DOI:
10.1039/D0RA07245A
(Paper)
RSC Adv., 2020,
10, 42413-42422
Targeting intracranial patient-derived glioblastoma (GBM) with a NIR-I fluorescent immunoconjugate for facilitating its image-guided resection†
Received
23rd August 2020
, Accepted 26th October 2020
First published on 23rd November 2020
Abstract
Glioblastoma multiforme (GBM) is the most aggressive form of primary brain tumor type and is associated with a high mortality rate borne out of such affording a survival rate of only 15 months. GBM aggressiveness is associated with the overexpression of epidermal growth factor receptor (EGFR) and its mutants. Targeting GBM with therapeutics is challenging because the blood-brain barrier (BBB) permits primarily select small-molecule entities across its semipermeable blockade. However, recent preclinical data suggest that large biomolecules, such as the anti-EGFR antibody therapeutic, cetuximab, could be capable of bypassing the BBB despite the relative enormity of its size. As such, we set forth to establish the feasibility of utilizing an EGFR-targeting near-infrared-I (NIR-I) fluorescent construct in the form of an immunoconjugate (cetuxmimab-IRDye800) to achieve visual differentiation between diseased brain tissue arising from a low-passage patient-derived GBM cell line (GBM39) and healthy brain tissue via utilizing orthotopic intracranial murine GBM39 tumor models for in vivo and ex vivo evaluation such that by doing so would establish proof of concept for ultimately facilitating its in vivo fluorescence-guided resection and ex vivo surgical back-table pathological confirmation in the clinic. As anticipated, we were not capable of distinguishing between malignant tumor tissue and healthy tissue in resected intact and slices of whole brain ex vivo under white-light illumination (WLI) due to both the diseased tissue and healthy tissue appearing virtually identical to the unaided eye. However, we readily observed over an average 6-fold enhancement in the fluorescence emission in the resected intact whole brain ex vivo when performing NIR-I fluorescence imaging (FLI) on the cohort of GBM39 tumor models that were administered the immunoconjugate compared to controls. In all, we laid the initial groundwork for establishing that NIR-I fluorescent immunoconjugates (theranostics) such as cetuximab–IRDye800 can bypass the BBB to visually afford GBM39 tumor tissue differentiation for its image-guided surgical removal.
1. Introduction
GBM is a primary malignant tumor of the central nervous system (CNS) that is the most common type of brain tumor as well as the most aggressive form of primary brain tumor type.1–3 GBM is associated with high morbidity, poor prognosis, high mortality, and thereby extremely low survival rates because such cancer type is highly invasive and neurologically destructive, especially as gliomas which include GBM can arise from a broad range of brain cell types.4 Accordingly, the median survival rate is only an average mere 15 months despite pre-/postoperative treatment (i.e., chemo-/radiotherapy) in conjunction with the standard form of intraoperative treatment (i.e., tumor tissue resection under white-light illumination, WLI).3,5 Malignant gliomas rapidly infiltrate healthy brain tissue with diseased tissue of very low cell concentration extending many centimeters beyond the amorphic localized hardened tumor mass, which significantly confounds the efficacy of any form of treatment especially surgery because the operating surgeon also relies on palpation (if feasible) to help distinguish between diseased and healthy tissue upon identifying such general areas from image-delayed pre- and intraoperative imaging modalities (i.e., images not acquired in real time). Moreover, GBM exhibits considerable intra- and intertumoral heterogeneity that also biologically adapts to become progressively chemo-/radioresistant or quiescent.6 Targeting brain diseases, such as GBM, with small-molecular or biological therapeutics is challenging because of the presence of the finicky semi-permeable blood-brain barrier (BBB). The BBB demonstrates extremely low solute permeability, which helps to maintain brain homeostasis. Accordingly, pre- and postoperative treatment options are limited and revolve around the use of small-molecule alkylating agents due to their physicochemical properties, such as orally-delivered temozolomide, because of extremely inefficient drug delivery of other molecule types across the BBB that are primarily due to (i) it predominantly affording only small molecules of low molecular weight (≤∼400–500 g mol−1) with other distinctive molecular properties (e.g., Lipinski's “rule of five”) to traverse such and (ii) the presence of active efflux pumps at the luminal side of the BBB that export foreign molecules out of the brain to maintain brain homeostasis.6–8 In the case of brain tumors, the BBB faces some abnormalities where, besides the morphological changes to the barrier, its permeability increases because of compromised, disrupted junctions in the layer of endothelial, ependymal, and tanycytic cells that comprise it.8 However, though pathological processes can increase BBB permeability, the extent of junction disruption typically remains insufficient for the passage of larger entities such as unchaperoned proteins. The ability of molecules of greater size to cross the BBB would facilitate larger therapeutics, such as anticancer antibodies, to reach their targets and exert their curative action.9 Accordingly, immunotherapy is one of the fastest developing approaches in clinical oncology with successful treatment towards many different cancers that are not within the brain. As such, the BBB and the unique immune environment of the CNS need special consideration when pursuing immunotherapeutic approaches for malignant gliomas such as GBM.
