
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential metabolism determinants and drug–drug interactions of a natural flavanone bavachinin†
Xinqiang Lia,
Han Xingb,
Zifei Qin *b,
Jing Yangb,
Peile Wangb,
Xiaojian Zhangb,
Zhihong Yao*cd and
Xinsheng Yaocd
*b,
Jing Yangb,
Peile Wangb,
Xiaojian Zhangb,
Zhihong Yao*cd and
Xinsheng Yaocd
aDepartment of Pathology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
bDepartment of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: qzf1989@163.com
cInternational Cooperative Laboratory of Traditional Chinese Medicine Modernization and Innovative Drug Development Ministry of P. R. China, Jinan University, Guangzhou 510632, China. E-mail: yaozhihong_jnu@163.com
dCollege of Pharmacy, Jinan University, Guangzhou 510632, China
First published on 23rd September 2020
Abstract
Bavachinin, a natural bioactive flavanone, is reported to have many pharmacological proprieties, especially anti-osteoporosis activity. Here we aim to determine the roles of cytochrome P450s (CYP), UDP-glucuronosyltransferases (UGT), and efflux transporters in metabolism and drug–drug interactions (DDI) of bavachinin. Phase I metabolism and glucuronidation were performed by human liver microsomes (HLM) and human intestine microsomes (HIM). Reaction phenotyping was used to identify the main CYPs and UGTs. Gene silencing methods were employed to investigate the roles of breast cancer resistance protein (BCRP) and multidrug resistance-associated proteins (MRPs) in HeLa1A1 cells. Inhibition mechanisms towards CYPs and UGTs were explored through kinetic modeling. Three phase I metabolites (M1–M3) and one glucuronide (G1) were detected after incubation of bavachinin with HLM and HIM. The intrinsic clearance (CLint) values of M1 and G1 by HLM were 89.4 and 270.2 μL min−1 mg−1, respectively, while those of M3 and G1 by HIM were 25.8 and 247.1 μL min−1 mg−1, respectively. CYP1A1, 1A2, 1B1, 2C8, 2C19, and UGT1A1, 1A8 participated more in bavachinin metabolism. The metabolism showed marked species difference. BCRP and MRP4 were identified as the main contributors. Bavachinin displayed potent inhibitory effects against several CYP and UGT isozymes (Ki = 0.28–2.53 μM). Bavachinin was subjected to undergo metabolism and disposition by CYPs, UGTs, BCRP, MRP4, and was also a potent non-selective inhibitor against several CYPs and UGTs.
Introduction
Bavachinin is a natural bioactive flavanone isolated from the dried seeds of Psoralea corylifolia which is widely used for the treatment of bone diseases.1 It accounts for about 389.32 μg g−1 in the dried seeds of Psoralea corylifolia.2 In addition, bavachinin shows many types of biological property. For example, it showed anti-tumor activity in several tumor cell lines (IC50 = 19.5–30.5 μM).3 Besides, it exhibited inhibitory effects against papain-like protease (PLpro) (Ki = 18.4 μM) which serves as an attractive target for antiviral drugs.4 Furthermore, it could generate beneficial effects in Alzheimer's disease (AD) prevention and treatment by inhibiting neuroinflammation, oxidative damage, and key AD-related protein targets.5,6 Recently, bavachinin was reported as a novel natural pan-peroxisome proliferator-activated receptor (PPAR) agonist with EC50 values of 0.74, 4.00 and 8.07 μM for PPAR-γ, PPAR-α and PPAR-β/δ in 293T cell, respectively.7–9These potential therapeutic effects stimulate increasing interests in the absorption, distribution, metabolism, and excretion (ADME) of bavachinin. Of these, metabolism (phase I and II reactions) participated more in determining drug exposure at target sites (pharmacodynamics) and toxicity profiles (detoxification).10,11 Isomerization, glucuronidation, sulfonation, hydroxylation, and reduction were considered as the main metabolic pathways in rat samples and rat liver microsomes.12,13 Poor bioavailability (5.27–36.39%)13,14 and low plasma concentration (2.13–908.8 ng mL−1)13–18 were mainly attributed to massive metabolism after oral administration of bavachinin or bavachinin-containing extracts. However, so far, UDP-glucuronosyltransferase (UGT) enzymes including UGT1A1, 1A3, 1A8 and 1A10 were the main contributors responsible for the glucuronidation of bavachinin.19,20 Furthermore, little is known about its metabolism involving in human cytochrome P450s (CYP).
Traditionally, UGT-mediated glucuronide conjugates cannot be transported from intracellular to extracellular by passive transport due to high hydrophilicity of glucuronides.21 Excretion of these metabolites were performed by efflux transporters (i.e., breast cancer resistance protein, BCRP/ABCG2; multidrug resistance-associated proteins, MRPs/ABCCs) following with ATP hydrolysis.21 In our previous works, many glucuronides of natural compounds were the substrates of BCRP or MRPs transporters.21–25 In addition, the glucuronidation activity would be altered when the function of BCRP or MRPs transporters were inhibited due to the presence of “glucuronidation-transport interplay”.21 However, the effects of BCRP and MRPs transporters on the metabolism and disposition of bavachinin still remains unknown.
Moreover, drug–drug interactions (DDI) are involved in clinical rational drug use, and are significant clinical safety concerns.26–28 Human CYP1A2 (9%), 2B6 (2%), 2C8, 2C9 (16%), 2C19 (12%), 2E1 (2%), 3A4 (46%), and UGT1A1 (15%), 1A9, 2B7 (35%) participated most in the metabolism or elimination of clinical drugs.29,30 Once the function of these CYP (or UGT) isozymes were inhibited (or induced) by bavachinin, the exposure (represented by AUC0–t values) of co-administrated drugs could be markedly increased (or decreased), which further brought several adverse reactions (or insufficient efficacy). However, the effects of bavachinin towards these CYP and UGT enzymes is not clear. A great deal of effort is needed to avoid in new drug of active ingredient research in avoiding the development of the active composition of natural medicine that will cause drug–drug interactions in clinical application.26–28 On the other hand, co-administration of herbal medicines is now a common therapeutic practice in patients with multiple complications and interactions are not avoidable.27 Thus, adequate preclinical studies are necessary for further clinical treatment.
In this study, we first characterized the phase I metabolism and glucuronidation by human tissue microsomes. Reaction phenotyping assay was used to identify the main contributors of CYP and UGT isozymes. Furthermore, HeLa1A1 cells was applied to evaluate the roles of BCRP and MRPs in excretion of bavachinin-O-glucuronide using gene silencing approach. Moreover, the inhibitory effect of bavachinin against several important CYP and UGT enzymes was investigated. This study aimed to understand the roles of CYP and UGT enzymes, BCRP and MRPs transporters in metabolism determinants and DDI of bavachinin.
Experimental
Chemicals and reagents
Bavachinin (purity over 98.0%) was purchased from Shanghai Winherb Medical Technology Co., Ltd (Shanghai, China). Magnesium chloride (MgCl2), alamethicin, D-saccharic-1, 4-lactone monohydrate, uridine 5′-diphosphoglucuronic acid (UDPGA) and nicotinamide adenine dinucleotide phosphate (NADPH) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). Bupropion, β-estradiol, chlorzoxazone, hydroxy-bupropion, 4-hydroxymephenytoin, 4-hydroxytolbutamide, mephenytoin, 6α-hydroxy-paclitaxel, 6-hydroxychlorzoxazone, nifedipine, oxidized nifedipine, paclitaxel, paracetamol, phenacetin, propofol, tolbutamide, and zidovudine were obtained from Aladdin Chemicals (Shanghai, China). β-Estradiol-3-O-glucuronide, propofol-O-glucuronide and zidovudine-N-glucuronide were purchased from Toronto Research Chemicals (North York, ON, Canada).Pooled human liver microsomes (HLM, 50 donors), pooled human intestine microsomes (HIM, 20 donors), rats' liver microsomes (RLM), mice liver microsomes (MLM), dog's liver microsomes (DLM), mini-pig liver microsomes (MpLM), human CYP isozymes (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5), and human UGT isoforms (UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 2B4, 2B7, 2B10, 2B15, 2B17) were all provided from Corning Biosciences (Corning, NY, USA). All other chemicals and reagents were used as received.
Incubation systems for phase I metabolism and glucuronidation
Phase I metabolism and glucuronidation are essentially incubation assays performed routinely in our laboratory.11,25,30 The incubation system for phase I metabolism included contained Tris–HCl buffer (50 mM, pH = 7.4), MgCl2 (5 mM), substrates (bavachinin, or specific substrate for each CYP enzyme), enzyme materials (HLM, HIM, animal liver microsomes, or CYP enzymes). The reaction was initiated by addition of NADPH (1 mM) after a pre-incubation at 37 °C for 5 min. All incubations were maintained at 37 °C. At the end of incubation, the reaction was terminated by ice-cold acetonitrile (100 μL). The supernatant was obtained by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g for 10 min. And 8 μL aliquots of the supernatant were injected into ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry (UHPLC/Q-TOF-MS) system (Waters, Manchester, UK) for metabolite identification or into UHPLC for metabolite quantification. The incubation mixtures without NADPH served was considered as control group to confirm that the metabolites were NADPH-dependent.
800g for 10 min. And 8 μL aliquots of the supernatant were injected into ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry (UHPLC/Q-TOF-MS) system (Waters, Manchester, UK) for metabolite identification or into UHPLC for metabolite quantification. The incubation mixtures without NADPH served was considered as control group to confirm that the metabolites were NADPH-dependent.
For glucuronidation assays, typical incubation mixture (100 μL) briefly contained Tris–HCl buffer (50 mM, pH = 7.4), solution B (including 125 μg mL−1 alamethicin, 25 mM D-saccharic-1,4-lactone monohydrate, and 5 mM MgCl2), substrates (bavachinin or specific substrates for UGT isozymes), enzyme materials (HLM, HIM, animal liver microsomes, or UGT enzymes), After preincubation at 37 °C for 5 min, the reaction was started by addition of solution A (12.5 mM UDPGA), and finally was terminated by adding 100 μL of cold acetonitrile. Then the incubation mixtures were vortexed and centrifuged at 13800g for 10 min to collect the supernatant for UHPLC analysis. Similarly, the incubation system without UDPGA served was considered as control group to confirm that the metabolites were UDPGA-dependent.
