
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCylindrical macrocyclic compounds synthesized by connecting two bowl-shaped calix[3]aramide moieties: structures and chiroptical properties†
Ryoko Sakagami,
Yuuki Saito ,
Ryuichi Mori,
Misa Satake,
Misaki Okayasu,
Shoko Kikkawa,
Hidemasa Hikawa and
Isao Azumaya*
,
Ryuichi Mori,
Misa Satake,
Misaki Okayasu,
Shoko Kikkawa,
Hidemasa Hikawa and
Isao Azumaya*
Faculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan. E-mail: isao.azumaya@phar.toho-u.ac.jp
First published on 18th September 2020
Abstract
Calix[3]aramide-based macrocycles 1 were successfully synthesized by a Glaser coupling reaction of two meta-calix[3]aramide moieties that have three ethynyl groups. The obtained macrocycles have stereoisomers: an enantiomeric pair and a meso form based on a combination of amide bond directions in the calix[3]aramide moieties at both ends of the molecule. Characteristic absorption spectra derived from the 1,4-diphenylbutadiyne structure were observed, while their ECD spectra were mirror-images. Single-crystal X-ray analysis revealed that each stereoisomer had a cylindrical rigid shape, and the absolute structure of the chiral-form was also assigned by comparing the Flack parameters. Mirror-image VCD spectra were observed for the enantiomeric chiral forms, and a VCD signal pattern of one enantiomer corresponded to that predicted by the relationship between the dihedral angle of the pair of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups.
O groups.
Introduction
Chiral molecules of medium molecular weight are expected as a new class of compound that can be applied to chiral recognition using the interior of the molecule1,2 or as chiral scaffolds using the exterior of molecule.3 These medium-molecular-weight compounds with macrocyclic structures,1–4 which consist of only covalent bonds, are robust and resistant to decomposition by solvent or high temperature conditions. Generally, synthetic strategies for such compounds, especially those with highly symmetric structures, have been a cyclized reaction of monomer1c,5,6 or a condensation of several partial structures.2a,2c,3 We focused on the condensation of two bowl-shaped compounds1b,2b,7 and attempted to synthesize chiral molecules with medium molecular weight using this synthetic strategy.In recent years, we have studied the synthesis of covalently bonded macrocyclic aromatic amides8,9 with lower molecular weights, such as a triple helicate10 and spherical aromatic amides,11 by using the cis-preference of tertiary aromatic amides.12 Subsequently, we synthesized cylindrical macrocyclic aromatic amides of medium molecular weight by a one-step cyclization reaction of a compound that had two sets of both alkylamino and carboxy groups as a monomer.5,6
In the present study, we successfully synthesized macrocyclic aromatic amides with three butadiynylene groups by connecting two equivalent meta-calix[3]aramides with three ethynyl groups using a Glaser coupling reaction13 (Fig. 1). The obtained macrocycles in the coupling reaction were dimeric products, which included stereoisomers (an enantiomeric pair and meso form) derived from the direction of the amide bond in each of the calix[3]aramide moieties (Fig. 2). This chirality was derived from stopping the flip of the phenylene rings in both end meta-calix[3]aramide moieties.9a These dimeric products had a cylindrical shape, and the absolute structure was assigned by X-ray structural analysis. The chiroptical properties of these macrocycles in solution were analyzed by electronic circular dichroism (ECD) and vibrational circular dichroism (VCD) spectroscopy measurements.
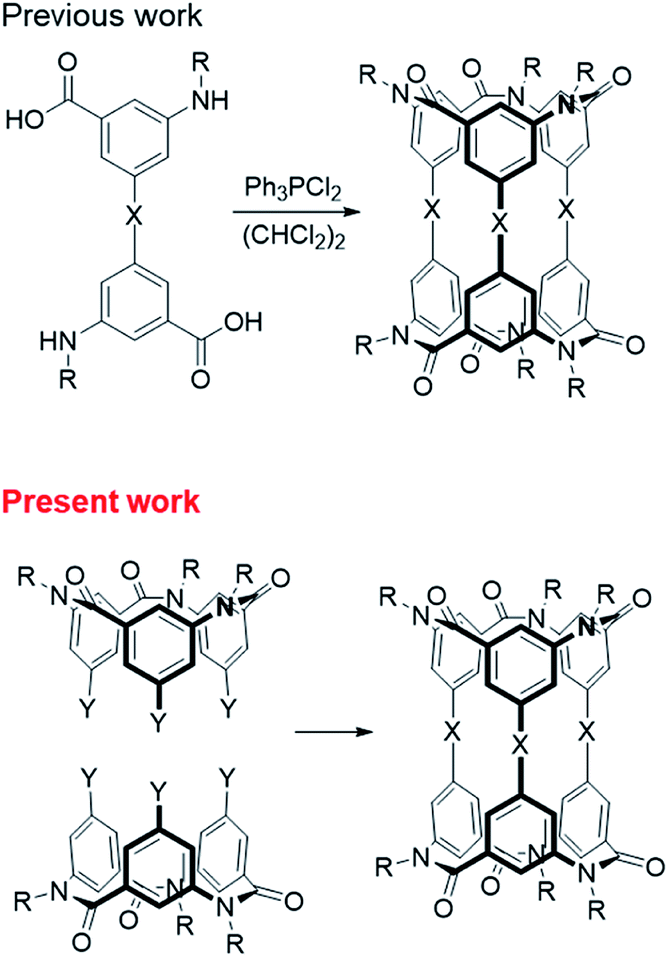 | ||