With respect to immunotherapeutic approaches, the epidermal growth factor receptor (EGFR) signaling pathways have been extensively investigated because such are considered to be associated with the extent of aggressiveness of GBM and its resistance to standard chemo-/radiotherapy.10,11 This receptor and its active mutants (e.g., EGFRvIII) are co-overexpressed in ∼60% of patients with GBM.11–13 As such, the role of EGFR-targeting therapies for treating GBM have been suggested based on the results from non-intracranial preclinical models of other cancer types (e.g., xenograft flank tumor models) expressing EGFR. Towards this end, cetuximab has been used because it is an efficacious anti-EGFR monoclonal antibody (i.e., inhibitor) that targets and binds to the extracellular domain of the EGFR and its mutants.14 More specifically, cetuximab is a recombinant, chimeric IgG1 antibody of ∼1152 kDa in molecular weight that inhibits the binding of EGF upon it doing so, and thereby such interaction ultimately inhibits tumor growth and induces apoptosis. It is believed that cetuximab is metabolized by the reticuloendothelial system with very little hepatic or renal metabolism, which is in contrast to most small-molecule drugs.15 The limited number of studies on the metabolism of cetuximab have demonstrated a limited extent of small amino acid sequences translocating from cetuximab onto other smaller proteins whose molecular weight thereafter were ∼11 kDa with their degradation to lower-molecular-weight metabolites of <5 kDa, and thereby presumably affording cetuximab to largely remain intact upon having obtained protein of a molecular weight > 1000 kDa.16 Such low metabolic activity and long systemic circulation half-life afford cetuximab and other antibodies the ability to exert their therapeutic effect at the tumor site, which could tentatively include those in the brain. Presumably, further studies towards investigating the metabolism of cetuximab itself or its fluorescent construct, cetuximab–IRDye800, have not occurred because such determination for gaining FDA approval are not required, which can be in contrast to many small-molecule drugs, because the FDA views antibody metabolites to be merely comprised of very low-molecular-weight, innocuous proteins that undergo rapid clearance. Due to its low metabolic breakdown, cetuximab has shown high activity towards various tumor models expressing EGFR and its mutants.14 In non-intracranial preclinical GBM tumor models, cetuximab also exerts an antitumor and radiosensitizing effect on GBM. Most importantly, in intracranial preclinical GBM models, systemic administration of cetuximab (alone) has shown via imaging to be an effective treatment towards GBM, thereby indirectly suggesting that therapeutic antibodies could be capable of bypassing the BBB presumably due to extensive BBB junction disruption. Therefore, cetuximab therapy is a plausible strategy for targeting and treating GBM tumors that overexpress EGFR and/or EGFRvIII, again presuming that cetuximab can bypass the BBB.
The clinical treatments for GBM include chemotherapy, radiotherapy, and tumor resection.17 Unfortunately, no nonsurgical treatments so far in the standard of care for GBM have been superior to its resection. Current treatment includes maximal surgical resection followed by adjuvant treatment with chemotherapy, starting with a low dose followed by a full dose of temozolomide for six months, also combined with radiotherapy. Tumor resection can improve the overall survival rate of patients afflicted with GBM, as the approach is to resect all cancer tissue/cells such that the patient would be cured of the cancer.18–22 Notably, the extent of resected malignant GBM tissue/cells entirely governs the outcome, and thereby the long-term survival rates. Thus, the capability of the operating surgeon to achieve complete resection of GBM tumor tissue is fundamentally vital to prevent recurrence from residual diseased tissue/cells.20 Cancer surgery, especially brain cancer surgery, accordingly entails the operating surgeon to have the capability of visually distinguishing between very subtle shades of white to pink or between very subtle shades of red to deep-red tissue at the macroscopic level under standard WLI in order to differentiate between diseased tissue and healthy tissue, wherein marginal differences between their colors exist (if even any at all). Thus, visually discriminating between the two tissue states is extraordinarily difficult because such appear virtually identical. In addition, the infiltrative nature of GBM further confounds the realization of its complete resection due to diseased tissue innervating healthy tissue. Moreover, the extent of resection must also be balanced with minimal impairment to brain function.20 Accordingly, enhancing the capability of the surgeon to visually distinguish between the two tissue states