panathur, U. Dalimba, P. G. Nayak and K. S. R. Pai, J. Heterocycl. Chem., 2016, 53, 513–524 CrossRef CAS.
- A. Imramovský, R. Jorda, K. Pauk, E. Řezníčková, J. Dušek, J. Hanusek and V. Kryštof, Eur. J. Med. Chem., 2013, 68, 253–259 CrossRef.
- R. Venkatesh, M. J. Ramaiah, H. K. Gaikwad, S. Janardhan, R. Bantu, L. Nagarapu, G. N. Sastry, A. R. Ganesh and M. Bhadra, Eur. J. Med. Chem., 2015, 94, 87–101 CrossRef CAS.
- M. F. Zayed, H. S. Rateb, S. Ahmed, O. A. Khaled and S. R. Ibrahim, Molecules, 2018, 23, 1699 CrossRef.
- S. Shinkaruk, M. Bayle, G. Lain and G. Deleris, Curr. Med. Chem. Anti Canc. Agents, 2003, 3, 95–117 CrossRef CAS.
- T. Veikkola, M. Karkkainen, L. Claesson-Welsh and K. Alitalo, Cancer Res., 2000, 60, 203–212 CAS.
- F.-W. Peng, J. Xuan, T.-T. Wu, J.-Y. Xue, Z.-W. Ren, D.-K. Liu, X.-Q. Wang, X.-H. Chen, J.-W. Zhang and Y.-G. Xu, Eur. J. Med. Chem., 2016, 109, 1–12 CrossRef CAS.
- X. Kang, J. Hu, Z. Gao, Y. Ju and C. Xu, RSC Med. Chem., 2012, 3, 1245–1249 RSC.
- R. Majeed, P. L. Sangwan, P. K. Chinthakindi, I. Khan, N. A. Dangroo, N. Thota, A. Hamid, P. R. Sharma, A. K. Saxena and S. Koul, Eur. J. Med. Chem., 2013, 63, 782–792 CrossRef CAS.
- A. Worachartcheewan, N. Songtawee, S. Siriwong, S. Prachayasittikul, C. Nantasenamat and V. Prachayasittikul, Med. Chem., 2019, 15, 328–340 CrossRef CAS.
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester and D. K. Hutchinson, J. Med. Chem., 2000, 43, 953–970 CrossRef CAS.
- N. G. Aher, V. S. Pore, N. N. Mishra, A. Kumar, P. K. Shukla, A. Sharma and M. K. Bhat, Bioorg. Med. Chem. Lett, 2009, 19, 759–763 CrossRef CAS.
- G. Le-Nhat-Thuy, T. Van Dinh, H. Pham-The, H. N. Quang, N. N. Thi, T. a. D. Thi, P. H. Thi, T. A. Le Thi, H. T. Nguyen and P. N. Thanh, Bioorg. Med. Chem. Lett, 2018, 28, 3741–3747 CrossRef CAS.
- P. Song, F. Cui, N. Li, J. Xin, Q. Ma, X. Meng, C. Wang, Q. Cao, Y. Gu and Y. Ke, Chin. J. Chem., 2017, 35, 1633–1639 CrossRef CAS.
- R. Gastpar, M. Goldbrunner, D. Marko and E. Von Angerer, J. Med. Chem., 1998, 41, 4965–4972 CrossRef CAS.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS.
- H. Hu, J. Wu, M. Ao, H. Wang, T. Zhou, Y. Xue, Y. Qiu, M. Fang and Z. Wu, Chem. Biol. Drug Des., 2016, 88, 766–778 CrossRef CAS.
- S. H. Watterson, T. M. Dhar, S. K. Ballentine, Z. Shen, J. C. Barrish, D. Cheney, C. A. Fleener, K. A. Rouleau, R. Townsend and D. L. Hollenbaugh, Bioorg. Med. Chem. Lett, 2003, 13, 1273–1276 CrossRef CAS.
- M. J. Mphahlele, M. M. Mmonwa, A. Aro, L. J. Mcgaw and Y. S. Choong, Int. J. Mol. Sci., 2018, 19, 2232 CrossRef.
- M. Fares, W. M. Eldehna, S. M. Abou-Seri, H. A. Abdel-Aziz, M. H. Aly and M. F. Tolba, Arch. Pharmazie, 2015, 348, 144–154 CrossRef CAS.
- V. Luxami, R. Rani, A. Sharma and K. Paul, J. Photochem. Photobiol., A, 2015, 311, 68–75 CrossRef CAS.
- J. Shao, E. Chen, K. Shu, W. Chen, G. Zhang and Y. Yu, Bioorg. Med. Chem., 2016, 24, 3359–3370 CrossRef CAS.
- A. Sharma, V. Luxami and K. Paul, Bioorg. Med. Chem. Lett, 2013, 23, 3288–3294 CrossRef CAS.