| Fig. 1 Synthetic strategy of calix[3]aramide-based cylindrical macrocycle. | ||
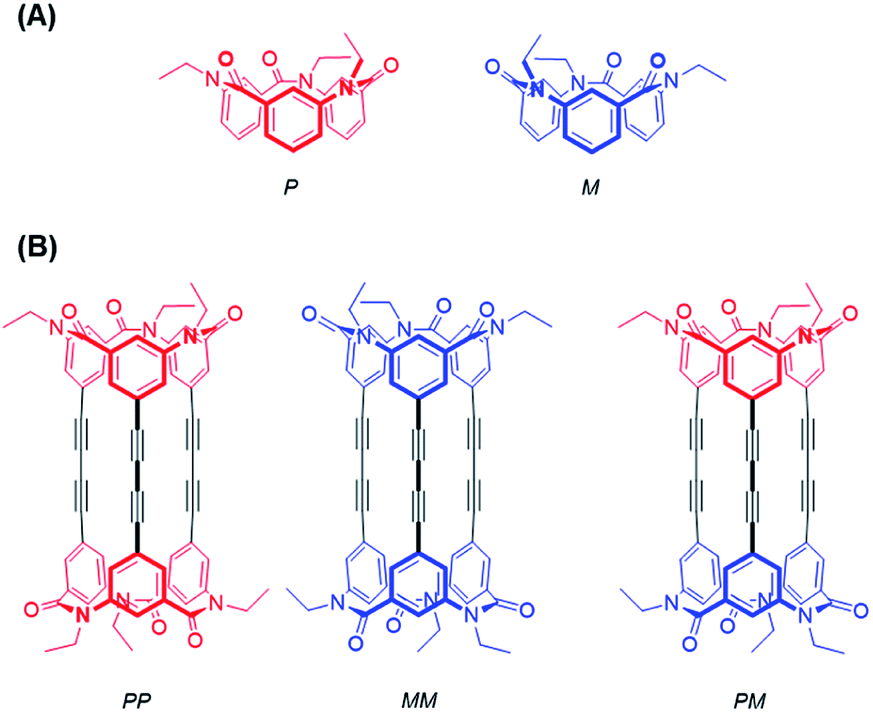 | ||
| Fig. 2 Stereostructures of calix[3]aramide-based macrocycles: (A) conformational enantiomers of meta-calix[3]aramide (P and M); (B) stereostructure of cylindrical macrocycle of enantiomers and meso-isomer. Colors: red: P conformation; blue: M conformation. | ||
Results and discussion
The synthetic route used to prepare 1 is shown in Scheme 1. Calix[3]aramide 2 was synthesized from 3-nitrobenzoic acid using a known procedure.6 A Sonogashira–Hagiwara cross coupling reaction of calix[3]aramide 2 with trimethylsilylacetylene in the presence of PdCl2(dppf) and copper(I) iodide gave calix[3]aramide 3 in 96% yield. By deprotection of the silyl group, calix[3]aramide 4 was obtained in 90% yield. Next, a Glaser coupling reaction using nickel(II) chloride hexahydrate and copper(I) iodide13 was performed with calix[3]aramide 4. The crude product of this coupling reaction was purified by silica gel column chromatography and gel-permeation chromatography (GPC). Atmospheric pressure chemical ionization high-resolution mass spectrometry (APCI-HRMS) of the isolated major product gave a peak at m/z 1021.3678 (Fig. S18 in the ESI†), which was assigned to the protonated adduct of dimeric products of 1. 1H NMR measurements after GPC (Fig. 3A) and stereochemical consideration (Fig. 2) suggested that the product was a mixture of diastereomers with a fixed calix[3]aramide structure because the aromatic protons were observed symmetrically, as in the case of the dimeric products linked by a phenylene group that we previously reported.6 The yield of dimeric products of 1 from calix[3]aramide 4 was 38%. A diastereomeric mixture of 1 was successfully separated by chiral HPLC using a ChiralPAK IA column, giving three components in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio in the order of retention time (Fig. S19†), as confirmed by 1H NMR analysis (Fig. 3B and C). The first and third eluted fractions in the chiral separation of 1 contained the same amount of compound and showed mirror-imaged Cotton effects (Fig. 4). Meanwhile, the second eluted fraction was silent in the ECD spectrum. These stereoisomers were generated due to different combinations of amide bond direction in the calix[3]aramide moieties at each end of the molecule, as in the case of the phenylene-linked derivatives.6 That is, the stereoisomers that were ECD active contained a homochiral combination of the calix[3]aramide moieties [1-chiral: 1-(PP) and 1-(MM)], while the stereoisomer that was ECD inactive contained heterochiral combination [1-meso: 1-(PM)]. The ratio of 1-chiral to 1-meso was 1:1, indicating that there was no energy difference between their reaction intermediates during the coupling reaction.
:2:1 ratio in the order of retention time (Fig. S19†), as confirmed by 1H NMR analysis (Fig. 3B and C). The first and third eluted fractions in the chiral separation of 1 contained the same amount of compound and showed mirror-imaged Cotton effects (Fig. 4). Meanwhile, the second eluted fraction was silent in the ECD spectrum. These stereoisomers were generated due to different combinations of amide bond direction in the calix[3]aramide moieties at each end of the molecule, as in the case of the phenylene-linked derivatives.6 That is, the stereoisomers that were ECD active contained a homochiral combination of the calix[3]aramide moieties [1-chiral: 1-(PP) and 1-(MM)], while the stereoisomer that was ECD inactive contained heterochiral combination [1-meso: 1-(PM)]. The ratio of 1-chiral to 1-meso was 1:1, indicating that there was no energy difference between their reaction intermediates during the coupling reaction.
