
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biocompatible supramolecular hydrogel with multivalent galactose ligands inhibiting Pseudomonas aeruginosa virulence and growth†
Shengnan
Liu
a,
Hang
Li
a,
Jikun
Zhang
a,
Xin
Tian
*b and
Xinming
Li
 *a
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: xinmingli@suda.edu.cn
bState Key Laboratory of Radiation Medicine and Protection, School for Radiological and Interdisciplinary Sciences (RAD-X), Soochow University, Suzhou, 215123, China. E-mail: xtian@suda.edu.cn
First published on 11th September 2020
Abstract
In recent years, peptide self-assembly proved to be an efficient strategy to create complex structures or functional materials with nanoscale precision. In this work, we designed and synthesized a novel glycopeptide molecule with a galactose moiety through peptide galactosylation. Then relying on peptide self-assembling strategies, we created a supramolecular hydrogel with multivalent galactose ligands on the surface of self-assembled nanofibers for molecular recognition and interactions. Because of multivalent galactose–LecA interactions, the self-assemblies of glycopeptide could target P. aeruginosa specifically, and acted as anti-virulence and antibacterial agents to inhibit biofilm formation and bacterial growth of P. aeruginosa. Moreover, in association with polymyxin B, a common antibiotic, the glycopeptide hydrogel exhibited a synergistic growth inhibition effect on biofilm colonization of P. aeruginosa.
Introduction
Carbohydrate–lectin interactions are ubiquitous in living systems and play an important role in both physiological and pathophysiological processes, including cell adhesion, growth, differentiation, inflammation, tumor metastasis, the immune response, and bacterial infections.1–3 The biomolecular interactions between an individual carbohydrate moiety and a carbohydrate-binding lectin are relatively weak and nonspecific.4,5 However, this weak binding-affinity and low specificity can be overcome in nature by multivalent displays of carbohydrates on cell surface in the forms of glycoproteins, glycolipids or glycocalyx.6,7 As a result, the concept of multivalency has received enormous attention and became a powerful approach to design multivalent carbohydrate clusters to control cell signal transduction or prevent pathogenic infection.8–10With the purpose of mimicking glycocalyx structures in nature, different scaffolds have been exploited to design architectures with multivalent carbohydrate ligands for studying the mechanisms involved in multivalent binding interactions.11–13 Some of them exhibited high potentials to work as decoys to block infection or as anti-biofilm agents to inhibit biofilm development by competitive binding with bacteria.6,14,15 However, because of the complex multivalent structures and complicated synthetic procedures, it is still desirable to design a facile way to prepare multivalent carbohydrate clusters that can interact with complementary proteins in biological systems.16,17 In recent years, supramolecular chemistry offered an interesting and alternative methodology for the design and fabrication of flexible and dynamic self-assembled glycol-architectures,18–22 which can display and orient carbohydrate ligands in appropriate positions for optimized binding with their targeted proteins.23–27 Owing to numerous advantages, such as easy synthesis, well-defined molecular structures, and inherent biocompatibility and biodegradability, self-assembling peptides became attractive scaffolds for constructing high-order glycol-structures through associated noncovalent interactions of small glycopeptides.28–30 This approach can offer an opportunity to mimic self-assembling processes in nature to form complex structures or functional materials with nanoscale precision.31
Pseudomonas aeruginosa (P. aeruginosa) is one of the most significant Gram-negative pathogens in hospitals that involved in acute and chronic lung infections.32 Recent studies revealed that one soluble lectin of P. aeruginosa, the galactose-binding lectin LecA (PA-IL), plays prominent roles in cellular adhesion and invasion, bacterial virulence, and biofilm formation.33,34 Consequently, developing P. aeruginosa inhibitors from multivalent scaffolds with various valences and spatial arrangement of glycosides to target PA-IL is a highly active research area, which provides efficient anti-infective agents against this pathogen.35,36
Therefore, in this work, we decided to take the advantage of peptide self-assembling strategies as an easy method to create novel multivalent architectures displaying galactoses on their surface for multivalent interactions with its targeted protein, PA-IL. The self-assembling peptide with a galactose moiety was designed to consist of a naphthyl group, a tetrapeptide segment (Phe-Phe-Ser-Tyr(H2PO3)) and a sugar moiety (D-galactose) linked to the side group of Ser (Scheme 1). The naphthyl group and Phe–Phe dipeptide worked as a rigid scaffold to intensify supramolecular self-assembling of the designed molecule via hydrophobic and π–π interactions. The incorporated tyrosine phosphate (Tyr(H2PO3)) residue endowed this molecule with good water solubility before supramolecular assembling and the ability for enzyme-triggered dephosphorylation reaction and supramolecular assembling. After supramolecular assembling induced by alkaline phosphatase (ALP), the attached galactose ligand worked as a bioactive cue to target PA-IL on P. aeruginosa via multivalent galactose–lectin interactions, and such interactions leaded to detrimental effects on P. aeruginosa growth and biofilm formation via cell membrane disruption. Therefore the rational integration of a self-assembling scaffold (e.g., NapFF) with a galactose residue offered a simple approach to prepare supramolecular architectures with galactose clusters targeting P. aeruginosa through multivalent binding with PA-IL.