Analytical conditions
Chromatographic separation was achieved on a BEH C18 column (2.1 mm × 50 mm, 1.7 mm, Waters, Ireland) using an Acquity UHPLC I-Class system (Waters Corporation, Manchester, UK). The mobile phases of water (A) and acetonitrile (B) both contained 0.1% formic acid. The gradient elution program was as follows. 0–1.0 min, 10% B; 1.0–8.0 min, 10–100% B; 8.0–10.0 min, 100 to 10% B. The flow rate was set as 0.5 mL min−1 and column temperature was set to 35 °C. The ultraviolet (UV) detection wavelengths were 254, 270, 315, and 335 nm. And the injection volume was 4 μL.UHPLC system was equipped with a Q-TOF tandem mass spectrometer (SYNAPT G2, Waters Corporation, Manchester, UK). The sample analysis was performed in positive ionization mode. The operating parameters were optimized as follows: capillary voltage, 3 kV; sample cone voltage, 35 V; extraction cone voltage, 4 V; source temperature, 100 °C; desolvation temperature, 300 °C; cone gas flow, 50 L h−1 and desolvation gas flow, 800 L h−1. In MSE mode, the trap collision energy for the low-energy function was set at 5 eV, while the ramp trap collision energy for the high-energy function was set at 30–50 eV. Argon was used as the collision gas for collision-induced dissociation (CID) in MSE mode. The full scan mass range was 50–1500 Da. The method used lock spray with leucine enkephalin (LE) (m/z 556.2771 in positive ion mode) to ensure the mass accuracy. All data were collected in centroid mode and processed using Masslynx 4.1 software (Waters Corporation, Manchester, UK).
Enzyme kinetic evaluation
The metabolic rates for phase I metabolism (or glucuronidation) were determined at a series of bavachinin concentrations based on the previous protocol.11,25,30 Preliminary experiments were performed to ensure that the metabolic rates were determined under a linear condition with respect to optimized incubation time and protein concentration. Kinetic model selection was based on the visual inspection of the Eadie–Hofstee plot. Michaelis–Menten equation (MM, eqn (1)), substrate inhibition equation (SI, eqn (2)) and Hill equation (eqn (3)) were fitted to the data of metabolic activities versus concentrations, respectively. Model fitting and parameter calculation were performed by Graphpad Prism V5 software (SanDiego, CA).
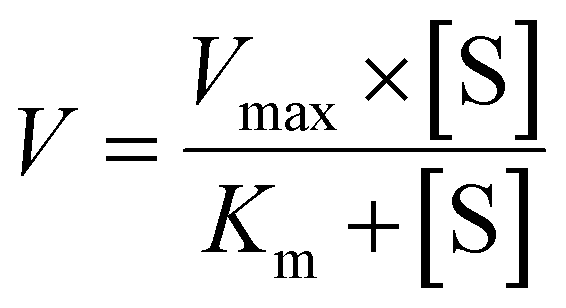 | (1) |
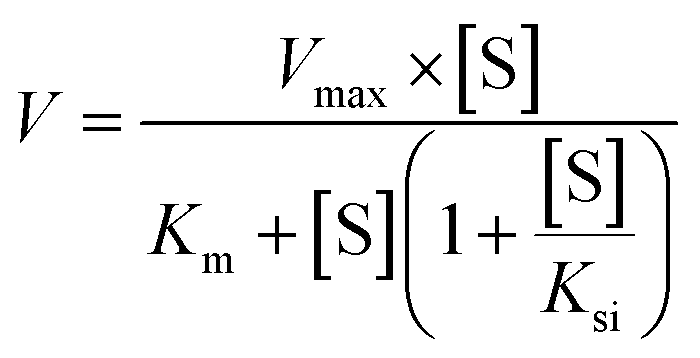 | (2) |
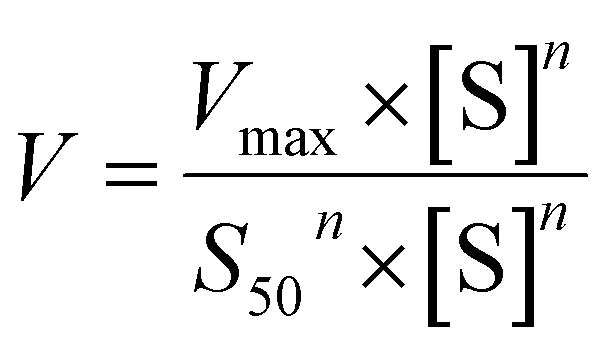 | (3) |
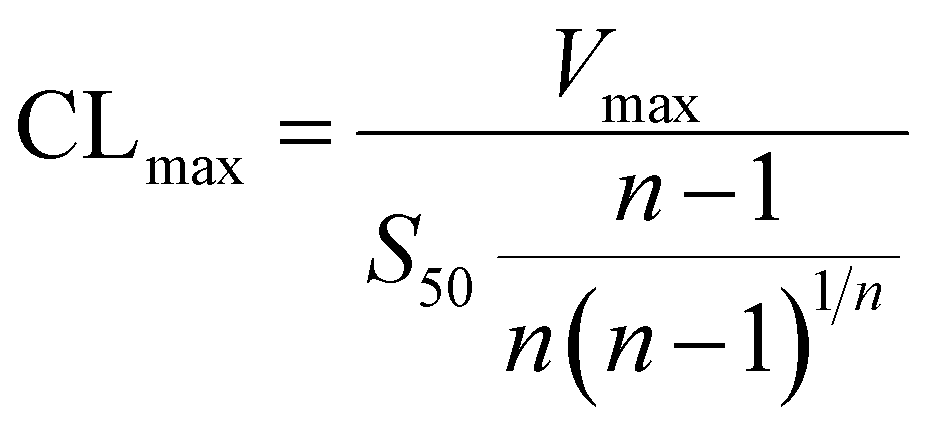 | (4) |
Species differences
The metabolic rates for phase I metabolism (or glucuronidation) were determined after serial of bavachinin solutions were incubated with NADPH-supplemented (UDPGA-supplemented) RLM, MLM, DLM and MpLM, respectively. Model fitting and parameter calculation were based on the protocol described in “Enzyme kinetic evaluation” above. The CLint values of bavachinin by HLM and four animal liver microsomes were used to estimate the species diversity.Excretion experiment
UGT1A1-overexpressing HeLa cell lines (also called HeLa1A1 cells) and shRNA fragments for BCRP, MRP1, MRP3, MRP4 transporters have been established and characterized in our laboratory.21 HeLa1A1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Firstly, HeLa1A1 cells were seeded in a 6-well plate at the density of 0.5 × 105 cells per well. After culture for 12 h, the plasmid construct containing scramble (or individual shRNA-transporter) (4 μg) was transfected into the cells by means of polyfectine according to the protocol (BioWit Technologies, Shenzhen). About 48 h later, the transfected HeLa1A1 cells were ready for excretion experiment.The transfected HeLa1A1 cells were washed twice with preheating HBSS (37 °C, pH = 7.4). Then, the cells were incubated at cell incubator (37 °C and 5% CO2) with HBSS (2 mL) containing bavachinin. At 0.5, 1.0, 1.5, and 2.0 h, extracellular fluid (200 μL) was sampled, and equal volume of dosing bavachinin solution was immediately added for replacement. Each sample was mixed with ice-cold acetonitrile (100 μL) followed by centrifugation at 13800g for 10 min. The supernatant was injected into UHPLC to determine the concentrations of bavachinin-O-glucuronide. At 2 h, the HeLa1A1 cells were collected and sonicated in 50% ice-cold methanol for 10 min. After centrifugation at 13800g and 4 °C for 10 min, the supernatant was analyzed by UHPLC to obtain intracellular amount of bavachinin-O-glucuronide. The total protein concentration in cell lysate was determined using Bio-Rad protein assay kit with bovine serum albumin as a standard. The excretion rates (ER), apparent clearance (CLapp) of bavachinin-O-glucuronide and the metabolized fraction of bavachinin (fmet) were estimated by eqn (5)–(7), respectively.21,24
 | (5) |
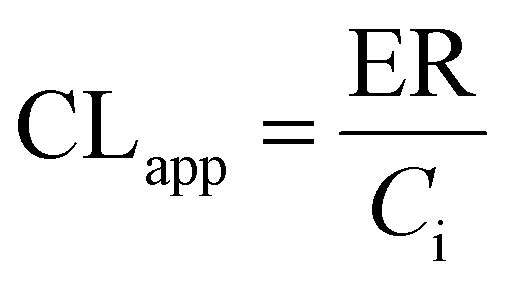 | (6) |
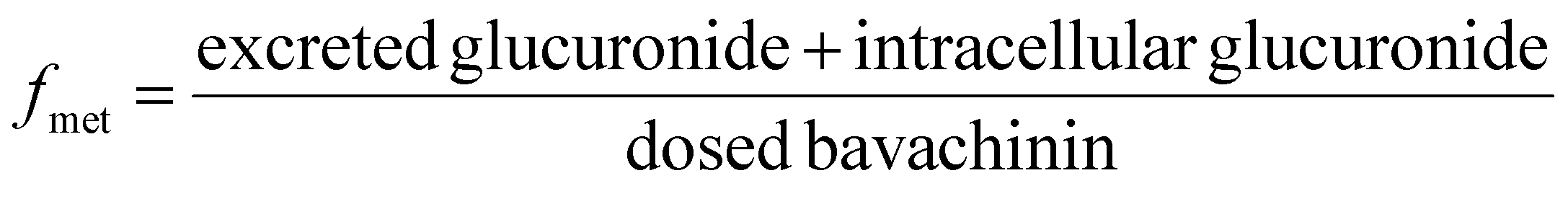 | (7) |
Inhibitory effects of bavachinin against CYP and UGT isozymes
As published previously, phenacetin (100 μM), bupropion (100 μM), paclitaxel (60 μM), tolbutamide (200 μM), mephenytoin (100 μM), chlorzoxazone (200 μM) and nifedipine (40 μM) have been well-accepted as the specific substrates for CYP1A2, 2B6, 2C8, 2C9, 2C19, 2E1, and 3A4, respectively.27,30 Similarly, β-estradiol (60 μM), propofol (40 μM) and zidovudine (500 μM) were typically used as the selective probe substrates for UGT1A1, 1A9, and 2B7, respectively.31,32 The substrates above were separately incubated with CYP isozyme (or UGT enzymes) in the absence (control) and presence of bavachinin (1, 10, 100 μM) at optimized incubation time and protein concentration as described previously.27,30–32The half-inhibition concentration (IC50) values of bavachinin towards individual CYP and UGT isozyme were obtained using the non-linear regression analysis. Traditionally, the inhibitory effects could be divided into four categories based on the IC50 values as follows: potent (less than 1 μM), moderate (between 1 and 10 μM), weak (over 10 μM), or no inhibition (over 100 μM).27,30 In this study, the inhibition kinetic of bavachinin against each CYP or UGT enzyme was further evaluated only when the IC50 values were less than 10 μM.