and accurately define the tumor margin is of paramount importance for obtaining complete resection whilst not damaging eloquent brain tissue. Current standard of care for preoperative tumor tissue delineation entails the use of non-optical imaging modalities such as computed tomography, magnetic resonance imaging, and positron emission tomography.23,24 Such modalities when separately used or configured for intraoperative use so as to enable non-optical image-guided surgery (via later co-registering prior images with the surgical field) have not improved tissue differentiation and especially surgical margin positivity rates.25 As of recent, optical imaging modalities such as FLI, have afforded the recent advent of fluorescence-guided surgery (FGS), which is utilized in conjunction with a non-/targeting fluorescent construct. FGS enhances the capability of the operating surgeon to visually discriminate between anatomical structures as well as between diseased and healthy tissue during surgery by color-coding the surgical field with overlaid contrasting pseudo colors that result from the extent of fluorescence emission intensities measured in real time that the accompanying fluorescent construct provides when displaying specific binding or preferential accumulation at the diseased site(s).26,27 Currently, there are only a very small number of fluorescent constructs available for intraoperative use that are FDA approved or undergoing clinical translation, such as fluorescein, indocyanine green, 5-aminolevulinic acid, and IRDye800, wherein all of such provide peak fluorescence emission in either the visible (ca. 405–700 nm) or NIR-I (ca. 700–900 nm) spectral region.28 Importantly, better tissue differentiation is progressively afforded using fluorescent constructs that display longer wavelengths of fluorescence emission, which would be those emitting in the NIR-I spectral region when compared to those emitting in the visible spectral region.29
Taken together, herein, we determine the feasibility of utilizing an EGFR-targeting fluorescent construct in the form of an immunoconjugate (cetuximab–IRDye800), which exhibits peak fluorescence emission in the NIR-I spectral region (∼795 nm), to achieve visual differentiation between diseased tissue (i) arising from a low-passage patient-derived GBM cell line (GBM39) and (ii) healthy tissue, via utilizing intracranial orthotopic murine GBM39 tumor models in in vivo and ex vivo settings such that by doing so such would establish proof of concept for ultimately facilitating its fluorescence-guided resection and its back-table pathological confirmation in the clinic (the molecular structure, physical, and photophysical properties of IRDye800 are provided in Table S1†). By achieving such, postoperative outcomes and long-term survival rates would have the potential to dramatically increase. Here, we prepared intracranial orthotopic murine tumor models of a low-passage patient-derived GBM39 cell line and administered the immunoconjugate to such GBM39 tumor models with exception of appropriate negative controls, wherein we administered either only saline or the unconjugated (free) IRDye800 fluorophore. We imaged daily the orthotopic intracranial murine GBM39 tumor models pre- and post-administration of the immunoconjugate using NIR-I FLI such that we could observe any fluorescence emission from targeted GBM tissue. On doing so, we determined the (i) in vivo bioluminescence imaging (BLI) total flux, (ii) in vivo NIR-I fluorescence emission, (iii) ex vivo intact whole brain NIR-I fluorescence emission, (iv) ex vivo intact whole brain BLI total flux, (v) NIR-I fluorescence emission from ex vivo slices of intact whole brain, and (vi) ex vivo biodistribution of the immunoconjugate via utilizing NIR-I FLI. In all, we established the feasibility of the cetuximab–IRDye800 immunoconjugate to bypass the BBB such that it can afford differentiation of GBM39 tumor tissue from healthy tissue by providing high tumor-to-background ratios (TBRs) via utilizing NIR-I FLI.
2. Results & discussion
In the information that follows, we acquired, analyzed, and reported on the relative BLI and/or relative localized NIR-I fluorescence emission intensities of an anticancer antibody–fluorophore conjugate (i.e., immunoconjugate) when applied to an orthotopic murine GBM39 tumor model by having measured (i) such within the brain in vivo, (ii) such within both the intact whole brain and whole brain slices ex vivo, and (iii) its biodistribution ex vivo in order to evaluate the feasibility of the cetuximab–IRDye800 immunoconjugate to bypass the BBB and afford identification of patient-derived GBM39 tumor tissue via recognizing any upregulated EGFR and/or EGFRvIII expression from such diseased tissue that its capability in doing so would facilitate in the visualization of the diseased tissue for its subsequent fluorescence-guided resection.