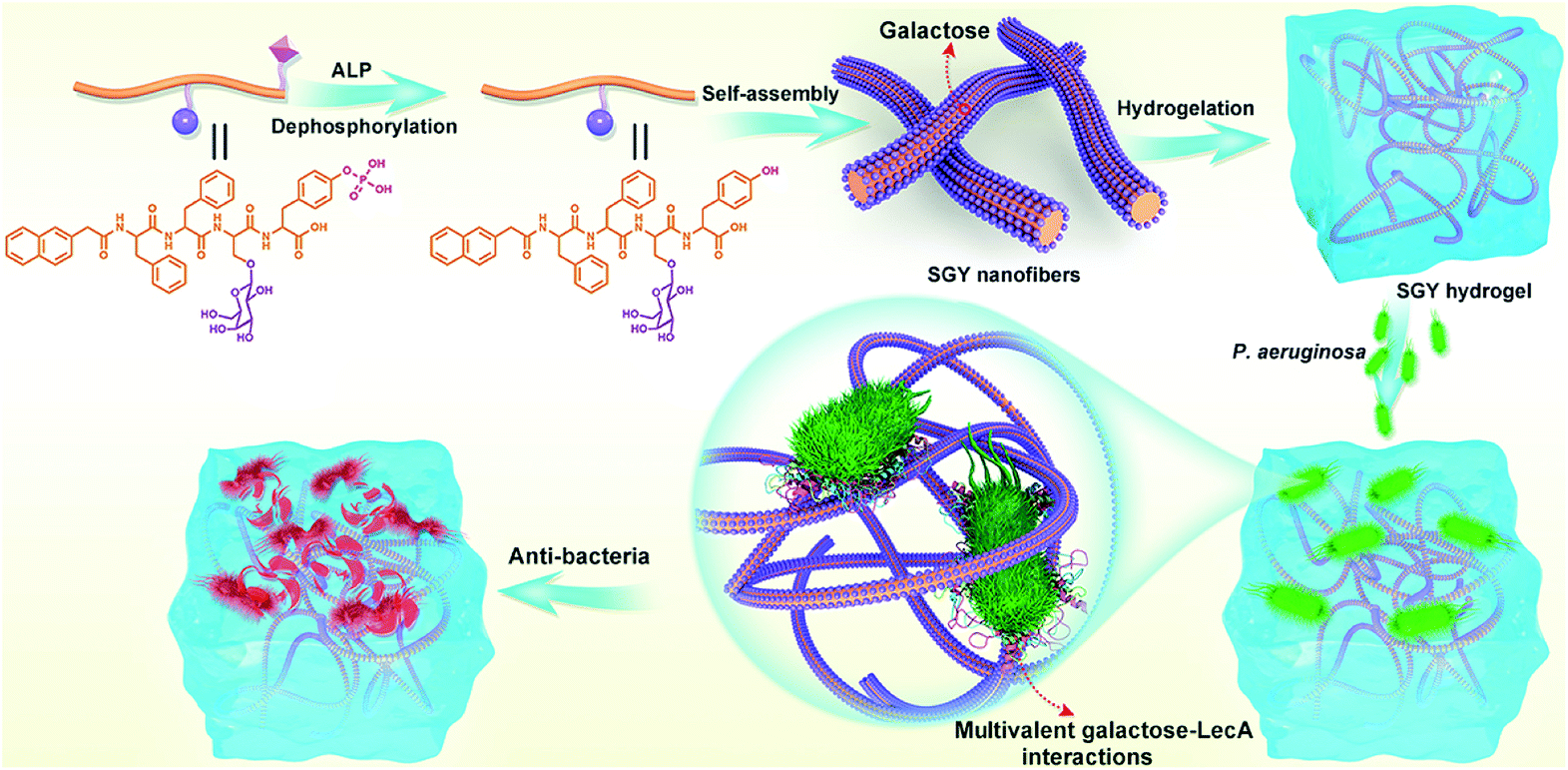 | ||
| Scheme 1 Illustration of the self-assembling process of SGY for the formation of supramolecular nanofibers and hydrogel with multivalent galactose ligands, and its specific targeting and antibacterial effects on P. aeruginosa via multivalent galactose–LecA interactions. | ||
Results and discussion
In order to build up new multivalent architectures targeting P. aeruginosa, we designed a short glycopeptide containing a galactose residue (NapFFSer(Gal)Yp), which was synthesized by following the standard solid-phase peptide synthesis methods from Fmoc-Tyr(H2PO3)-OH, Fmoc-Ser[Gal(OAc)4]-OH, Fmoc-Phe-OH and 2-(naphthalen-6-yl)acetic acid (Scheme 2 and Fig. S4–S6†). The molecule of Fmoc-Ser[Gal(OAc)4] was prepared from Fmoc-Ser-OH and β-D-galactose pentaacetate via glycosylation reaction in CH3CN by using BF3·Et2O as the Lewis acid (Fig. S1–S3†).37,38 After removal of the acetyl groups from NapFFS[Gal(OAc)4]Yp by NaOMe in methanol, we obtained the gelation precursor of NapFFSer(Gal)Yp (SGYp) (Fig. S7 and S8†). The precursor of SGYp showed a good solubility in water after the adjustment of pH to 7.4 (Fig. 1A), because of the presence of a Tyr(H2PO3) group. Followed by the addition of ALP, the solution of SGYp transformed to a stable hydrogel in 20 min, because of the molecular conversion of SGYp to SGY via enzyme-triggered dephosphorylation (Fig. 1B). During this process, the resulting SGY acted as a gelator to perform supramolecular self-assembling and gelation, relying on increased molecular hydrophobicity and extended intermolecular interactions, which resulted in the formation of a supramolecular hydrogel with minimum gelation concentration around 0.6 wt%.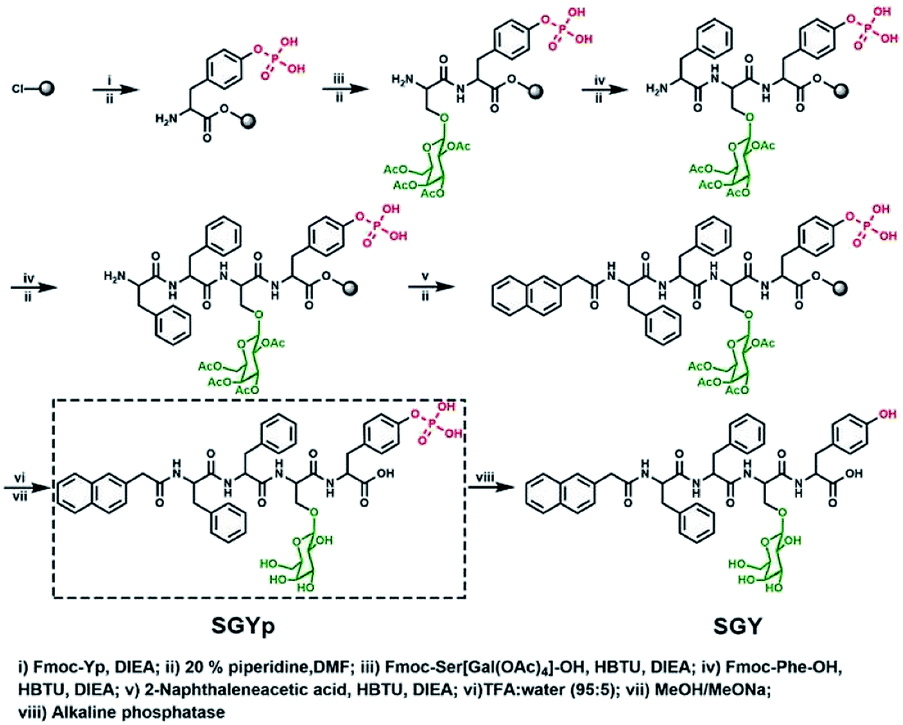 | ||
| Scheme 2 Synthetic processes for the preparation of SGYp together with its enzymatic conversion to SGY by ALP for supramolecular assembling and gelation. | ||
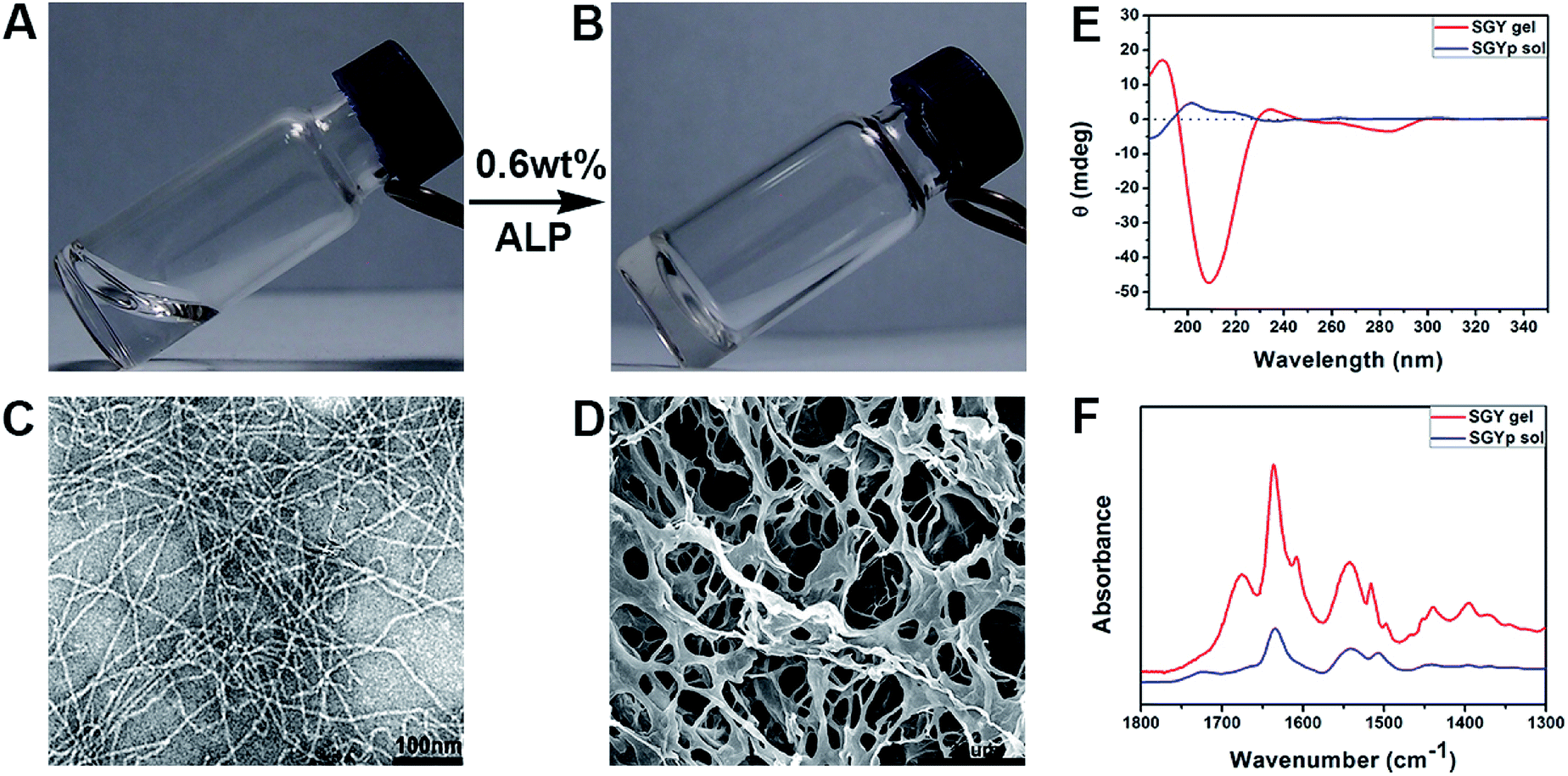 | ||
| Fig. 1 Optical images of the solution of (A) SGYp (0.6 wt%, pH = 7.4), and (B) the self-assembled SGY gel triggered by ALP (10 units per mL); (C) TEM and (D) SEM images of the SGY hydrogel shown in (B). (E) CD and (F) FTIR spectra of the SGYp solution and the SGY gel shown in (A) and (B). | ||
Transmission electron microscopy was used to characterize nanostructural morphologies of the self-assembled SGY gel. As shown in Fig. 1C, well-defined and flexible nanofibrous structures were observed within the matrices of the SGY gel, with 8–10 nm in width and several micrometers in length. These nanofibers tended to entangle with each other to form cross-linked nanofibrous networks within the SGY gel matrix, and encapsulate a large amount of water (99.4%). Scanning electron microscopy (SEM) analysis revealed the formation of fibrillar and interconnected porous structures (14 μm) self-assembled from SGY (Fig. 1D). The viscoelastic property of the SGY gel was assessed by dynamic oscillatory rheology. As shown in Fig. S13,† its storage modulus (G′ = 635.5 Pa) was much higher than the loss modulus (G′′ = 89.8 Pa) within tested oscillating frequency limit (0.1–10 Hz), indicating a dominantly elastic property of the SGY gel.