Inhibition kinetic evaluation
Competitive inhibition (eqn (8)), noncompetitive inhibition (eqn (9)), and mixed-type inhibition (eqn (10)) were used to estimate the Ki values by nonlinear regression analysis, respectively. And, the inhibition kinetic models with the smallest Akaike information criterion (AIC) and Schwartz information criterion (SC) values were considered as the appropriate model fitting.27,30,32
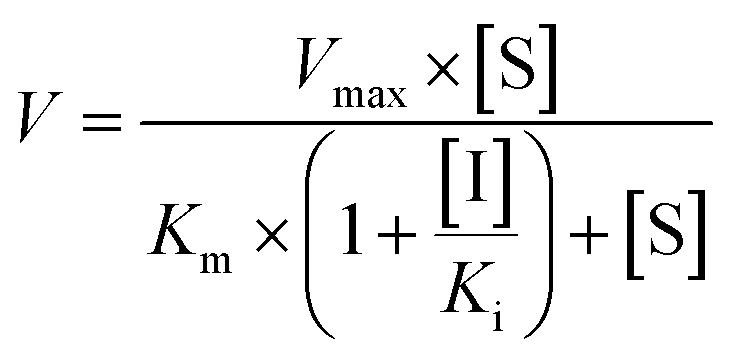 | (8) |
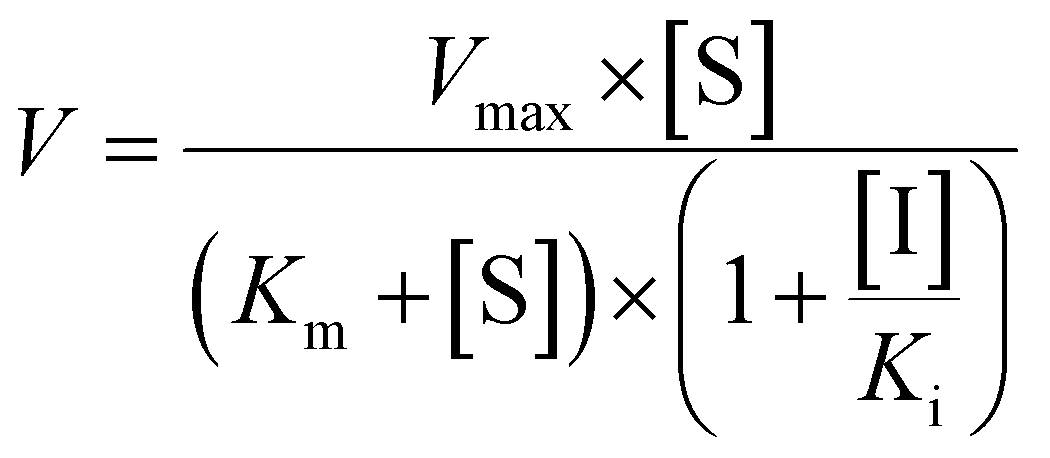 | (9) |
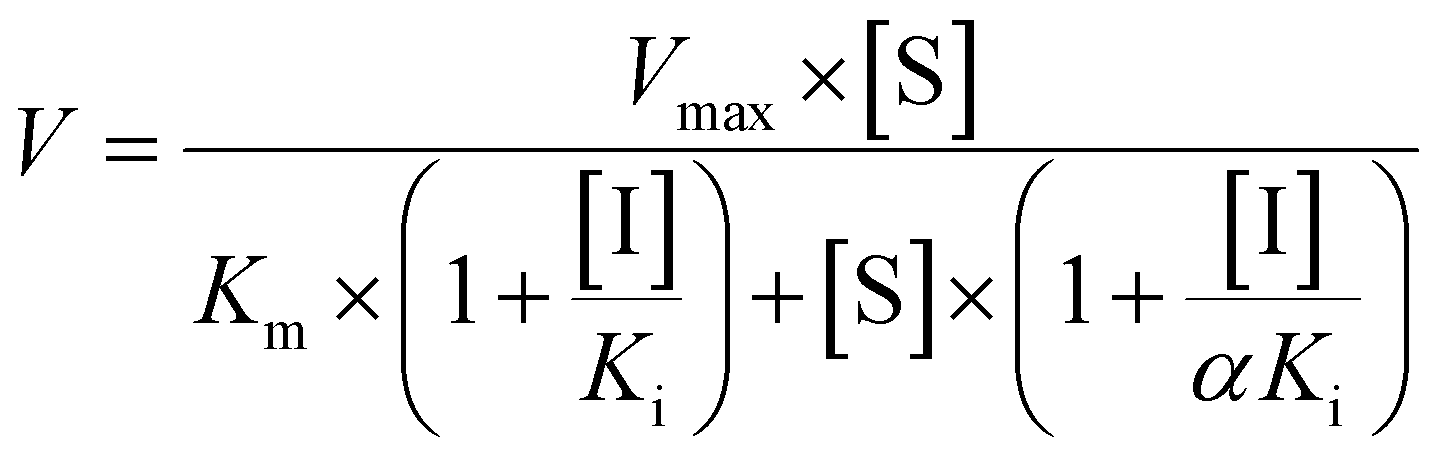 | (10) |
Statistical analysis
All experiments were performed in triplicate (n = 3). The differences among treatment and control groups were analyzed by Kruskal–Wallis test. The levels of difference were set at p < 0.05 (*) and p < 0.01 (**).Results
Characterization of bavachinin and related metabolites
Incubation of bavachinin (10 μM, P0, tR = 6.71 min, Fig. S1a†) with NADPH-supplemented HLM (or HIM) generated two mono-oxidated metabolite (M1, tR = 5.75 min; M2, tR = 6.09 min, Fig. S1b†) and one isomerized metabolite (M3, tR = 6.58 min, Fig. S1b†), while one glucuronide conjugate (G1, tR = 6.13 min, Fig. S1c†) was produced in UDPGA-supplemented HLM (or HIM) incubation system (Table S1†). P0 showed a [M + H]+ ion at m/z 339.161, following the fragment ions at m/z 283.096, 271.097, 219.103, and 147.049 (Fig. 1a). The ion at m/z 283.096 was generated by natural loss of a C4H8 group. The proposed fragmentation pathway of P0 was shown in Fig. 1b. M1 displayed the fragment ions at m/z 337.144, 235.107, 217.087, and 147.045 (Fig. 1c), suggesting that the oxidation unit was at the B ring of bavachinin. M2 was also characterized as mono-oxidated bavachinin with the oxidation position at the isopentenyl group due to the presence of m/z 283.096 (Fig. 1d). M3 was identified as isomerized bavachinin due to the fragment ions at m/z 287.088, 219.101, and 163.038 (Fig. 1e), which kept in line with previous in vivo and in vitro study.12,13 G1 (m/z 515.195) was identified as bavachinin-O-glucuronide due to the presence of only one phenolic hydroxyl group (Fig. 1f). | ||
| Fig. 1 The MSE spectra and proposed fragment pathway of bavachinin and its related metabolites. (a) The MSE spectra of bavachinin, P0; (b) the proposed fragment pathway of P0; (c) the MSE spectra of M1; (d) the MSE spectra of M2; (e) the MSE spectra of M3; (f) the MSE spectra of G1. | ||
Enzyme kinetics of bavachinin by HLM, HIM
The formation of M1 by HLM (Fig. S2a†), and the formation of M1 and M2 by HIM (Fig. S2d and e†) all followed the classical MM model because their corresponding Eadie–Hofstee plots were all straight lines. The kinetic profiles for M2 and M3 by HLM (Fig. S2b and c†), and M3 by HIM (Fig. S2f†) all showed SI equation. The maximal CLint values (representing the catalytic efficiency) of bavachinin by HLM and HIM were 89.4 and 25.8 μL min−1 mg−1, respectively (Table 1).| Enzyme | Metabolite | Vmax (pmol min−1 mg−1) | Km (S50) (μM) | Ki (μM) | n | CLint (CLmax) (μL min−1 mg−1) | Model |
|---|---|---|---|---|---|---|---|
| a N.A.: not available; SI: substrate inhibition model; MM: Michaelis–Menten model; Hill, Hill equation. | |||||||
| HLM | M1 | 544.0 ± 11.1 | 6.1 ± 0.4 | N.A. | N.A. | 89.4 ± 6.6 | MM |
| M2 | 138.1 ± 9.0 | 7.0 ± 0.8 | 79.2 ± 13.2 | N.A. | 19.9 ± 2.7 | SI | |
| M3 | 70.1 ± 10.5 | 12.1 ± 3.0 | 83.5 ± 29.7 | N.A. | 5.8 ± 1.7 | SI | |
| HIM | M1 | 161.6 ± 4.6 | 19.6 ± 1.5 | N.A. | N.A. | 8.2 ± 0.7 | MM |
| M2 | 92.8 ± 2.4 | 27.1 ± 1.7 | N.A. | N.A. | 3.4 ± 0.2 | MM | |
| M3 | 737.3 ± 110.9 | 28.6 ± 5.7 | 32.5 ± 7.4 | N.A. | 25.8 ± 6.5 | SI | |
| CYP1A1 | M1 | 836.9 ± 192.7 | 17.5 ± 4.9 | 6.8 ± 2.0 | N.A. | 48.0 ± 17.5 | SI |
| M2 | 17.9 ± 6.3 | 5.4 ± 3.5 | 37.9 ± 26.5 | N.A. | 3.3 ± 2.4 | SI | |
| M3 | 18.8 ± 2.6 | 0.4 ± 0.2 | 29.3 ± 12.6 | N.A. | 43.8 ± 19.7 | SI | |
| CYP1A2 | M1 | 122.0 ± 18.1 | 3.0 ± 0.8 | 15.9 ± 4.3 | N.A. | 41.1 ± 12.3 | SI |
| M3 | 13.3 ± 2.3 | 3.7 ± 1.2 | 32.0 ± 11.3 | N.A. | 3.6 ± 1.3 | SI | |
| CYP1B1 | M1 | 18.7 ± 0.3 | 0.6 ± 0.1 | N.A. | N.A. | 29.1 ± 2.1 | MM |
| M2 | 22.1 ± 1.7 | 11.8 ± 2.2 | N.A. | N.A. | 1.9 ± 0.4 | MM | |
| CYP2A6 | M1 | 14.8 ± 0.3 | 12.8 ± 0.6 | N.A. | N.A. | 1.2 ± 0.1 | MM |
| CYP2B6 | M3 | 34.7 ± 3.1 | 2.4 ± 0.4 | 27.1 ± 5.7 | N.A. | 14.5 ± 2.9 | SI |
| CYP2C8 | M1 | 218.1 ± 7.4 | 1.2 ± 0.2 | N.A. | N.A. | 182.4 ± 26.4 | MM |
| CYP2C19 | M1 | 143.3 ± 14.2 | 1.1 ± 0.3 | 34.6 ± 10.4 | N.A. | 133.2 ± 34.4 | SI |
| M3 | 65.6 ± 5.6 | 4.6 ± 0.7 | 32.4 ± 6.1 | N.A. | 14.4 ± 2.4 | SI | |
| CYP2D6 | M1 | 27.2 ± 0.8 | 2.6 ± 0.3 | N.A. | N.A. | 10.3 ± 1.1 | MM |
| CYP3A4 | M1 | 20.8 ± 0.7 | 4.1 ± 0.6 | N.A. | N.A. | 5.0 ± 0.7 | MM |
| M2 | 32.8 ± 4.6 | 3.8 ± 1.1 | 49.9 ± 16.6 | N.A. | 8.6 ± 2.7 | SI | |
| CYP3A5 | M1 | 20.3 ± 0.9 | 3.8 ± 0.7 | N.A. | N.A. | 5.3 ± 0.9 | MM |
| M2 | 13.4 ± 0.5 | 5.9 ± 0.7 | N.A. | N.A. | 2.3 ± 0.3 | MM | |
| M3 | 6.0 ± 0.1 | 3.2 ± 0.3 | N.A. | N.A. | 1.9 ± 0.1 | MM | |
| RLM | M1 | 28.3 ± 1.2 | 8.6 ± 1.2 | N.A. | N.A. | 3.3 ± 0.5 | MM |
| M2 | 172.3 ± 19.6 | 13.3 ± 2.4 | 56.8 ± 12.9 | N.A. | 13.0 ± 2.8 | SI | |
| M3 | 132.8 ± 8.8 | 17.8 ± 3.2 | N.A. | N.A. | 7.5 ± 1.4 | MM | |
| MLM | M1 | 1678.0 ± 294.7 | 20.2 ± 5.2 | 60.2 ± 20.3 | N.A. | 83.3 ± 26.2 | SI |