- B. Banerji, K. Chandrasekhar, K. Sreenath, S. Roy, S. Nag and K. D. Saha, ACS Omega, 2018, 3, 16134–16142 CrossRef CAS.
- F.-W. Peng, T.-T. Wu, Z.-W. Ren, J.-Y. Xue and L. Shi, Bioorg. Med. Chem. Lett, 2015, 25, 5137–5141 CrossRef CAS.
- J. Drews, Science, 2000, 287, 1960–1964 CrossRef CAS.
- C. T. Supuran, A. Casini, A. Mastrolorenzo and A. Scozzafava, Mini Rev. Med. Chem., 2004, 4, 625–632 CrossRef CAS.
- F. Abbate, A. Casini, T. Owa, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett, 2004, 14, 217–223 CrossRef CAS.
- M. M. Ghorab, M. S. Alsaid, M. S. Al-Dosari, M. G. El-Gazzar and M. K. Parvez, Molecules, 2016, 21, 189 CrossRef.
- M. Dinari, F. Gharahi and P. Asadi, J. Mol. Struct., 2018, 1156, 43–50 CrossRef CAS.
- R. Romagnoli, P. G. Baraldi, F. Prencipe, J. Balzarini, S. Liekens and F. Estévez, Eur. J. Med. Chem., 2015, 101, 205–217 CrossRef CAS.
- S. Gatadi, J. Gour, M. Shukla, G. Kaul, S. Das, A. Dasgupta, S. Malasala, R. S. Borra, Y. Madhavi and S. Chopra, Eur. J. Med. Chem., 2018, 157, 1056–1067 CrossRef CAS.
- V. Veeramreddy, T. Allaka, J. Anireddy and R. Varala, Org. Commun., 2015, 8, 98 CAS.
- X.-M. Peng, K. V. Kumar, G. L. Damu and C.-H. Zhou, Sci. China Chem., 2016, 59, 878–894 CrossRef CAS.
- C. H Zhou and Y. Wang, Curr. Med. Chem., 2012, 19, 239–280 CrossRef.
- X.-M. Peng, L.-P. Peng, S. Li, S. R. Avula, V. K. Kannekanti, S.-L. Zhang, K. Y. Tam and C.-H. Zhou, Future Med. Chem., 2016, 8, 1927–1940 CrossRef CAS.
- N. Desai, A. M. Dodiya and P. N. Shihora, Med. Chem. Res., 2012, 21, 1577–1586 CrossRef CAS.
- N. Desai, H. Vaghani and P. Shihora, J. Fluorine Chem., 2013, 153, 39–47 CrossRef CAS.
- H. Patel, A. Shirkhedkar, S. Bari, K. Patil, A. Arambhi, C. Pardeshi, A. Kulkarni, and S. Surana, Bulletin of Faculty of Pharmacy, Cairo University, 2018, vol. 56, pp. 83–90 Search PubMed.
- U. Pandit and A. Dodiya, Med. Chem. Res., 2013, 22, 3364–3371 CrossRef CAS.
- N. K. Maddali, I. K. Viswanath, Y. Murthy, R. Bera, M. Takhi, N. S. Rao and V. Gudla, Med. Chem. Res., 2019, 28, 559–570 CrossRef CAS.
- D. R. Shah, R. P. Modh, D. D. Desai and K. H. Chikhalia, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2014, 53B, 1169–1177 CAS.
- Q.-S. Long, L.-W. Liu, Y.-L. Zhao, P.-Y. Wang, B. Chen, Z. Li and S. Yang, J. Agric. Food Chem., 2019, 67, 11005–11017 CrossRef CAS.
- Z. Fan, J. Shi, N. Luo, M. Ding and X. Bao, J. Agric. Food Chem., 2019, 67, 11598–11606 CrossRef CAS.
- Z. Fan, J. Shi and X. Bao, Mol. Divers., 2018, 22, 657–667 CrossRef CAS.
- X. Zhu, K. S. Van Horn, M. M. Barber, S. Yang, M. Z. Wang, R. Manetsch and K. A. Werbovetz, Bioorg. Med. Chem., 2015, 23, 5182–5189 CrossRef CAS.
- Q. Ji, D. Yang, X. Wang, C. Chen, Q. Deng, Z. Ge, L. Yuan, X. Yang and F. Liao, Bioorg. Med. Chem., 2014, 22, 3405–3413 CrossRef CAS.
- P. M. Bedi, V. Kumar and M. P. Mahajan, Bioorg. Med. Chem. Lett, 2004, 14, 5211–5213 CrossRef CAS.
- A. Kumar, P. Sharma, P. Kumari and B. L. Kalal, Bioorg. Med. Chem. Lett, 2011, 21, 4353–4357 CrossRef CAS.