Circular dichroism spectra (CD) showed the molecular conformations of SGYp and SGY before and after supramolecular self-assembling (Fig. 1E). For example, the solution of SGYp showed a negative peak at 185 nm, a positive peak at 200 nm and a shoulder peak at 219 nm, indicating the lack of regular secondary structures of SGYp in water. However, the assembled SGY gel displayed a predominant positive peak at 190 nm and a negative peak at 209 nm, which corresponded to the formation of typical β-sheet structures.39 In addition, Fourier transform infrared spectroscopy (FTIR) was performed to analyze secondary structures of the SGY gel. As shown in Fig. 1F, the SGYp solution showed a absorption peak near 1634 cm−1 and a broad peak near 1540 cm−1, which were attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of amide, and N–H bending vibration/C–N stretching vibration, respectively.40,41 In comparison, the SGY gel displayed two amide I bands at 1636 and 1675 cm−1, and an amide II absorption peak around 1542 cm−1, which can be ascribed to the formation of antiparallel β-sheet structures, which were consistent with the results of CD.41,42 In addition, the peak at 1602 cm−1 and 1514 cm−1 belonged to the aromatic skeleton vibrations of the tyrosine amino acid.43,44
O stretching vibrations of amide, and N–H bending vibration/C–N stretching vibration, respectively.40,41 In comparison, the SGY gel displayed two amide I bands at 1636 and 1675 cm−1, and an amide II absorption peak around 1542 cm−1, which can be ascribed to the formation of antiparallel β-sheet structures, which were consistent with the results of CD.41,42 In addition, the peak at 1602 cm−1 and 1514 cm−1 belonged to the aromatic skeleton vibrations of the tyrosine amino acid.43,44
In order to prove that the galactose moieties on the self-assembled nanostructures of SGY maintained its activity to target galactose-binding lectins, we performed turbidity assays with peanut agglutinin (PNA) and concanavalin (ConA). The multivalent galactose–PNA binding interactions were tested by examining the absorbance changes of samples at the wavelength of 420 nm (Fig. 2A). As shown in Fig. 2B, when the solution containing self-assembled nanostructures of SGY (800 μM) which derived from SGYp and ALP was mixed with PNA, a remarkable absorption at 420 nm was observed, and its intensity increased with time, suggesting the occurrence of binding interactions and agglutination of SGY with PNA. However, under the same conditions, no obvious absorption was identified at 420 nm for the sample containing SGYp (800 μM) and PNA, due to the low binding affinity between single molecule of SGYp and PNA. The solution of SGY at a much lower concentration of 400 μM, less than its critical aggregation concentration at 583 μM (Fig. S14†), was also used for turbidity assays, and no obvious absorbance changes were observed with time at 420 nm, indicating the important role of formation of multivalent galactose clusters on the self-assembled structures of SGY for PNA agglutination. In addition, the molecule of SY without the galactose residue was also incapable of inducing PNA aggregation under the same conditions (Fig. S9–S12†). In order to investigate the specific lectin binding properties of SGY, we performed additional turbidity assays with ConA under the same conditions, no obvious signals were observed for all samples (Fig. 2C), because ConA is a glucose/mannose-specific lectin.45 These results indicated that the formation of multivalent galactose clusters on the self-assembled structures of SGY can intensify the specific binding interactions with PNA, and result in PNA agglutination.