| M2 | 54.1 ± 2.9 | 13.5 ± 2.1 | N.A. | N.A. | 4.0 ± 0.7 | MM | |
| M3 | 291.3 ± 50.7 | 15.2 ± 4.2 | 81.2 ± 31.9 | N.A. | 19.1 ± 6.3 | SI | |
| DLM | M1 | 233.1 ± 4.4 | 8.6 ± 0.52 | N.A. | 1.7 ± 0.1 | 13.7 ± 1.5 | Hill |
| M2 | 140.3 ± 5.3 | 21.0 ± 2.1 | N.A. | N.A. | 6.7 ± 0.7 | MM | |
| M3 | 211.0 ± 37.4 | 18.6 ± 4.7 | 35.3 ± 10.3 | N.A. | 11.4 ± 3.5 | SI | |
| MpLM | M1 | 2640.0 ± 583.9 | 34.0 ± 9.8 | 31.2 ± 10.2 | N.A. | 77.6 ± 28.2 | SI |
| M2 | 53.7 ± 1.0 | 11.9 ± 0.7 | N.A. | N.A. | 4.5 ± 0.3 | MM | |
| M3 | 130.5 ± 3.9 | 5.2 ± 0.6 | N.A. | N.A. | 25.1 ± 2.8 | MM | |
Glucuronidation kinetic profiles of bavachinin with HLM also followed classical MM model (Fig. S3a†), whereas the formation of G1 by HIM followed SI kinetic (Fig. S3b†). Their corresponding CLint values were 270.2 and 247.1 μL min−1 mg−1, respectively (Table 2).
| Enzyme | Metabolite | Vmax (pmol min−1 mg−1) | Km (μM) | Ki (μM) | CLint (μL min−1 mg−1) | Model |
|---|---|---|---|---|---|---|
| a N.A.: not available; SI: substrate inhibition model; MM: Michaelis–Menten model. | ||||||
| HLM | G1 | 1576.0 ± 68.0 | 5.8 ± 0.8 | N.A. | 270.2 ± 38.4 | MM |
| HIM | G1 | 3086.0 ± 726.1 | 12.5 ± 4.2 | 20.6 ± 7.7 | 247.1 ± 101.2 | SI |
| UGT1A1 | G1 | 569.6 ± 18.0 | 3.0 ± 0.3 | 165.0 ± 23.4 | 191.2 ± 16.1 | SI |
| UGT1A3 | G1 | 14.4 ± 0.4 | 3.5 ± 0.4 | N.A. | 4.1 ± 0.5 | MM |
| UGT1A8 | G1 | 685.0 ± 21.8 | 6.3 ± 0.6 | N.A. | 109.0 ± 10.8 | MM |
| UGT2B7 | G1 | 119.1 ± 52.8 | 14.1 ± 8.2 | 10.4 ± 6.2 | 8.4 ± 6.2 | SI |
| RLM | G1 | 115.6 ± 2.5 | 7.7 ± 0.5 | N.A. | 15.0 ± 1.0 | MM |
| MLM | G1 | 9634.0 ± 388.4 | 15.6 ± 1.5 | N.A. | 617.6 ± 64.3 | MM |
| DLM | G1 | 4994.0 ± 213.8 | 22.7 ± 2.1 | N.A. | 220.4 ± 22.2 | MM |
| MpLM | G1 | 8305.0 ± 395.1 | 14.9 ± 1.8 | N.A. | 558.9 ± 69.6 | MM |
Enzyme kinetics of bavachinin by expressed CYP and UGT enzymes
To determine the contribution of expressed CYP and UGT enzymes, twelve CYP isozymes and twelve UGT isoforms were tested, respectively. Among them, CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C19, 2D6, 3A4, and 3A5 participated in phase I metabolism (Fig. S4a†), while UGT1A1, 1A3, 1A8 and 2B7 can catalyze glucuronidation of bavachinin (Fig. S4b†).The kinetic profiles of M1 by these active CYP enzymes were shown in Fig. S5.† Their activities followed the order of CYP2C8 (182.4 μL min−1 mg−1) > CYP2C19 (133.2 μL min−1 mg−1) > CYP1A1 (48.0 μL min−1 mg−1) > CYP1A2 (41.1 μL min−1 mg−1) > CYP1B1 (29.1 μL min−1 mg−1) > CYP2D6 (10.3 μL min−1 mg−1) > CYP3A5 (5.3 μL min−1 mg−1) > CYP3A4 (5.0 μL min−1 mg−1) > CYP2A6 (1.2 μL min−1 mg−1) (Table 1). The best kinetic models were selected according to the Eadie–Hofstee plots for M2 by active CYP enzymes (Fig. S6†). The CLint values for M2 by CYP1A1, 1B1, 3A4, and 3A5 were all less than 10.0 μL min−1 mg−1 (Table 1). Similarly, SI model was used to fit the data for M3 generated by these functional enzymes except CYP3A5 (Fig. S7†). CYP3A5 kinetics was best described by the MM kinetic model (Fig. S7e†). The CLint values for M2 by CYP1A1, 1A2, 2B6, 2C19, and 3A5 were 43.8, 3.6, 14.5, 14.4 and 1.9 μL min−1 mg−1, respectively (Table 1). The total CLint values for phase I metabolism of bavachinin were shown in Fig. 2a.
 | ||
| Fig. 2 The intrinsic clearance (CLint) values for metabolic activities of bavachinin by enzyme materials. (a) Comparison of CLint values by expressed CYP enzymes; (b) comparison of CLint values by expressed UGT isozymes; (c) comparison of CLint values for phase I metabolism by HLM, HIM and animal liver microsomes; (d) comparison of CLint values for glucuronidation by HLM, HIM and animal liver microsomes; the data were presented as mean ± SD (n = 3). (*p < 0.05, **p < 0.01). | ||
As shown in Fig. S8,† the formation of G1 by UGT1A1 and 2B7 both followed SI model, and G1 by UGT1A3 and 1A8 followed the classical MM kinetic equation. Of note, UGT1A1 and 1A8 were more efficient in bavachinin glucuronidation with CLint values of 191.2 and 109.0 μL min−1 mg−1, respectively (Table 2). In contrast, UGT1A3 and 2B7 participated less in the glucuronidation with the CLint values less than 10.0 μL min−1 mg−1 (Table 2). The comparison of CLint values for G1 was displayed in Fig. 2b.
Species differences
The kinetic profiles of M1–M3 by NADPH-supplemented RLM (Fig. S9a–c†), MLM (Fig. S9d–f†), DLM (Fig. S10a–c†) and MpLM (Fig. S10d–f†) were separately characterized. And the apparent kinetic parameters including Km, Vmax and CLint values were estimated (Table 1). As shown in Fig. 2c, the CLint values by MLM and MpLM were closer to that by HLM, suggesting that mice and mini-pig were appropriate animals to investigate phase I metabolism of bavachinin in vivo.Glucuronidation kinetic profiles of bavachinin by RLM (Fig. S11a†), MLM (Fig. S11b†), DLM (Fig. S11c†), and MpLM (Fig. S11d†) all followed the classical Michaelis–Menten model. The CLint values for G1 followed the order of MLM (617.6 μL min−1 mg−1) > MpLM (558.9 μL min−1 mg−1) > DLM (220.4 μL min−1 mg−1) > RLM (15.0 μL min−1 mg−1), respectively (Table 2). Their CLint values showed significant species differences. And dogs were appropriate animals to explore the glucuronidation of bavachinin in vivo (Fig. 2d).