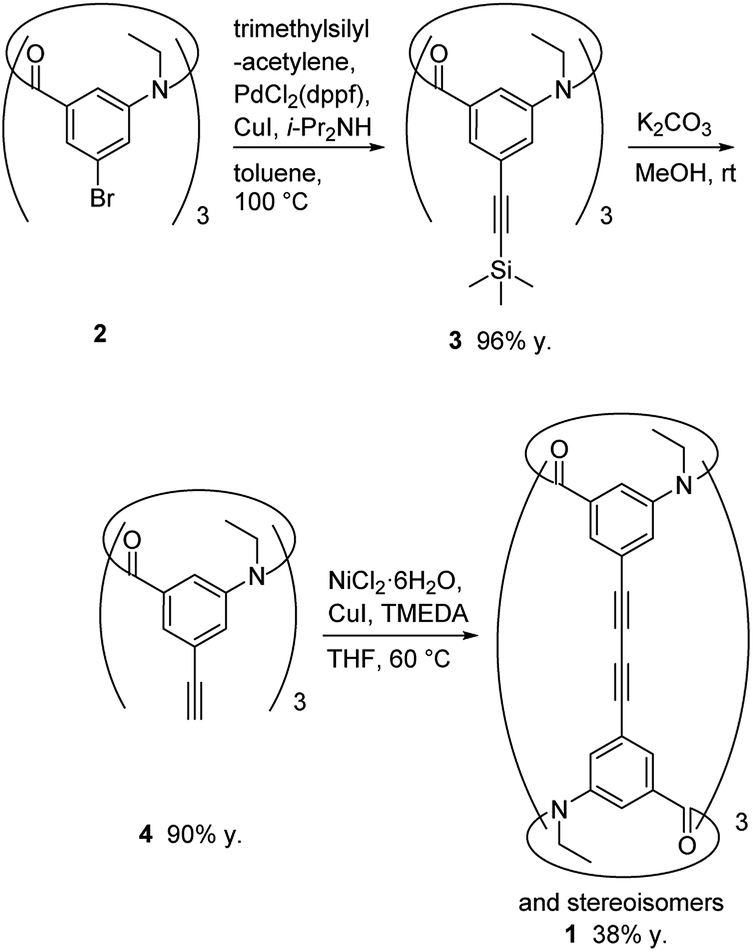 | ||
| Scheme 1 Synthesis of cylindrical macrocycle 1. | ||
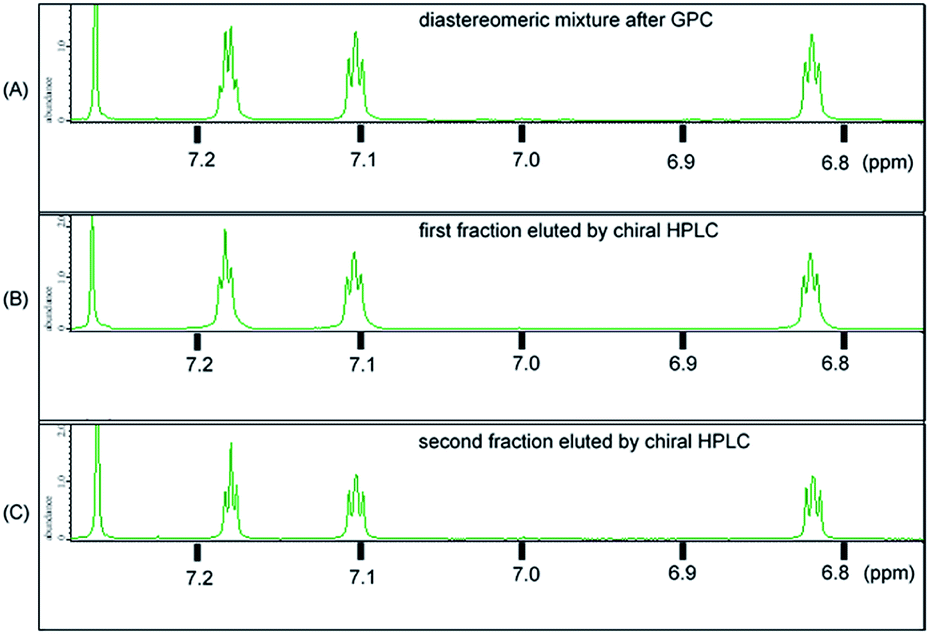 | ||
| Fig. 3 Magnified aromatic region of 1H NMR spectrum of 1 (CDCl3, 296 K): (A) diastereomeric mixture after GPC; (B) the first fraction eluted by chiral HPLC; (C) the second fraction eluted by chiral HPLC. | ||
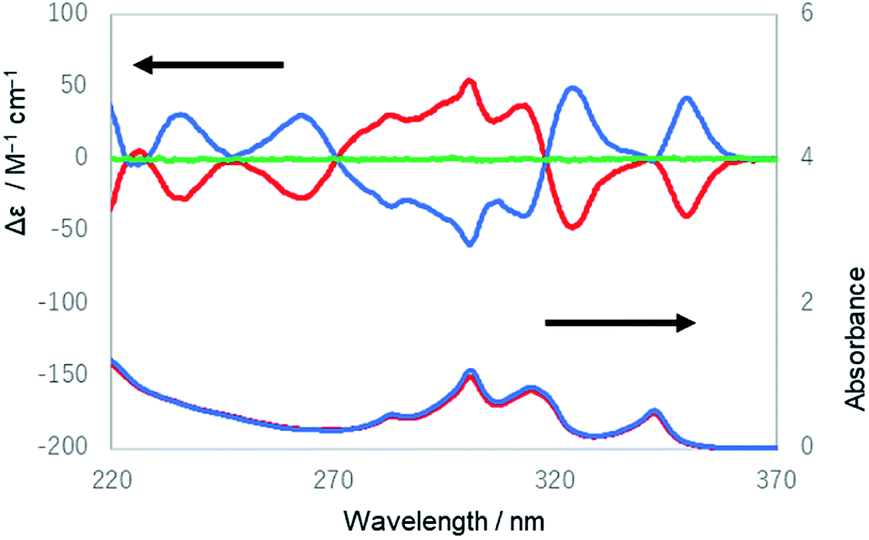 | ||
| Fig. 4 UV and ECD spectra of the first (red lines) and the third (blue lines) eluted compounds of 1 from HPLC and the ECD spectrum of the second eluted compound of 1 (green line) in ethanol. | ||
Next, we examined 1H and 13C NMR spectra of the separated fractions (1-chiral: the first and the third eluted compounds; 1-meso: the second eluted compound) in detail. In the 1H NMR spectra of 1-chiral and 1-meso, three proton signals were observed in the aromatic region, which were fully assigned using 1H–13C heteronuclear multiple bond correlation (HMBC) (Fig. S9 and S15†). Furthermore, prochiral protons (N–CH2–) were nonequivalently observed, which indicates that the conformation of the meta-calix[3]aramide moieties was fixed by covalent bonds.