- P. Zoumpoulakis, C. Camoutsis, G. Pairas, M. Soković, J. Glamočlija, C. Potamitis and A. Pitsas, Bioorg. Med. Chem., 2012, 20, 1569–1583 CrossRef CAS.
- S. M. Hashemi, H. Badali, H. Irannejad, M. Shokrzadeh and S. Emami, Bioorg. Med. Chem., 2015, 23, 1481–1491 CrossRef CAS.
- Y. Jiang, J. Zhang, Y. Cao, X. Chai, Y. Zou, Q. Wu, D. Zhang, Y. Jiang and Q. Sun, Bioorg. Med. Chem. Lett, 2011, 21, 4471–4475 CrossRef CAS.
- Y. Wang, G. L. Damu, J.-S. Lv, R.-X. Geng, D.-C. Yang and C.-H. Zhou, Bioorg. Med. Chem. Lett, 2012, 22, 5363–5366 CrossRef CAS.
- L. Yang, S. Ge, J. Huang and X. Bao, Mol. Divers., 2018, 22, 71–82 CrossRef CAS.
- V. Gupta, S. K. Kashaw, V. Jatav and P. Mishra, Med. Chem. Res., 2008, 17, 205–211 CrossRef CAS.
- J. Jampilek, R. Musiol, J. Finster, M. Pesko, J. Carroll, K. Kralova, M. Vejsova, J. O'mahony, A. Coffey and J. Dohnal, Molecules, 2009, 14, 4246–4265 CrossRef CAS.
- L. Yang and X.-P. Bao, RSC Adv., 2017, 7, 34005–34011 RSC.
- C. Li, J.-C. Liu, Y.-R. Li, C. Gou, M.-L. Zhang, H.-Y. Liu, X.-Z. Li, C.-J. Zheng and H.-R. Piao, Bioorg. Med. Chem. Lett, 2015, 25, 3052–3056 CrossRef CAS.
- A. Vijesh, A. M. Isloor, P. Shetty, S. Sundershan and H. K. Fun, Eur. J. Med. Chem., 2013, 62, 410–415 CrossRef CAS.
- D. Shah, H. Lakum and K. Chikhalia, Russ. J. Bioorg. Chem., 2015, 41, 209–222 CrossRef CAS.
- S. S. Chauhan, S. Pandey, R. Shivahare, K. Ramalingam, S. Krishna, P. Vishwakarma, M. Siddiqi, S. Gupta, N. Goyal and P. M. Chauhan, RSC Med. Chem., 2015, 6, 351–356 RSC.
- M. Sharma, K. Chauhan, R. Shivahare, P. Vishwakarma, M. K. Suthar, A. Sharma, S. Gupta, J. K. Saxena, J. Lal and P. Chandra, J. Med. Chem., 2013, 56, 4374–4392 CrossRef CAS.
- V. Gurram, R. Garlapati, C. Thulluri, N. Madala, K. S. Kasani, P. K. Machiraju, R. Doddapalla, U. Addepally, R. Gundla and B. Patro, Med. Chem. Res., 2015, 24, 2227–2237 CrossRef CAS.
- K. Javaid, S. M. Saad, S. Rasheed, S. T. Moin, N. Syed, I. Fatima, U. Salar, K. M. Khan, S. Perveen and M. I. Choudhary, Bioorg. Med. Chem., 2015, 23, 7417–7421 CrossRef CAS.
- M. Wei, W.-M. Chai, R. Wang, Q. Yang, Z. Deng and Y. Peng, Bioorg. Med. Chem., 2017, 25, 1303–1308 CrossRef CAS.
- F. Jabeen, S. A. Shehzadi, M. Q. Fatmi, S. Shaheen, L. Iqbal, N. Afza, S. S. Panda and F. L. Ansari, Bioorg. Med. Chem. Lett, 2016, 26, 1029–1038 CrossRef CAS.
- Y. Chinthala, A. K. Domatti, A. Sarfaraz, S. P. Singh, N. K. Arigari, N. Gupta, S. K. Satya, J. K. Kumar, F. Khan and A. K. Tiwari, Eur. J. Med. Chem., 2013, 70, 308–314 CrossRef CAS.
- S. Iqbal, M. A. Khan, K. Javaid, R. Sadiq, S. Fazal-Ur-Rehman, M. I. Choudhary and F. Z. Basha, Bioorg. Chem., 2017, 74, 72–81 CrossRef CAS.
- M. Saeedi, M. Mohammadi-Khanaposhtani, P. Pourrabia, N. Razzaghi, R. Ghadimi, S. Imanparast, M. A. Faramarzi, F. Bandarian, E. N. Esfahani and M. Safavi, Bioorg. Chem., 2019, 83, 161–169 CrossRef CAS.
- S. Jangam and S. Wankhede, Russ. J. Gen. Chem., 2019, 89, 1029–1041 CrossRef CAS.