2.1 In vivo bioluminescence imaging
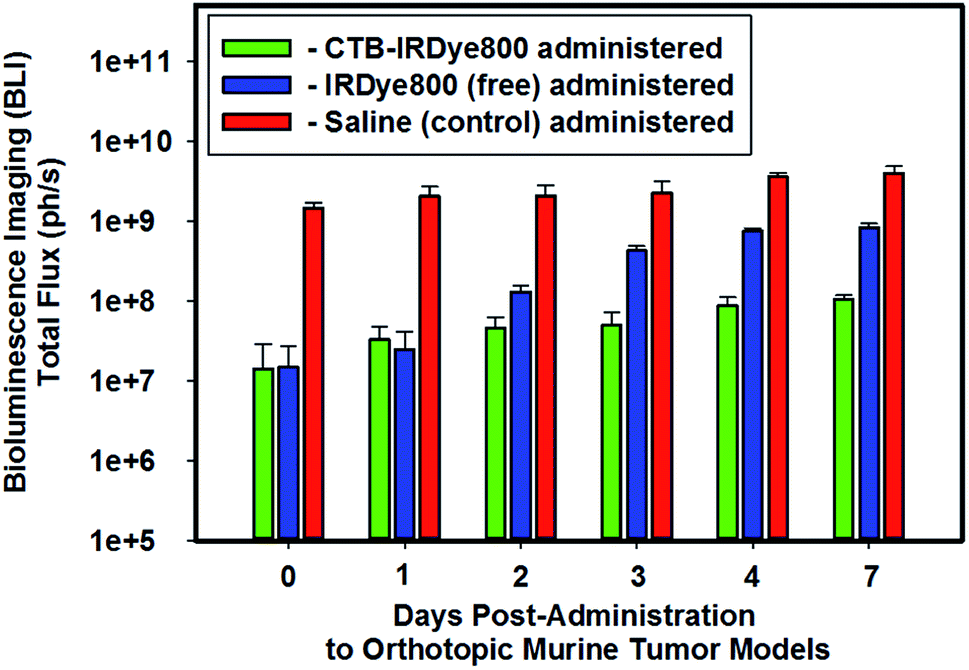
We performed BLI imaging to evaluate the growth of the intracranially-implanted GBM39 cells routinely throughout the (i) 5–6 weeks of their proliferation post-implantation and (ii) pre- and post-administration of either saline (negative control), the unconjugated (free), or the cetuximab–IRDye800 immunoconjugate (Fig. 1). The molar ratio of free IRDye800 to the conjugated form of IRDye800 (i.e., cetuximab–IRDye800) was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. We stratified the orthotopic murine GBM39 tumor models into the following three separate cohorts based on the total flux that we obtained from each such model via using BLI: high-, mid-, and low-total flux cohorts.
:1. We stratified the orthotopic murine GBM39 tumor models into the following three separate cohorts based on the total flux that we obtained from each such model via using BLI: high-, mid-, and low-total flux cohorts.
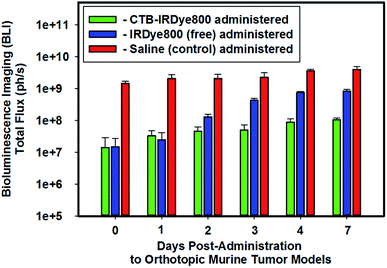 |
| | Fig. 1 In vivo bioluminescence imaging. Representative relative averaged total flux (photons per s) of intracranial GBM tumor tissue from cohorts of orthotopic murine GBM39 tumor models pre- and post-administration of either saline (200 μL), IRDye800 (200 μL, 27.44 μM, 6.40 μg, 5.49 nanomoles of unconjugated IRDye800), or the cetuximab–IRDye800 immunoconjugate (200 μL, 2 mg mL−1, 400 μg, 5.49 nanomoles of conjugated IRDye800) via visualizing the luciferase activity upon injecting such models intraperitoneally with a prepared (15 mg mL−1 in PBS) D-luciferin (firefly) potassium salt solution (150 μL) and imaging 10 minutes later with an exposure time of 0.3 s. The bioluminescence emission was collected from 430–650 nm. | |
We intentionally assigned the cohort of orthotopic murine GBM39 tumor models that demonstrated the lowest relative averaged BLI total flux to be administered the immunoconjugate so as to ensure that we would not suffer from the fallacy of utilizing the cohort of orthotopic murine GBM39 tumor models that demonstrated the highest in vivo relative averaged BLI total flux to demonstrate proof of concept, whereby if such were to occur the results could be subject to multiple interpretations and not be necessarily conclusive. A case in point is if we had done otherwise and had also obtained the highest fluorescence emission intensities from such when measuring the fluorescence emission intensities from the brain ex vivo; any such positive results could be indicative of the fact that the higher measured fluorescence emission intensities were simply due to the cohort of orthotopic murine GBM39 tumor models having the greatest proliferation of GBM39 tumor tissue, which affords the potential for a higher expression of EGFR and EGFRvIII to which the cetuximab–IRDye800 could target. On the other hand, such a result could also simply be interpreted as being that the BBB is now more disrupted due to the extensive proliferation of GBM39 tumor tissue in which the results simply reflect. In short, we intentionally wanted to add negative experimental bias to our study by doing as per above such that if we obtained positive results, the potential for the former interpretation would be minimized. Accordingly, we intentionally opted to utilize the cohort of orthotopic murine GBM39 tumor models that demonstrated the lowest averaged relative BLI total flux to address the capability of whether the immunoconjugate can circumvent the BBB. As shown in Fig. 1, the averaged relative BLI total flux for the three cohorts of orthotopic murine GBM39 tumor models essentially progressively increased in succession from day 0 to day 7 post-administration of the select agent that a cohort was assigned. The high-total flux cohort that was administered saline had approximately 100-10-fold greater proliferation fo GBM39 tumor tissue over day 0 to day 7, respectively, when compared that of the low-total flux cohort that was administered the immunoconjugate.