The UV and ECD spectra of 1 in ethanol are shown in Fig. 4. The UV spectrum of the third eluted compound of 1 exhibited characteristic absorption peaks with maxima at 283, 301, 314, and 342 nm, which were similar to the spectrum of 1,4-diphenylbutadiynylene derivatives,14 while its ECD spectrum showed positive Cotton effects at 235, 263, 324, and 350 nm and negative Cotton effects at 283, 301, and 313 nm. The first and third eluted compounds showed mirror-imaged spectra, and the second eluted compound showed a silent spectrum in the ECD measurement. These results mean that the first and third eluted compounds were 1-chiral and enantiomeric to each other, and the second eluted compound was 1-meso [1-(PM)]. The dissymmetry factor for the ECD (|gabs|) was 7.2 × 10−3 (at 353 nm). Next, the temperature dependence of the ECD spectra of 1 was measured (Fig. S20–S23†). The intensity of the peak at 301 nm slightly increased as the temperature decreased, which is considered to be derived from a shift of the equilibrium to be more twisted in the twist of the molecule.
Single-crystal X-ray analysis of 1-(PM) of the crystal obtained by slow evaporation of solvent of 1-(PM) in chloroform/methanol was conducted successfully (Fig. 5). The crystal was conglomerate and belonged to the space group P212121. The molecule has a cylindrical shape, as expected (length, 16 Å; diameter, ca. 11 Å), and an interior space surrounded by three butadiynylene linkers (length, ca. 8 Å; diameter, ca. 1 Å). The crystal may include some disordered solvent molecules. Removal of the solvent molecules and subsequent refinement were executed using SQUEEZE, part of the PLATON program.15 The single crystal of 1-(PM) was optically active due to a twist of three butadiynylene moieties of symmetry-independent molecules in the unit cell in the same direction (Table 1).
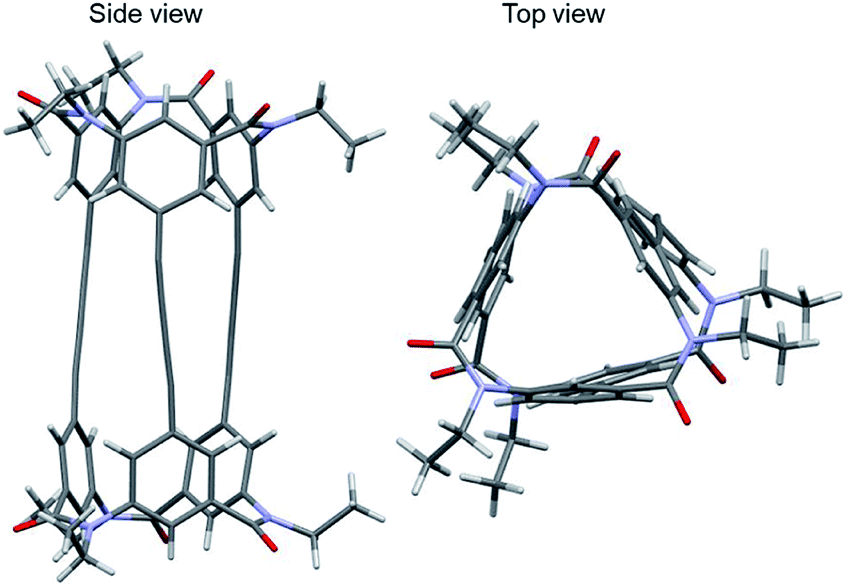 | ||
| Fig. 5 Crystal structure of 1-(PM) (CCDC no. 1992363†). Colors: H, white spheres; C, gray spheres; N, blue spheres; O, red spheres. | ||
Next, a crystal of the third eluted compound was obtained by slow evaporation of the solvent of the third eluted compound in toluene/methanol (Fig. 6). The crystal belonged to the space group P21. The molecule has a cylindrical shape (length, 17 Å; diameter, ca. 10 Å) and an interior space surrounded by three butadiynylene linkers (length, ca. 9 Å; diameter, ca. 1 Å). Then, the final Flack parameter of this crystal was 0.04(5).16 As a result, the third eluted compound of 1 was assigned to be an MM configuration.