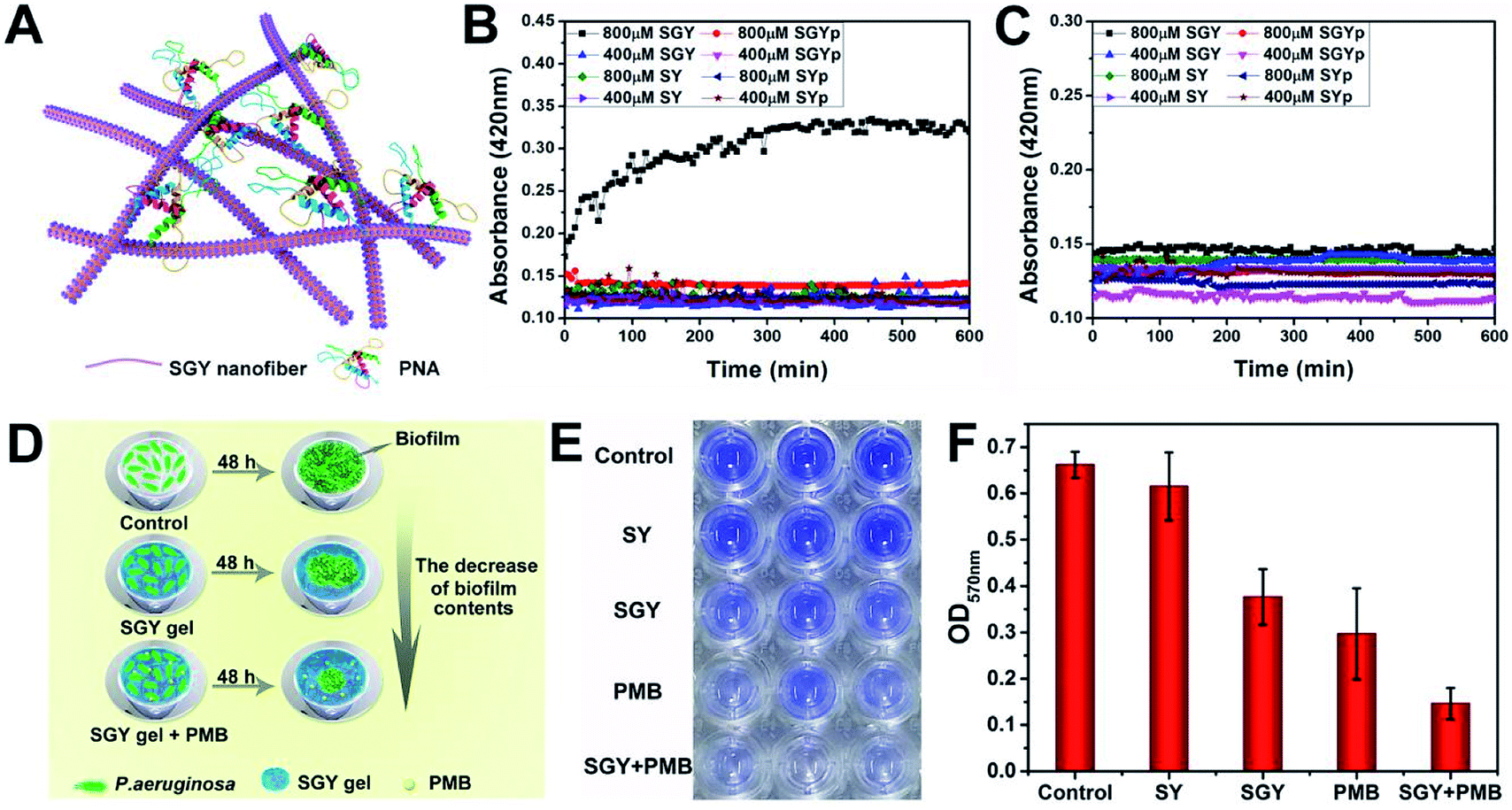 | ||
| Fig. 2 (A) Illustration of the agglutination of PNA with SGY through multivalent galactose–lectin binding interactions. Turbidimetric assays of the binding interactions of (B) PNA (2 mg mL−1) and (C) ConA (2 mg mL−1) with different concentrations of SGYp (400 and 800 μM), SGY (400 and 800 μM), SYp (400 and 800 μM) and SY (400 and 800 μM) in HEPES buffer. (D) Illustration of the effects of the SGY gel, PMB and SGY gel + PMB on inhibiting biofilm growth of P. aeruginosa. (E) Crystal violet staining P. aeruginosa (105 CFU mL−1) in a 96-well microplate with the presence of the SY gel (1 wt%), SGY gel (1 wt%), PMB (20 μg mL−1), and SGY gel (1 wt%) encapsulating 20 μg mL−1 of PMB. (F) The relative biofilm content of P. aeruginosa shown in Fig. 2E was determined by the absorbance at 570 nm after cultivation at 37 °C for 48 h. | ||
The galactose-specific lectin LecA (PA-IL) of P. aeruginosa plays prominent roles in biofilm development via lectin-mediated molecular recognition and aggregation.46–48 Therefore, inhibition of PA-IL activities by multivalent architectures of galactose was useful for preventing biofilm development.49,50 Because of the presence of multivalent galactose ligands on the self-assembled structures of SGY, we decided to examine its potentials to inhibit biofilm formation of P. aeruginosa (ATCC 27853). After incubating bacteria with the SGY gel for 48 h, we performed crystal violet staining and determined the fluorescence intensity at 570 nm, which reflected the amount of residual biofilms.51,52 The inhibition effects of SGY gels on biofilm growth of P. aeruginosa were illustrated in Fig. 2D. As shown in Fig. 2E, similar to the control group, the SY gel without galactose ligands had no noticeable inhibitory effect on P. aeruginosa for biofilm formation, as exemplified by the high intensities of fluorescence at 570 nm. In comparison, the SGY gel exhibited a potent inhibition of biofilm formation by 43%. Since bacteria within a biofilm were more resistant to antibiotics, combinatorial therapy of anti-biofilm agents with antibiotics offered a promising strategy to combat antibiotic resistance and biofilm-associated infection.53 In order to investigate the abilities of SGY to work synergistically with a common antibiotic to inhibit biofilm colonization, we prepared a mixed SGY gel in combination with a small amount of polymyxin B (PMB) antibiotic (20 μg mL−1), and incubated with P. aeruginosa. The sample containing PMB alone can inhibit P. aeruginosa biofilm formation by 55%, while the sample containing both the SGY gel and PMB showed synergistically inhibitory effects on biofilm formation by 80% (Fig. 2E and F).