2.2 In vivo tumor NIR-I fluorescence imaging analysis
Next, we imaged the three cohorts of orthotopic murine GBM39 tumor models via using NIR-I FLI in attempt to visualize intracranial GBM39 tumor tissue in vivo pre- and post-administration of either saline, IRDye800 (free), and the cetuximab–IRDye800 immunoconjugate (Fig. 2). We did not visualize any NIR-I fluorescence emission from intracranial tumor tissue of the cohorts of orthotopic murine GBM39 tumor models that separately received either saline or IRDye800 in its unconjugated form. We anticipated such results. However, we believe we could visualize NIR-I fluorescence emission from intracranial tumor tissue from the cohort of orthotopic murine GBM39 tumor models that separately received the cetuximab–IRDye800 immunoconjugate. We believe we visualized such during days 0–4 post-administration due to the localized NIR-I fluorescence emission at/near the sight of inoculation, wherein the NIR-I fluorescence emission intensity progressively decreased each day. On day 7 post-administration, we did not observe any NIR-I fluorescence emission from the cohort of orthotopic murine GBM tumor models that were administered the cetuximab–IRDye800 immunoconjugate.
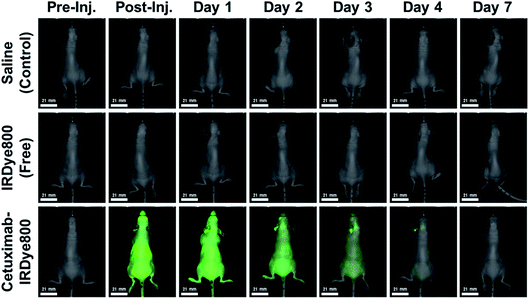 |
| | Fig. 2 Representative images using NIR-I fluorescence imaging in attempt to visualize intracranial GBM39 tumor tissue in vivo from the three cohorts of orthotopic murine GBM39 tumor models. The representative images of such models from the three cohorts are pre- and post-administration of either saline, IRDye800 (200 μL, 27.44 μM, 6.40 μg, 5.49 nanomoles of unconjugated IRDye800), or the cetuximab–IRDye800 immunoconjugate (200 μL, 2 mg mL−1, 400 μg, 5.49 nanomoles of conjugated IRDye800). The excitation source was a 785 nm solid-state laser diode. Fluorescence emission in the NIR-I spectral region was collected at 820 ± 10 nm using a bandpass filter. Thus, for NIR-I FLI, all tumor and control murine models were excited at 785 nm and the resultant fluorescence emission was collected from 810–830 nm. The scan time was 500 ms and the resolution was set at 170 μm. | |