Effects of gene silencing of efflux transporters on excretion of G1
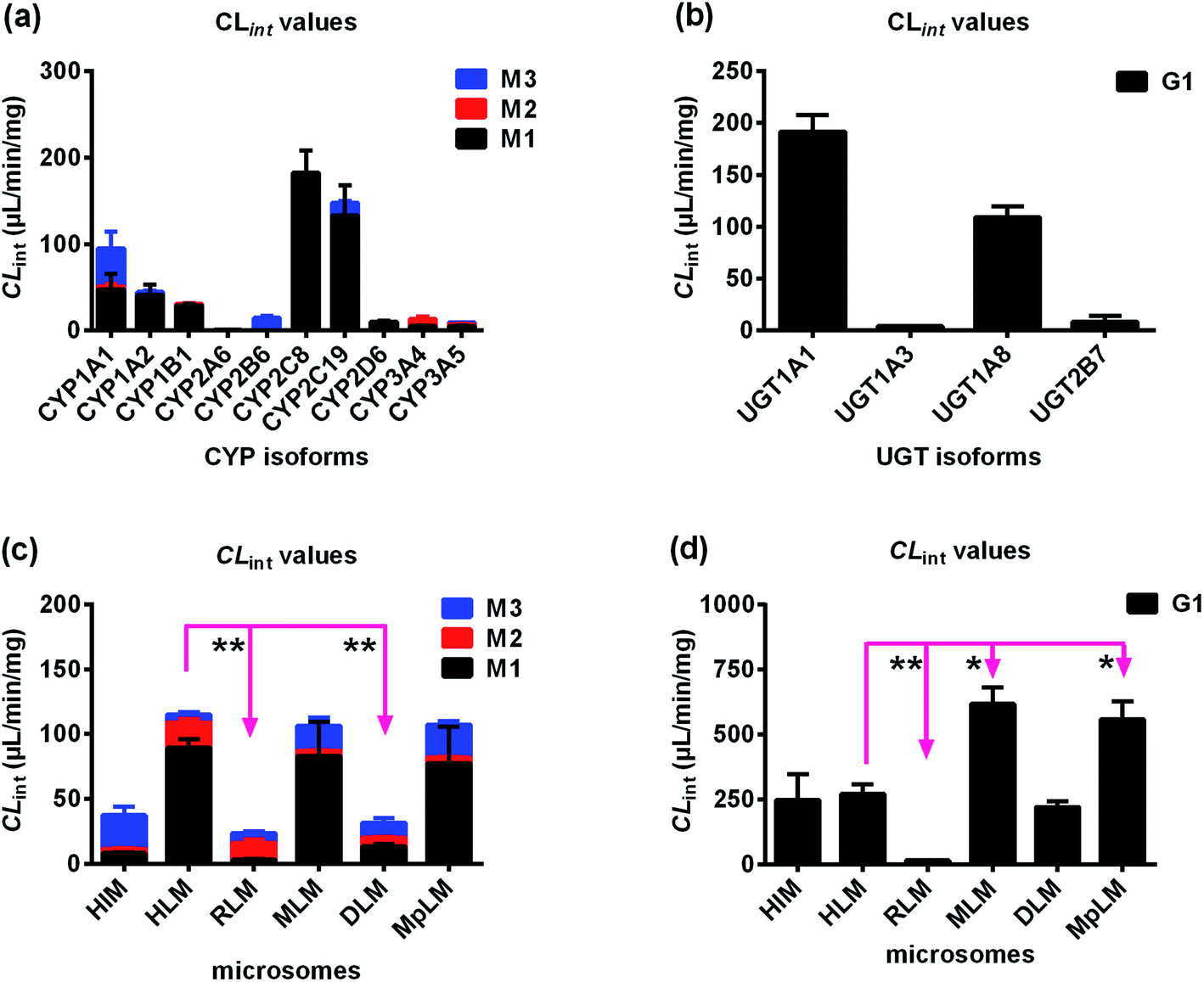
After treated bavachinin with Hela1A1 cells, the glucuronide conjugate G1 could be detected. BCRP transporters silencing could lead to 30.1–58.8% reductions in cellular excretion of G1 (Fig. 3a, p < 0.05), 31.0% decrease of the CLapp value (representing the efflux efficiency of glucuronide) (Fig. 3c, p < 0.05). Consistently, it also resulted in a significant reduction in the fmet value (Fig. 3e, p < 0.05). In contrast, partial knock-down of BCRP transporters did not alter the intracellular accumulation of G1 (Fig. 3d, p > 0.05). These data demonstrated that BCRP was an important contributor to excretion of G1.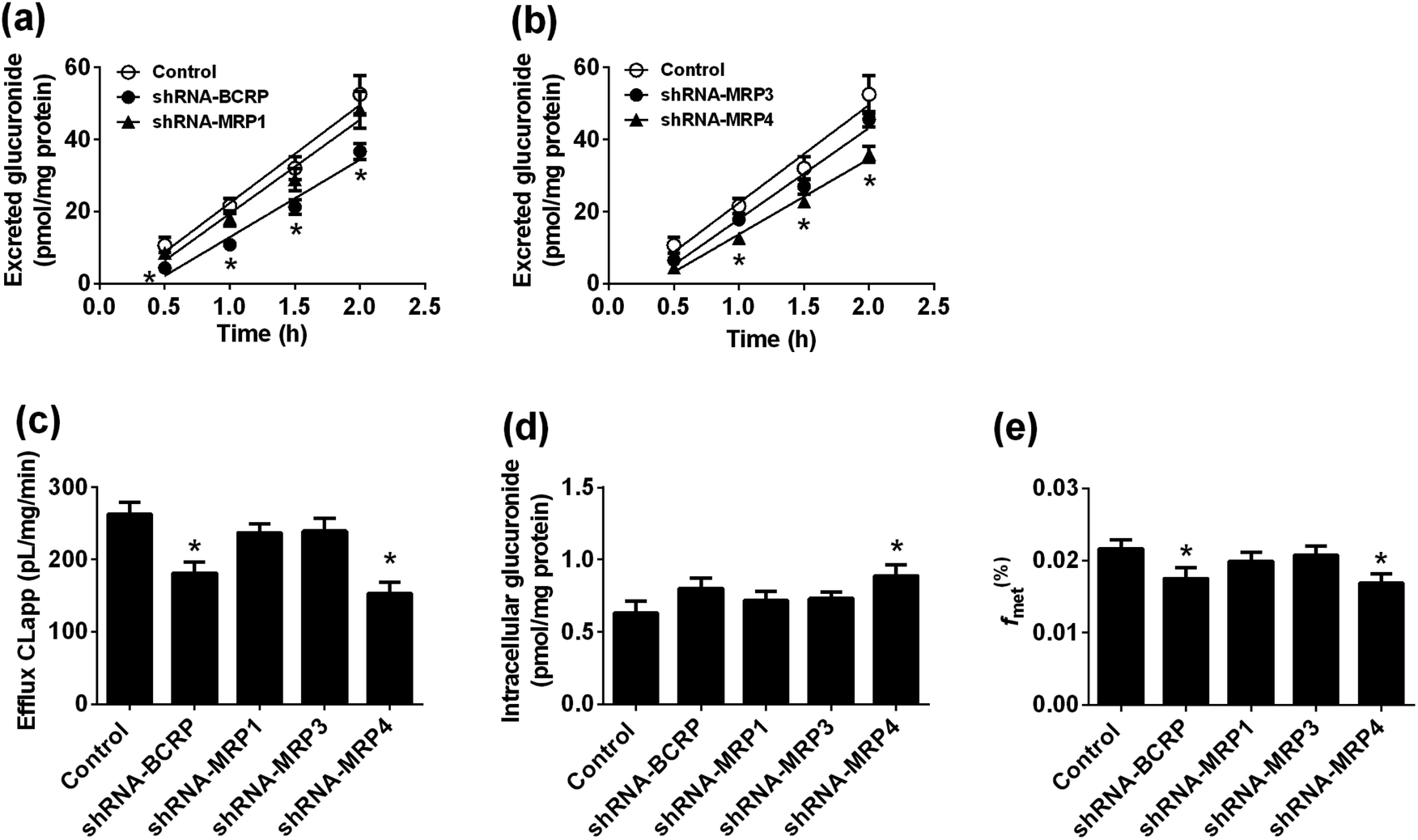 | ||
| Fig. 3 Effects of gene silencing in bavachinin-O-glucuronide (G1) excretion in HeLa1A1 cells. (a) Effects of BCRP and MRP1 transporters silencing in excretion of G1; (b) effects of MRP3 and MRP4 transporters knock-down in excretion of G1; (c) effects of efflux transporters silencing in excretion CLapp of G1; (d) effects of efflux transporters silencing in intracellular levels of G1; (e) effects of efflux transporters knock-down in fmet value of bavachinin; the data were presented as mean ± SD. All experiments were performed in triplicate (n = 3). (*p < 0.05). | ||
In addition, silencing of MRP1 transporter did not change cellular excretion of G1 (8.1–20.5%, Fig. 3a), CLapp value (9.6%, Fig. 3c), intracellular accumulation of G1 (13.8%, Fig. 3d), and fmet value (8.0%, Fig. 3e). Furthermore, similar results were observed when MRP3 transporters were partially knock-down (Fig. 3). When MRP4 transporter was silenced, the excretion of G1 (28.9–58.3%, Fig. 3b) and CLapp value (41.6%, Fig. 3c) both significantly decreased (p < 0.05). Meanwhile, it resulted in elevated intracellular accumulation of glucuronide (40.8%, Fig. 3d), and a significant reduction in the fmet value (21.9%, Fig. 3e). These results suggested that cellular metabolism of bavachinin significantly reduced due to MRP4 inhibition. These findings also meant MRP4 participated more in the excretion of G1 than MRP1 and MRP3.