Contrary to conventional antibiotics, most of anti-biofilm compounds only worked as anti-virulence agents as they did not exhibited toxic effects on bacterial growth.54 Because of the hydrophobic nature of SGY with predominant aromatic groups in its structure, we decided to investigate its efficacy to disrupt cell membranes by physical insertion of SGY into the hydrophobic regions of lipid membranes.55 After P. aeruginosa was incubated to the exponential growth stage, diluted suspension of bacteria (1.0 × 105 CFU mL−1) was added to the surface of the SGY gel (1.0 wt%) in Luria–Bertani (LB) media. After incubation at 37 °C for 24 h, the bacteria viability was measured by colony counting methods. Under the same conditions, bacteria cultured on the surface of TCPS plates or the surface of the SY gel were used as controls. As shown in Fig. 3A and B, the SY gel of (1.0 wt%) without galactose ligands did not showed any inhibitory effect on the viability of P. aeruginosa, as exemplified by the high survival rates of the cells (95%). However, when P. aeruginosa was cultured on the surface of the SGY gel, noticeable inhibitory activity against the growth of P. aeruginosa was observed from the much lower survival rates of the cells (47%). In order to further confirm the cell viability of P. aeruginosa treated by SGY and SY gels, we performed live/dead assays. The living cells with intact cell membranes show green fluorescence, whereas dead cells or cells with damaged membranes exhibit significantly red fluorescence under fluorescence microscope. As shown in Fig. 3C, most of bacteria in the control group or cultured on the surface of the SY gel exhibited green fluorescence, indicating the high viability of the cells. However, bacteria incubation with the SGY gel showed very low viability, as exemplified by remarkable red fluorescence. Furthermore, the z-stack confocal images (3D) further revealed that most of dead bacteria adhered on surface of SGY hydrogel and exhibited red fluorescence. These results indicated that the supramolecular hydrogel of SGY with galactose ligands displayed noticeable inhibitory effects on P. aeruginosa by inducing bacterial death.
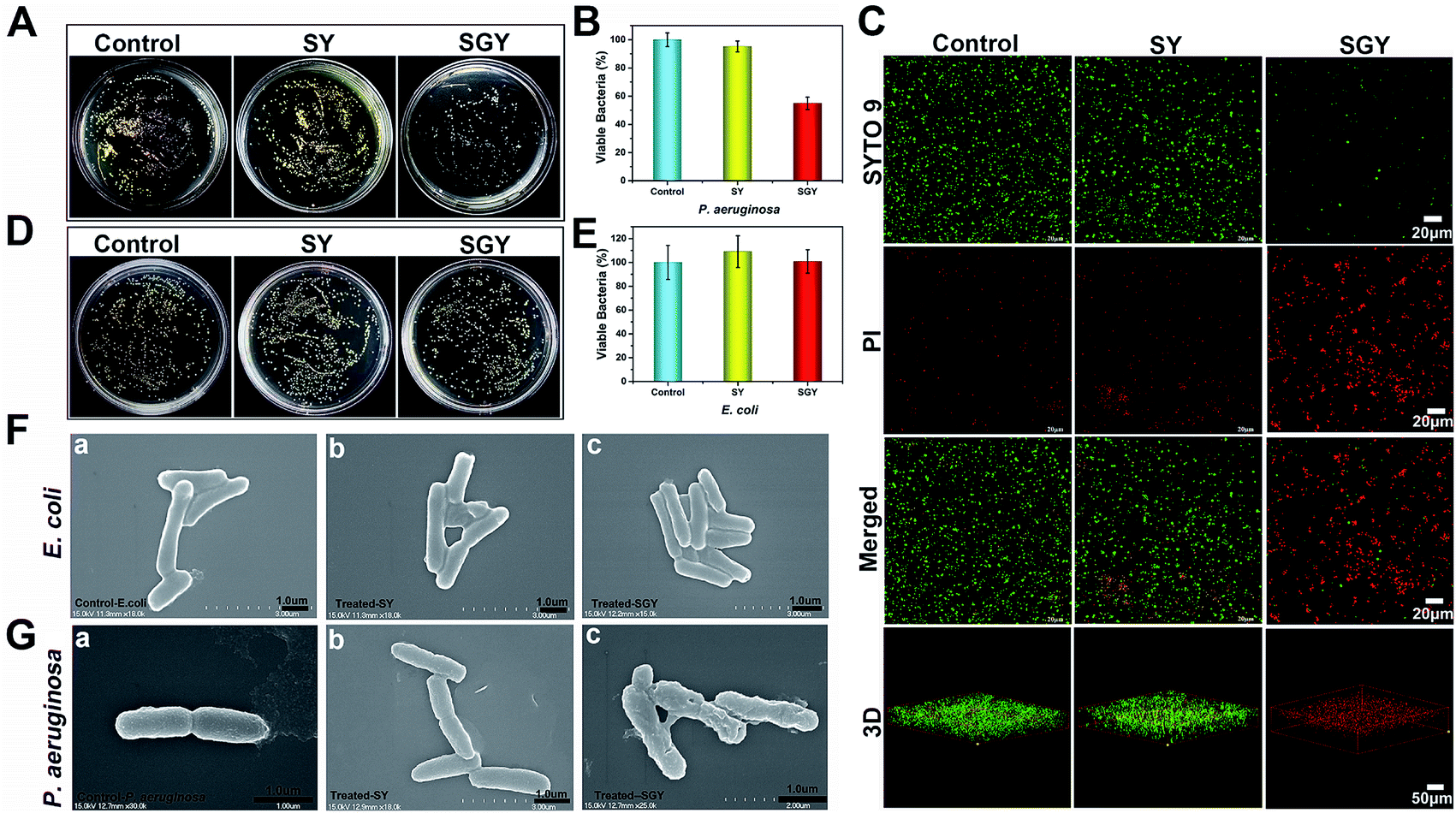 | ||
| Fig. 3 (A) Optical images of bacterial colonies formed on agar plated at 37 °C for 24 h, after P. aeruginosa (1 × 105 CFU mL−1) was treated by 1 wt% of SGY and 1 wt% of SY hydrogels, respectively. (B) The survival rates of P. aeruginosa shown in Fig. 3A. (C) Fluorescence microscopy images of P. aeruginosa (1 × 105 CFU mL−1) in the control group or incubated on the surface of the SGY (1 wt%) and SY (1 wt%) hydrogels for 24 h, followed by live/dead staining. (D) Optical images of bacteria colonies formed on agar plated at 37 °C for 24 h, after E. coli (1 × 105 CFU mL−1) was treated by 1 wt% of SGY and 1 wt% of SY hydrogels. (E) The survival rates of E. coli shown in Fig. 3D. SEM images of (F) E. coli and (G) P. aeruginosa in the control group or treated by the SY and SGY hydrogels for 24 h, respectively. | ||
To confirm that the antibacterial activity of SGY towards P. aeruginosa was related with specific and multivalent galactose–LecA interactions, another kind of Gram negative bacteria, Escherichia coli (E. coli) (ATCC 25922), was selected for bacterial growth tests. As shown in Fig. 3D and E, E. coli cultured on the surface of the SGY gel showed extremely high viability. SEM analysis revealed that E. coli in contact with the SGY gel showed a smooth and intact cell surface, similar to the cells in the control group (Fig. 3F). In comparison, P. aeruginosa treated by the SGY gel displayed a wrinkled cell surface with membrane deformation and breakage (Fig. 3G). These results suggested that the bactericidal activity of the SGY gel towards P. aeruginosa stems from the specific and multivalent galactose–LecA interactions, which may cause the membrane damage and cell death.