- T. S. Patel, S. F. Vanparia, U. H. Patel, R. B. Dixit, C. J. Chudasama, B. D. Patel and B. C. Dixit, Eur. J. Med. Chem., 2017, 129, 251–265 CrossRef CAS.
- T. S. Patel, J. D. Bhatt, R. B. Dixit, C. J. Chudasama, B. D. Patel and B. C. Dixit, Arch. Pharmazie, 2019, 352, 1900099 CrossRef.
- T. S. Patel, J. D. Bhatt, S. F. Vanparia, U. H. Patel, R. B. Dixit, C. J. Chudasama, B. D. Patel and B. C. Dixit, Bioorg. Med. Chem., 2017, 25, 6635–6646 CrossRef CAS.
- T. S. Patel, J. D. Bhatt, R. B. Dixit, C. J. Chudasama, B. D. Patel and B. C. Dixit, Bioorg. Med. Chem., 2019, 27, 3574–3586 CrossRef CAS.
- S. Malik, R. S. Bahare and S. A. Khan, Eur. J. Med. Chem., 2013, 67, 1–13 CrossRef CAS.
- S. S. Jangam, S. B. Wankhede and S. S. Chitlange, Res. Chem. Intermed., 2019, 45, 471–486 CrossRef CAS.
- T. FröHlich, C. Reiter, M. M. Ibrahim, J. Beutel, C. Hutterer, I. ZeitträGer, H. Bahsi, M. Leidenberger, O. Friedrich and B. Kappes, ACS Omega, 2017, 2, 2422–2431 CrossRef.
- E. Giovannucci, D. M. Harlan, M. C. Archer, R. M. Bergenstal, S. M. Gapstur, L. A. Habel, M. Pollak, J. G. Regensteiner and D. Yee, Ca-Cancer J. Clin., 2010, 60, 207–221 CrossRef.
- M.-Y. Lee, K.-D. Lin, P.-J. Hsiao and S.-J. Shin, Metabolism, 2012, 61, 242–249 CrossRef CAS.
- Y. Zhang, H. Gao, R. Liu, J. Liu, L. Chen, X. Li, L. Zhao, W. Wang and B. Li, Bioorg. Med. Chem. Lett, 2017, 27, 4309–4313 CrossRef CAS.
- R. P. Modh, E. De Clercq, C. Pannecouque and K. H. Chikhalia, J. Enzym. Inhib. Med. Chem., 2014, 29, 100–108 CrossRef CAS.
- G. D. Goss, E. Felip, M. Cobo, S. Lu, K. Syrigos, K. H. Lee, E. Göker, V. Georgoulias, W. Li, S. Guclu, D. Isla, Y. J. Min, A. Morabito, A. Ardizzoni, S. M. Gadgeel, A. Fölöp, C. Böhnemann, N. Gibson, N. Krämer, F. Solca, A. Cseh, E. Ehrnrooth and J. C. Soria, JAMA Oncol., 2018, 4, 1189–1197 CrossRef.
- A. Drilon, S. Fu, M. R. Patel, M. Fakih, D. Wang, A. J. Olszanski, D. Morgensztern, S. V. Liu, B. C. Cho, L. Bazhenova, C. P. Rodriguez, R. C. Doebele, A. Wozniak, K. L. Reckamp, T. Seery, P. Nikolinakos, Z. Hu, J. W. Oliver, D. Trone, K. McArthur, R. Patel, P. S. Multani and M. J. Ahn, Cancer Discovery, 2019, 9, 384–395 CrossRef CAS.
- J. Qu, Y. N. Wang, P. Xu, D. X. Xiang, R. Yang, W. Wei and Q. Qu, Oncotarget, 2017, 8, 33961–33971 CrossRef.
- T. S. Mok, Y. Cheng, X. Zhou, K. H. Lee, K. Nakagawa, S. Niho, M. Lee, R. Linke, R. Rosell, J. Corral, M. R. Migliorino, A. Pluzanski, E. I. Sbar, T. Wang, J. L. White and Y. L. Wu, J. Clin. Oncol., 2018, 36, 2244–2250 CrossRef CAS.
- S. Pallesen, H. S. Hamre, N. Lang and B. Bjorvatn, SAGE Open Med. Case Rep., 2020, 8, 2050313X2093607 CrossRef.
- P. Lischka, D. Michel and H. Zimmermann, J. Infect. Dis., 2016, 213, 23–30 CrossRef CAS.
Footnotes |
| † Equal contribution. |
| ‡ Present address: Laboratory of Natural Product Chemistry, Department of Pharmacy, Birla Institute of Technology and Science, Pilani (BITS Pilani), Pilani Campus, Pilani – 333031, Rajasthan, India. |
| This journal is © The Royal Society of Chemistry 2020 |