Inhibitory effects of bavachinin towards CYP and UGT enzymes
As shown in Fig. 4, when treated with bavachinin (100 μM), the residual activities of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2E1, 3A4, and UGT1A1, 1A9, 2B7 were 91.5%, 0, 21.5%, 0, 0, 51.5%, 24.7%, 23.8%, 3.0%, and 0 of negative control, respectively. Furthermore, concentration-dependent inhibitory curves of bavachinin towards CYP2B6 (Fig. S12a†), CYP2C9 (Fig. S12b†), CYP2C19 (Fig. S12c†), UGT1A1 (Fig. S12d†), UGT1A9 (Fig. S12e†), UGT2B7 (Fig. S12f†) were depicted, respectively. In addition, the inhibitory data were fitted to log(bavachinin) and normalized response equations to estimate the respective IC50 values. The inhibitory effects of bavachinin against these six CYP and UGT enzymes followed a dose-dependent manner (Fig. S13†). Their IC50 values were 1.68, 0.28, 1.38, 20.66, 2.34 and 2.53 μM for CYP2B6, 2C9, 2C19 and UGT1A1, 1A9, 2B7, respectively (Table 3).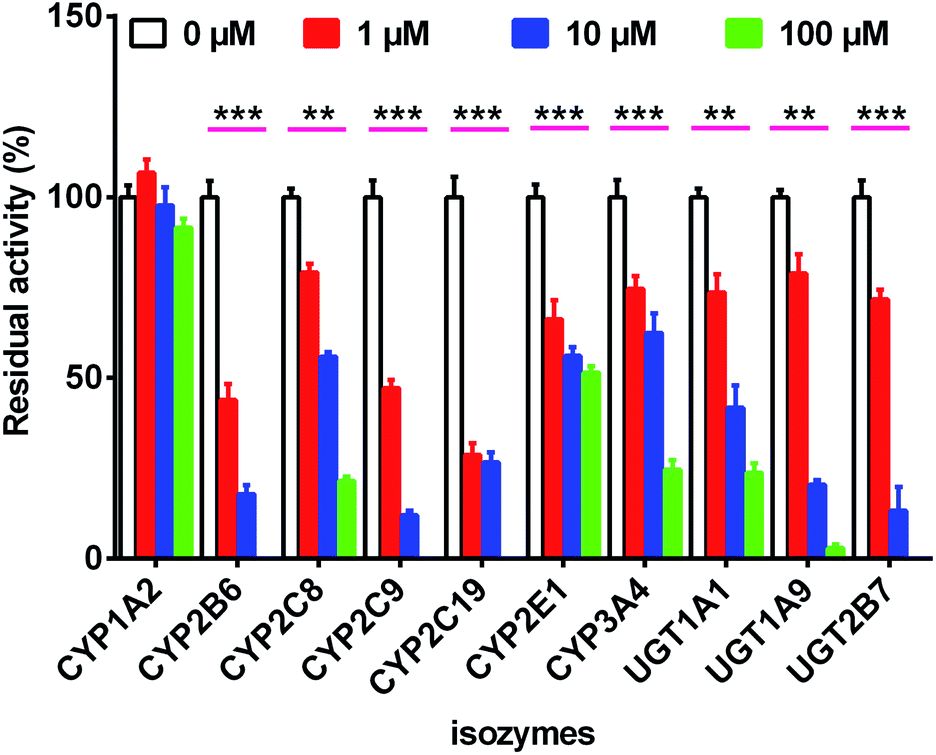 | ||
| Fig. 4 The inhibitory effects of bavachinin towards several expressed CYP and UGT isozymes. The selective substrates were incubated at 37 °C in the absence (control) and presence of bavachinin (1, 10 and 100 μM). The data were presented as mean ± SD (n = 3). (**p < 0.01, ***p < 0.001). | ||
| Isozymes | Substrate | IC50 (μM) | Type of inhibition | Ki (μM) | α | R2 | AIC | SC | Selection |
|---|---|---|---|---|---|---|---|---|---|
| CYP2B6 | Bupropion | 1.68 ± 0.63 | Competitive | 0.42 ± 0.09 | — | 0.9456 | 19.53 | 22.52 | |
| Non-competitive | 1.51 ± 0.12 | — | 0.9790 | 0.55 | 3.53 | ✓ | |||
| Mixed-type | 1.28 ± 0.44 | 1.30 ± 0.72 | 0.9792 | 2.26 | 6.24 | ||||
| CYP2C9 | Tolbutamide | 0.28 ± 0.07 | Competitive | 0.19 ± 0.03 | — | 0.9728 | −117.07 | −114.08 | ✓ |
| Non-competitive | 0.42 ± 0.05 | — | 0.9709 | −115.69 | −112.70 | ||||
| Mixed-type | 0.25 ± 0.08 | 4.48 ± 5.98 | 0.9746 | −116.44 | −112.46 | ||||
| CYP2C19 | Mephenytoin | 1.38 ± 0.53 | Competitive | 0.89 ± 0.14 | — | 0.9703 | −116.62 | −113.63 | |
| Non-competitive | 1.86 ± 0.16 | — | 0.9816 | −126.24 | −123.25 | ✓ | |||
| Mixed-type | 1.90 ± 0.66 | 0.95 ± 0.68 | 0.9816 | −124.24 | −120.26 | ||||
| UGT1A1 | Estradiol | 20.66 ± 10.63 | — | — | — | — | — | — | — |
| UGT1A9 | Propofol | 2.34 ± 0.61 | Competitive | 0.37 ± 0.04 | — | 0.9891 | 97.16 | 100.14 | ✓ |
| Non-competitive | 0.81 ± 0.06 | — | 0.9861 | 102.04 | 105.02 | ||||
| Mixed-type | 0.47 ± 0.09 | 5.33 ± 4.78 | 0.9902 | 96.93 | 100.91 | ||||
| UGT2B7 | Zidovudine | 2.53 ± 0.62 | Competitive | 0.47 ± 0.11 | — | 0.9368 | 227.79 | 230.78 | |
| Non-competitive | 1.13 ± 0.12 | — | 0.9683 | 214.01 | 217.00 | ✓ | |||
| Mixed-type | 1.91 ± 1.01 | 0.41 ± 0.32 | 0.9708 | 214.34 | 218.32 |
Inhibition kinetics of bavachinin against CYP and UGT isozymes
The inhibitory kinetic data were fitted by competitive (or noncompetitive, mixed-type) inhibition models to calculate their respective AIC and SC values. The best models were described in Table 3 according to the smallest AIC and SC values. Bavachinin exhibited competitive inhibition against CYP2C9 and UGT1A9, while it displayed noncompetitive inhibitory effects towards CYP2B6, CYP2C19 and UGT2B7 (Table 3). The corresponding Dixon plots also provided strong evidences to support these judgments (Fig. 5). Their Ki values ranged from 0.28 to 2.53 μM (Table 3). These findings indicated that bavachinin was a potent non-selective inhibitor against these five CYP and UGT enzymes.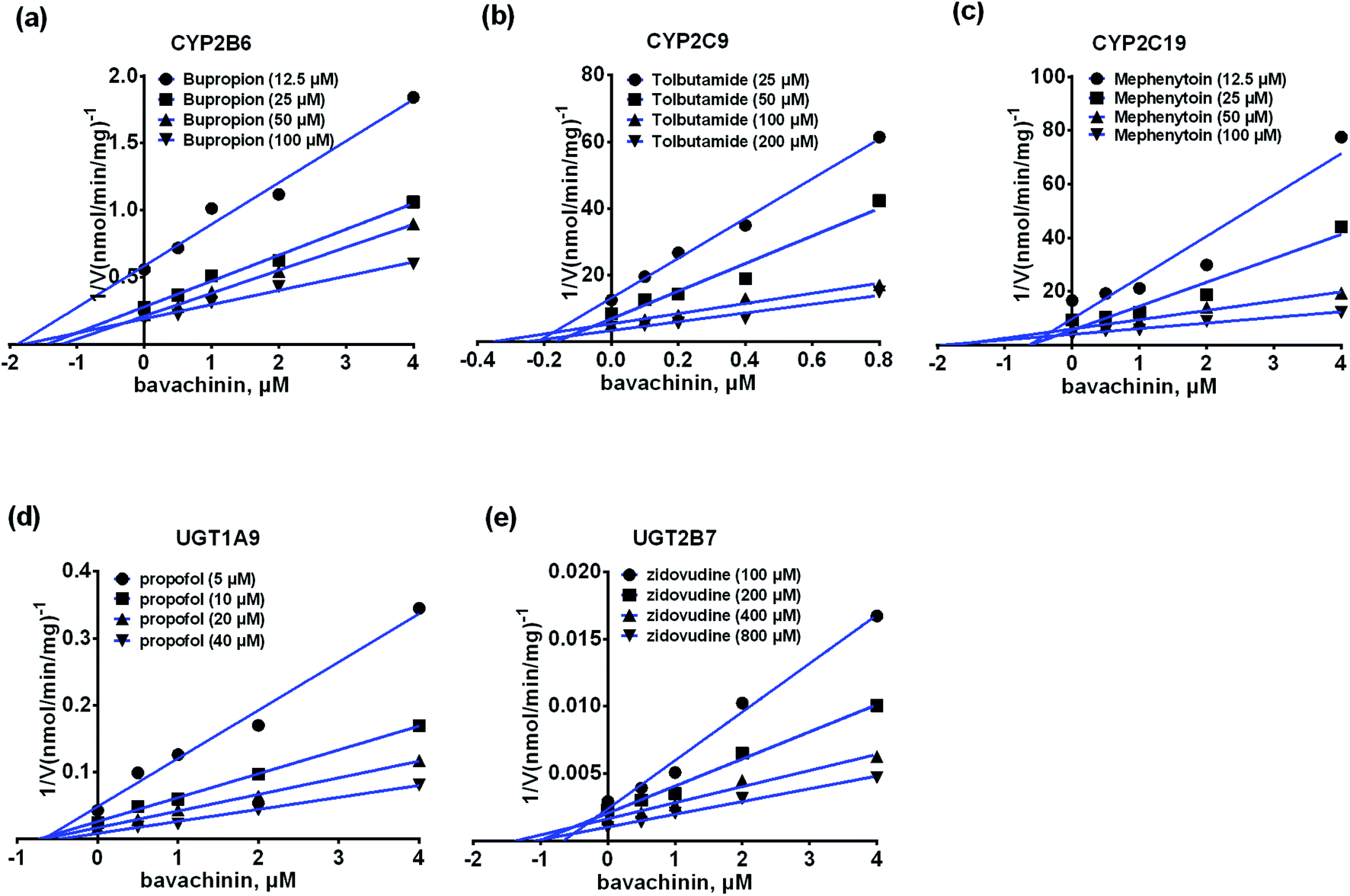 | ||
| Fig. 5 The Dixon plots for the inhibitory effects of bavachinin towards each CYP and UGT enzyme. (a) The Dixon plots against bupropion-hydroxylation; (b) the Dixon plots towards tolbutamide-4-hydroxylation; (c) the Dixon plots against mephenytoin-4-hydroxylation; (d) the Dixon plots towards propofol-O-glucuronidation; (e) the Dixon plots against zidovudine-N-glucuronidation; bupropion, tolbutamide, mephenytoin, propofol and zidovudine were used as the probe substrates of CYP2B6, 2C9, 2C19, and UGT1A9, 2B7 enzymes, respectively. All experiments were performed in triplicate determinations (n = 3). | ||
Discussion
Nature has been a source of medicinal products for thousands of years, and many useful drugs have been developed from plants. And the combination of natural products will play a positive role in the treatment of the disease, for example diminution of liver steatosis.33 In this study, CYP450 systems are the main pathways that induce reactive oxygen species and toxic metabolites leading to oxidative stress.34 For example, CYP2E1 participates in alcohol-mediated oxidative stress leading to cancers or liver injury.34 Similarly, CYP2C9 is involved in hydroxylation of many natural products.35 Traditionally, hepatic steatosis and mitochondrial function are closely related to oxidative stress.36,37 Therefore, if the pharmacokinetic properties of the active ingredients of natural drugs are fully understood before combine use, the therapeutic effect can be increased.Bavachinin is a bioactive natural compound isolated from Psoralea corylifolia.1,4,7–9 However, poor bioavailability (5.27–36.39%)13,14 limited its pharmacological effects. Also, active ingredients of herbal induction or inhibition of one or more drug-metabolizing enzymes including UGTs and CYPs can lead to changes in pharmacokinetics or toxicity of co-administered drug.38 In this study, we demonstrated that bavachinin could undergo massive phase I metabolism and glucuronidation by HLM and HIM (Tables 1 and 2). And glucuronidation activity was much more efficient than phase I metabolism of bavachinin (Fig. 2). In addition, CYP1A1, 1A2, 2C8, 2C19, and UGT1A1, 1A8 were identified as the main isozymes responsible for the metabolism of bavachinin (Fig. 2). The common pharmacokinetic behaviors of flavonoids with one or more hydroxyl group were poor oral bioavailability and low in vivo exposure because of extensive metabolism in liver and intestine.38,39 Accordingly, its metabolic characteristics were summarized (Fig. 6). Obviously, its metabolism was similar to the metabolism of its prenylflavonoid analogues derived from Psoralea corylifolia.11,25,30,40,41
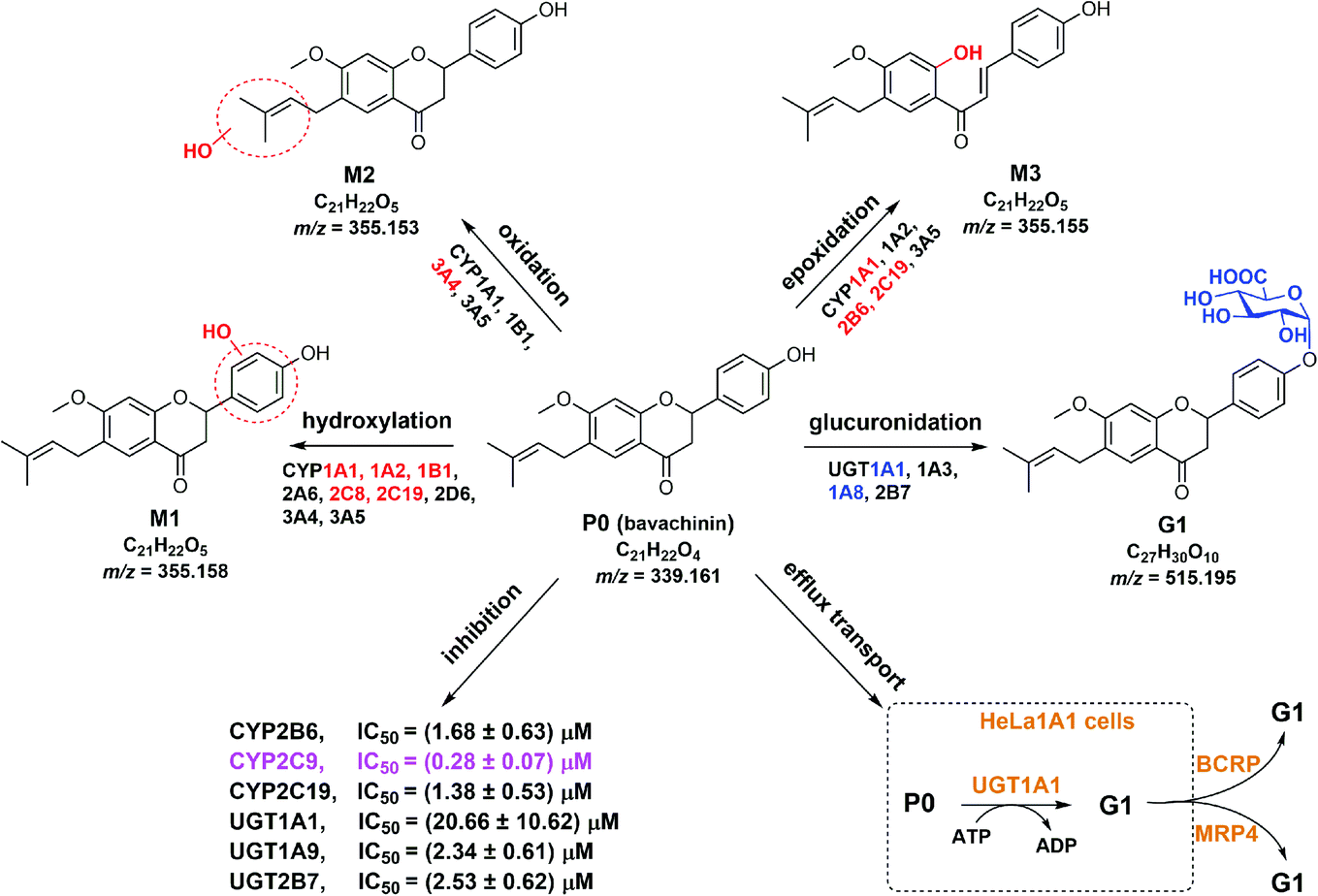 | ||
| Fig. 6 Metabolic fates, isozyme contribution, potential DDI of bavachinin, and BCRP and MRP4 as the main contributors for bavachinin-O-glucuronide excretion in HeLa1A1 cells. | ||