In order to further understand the bactericidal mode action of SGY towards P. aeruginosa instead of E. coli, we examined its ability to interact with cell membranes of P. aeruginosa and E. coli for membrane permeation and depolarization (Fig. 4A and B).56 The outer membrane permeation of bacteria was tested by following the 8-anilino-1-naphthalenesulfonic acid (ANS) uptake assay. After treatment with SGY and ANS, the fluorescent properties of ANS inside bacteria were analysed. As shown in Fig. 4C, P. aeruginosa treated by the SGY solution or hydrogel showed significant enhancement of fluorescence of ANS, indicating the occurrence of outer membrane permeation of bacteria. In comparison, the bacteria in contact with SGYp or in control group did not show obvious fluorescent changes of ANS. Moreover, the inner membrane depolarization properties of SGY was also examined by using 3-3′-dipropylthiadicarbocyanine iodide (disC3(5)). Based on the disC3(5) uptake assay of P. aeruginosa, SGY can also depolarize the inner membrane of P. aeruginosa, as exemplified by the significant increase of fluorescence intensities of disC3(5) with time in the SGY solution or hydrogel group, in contrast to the SGYp or control group (Fig. 4D). The membrane permeation and depolarization properties of SGY towards E. coli were also examined under the same conditions. Based on the results shown in Fig. 4E, we found that SGY can induce the outer membrane permeation of E. coli, but its disruptive effects was less than that on P. aeruginosa. The disC3(5) uptake tests of E. coli revealed that SGY had no damaging effect on the inner membrane of E. coli from the low intensity of disC3(5) fluorescence (Fig. 4F). These results indicated that the bacterial cell death of P. aeruginosa induced by SGY was associated with permeation and depolarization of its outer and inner membrane. In contrast to E. coli, P. aeruginosa can intensify the interactions between the self-assembled SGY and bacteria for cell membrane disruption via specific and multivalent binding between galactose ligands and LecA.
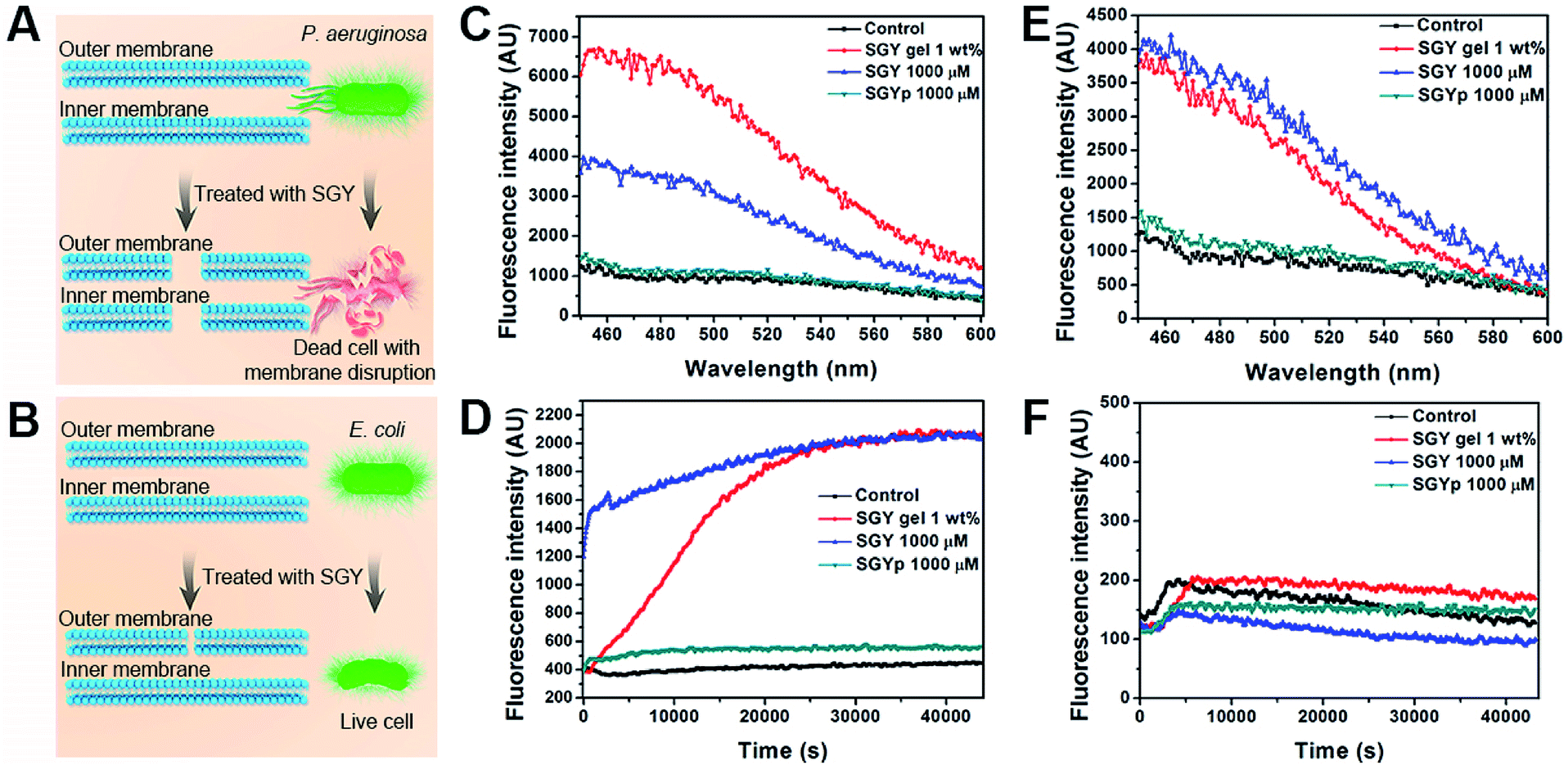 | ||
| Fig. 4 Illustration of membrane disruptive effect of SGY on (A) P. aeruginosa and (B) E. coli. Fluorescence spectra of (C) P. aeruginosa and (E) E. coli from ANS uptake assays. The enhanced fluorescence indicated outer membrane permeation of bacteria induced by different concentrations of SGY. Fluorescence spectra of (D) P. aeruginosa and (F) E. coli from disC3(5) uptake assays. The increased fluorescence observed in the samples of P. aeruginosa suggested the depolarization of inner membrane of P. aeruginosa with the presence of SGY, in contrast to E. coli. | ||
While antimicrobial agents had excellent antimicrobial activity, most of them also exhibited cytotoxicity toward human cells, limiting their potential clinical applications.57 Thus, it is essential that antimicrobial agents are not only potentially effective in combating pathogens but also demonstrate good biocompatibility with human cells. To examine the biocompatibility of SGY, we conducted cell viability tests based on CCK-8 assays. As shown in Fig. S15,† the HUVEC cells incubating in the solution of SGYp or on the surface of the SGY gel can maintained good cell viability more than 90% over the course of 72 h culture, indicating the good biocompatibility of the SGYp molecule and the SGY gel.