2.3 Ex vivo intact whole brain NIR-I fluorescence imaging analysis
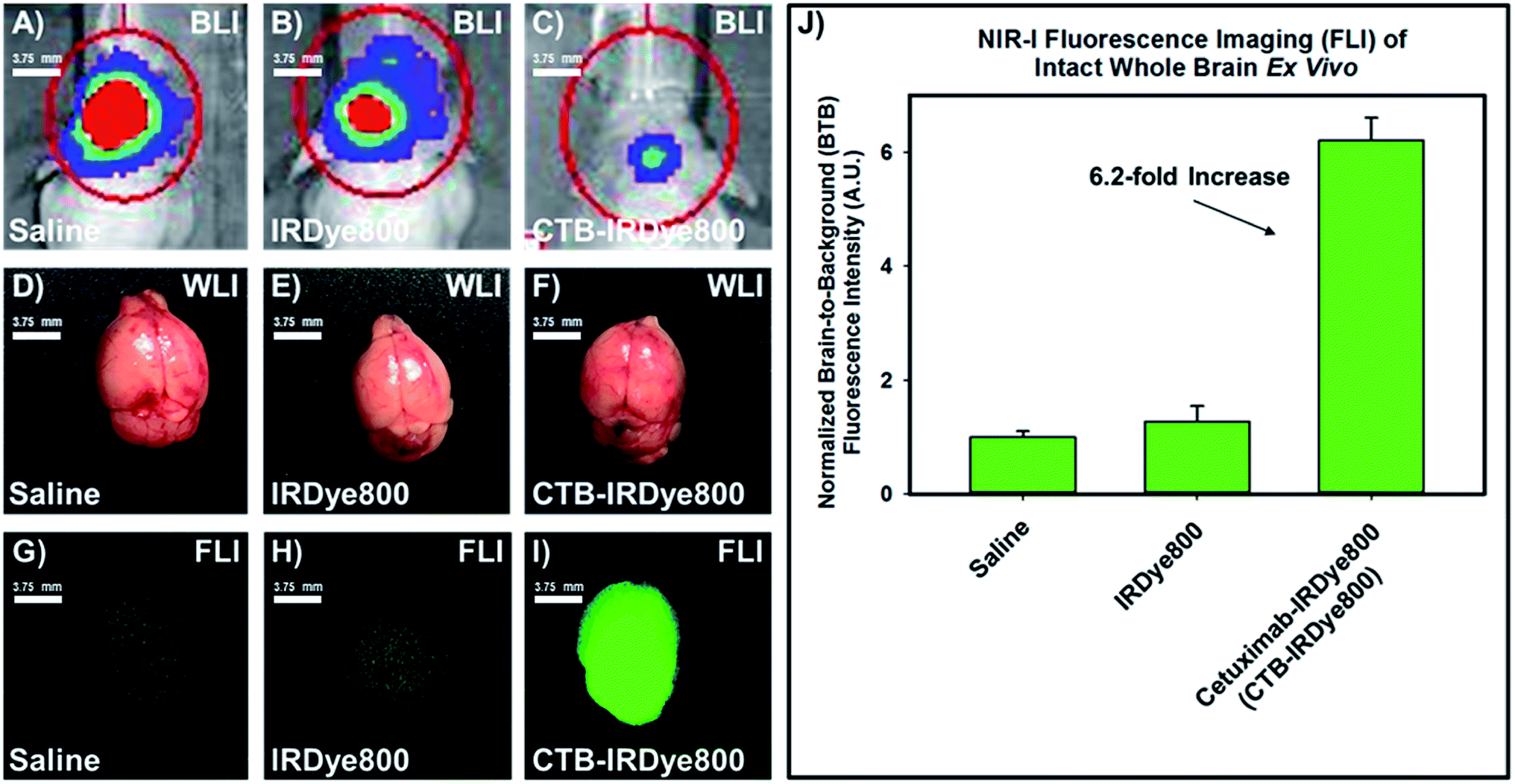
Next, we resected the whole brain while maintaining it intact from the three separate cohorts of orthotopic murine GBM39 tumor models and visualized them under standard WLI (Fig. 3D–F). A visual inspection revealed no differences could be discerned between the diseased tissue and the healthy tissue under standard WLI. However, a visual inspection revealed a drastic distinction between the diseased tissue and the healthy tissue for each cohort of orthotopic murine GBM39 tumor model using NIR-I FLI. Here, the resected intact whole brains from the cohorts that were administered saline and free IRDye800 displayed marginal fluorescence emission intensity (Fig. 3G and H), despite comprising the cohorts that demonstrated the highest total flux via utilizing BLI, respectively (Fig. 3A and B). Resected intact whole brain from the cohort that was administered the cetuximab–IRDye800 immunoconjugate displayed pronounced relative average NIR-I fluorescence emission intensity (Fig. 3I), despite comprising the cohort that displayed the lowest total flux via utilizing BLI (Fig. 3C). The fluorescence emission intensities of the brain from all three cohorts were quantified. The quantified average fluorescence emission intensities from the high-, mid, and low-total flux cohorts were compared and subsequently normalized to the fluorescence emission intensity from the high-total flux, wherein such was administered saline (Fig. 3J). The relative normalized averaged fluorescence emission intensity between to the high- and mid-total flux cohorts were essentially comparable. However, the relative normalized average fluorescence emission intensity between the low-total flux cohort and the high-total flux was 6.2-fold higher, wherein each were administered the cetuximab–IRDye800 immunoconjugate and saline, respectively.