The metabolism of bavachinin was much more efficient in HLM than that in HIM (Fig. 2), indicating a more important role of the liver versus the intestine. This is supported by the fact that the liver-specific enzymes CYP1A2, 2C8, 2C19 and UGT1A1 are also much more active in bavachinin metabolism (Tables 1 and 2). In addition, the intestine-specific enzymes CYP1A1, UGT1A8 and 1A10 have moderate abilities in catalyzing bavachinin metabolism (Fig. 2), which agreed with previous studies.19,20 Considering that intestine is the first organ that the drugs (or natural compounds) encounter after oral uptake, the role of intestine in in vivo metabolism of bavachinin cannot be neglected.
Additionally, our findings suggested BCRP and MRP4 transporters as the important determinants in excretion of bavachinin-O-glucuronide (Fig. 3). This was consistent with our previous reports in which BCRP and MRP4 participated in excretion of conjugated glucuronides of several prenylated flavonoids (or flavanones).21,22,25,41 Another data showed that when the function of BCRP or MRP4 transporters was inhibited, significant down-regulation of cellular glucuronidation of bavachinin in HeLa1A1 cells was observed (Fig. 3e). This provided strong support to the interplay between UGT enzymes and efflux transporters as noted previously.21,42,43 This phenomenon was mainly attributed the function of β-glucuronidase. It could mediate the de-glucuronidation of intracellular glucuronides, and thereby facilitate the conversion of glucuronides back to parent compound and reduce the total cellular glucuronidation.42
As described previously, massive glucuronide (G1) was detected in rat bile.13,44 Traditionally, BCRP and MRP2 transporters participated more in transport of glucuronides from liver to bile.43 A remarkable limitation is the unexplained role of MRP2 in excretion of G1 due to the absence of MRP2 transporters in HeLa1A1 cells.21–23,25,41,43 Furthermore, MDCKII-MRP2-UGT1A1 cells could well characterize the glucuronidation by UGT1A1 and glucuronides excretion by MRP2 transporters.45 In addition, Mrp2 (Abcc2) knock-out mice were usually used to evaluate the roles of Mrp2 according to the pharmacokinetic behaviors.46
It was noteworthy that there were several bavachinin-O-sulfate in rat samples.13,44 And the present study did not explore the roles of bavachinin sulfation mediated by sulfotransferases (SULTs). This was because glucuronidation reaction of flavonoids was a predominant in vivo metabolic pathway, whereas the roles of sulfation were relatively minor.47 However, it remains unknown that which SULT enzyme catalyze the sulfation of bavachinin. Furthermore, sulfation by 3′-phosphoadenosine-5′-phosphosulfate (PAPS)-supplemented SULT enzyme could be used to determine the sulfation activity. In addition, Caco-2 TC-7 cells could be applied to evaluate the sulfation by SULT1A3 enzyme due to the most abundant expression of SULT1A3 in these cells.48 Besides, a developed HEK293 cell overexpressing SULT1A3 (or other SULT isozymes) model can be a simple and practical tool to investigate excretion of bavachinin-O-sulfate and sulfation-transport interplay.49,50 Further investigations are needed to answer these question.
Moreover, our results demonstrated that bavachinin exhibited potent inhibitory effects against CYP2B6, 2C9, 2C19 and UGT1A9, 2B7 isozymes (IC50 = 0.28–2.53 μM) (Table 3). Notably, the IC50 value of bavachinin towards UGT1A1 in this study was 20.66 μM, which was significantly different with previous study (IC50 = 1.27 μM).51 This may be because different substrates of UGT1A1 enzyme, such as N-(3-carboxy propyl)-4-hydroxy-1,8-naphthalimide, and β-estradiol, were used in these two independent assays.51 Besides, bavachinin displayed remarkable inhibitory effects against human carboxylesterase 1 and 2 with respective IC50 values of 0.5 and 4.31 μM.52–54 Considering that these active enzymes involved more in metabolism and clearance of clinical drugs,29,30 these significant inhibitions by bavachinin may trigger potential DDI when co-administrated with substrates or clinical drugs of these active enzymes.28,51
In clinics, bavachinin and bavachinin-containing herbal products are usually co-administrated with several anti-osteoporosis drugs, such as tibolone, raloxifene, alendronate, calcitonin and so on. For instance, Xian-Ling-Gu-Bao capsule is widely used for the prevention and treatment of osteoporosis by co-administration with calcium regulators in China.55 Based on our results, UGT1A1, 1A8 participated more in bavachinin metabolism. However, metabolites of raloxifene are also produced by UGT1A1, 1A8 and 1A10 in liver and intestines.56,57 Therefore, special attention needs to be paid to the possible interaction when co-administration.
Conclusions
In summary, bavachinin could undergo efficient phase I metabolism and glucuronidation by HLM and HIM. We further identified the individual CYP (CYP1A1, 1A2, 2C8 and 2C19) and UGT (UGT1A1 and 1A8) enzymes as the important contributors. In addition, the metabolism of bavachinin showed remarkable species differences. Besides, BCRP and MRP4 transporters was involved in excretion of bavachinin glucuronide. And partial inhibition of active transporters resulted in decreased cellular glucuronidation. Moreover, bavachinin was identified as a potent non-selective inhibitor against several CYP (CYP2B6, 2C9, and 2C19) and UGT (UGT1A9 and 2B7) enzymes. Taken together, our data revealed the potential metabolism determinants and DDI of bavachinin.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81903704 & 81703799), Guangdong Basic and Applied Basic Research Foundation (2019A1515011285), the State Key Laboratory of Drug Research (SIMM1903KF-07), and Foundation of He’nan Educational Committee (20A350012).References
- B. Chopra, A. K. Dhingra and K. L. Dhar, Fitoterapia, 2013, 90, 44–56 CrossRef CAS.
- K. Li, N. Zhou, X. K. Zheng, W. S. Feng, F. Li, Z. L. Zhang and Y. Q. Lu, Sci. Rep., 2019, 9, 661 CrossRef.
- N. Gupta, A. Qayum, A. Raina, R. Shankar, S. Gairola, S. Singh and P. L. Sangwan, Eur. J. Med. Chem., 2018, 145, 511–523 CrossRef CAS.
- D. W. Kim, K. H. Seo, M. J. Curtis-Long, K. Y. Oh, J. W. Oh, J. K. Cho, K. H. Lee and K. H. Park, J. Enzyme Inhib. Med. Chem., 2014, 29, 59–63 CrossRef CAS.
- Q. X. Xu, Y. Hu, G.-Y. Li, W. Xu, Y. T. Zhang and X. W. Yang, Molecules, 2018, 23, 614 CrossRef.
- X. Chen, Y. Yang and Y. Zhang, FEBS Lett., 2013, 587, 2930–2935 CrossRef CAS.