Conclusions
In summary, we reported the synthesis of a new type of glycopeptide from a self-assembling peptide scaffold and a galactose moiety. Relying on extended noncovalent interactions of peptide scaffolds, this molecule can undergo supramolecular self-assembling to form self-assembled nanofibers and hydrogels under physiological conditions by alkaline phosphatase. The galactose ligands exposed on the surface of self-assembled nanofibers and hydrogels can form multivalent galactose clusters, and work as bioactive cues to target galactose-binding proteins, resulting in inhibition of biofilm formation and bacterial growth of P. aeruginosa. Moreover, in association with polymyxin B, a common antibiotic, the glycopeptide hydrogel also exhibited a synergistic effect on inhibiting biofilm growth and colonization of P. aeruginosa.Experimental
TEM and SEM characterizations
10 μL of the SGY gel was placed on the copper grid, and then the grid was stained by 10 μL of phosphotungstic acid (1.0%) for 15 min. After removal of excess phosphotungstic acid, the sample was dried under an infrared lamp for 3–5 min. The nanostructures within the sample were visualized by TEM (Hitachi HT7700). SEM characterization was conducted on a scanning electron microscope (Hitachi S-4700). The sample was prepared by loading 5 μL of the SGY gel on a clean silicon wafer, and then sprayed with a thin layer of gold.Rheological tests
200 μL of the SGY gel were placed on a parallel plate of 20 mm in diameter, and its rheological properties were examined on a rheometer of Thermo Scientific HAAKE ReoStress 6000 at 25 °C. The dynamic strain sweeps were carried out from 0.1% to 10% strain at a fixed frequency of 6.28 rad s−1. Dynamic frequency sweeps were performed from 0.1 to 100 Hz at a fixed strain of 0.5%.Circular dichroism and FTIR characterizations
20 μL of the SGY gel were loaded into a 1 mm thick quartz cuvette and scanned from 185 nm to 400 nm on a Jasco J-810 spectrometer under N2 atmosphere. The hydrogel for FTIR characterization was prepared by using deuterium oxide (D2O) as a solvent and loaded into a KBr cuvette. FTIR spectra were collected on a PerkinElmer spectrophotometer.Turbidimetric assays
Time-dependent turbidimetric assays were conducted in a 96-well microtiter plate with SGY (400 and 800 μM), SGYp (400 and 800 μM), SY (400 and 800 μM) and SYp (400 and 800 μM). After addition of 100 μL of PNA (2 mg mL−1) or ConA (2 mg mL−1) solution in 25 mm HEPES buffer (pH = 7.4, 10 mm HEPES, 150 mm NaCl, 1 mm CaCl2 and 1 mm MnCl2) to each group of samples (100 μL), the samples were incubated at room temperature, and the absorbance at 420 nm was measured.Bacterial growth assays
The antibacterial activity of the SGY gel towards P. aeruginosa was assessed in a 96-well TCP plate by using colony counting methods. 80 μL of the SGY gel were added in each well (three parallel wells per sample), and 200 μL of LB medium was loaded on the surface of the gels. After equilibrating at 37 °C for 12 h, the LB medium was removed and 100 μL of bacterial suspension (1 × 105 CFU mL−1) of P. aeruginosa were added. After incubation at 37 °C for 24 h, 100 μL of diluted bacterial solution above gels were spread on agar plates. The ager plates were inverted and incubated at 37 °C for 24 h.Morphologies of bacterial cells
The morphologies of bacteria before and after treatment by the SGY gel were examined by SEM. After treatment by the SGY gel at 37 °C for 24 h, the bacterial suspension was centrifuged at 5000 rpm for 5 min, and fixed by glutaraldehyde (4%) for 2 h at room temperature. Then the samples were dehydrated with increasing concentrations of ethanol (30%, 50%, 75%, 95%, and 100%). Finally, the samples were loaded on clean silicon wafers, sprayed with a thin layer of gold and visualized by SEM (Hitachi S-4700).Live/dead staining assay
80 μL aliquots of the SGY gel were prepared in an eight-well borosilicate glass, and then 400 μL of LB media were introduced to each well. After equilibrating at 37 °C for 12 h, the medium was removed, followed by the addition of 100 μL of bacterial suspension (1 × 105 CFU mL−1) of P. aeruginosa. After cultivation at 37 °C for 24 h, 100 μL of live/dead BacLight solution were added in each well and stained for 15 min in dark. The samples were directly observed by using a Zeiss NLO510 laser scanning confocal microscope.Outer membrane permeability assays
The bacteria suspension (1 × 105 CFU mL−1) of P. aeruginosa was washed by Tris buffer (10 mM Tris, 150 mM NaCl, pH = 7.4), and then re-suspended in the ANS solution (10 mM) in 10 mM Tris buffer (10 mM Tris, 150 mM NaCl, pH = 7.4) for staining. After 10 min in dark, the bacteria solutions were added to the samples in 96-well plates. The changes of fluorescence emission were measured by a multi-functional enzyme analyzer between 450–600 nm with excitation at 380 nm.Inner membrane depolarization assays
The bacteria suspension (1 × 105 CFU mL−1) of P. aeruginosa was washed and re-suspended in HEPES buffer (5 mM HEPES, 2 mM EDTA). After incubation with 1 μM disC3(5) for 60 min in dark, the solution was added to the sample in 96-well plates, and the fluorescent changes were measured by a multi-functional enzyme analyzer (excitation, 622 nm; emission, 670 nm).Crystal violet staining
80 μL of the SGY gel (1%), SY gel (1%), 20 μg mL−1 PMB or SGY gel encapsulating 20 μg mL−1 PMB were added to the wells in a 96-well TCP plate, and 200 μL of LB medium was loaded. After equilibrating at 37 °C for 12 h, the LB medium was removed and 100 μL of bacterial suspension (1 × 105 CFU mL−1) of P. aeruginosa were added. After incubation at 37 °C for 48 h, the bacteria solution was carefully removed and the wells were washed for 3 times by PBS (0.1 M, pH = 7.4). The wells were air-dried, and 200 μL of crystal violet dye (0.4%) were added. After staining for 15 min in dark, the redundant crystal violet dye was removed and washed gently for 3 times by PBS (0.1 M, pH 7.4). The wells were air-dried, and 200 μL of 33% acetic acid were introduced to each well. Finally, the relative content of biofilm was determined by measuring the absorbance at 570 nm. Each measurement was performed in triplicate.Cytocompatibility tests in vitro
The cytocompatibility of SGYp and the SGY gel was assessed with HUVEC by following CCK-8 (cell counting kit-8) protocols. HUVEC cells (2 × 104 cells per mL) were planted in a 96-well plate and cultivated for 1, 2, and 3 days. The culture medium was replaced with complete culture medium supplemented with 500 μM of SGYp. After incubation for 1, 2, and 3 days, the fresh medium with 10% CCK-8 solution was added and incubated for 2 h, and the absorbance at 450 nm was detected.HUVEC cells (2 × 104 cells per mL) were planted on the surface of the SGY gel (1 wt%) in a 96-well plate and incubated at 37 °C in an incubator with 5% CO2. The culture medium was replaced with fresh one every day. After incubation for 1, 2, and 3 days, the fresh medium with 10% CCK-8 solution was added and incubated for 2 h, and the absorbance at 450 nm was detected. Cell viability of the blank group (untreated) was set as 100%. All measurements were performed in triplicate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Ministry of Science and Technology of China, 2016YFC1100100) and the National Natural Science Foundation of China (51673142).Notes and references
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Camara, A. Stocker, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed. Engl., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- A. Imberty and A. Varrot, Curr. Opin. Struct. Biol., 2008, 18, 567–576 CrossRef CAS PubMed.