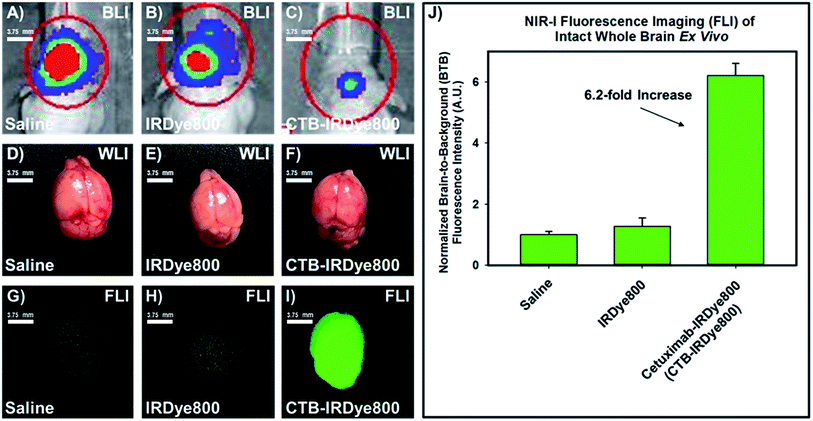 |
| | Fig. 3 Representative images of the resected (on day 7) intact whole brains from the three separate cohorts of orthotopic murine GBM39 tumor models via utilizing standard white-light illumination and NIR-I fluorescence imaging as well as their corresponding normalized relative average brain-to-background ratio when comparing their NIR-I fluorescence emission intensity (panel J). Panels (A–C) represent the in vivo BLI of the high-, mid-, and low-total flux cohort, respectively, and the assigned agent administered to a cohort that includes saline, free IRDye800, and cetuximab–IRDye800 immunoconjugate, respectively. Panels (D–F) represent the resected intact whole brain under WLI from the corresponding high-, mid-, and low-total flux cohort, respectively, wherein each cohort was separately administered saline, IRDye800 (free), or cetuximab–IRDye800 immunoconjugate, respectively. Panels (G–I) represent the resected intact whole brain under NIR-I FLI from the corresponding high-, mid-, and low-total flux cohort, wherein each cohort was separately administered saline, IRDye800 (free), or cetuximab–IRDye800 immunoconjugate, respectively. The bar graph comprising panel (J) is normalized to the average fluorescence emission intensity of the resected intact whole brain from the cohort of orthotopic murine GBM39 tumor models that were administered saline. BLI (luciferase activity) was visualized upon injecting such models intraperitoneally with a prepared (15 mg mL−1 in PBS) D-luciferin (firefly) potassium salt solution (150 μL) and imaging 10 minutes later with an exposure time of 0.3 s. The BLI emission was collected from 430–650 nm. Fluorescence emission in the NIR-I spectral region was collected at 820 ± 10 nm using a bandpass filter. Thus, for NIR-I FLI, all tumor and control murine models as well as resected brain were excited at 785 nm and the resultant fluorescence emission was collected from 810–830 nm. The scan time was 500 ms and the resolution was set at 170 μm. | |
2.4 BLI of intact whole brain via topical application of luciferin–ATP
To compare the results that we obtained via utilizing NIR-I FLI ex vivo, we liberally applied luciferin–ATP to the intact whole brain ex vivo from each cohort of orthotopic murine GBM39 tumor models (Fig. 4). On doing so, we confirmed the results we obtained when having performed BLI in vivo, wherein the high-, mid-, and low-total flux cohorts of orthotopic murine GBM39 tumor models that were administered saline, free IRDye800, and the cetuximab–IRDye800 immunoconjugate, were in line with all earlier results (Fig. 4A–C). As such, the relative values of the BLI total flux were in sync with those obtained when performing BLI in vivo. Accordingly, the cohort that saline was administered expressed over an 8-fold GBM39 tumor tissue when compared to the tumor model that was administered the immunoconjugate.