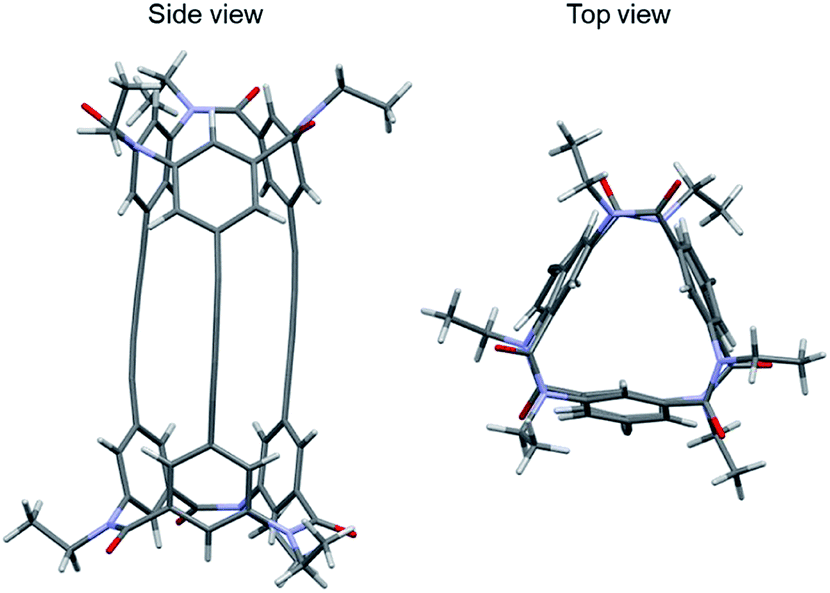 | ||
| Fig. 6 Crystal structure of 1-(MM) (CCDC no. 1992335†). Colors: H, white spheres; C, gray spheres; N, blue spheres; O, red spheres. | ||
Furthermore, a crystal of 1-(racemic mixture) was obtained by slow evaporation of its solution in toluene/ethyl acetate (Fig. 7). The crystal was a racemic compound and belonged to the space group P21/c. In the crystals of 1-(MM) and 1-(racemic mixture), the molecules were not twisted as observed in the crystal of 1-(PM). Meanwhile, the butadiynylene moieties were bent at 18.83–33.59° in all the crystals (Table 2).
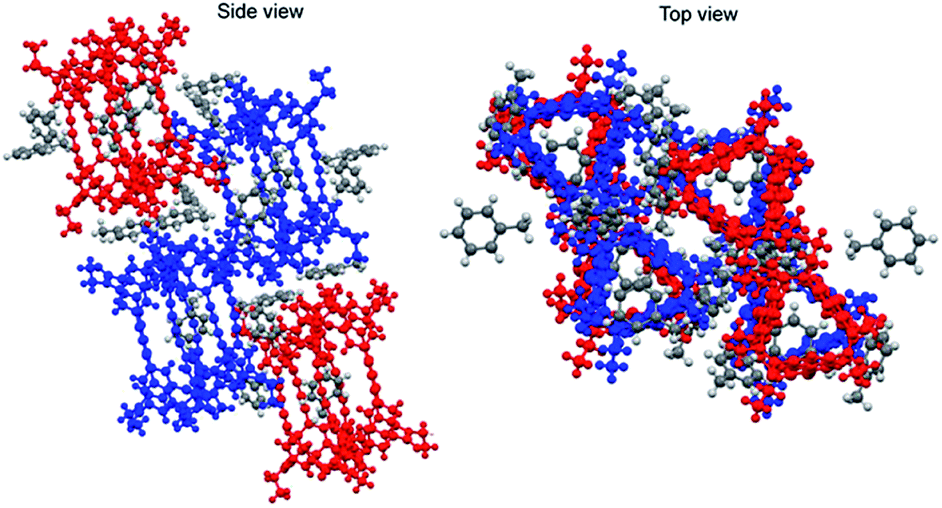 | ||
| Fig. 7 Crystal structure of 1-(racemic mixture) (CCDC no. 1992336†). Colors: MM conformation, blue; PP conformation, red. | ||
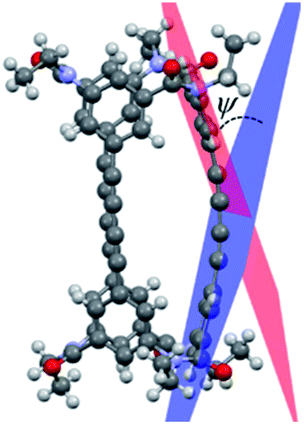
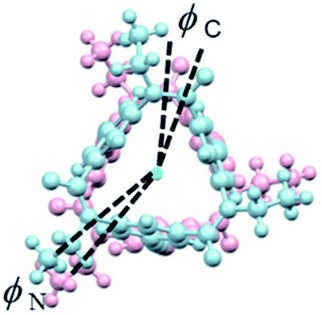
Lastly, IR and VCD spectra of 1-chiral were measured in chloroform-d (Fig. 8). The VCD spectra of 1-chiral showed two dispersion-type signals at 1586 and 1651 cm−1. The former and latter signals were associated with the CC stretching of benzene rings and CO stretching of carbonyl groups, respectively. The first and third eluted compounds of 1 showed mirror-imaged spectra. The VCD spectra patterns were similar to those of cylindrical macrocycles previously reported5,6 and were associated with the planar chirality of meta-calix[3]aramides. The absolute configuration, which was predicted by the relationship between the dihedral angle (θ: defined by the orientation of dipole moment of the two CO bonds) and the sign of the VCD spectra, was in agreement with the absolute configuration assigned by X-ray structural analysis17 (Fig. S24†).