- P. Bojarova and V. Kren, Biomater. Sci., 2016, 4, 1142–1160 RSC.
- H. Ghazarian, B. Idoni and S. B. Oppenheimer, Acta Histochem., 2011, 113, 236–247 CrossRef CAS.
- C. N. Spaulding, R. D. Klein, S. Ruer, A. L. Kau, H. L. Schreiber, Z. T. Cusumano, K. W. Dodson, J. S. Pinkner, D. H. Fremont, J. W. Janetka, H. Remaut, J. I. Gordon and S. J. Hultgren, Nature, 2017, 546, 528–532 CrossRef CAS PubMed.
- K. Hatano, K. Matsuoka and D. Terunuma, Chem. Soc. Rev., 2013, 42, 4574–4598 RSC.
- Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC.
- M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693–1752 CrossRef CAS.
- X. Yan, A. Sivignon, N. Yamakawa, A. Crepet, C. Travelet, R. Borsali, T. Dumych, Z. Li, R. Bilyy, D. Deniaud, E. Feury, N. Barnich, A. Darfeuille-Michaud, S. G. Gouin, J. Bouckaert and J. Bernard, Biomacromolecules, 2015, 16, 1827–1836 CrossRef CAS PubMed.
- E. L. Dane, A. E. Ballok, G. A. O'Toole and M. W. Grinstaff, Chem. Sci., 2014, 5, 551–557 RSC.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2011, 2, 2917 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- J. Huang, C. Bonduelle, J. Thevenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119–122 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- K.-S. Krannig and H. Schlaad, J. Am. Chem. Soc., 2012, 134, 18542–18545 CrossRef CAS.
- R. Roytman, L. Adler-Abramovich, K. S. A. Kumar, T.-C. Kuan, C.-C. Lin, E. Gazit and A. Brik, Org. Biomol. Chem., 2011, 9, 5755–5761 RSC.
- X. Du, J. Zhou, O. Guvench, F. O. Sangiorgi, X. Li, N. Zhou and B. Xu, Bioconjugate Chem., 2014, 25, 1031–1035 CrossRef CAS.
- S. W. Liao, T.-B. Yu and Z. Guan, J. Am. Chem. Soc., 2009, 131, 17638–17646 CrossRef CAS.
- Y.-B. Lim, S. Park, E. Lee, J.-H. Ryu, Y.-R. Yoon, T.-H. Kim and M. Lee, Chem.–Asian J., 2007, 2, 1363–1369 CrossRef CAS.
- Y. Ogawa, C. Yoshiyama and T. Kitaoka, Langmuir, 2012, 28, 4404–4412 CrossRef CAS PubMed.
- W. Wang, H. Wang, C. Ren, J. Wang, M. Tan, J. Shen, Z. Yang, P. G. Wang and L. Wang, Carbohydr. Res., 2011, 346, 1013–1017 CrossRef CAS.
- D. J. Welsh, P. Posocco, S. Pricl and D. K. Smith, Org. Biomol. Chem., 2013, 11, 3177–3186 RSC.
- F. Zhao, C. S. Weitzel, Y. Gao, H. M. Browdy, J. Shi, H. C. Lin, S. T. Lovett and B. Xu, Nanoscale, 2011, 3, 2859–2861 RSC.
- G. Tao, T. Ji, N. Wang, G. Yang, X. Lei, W. Zheng, R. Liu, X. Xu, L. Yang, G.-Q. Yin, X. Liao, X. Li, H.-m. Ding, X. Ding, J. Xu, H.-B. Yang and G. Chen, ACS Macro Lett., 2019, 9, 61–69 CrossRef PubMed.
- M. J. Grogan, M. R. Pratt, L. A. Marcaurelle and C. R. Bertozzi, Annu. Rev. Biochem., 2002, 71, 593–634 CrossRef CAS PubMed.
- V. E. Wagner and B. H. Iglewski, Clin. Rev. Allergy Immunol., 2008, 35, 124–134 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerova, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS.
- N. Garber, U. Guempel, A. Belz, N. Gilboagarber and R. J. Doyle, Biochim. Biophys. Acta, 1992, 1116, 331–333 CrossRef CAS.
- J.-L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Camara, A. Stocker, T. Darbre and J.-L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- Y. Zhang, S. M. Muthana, D. Farnsworth, O. Ludek, K. Adams, J. J. Barchi, Jr. and J. C. Gildersleeve, J. Am. Chem. Soc., 2012, 134, 6316–6325 CrossRef CAS.
- M. Liu, A. Borgert, G. Barany and D. Live, Biopolymers, 2008, 90, 358–368 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3095–3103 CrossRef.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS.
- J. Kubelka and T. A. Keiderling, J. Am. Chem. Soc., 2001, 123, 12048–12058 CrossRef CAS PubMed.
- S. Ganesh, S. Prakash and R. Jayakumar, Biopolymers, 2003, 70, 346–354 CrossRef CAS.
- Y.-b. Lim, E. Lee, Y.-R. Yoon, M. S. Lee and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 4525–4528 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, 2007, 1767, 1073–1101 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. Escale, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- S. Zheng, T. Eierhoff, S. Aigal, A. Brandel, R. Thuenauer, S. de Bentzmann, A. Imberty and W. Romer, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 1236–1245 CrossRef CAS.
- H. Mikkelsen, M. Sivaneson and A. Filloux, Environ. Microbiol., 2011, 13, 1666–1681 CrossRef CAS PubMed.
- A. Novoa, T. Eierhoff, J. Topin, A. Varrot, S. Barluenga, A. Imberty, W. Romer and N. Winssinger, Angew. Chem., Int. Ed. Engl., 2014, 53, 8885–8889 CrossRef CAS.
- M. Vallet-Regi, B. Gonzalez and I. Izquierdo-Barba, Int. J. Mol. Sci., 2019, 20, 3806 CrossRef CAS.
- G. Sharma, S. Rao, A. Bansal, S. Dang, S. Gupta and R. Gabrani, Biologicals, 2014, 42, 1–7 CrossRef CAS.
- E. Burton, N. Yakandawala, K. LoVetri and M. S. Madhyastha, J. Ind. Microbiol. Biotechnol., 2007, 34, 1–4 CrossRef CAS.
- E. L. Dane, A. E. Ballok, G. A. O'Toole and M. W. Grinstaff, Chem. Sci., 2014, 5, 551–557 RSC.
- R. Roy, M. Tiwari, G. Donelli and V. Tiwari, Virulence, 2018, 9, 522–554 CrossRef CAS PubMed.
- B. Parrino, D. Schillaci, I. Carnevale, E. Giovannetti, P. Diana, G. Cirrincione and S. Cascioferro, Eur. J. Med. Chem., 2019, 161, 154–178 CrossRef CAS.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. DeGrado and E. Gazit, Nat. Commun., 2017, 8, 1365 CrossRef PubMed.
- G. Mueller and A. Kramer, J. Antimicrob. Chemother., 2008, 61, 1281–1287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of SGYp and its corresponding NMR and MS spectra. See DOI: 10.1039/d0ra06718k |
| This journal is © The Royal Society of Chemistry 2020 |