- L. Feng, H. Luo, Z. Xu, Z. Yang, G. Du, Y. Zhang, L. Yu, K. Hu, W. Zhu, Q. Tong, K. Chen, F. Guo, C. Huang and Y. Li, Diabetologia, 2016, 59, 1276–1286 CrossRef CAS.
- G. Du, L. Feng, Z. Yang, J. Shi, C. Huang, F. Guo, B. Li, W. Zhu and Y. Li, Bioorg. Med. Chem. Lett., 2015, 25, 2579–2583 CrossRef CAS.
- L. N. Ge, L. Yan, C. Li and K. Cheng, Mol. Med. Rep., 2019, 20, 2805–2811 CAS.
- L. Wang, X. Hong, Z. Yao, Y. Dai, G. Zhao, Z. Qin, B. Wu, F. J. Gonzalez and X. Yao, Xenobiotica, 2018, 48, 357–367 CrossRef CAS.
- Z. Qin, S. Li, Z. Yao, X. Hong, J. Xu, P. Lin, G. Zhao, F. J. Gonzalez and X. Yao, J. Pharm. Biomed. Anal., 2018, 155, 157–168 CrossRef CAS.
- F. Xie, G. Du, S. Ma, Y. Li, R. Wang and F. Guo, Xenobiotica, 2016, 46, 296–306 CrossRef CAS.
- J. Qian, F. Xie, Y. Shi, J. Li, L. Zhang, Y. Li, F. Guo and R. Wang, Biomed. Chromatogr., 2018, 32, e4293 CrossRef.
- L. Liu, K. N. Liu, Y. B. Wen, H. W. Zhang, Y. X. Lu and Z. Yin, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 893–894, 21–28 CAS.
- Z. X. Zhou, L. Yang, L. Y. Cheng, Y. L. Yu, L. Song, K. Zhou, Y. L. Wu and Y. Zhang, J. Sep. Sci., 2020, 43, 2804–2816 CrossRef CAS.
- Z. H. Yao, Z. F. Qin, L. L. He, X. L. Wang, Y. Dai, L. Qin, F. J. Gonzalez, W. C. Ye and X. S. Yao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1041–1042, 104–112 CrossRef CAS.
- Y. F. Yang, Y. B. Zhang, Z. J. Chen, Y. T. Zhang and X. W. Yang, Phytomedicine, 2018, 38, 166–174 CrossRef CAS.
- X. Y. Tang, Z. Q. Dai, Q. C. Wu, J. X. Zeng, M. X. Gao, H. H. Xiao, Z. H. Yao, Y. Dai and X. S. Yao, J. Pharm. Biomed. Anal., 2020, 177, 112836 CrossRef CAS.
- X. Lv, J. Hou, Y. L. Xia, J. Ning, G. Y. He, P. Wang, G. B. Ge, Z. L. Xiu and L. Yang, Drug Metab. Pharmacokinet., 2015, 30, 358–365 CrossRef CAS.
- J. Troberg, E. Järvinen, G.-B. Ge, L. Yang and M. Finel, Mol. Pharm., 2017, 14, 2875–2883 CrossRef CAS.
- Z. Qin, S. Li, Z. Yao, X. Hong, B. Wu, K. W. Krausz, F. J. Gonzalez, H. Gao and X. Yao, Food Funct., 2018, 9, 1410–1423 RSC.
- S. Li, J. Xu, Z. Yao, L. Hu, Z. Qin, H. Gao, K. W. Krausz, F. J. Gonzalez and X. Yao, Chem.-Biol. Interact., 2018, 296, 45–56 CrossRef CAS.
- J. Yang, B. Zhang, Z. Qin, S. Li, J. Xu, Z. Yao, X. Zhang, F. J. Gonzalez and X. Yao, Biofactors, 2018, 44, 558–569 CrossRef CAS.
- Z. Qin, B. Zhang, J. Yang, S. Li, J. Xu, Z. Yao, X. Zhang, F. J. Gonzalez and X. Yao, Front. Pharmacol., 2019, 10, 496 CrossRef CAS.
- Y. Li, C. Xu, J. Xu, Z. Qin, S. Li, L. Hu, Z. Yao, F. J. Gonzalez and X. Yao, J. Pharm. Pharmacol., 2020 DOI:10.1111/jphp.13324.
- X. Lv, Y. Xia, M. Finel, J. Wu, G. Ge and L. Yang, Acta Pharm. Sin. B, 2019, 9, 258–278 CrossRef.
- Z. Qin, M. Jia, J. Yang, H. Xing, Z. Yin, Z. Yao, X. Zhang and X. Yao, Chin. Med., 2020, 15, 69 CrossRef CAS.
- Q. H. Zhou, Y. D. Zhu, F. Zhang, Y. Q. Song, S. N. Jia, L. Zhu, S. Q. Fang and G. B. Ge, Chin. J. Nat. Med., 2019, 17, 858–870 Search PubMed.
- F. P. Guengerich, AAPS J., 2006, 8, E101–E111 CrossRef CAS.
- H. Xing, J. Yang, K. Ren, Z. Qin, P. Wang, X. Zhang, Z. Yao, F. J. Gonzalez and X. Yao, J. Pharm. Pharmacol., 2020 DOI:10.1111/jphp.13337.
- Z. Yao, S. Li, Z. Qin, X. Hong, Y. Dai, B. Wu, W. Ye, F. J. Gonzalez and X. Yao, RSC Adv., 2017, 7, 52661–52671 RSC.
- L. Gao, Z. Qin, B. Zhang, Z. Yin, X. Zhang and J. Yang, RSC Adv., 2020, 10, 9610–9622 RSC.
- R. Valenzuela and L. A. Videla, Nutrients, 2020, 12, 499 CrossRef CAS.
- N. Nieto, S. L. Friedman and I. Cederbaum, Hepatology, 2002, 35, 62–73 CrossRef CAS.
- A. M. Posadino, A. Cossu, R. Giordo, A. Zinellu, S. Sotgia, A. Vardeu, P. T. Hoa, L. Deiana, C. Carru and G. Pintus, Cardiovasc. Toxicol., 2013, 13, 301–306 CrossRef CAS.
- R. Valenzuela, P. Illesca, F. Echeverría, A. Espinosa, M. Á. Rincón-Cervera, M. Ortiz, M. C. Hernandez-Rodas, A. Valenzuela and L. A. Videla, Food Funct., 2017, 8, 1526–1537 RSC.
- C. Barrera, R. Valenzuela, M. Á. Rincón, A. Espinosa, F. Echeverria, N. Romero, D. Gonzalez-Mañan and L. A. Videla, Free Radical Biol. Med., 2018, 126, 313–321 CrossRef CAS.
- B. Wu, K. Kulkarni, S. Basu, S. Zhang and M. Hu, J. Pharm. Sci., 2011, 100, 3655–3681 CrossRef CAS.
- B. Wu, D. Dong, M. Hu and S. Zhang, Curr. Top. Med. Chem., 2013, 13, 1343–1352 CrossRef CAS.
- J. Xu, M. Li, Z. Yao, Y. Zhang, S. Li, L. Hu, Z. Qin, F. J. Gonzalez and X. Yao, J. Pharm. Biomed. Anal., 2018, 158, 351–360 CrossRef CAS.
- Y. Li, J. Xu, C. Xu, Z. Qin, S. Li, L. Hu, Z. Yao, F. J. Gonzalez and X. Yao, Xenobiotica, 2020, 50, 997–1008 CrossRef CAS.
- B. Wu, J. Pharm. Sci., 2012, 101, 381–393 CrossRef CAS.
- E. Quan, H. Wang, D. Dong, X. Zhang and B. Wu, Drug Metab. Dispos., 2015, 43, 433–443 CrossRef CAS.
- P. L. Wang, Z. H. Yao, F. X. Zhang, X. Y. Shen, Y. Dai, L. Qin and X. S. Yao, J. Pharm. Biomed. Anal., 2015, 112, 23–35 CrossRef CAS.
- M. Wang, G. Yang, Y. He, B. Xu, M. Zeng, S. Ge, T. Yin, S. Gao and M. Hu, Mol. Nutr. Food Res., 2016, 60, 1967–1983 CrossRef CAS.
- M. L. H. Vlaming, K. Mohrmann, E. Wagenaar, D. R. de Waart, R. P. J. O. Elferink, J. S. Lagas, O. van Tellingen, L. D. Vainchtein, H. Rosing, J. H. Beijnen, J. H. M. Schellens and A. H. Schinkel, J. Pharmacol. Exp. Ther., 2006, 318, 319–327 CrossRef CAS.
- B. Wu, S. Basu, S. Meng, X. Wang and M. Hu, Curr. Drug Metab., 2011, 12, 900–916 CrossRef CAS.
- S. Meng, B. Wu, R. Singh, T. Yin, J. K. Morrow, S. Zhang and M. Hu, Mol. Pharm., 2012, 9, 862–873 CrossRef CAS.
- M. Zhao, S. Wang, F. Li, D. Dong and B. Wu, Drug Metab. Dispos., 2016, 44, 1441–1449 CrossRef CAS.
- H. Sun, X. Wang, X. Zhou, D. Lu, Z. Ma and B. Wu, Drug Metab. Dispos., 2015, 43, 1430–1440 CrossRef CAS.
- X. X. Wang, X. Lv, S. Y. Li, J. Hou, J. Ning, J. Y. Wang, Y. F. Cao, G. B. Ge, B. Guo and L. Yang, Toxicol. Appl. Pharmacol., 2015, 289, 70–78 CrossRef CAS.
- D. X. Sun, G. B. Ge, P. P. Dong, Y. F. Cao, Z. W. Fu, R. X. Ran, X. Wu, Y. Y. Zhang, H. M. Hua, Z. Zhao and Z. Z. Fang, Xenobiotica, 2016, 46, 503–510 CrossRef CAS.
- Y. G. Li, J. Hou, S. Y. Li, X. Lv, J. Ning, P. Wang, Z. M. Liu, G. B. Ge, J. Y. Ren and L. Yang, Fitoterapia, 2015, 101, 99–106 CrossRef CAS.
- W. Lei, D. D. Wang, T. Y. Dou, J. Hou, L. Feng, H. Yin, Q. Luo, J. Sun, G. B. Ge and L. Yang, Toxicol. Appl. Pharmacol., 2017, 321, 48–56 CrossRef CAS.
- C. H. Chen and R. O'Keefe, J. Orthop. Translat., 2018, 12, A3–A4 CrossRef.
- T. T. Lusin, J. Trontelj and A. Mrhar, Drug Metab. Dispos., 2011, 39, 2347–2354 CrossRef CAS.
- T. Mizuma, Int. J. Pharm., 2009, 378, 140–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06961b |
| This journal is © The Royal Society of Chemistry 2020 |