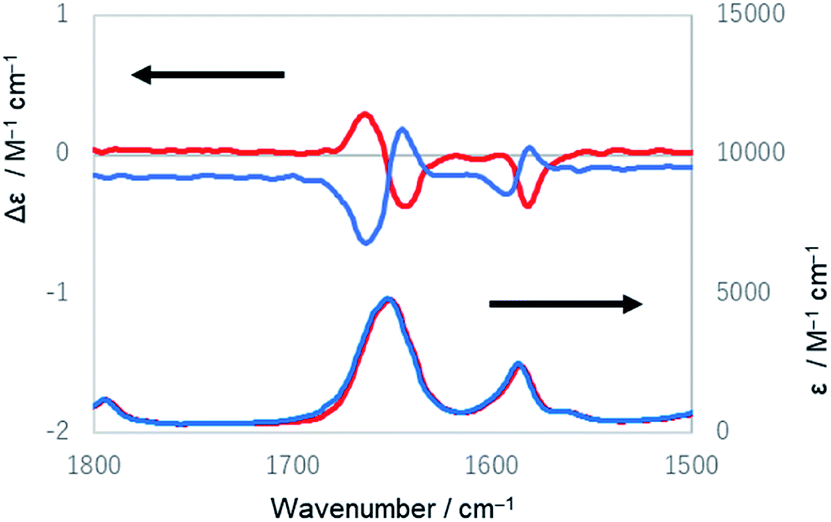 | ||
| Fig. 8 IR and VCD spectra of 1-(PP) (red lines) and 1-(MM) (blue lines) in chloroform-d. | ||
Conclusions
We successfully synthesized macromolecules of medium molecular weight by the coupling reaction of two calix[3]aramides that has three ethynyl groups. The major products were stereoisomers of dimers. The chirality was topologically fixed by connecting two meta-calix[3]aramide moieties with three butadiynylene linkers. The compounds were shown to be a cylindrical shape, and their absolute configurations were assigned by X-ray structural analysis. The enantiomeric pairs of the stereoisomers were distinguished by mirror-imaged ECD and VCD spectra. Our results provide new possibilities for the synthesis of medium-molecular-weight molecules with unique chiroptical properties. These series of medium molecular weight compounds, whose structures were confirmed, are expected to be widely used for the structural core of various functional molecules. Due to the fixed chiral structure with conjugated system, these cylindrical cage compounds can be utilized as characteristic chiroptical materials which may work as host molecules for metal cations.Experimental section
General
All of the starting materials and solvents were purchased from Kanto Chemical Co., Inc., TCI, FUJIFILM Wako. The calix[3]aramide 3 was synthesized following the known procedure.6 1H and 13C NMR spectra, 1H–1H correlation spectroscopy (COSY), 1H–13C heteronuclear single-quantum correlation spectroscopy (HSQC), and 1H–13C heteronuclear multiple bond coherence (HMBC) were recorded on a JEOL JNM-ECS400 spectrometer (400 MHz for 1H, 100 MHz for 13C). The internal standard for the 1H NMR spectra was tetramethylsilane (0.00 ppm), and for the 13C NMR spectra was CDCl3 (77.16 ppm). The following abbreviations are used: s = singlet, t = triplet, sext = sextet, m = multiplet. Infrared (IR) spectra were recorded on a JASCO FT-IR-4100 spectrophotometer. Fast atom bombardment mass spectra (FAB-MS) was performed on a JEOL JMS700 MStation mass system. Atmospheric pressure chemical ionization mass spectra (APCI-MS) were obtained by a Thermo Fisher Scientific LTQ Orbitrap XL mass system. Column chromatography was performed on silica gel (Silica gel PSQ 100B, spherical, 3–100 nm, Fuji Silysia) with a specified solvent. Purifications with preparative gel permeation chromatography (GPC) were carried out on a Japan analytical industry LC-9210 II NEXT system using tandem JAIGEL 2HH columns (CHCl3 as an eluent, flow rate = 5.0 mL min−1) equipped with an ultraviolet (UV) detector monitored at 254 nm. The chiral high performance liquid chromatography (HPLC) analysis was performed on a JASCO PU-2080 liquid chromatograph equipped with a UV detector (JASCO UV-2075) using a CHIRALPAK IA column (Daicel Chemical Industries, Ltd) (0.46 cm (i.d.) × 25 cm). UV-Vis spectra of ∼1.0 × 10−4.5 M solutions in ethanol were recorded in a 10 mm quartz cell on a HITACHI U-3010 spectrophotometer equipped. The electronic circular dichroism (ECD) spectra of 1.0 × 10−5 M solutions in ethanol were measured in 10 mm quartz cells on a JASCO J-1500 spectrophotometer. The temperature was controlled with a liquid nitrogen-controlled 10 mm quartz cell in a cryostat from +20 °C to −90 °C. The vibrational CD (VCD) spectra were recorded with a JASCO FVS-6000 spectrometer, using a 150 μm BaF2 cell. The absorption signals were detected using an MCT infrared detector. The spectra of 1.2 × 10−2 M solutions in CDCl3 were collected for ca. 1 h with 4 cm−1 resolution. Signals were accumulated for 6000 scans.Calix[3]aramide 3
To the degassed solution of calix[3]aramide 2 (0.57 g, 0.83 mmol) in dry degassed toluene (12.5 mL) and dry degassed diisopropylamine (4.0 mL, 28 mmol, 33 eq.) was added PdCl2(dppf) (0.25 g, 0.30 mmol, 0.36 eq.), copper(I) iodide (60 mg, 0.30 mmol, 0.36 eq.) and trimethylsilylacetylene (1.7 mL, 13 mmol, 15 eq.) and stirred at 80 °C for 2.5 h under argon atmosphere. After cooling, the reaction mixture was poured into water, and extracted with dichloromethane. The organic layer was washed with sat. NH4Cl, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (acetonitrile:chloroform = 1:8), followed by preparative GPC (chloroform as an eluent) to give calix[3]aramide 3 as a light yellow solid (0.59 g, 0.80 mmol, 96% y.). Mp 134–136 °C. FT-IR (KBr): 2159, 1656, 1585 cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm): 0.24 (s, 27H), 1.16 (t, 9H, J = 6.4 Hz), 3.71–3.87 (m, 6H), 6.28 (s, minor 3H), 6.83 (s, major 3H), 7.02 (s, major 3H), 7.16 (s, major 3H), 7.33 (s, minor 3H), 7.60 (s, minor 3H); 13C NMR (100 MHz, CDCl3) δ (ppm): −0.99, 12.9, 45.1, 97.3, 102.5, 125.3, 127.1, 130.1, 132.8, 139.2, 141.9, 169.2; HRMS (FAB): calcd for C42H52N3O3Si3 [M + H]+: 730.3303, found: 730.3314.
Calix[3]aramide 4
A mixture of calix[3]aramide 3 (0.59 g, 0.80 mmol) and potassium carbonate (0.89 g, 6.4 mmol, 8.0 eq.) in methanol (23 mL) was stirred at room temperature for 15 min under argon atmosphere. The reaction mixture was poured into water, and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (acetonitrile:chloroform = 1:8), followed by preparative GPC (chloroform as an eluent) to give calix[3]aramide 4 as a white powder (0.34 g, 0.72 mmol, 90% y.). Mp 140–142 °C. FT-IR (KBr): 3285, 2362, 1652, 1585 cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.17 (t, 9H, J = 6.6 Hz), 3.12 (s, 3H), 3.66–3.92 (m, 6H), 6.33 (s, minor 3H), 6.87 (s, major 3H), 7.04 (s, major 3H), 7.17 (s, major 3H), 7.37 (s, minor 3H), 7.63 (s, minor 3H); 13C NMR (100 MHz, CDCl3) δ (ppm): 12.8, 45.0, 79.9, 81.2, 124.3, 127.4, 130.3, 133.3, 139.3, 142.0, 169.0; HRMS (FAB): calcd for C33H28N3O3 [M + H]+: 514.2124, found: 514.2131.
Cylindrical macrocycle 1
A mixture of calix[3]aramide 4 (26 mg, 50 μmol), nickel(II) chloride hexahydrate (1.8 mg, 7.6 μmol, 0.15 eq.) and copper(I) iodide (51.5 mg, 7.6 μmol, 0.15 eq.) in TMEDA (4.5 μL, 30 μmol, 0.60 eq.) and THF (1.0 mL) was stirred at 60 °C for 24 h under argon atmosphere. After cooling, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (acetonitrile:chloroform = 1:8), followed by preparative GPC (chloroform as an eluent) to give compound 1 as a white powder of diastereomeric mixture (9.8 mg, 9.6 μmol, 38% y.). Optical resolution was carried out by chiral high performance liquid chromatography with a Daicel CHIRALPAK IA column [0.46 cm (i.d.) × 25 cm] using dichloromethane as an eluent at a flow rate of 0.5 mL min−1 (elution time 6.5 min, 10.1 min, 14.3 min). HRMS (APCI) calcd for C66H49N6O6 [M + H]+: 1021.3702, found: 1021.3678.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Katsuyoshi Mitsunaga (Faculty of Pharmaceutical Sciences, Toho University) and Dr Hyuma Masu and Ms Makiko Fujinami (Center for Analytical Instrumentation, Chiba University) for assistance with mass spectrometry.Notes and references
- (a) J. Canceill, L. Lacombe and A. Collet, J. Am. Chem. Soc., 1985, 107, 6993–6996 CrossRef CAS; (b) D. J. Cram, M. E. Tanner, S. J. Keipert and C. B. Knobler, J. Am. Chem. Soc., 1991, 113, 8909–8916 CrossRef CAS; (c) T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS; (d) G. Huber, T. Brotin, L. Dubois, H. Desvaux, J.-P. Dutasta and P. Berthault, J. Am. Chem. Soc., 2006, 128, 6239–6246 CrossRef CAS; (e) T. Brotin, E. Jeanneau, P. Berthault, E. Léonce, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2018, 83, 14465–14471 CrossRef CAS.
- (a) H. Takahagi, S. Fujibe and N. Iwasawa, Chem.–Eur. J., 2009, 15, 13327–13330 CrossRef CAS; (b) S. Míguez-Lago, A. L. Llamas-Saiz, M. M. Cid and J. L. Alonso-Gómez, Chem.–Eur. J., 2015, 21, 18085–18088 CrossRef; (c) A. U. Malik, F. Gan, C. Shen, N. Yu, R. Wang, J. Crassous, M. Shu and H. Qiu, J. Am. Chem. Soc., 2018, 140, 2769–2772 CrossRef CAS.
- (a) T. Hasell, X. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS; (b) A. G. Slater, M. A. Little, A. Pulido, S. Y. Chong, D. Holden, L. Chen, C. Morgan, X. Wu, G. Cheng, R. Clowes, M. E. Briggs, T. Hasell, K. E. Jelfs, G. M. Day and A. I. Cooper, Nat. Chem., 2017, 9, 17–25 CrossRef CAS; (c) C. J. Pugh, V. Santolini, R. L. Greenaway, M. A. Little, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Cryst. Growth Des., 2018, 18, 2759–2764 CrossRef CAS.
- (a) R. Shomura, S. Higashibayashi, H. Sakurai, Y. Matsushita, A. Sato and M. Higuchi, Tetrahedron Lett., 2012, 53, 783–785 CrossRef CAS; (b) T. Šolomek, N. E. Powers-Riggs, Y. Wu, R. M. Young, M. D. Krzyaniak, N. E. Horwitz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 3348–3351 CrossRef; (c) D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS; (d) T. Matsushima, S. Kikkawa, I. Azumaya and S. Watanabe, ChemistryOpen, 2018, 7, 278–281 CrossRef CAS.
- R. Yamakado, K. Mikami, K. Takagi, I. Azumaya, S. Sugimoto, S. Matsuoka, M. Suzuki, K. Katagiri, M. Uchiyama and A. Muranaka, Chem.–Eur. J., 2013, 19, 11853–11857 CrossRef CAS.
- Y. Saito, M. Satake, R. Mori, M. Okayasu, H. Masu, M. Tominaga, K. Katagiri, K. Yamaguchi, S. Kikkawa, H. Hikawa and I. Azumaya, Org. Biomol. Chem., 2020, 18, 230–236 RSC.
- (a) D. O. Krongly, S. R. Denmeade, M. Y. Chiang and R. Breslow, J. Am. Chem. Soc., 1985, 107, 5544–5545 CrossRef; (b) B. P. Friedrichsen and H. W. Whitlock, J. Am. Chem. Soc., 1989, 111, 9132–9134 CrossRef CAS; (c) Y. Rubin, T. C. Parker, S. I. Khan, C. L. Holliman and S. W. McElvany, J. Am. Chem. Soc., 1996, 118, 5308–5309 CrossRef CAS; (d) Y. Tobe, N. Nakagawa, K. Naemura, T. Wakabayashi, T. Shida and Y. Achiba, J. Am. Chem. Soc., 1998, 120, 4544–4545 CrossRef CAS; (e) M. Tominaga, K. Ohara, K. Yamaguchi and I. Azumaya, J. Org. Chem., 2014, 79, 6738–6742 CrossRef CAS.
- (a) I. Azumaya, T. Okamoto, F. Imabeppu and H. Takayanagi, Tetrahedron, 2003, 59, 2325–2331 CrossRef CAS; (b) K. Katagiri, T. Tohaya, H. Masu, M. Tominaga and I. Azumaya, J. Org. Chem., 2009, 74, 2804–2810 CrossRef CAS; (c) K. Katagiri, T. Ikeda, A. Muranaka, M. Uchiyama, M. Tominaga and I. Azumaya, Tetrahedron: Asymmetry, 2009, 20, 2646–2650 CrossRef CAS; (d) N. Fujimoto, M. Matsumura, I. Azumaya, S. Nishiyama, H. Masu, H. Kagechika and A. Tanatani, Chem. Commun., 2012, 48, 4809–4811 RSC; (e) S. Nishiyama, K. Urushibara, H. Masu, I. Azumaya, H. Kagechika and A. Tanatani, Chirality, 2015, 27, 487–491 CrossRef CAS; (f) K. Katagiri, S. Komagawa, M. Uchiyama, K. Yamaguchi and I. Azumaya, Org. Lett., 2015, 17, 3650–3653 CrossRef CAS.
- (a) I. Azumaya, H. Kagechika, K. Yamaguchi and K. Shudo, Tetrahedron Lett., 1996, 37, 5003–5006 CrossRef CAS; (b) A. Yokoyama, T. Maruyama, K. Tagami, H. Masu, K. Katagiri, I. Azumaya and T. Yokozawa, Org. Lett., 2008, 10, 3207–3210 CrossRef CAS; (c) H. Kakuta, I. Azumaya, H. Masu, M. Matsumura, K. Yamaguchi, H. Kagechika and A. Tanatani, Tetrahedron, 2010, 66, 8254–8260 CrossRef CAS; (d) K. Takagi, S. Sugimoto, R. Yamakado and K. Nobuke, J. Org. Chem., 2011, 76, 2471–2478 CrossRef CAS; (e) R. Yamakado, S. Matsuoka, M. Suzuki, D. Takeuchi, H. Masu, I. Azumaya and K. Takagi, RSC Adv., 2014, 4, 6752–6760 RSC; (f) R. Yamakado, S. Matsuoka, M. Suzuki, D. Takeuchi, H. Masu, I. Azumaya and K. Takagi, Chem. Commun., 2015, 51, 5710–5713 RSC.
- M. Tominaga, H. Masu, K. Katagiri, T. Kato and I. Azumaya, Org. Lett., 2005, 7, 3785–3787 CrossRef CAS.
- (a) H. Masu, K. Katagiri, T. Kato, H. Kagechika, M. Tominaga and I. Azumaya, J. Org. Chem., 2008, 73, 5143–5146 CrossRef CAS; (b) H. Masu, Y. Sagara, F. Imabeppu, H. Takayanagi, K. Katagiri, M. Kawahata, M. Tominaga, H. Kagechika, K. Yamaguchi and I. Azumaya, CrystEngComm, 2011, 13, 406–409 RSC.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177–6180 CrossRef CAS.
- W. Yin, C. He, M. Chen, H. Zhang and A. Lei, Org. Lett., 2009, 11, 709–712 CrossRef CAS.
- (a) Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, J. Am. Chem. Soc., 2003, 124, 14103–14112 CrossRef; (b) M. Wierzbicka, I. Bylińska, A. Sikorski, C. Czaplewski and W. Wiczk, Photochem. Photobiol. Sci., 2015, 14, 2251–2260 RSC.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS.
- (a) H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908–915 CrossRef; (b) H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CrossRef CAS.
- (a) H.-Z. Tang, B. M. Novak, J. He and P. L. Polavarapu, Angew. Chem., Int. Ed., 2005, 44, 7298–7301 CrossRef CAS; (b) T. Kawauchi, J. Kumaki, A. Kitaura, K. Okoshi, H. Kusanagi, K. Kobayashi, T. Sugai, H. Shinohara and E. Yashima, Angew. Chem., Int. Ed., 2008, 47, 515–519 CrossRef CAS; (c) T. Taniguchi and K. Monde, J. Am. Chem. Soc., 2012, 134, 3695–3698 CrossRef CAS; (d) F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour, Angew. Chem., Int. Ed., 2013, 52, 1796–1800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1992363, 1992335 and 1992336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07060b |
| This journal is © The Royal Society of Chemistry 2020 |