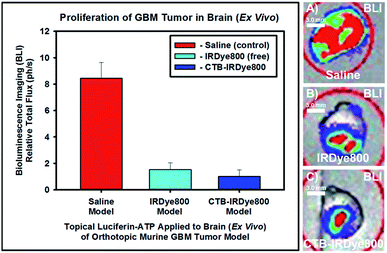 |
| | Fig. 4 Bioluminescence of resected intact whole brain. Topical luciferin–ATP was liberally applied, 6 minutes elapsed, and the total flux was obtained upon imaging: (A)–(C) are representative bioluminenscence images of the resected intact whole brain of the cohort of otrhotopic murine GBM39 tumor models that received saline. IRDye800, or cetuximab-IRDye800 model. respectively. The total flux was normalized to the cohort of orthotopic murine GBM39 tumor models that were administered cetuximab–IRDye800 immunoconjugate, as such exhibited the lowest average total flux. BLI (luciferase activity) was visualized upon injecting such models intraperitoneally with a prepared (15 mg mL−1 in PBS) D-luciferin (firefly) potassium salt solution (150 μL) and imaging 10 minutes later with an exposure time of 0.3 s. The BLI emission was collected from 430–650 nm. | |
2.5 Ex vivo whole-brain slices fluorescence imaging analysis
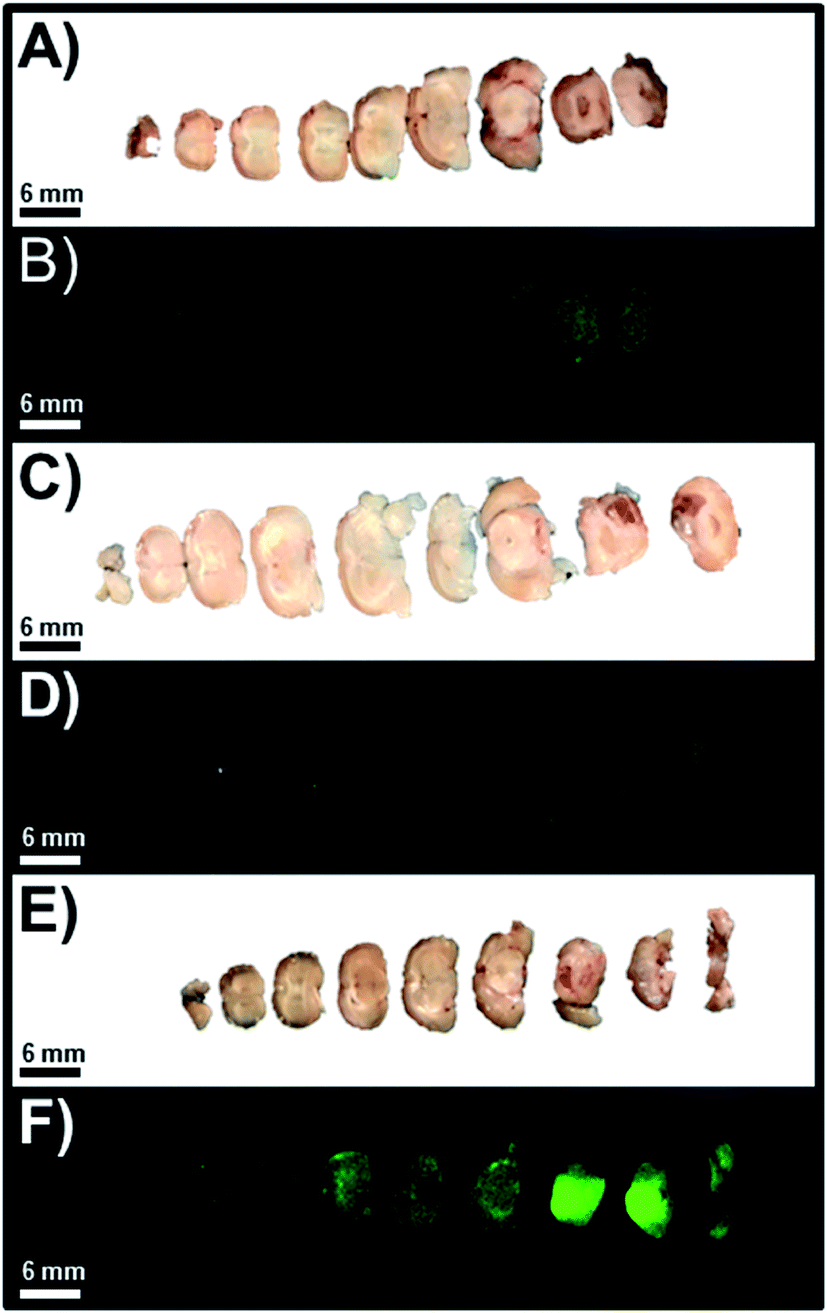
As anticipated and in accordance with the quantitative analysis of the resected intact whole brain NIR-I fluorescence images, we observed that the slices of the intact whole brain from the high-, mid-, and low-total flux cohorts of orthotopic murine GBM39 tumor models were consistent with previous results (Fig. 5). As such, we observed that the slices of intact whole brains from the high- and mid-total flux cohorts of orthotopic murine GBM39 tumor models displayed comparable NIR-I fluorescence emission intensity at marginal levels (Fig. 5A–D). However, we observed that the slices of the intact whole brain from the low-total flux cohort of orthotopic murine GBM39 tumor models displayed a pronounced NIR-I fluorescence emission intensity (Fig. 5E and F). Upon further inspection, it appeared that such NIR-I fluorescence emission was being unevenly elicited throughout the brain, which aligns well with the propensity of GBM39 to infiltrate and proliferate nonuniformly via innervating throughout the brain. Also, it appeared that the fluorescence emission was near the site of inoculation, which would be consistent with our implantation procedure.
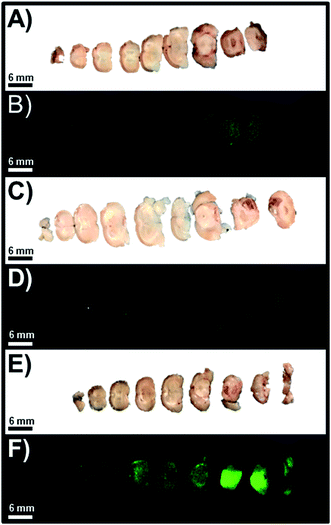 |
| | Fig. 5 Representative images of slices of resected whole brain that were separately visualized using standard WLI and FLI for the high-, mid-, and low-total flux cohorts of orthotopic murine GBM39 tumor models, which correspond to image set A and B, image set C and D, and image set E and F, respectively. The excitation source was a 785 nm solid-state laser diode. Fluorescence emission in the NIR-I spectral region was collected at 820 ± 10 nm using a bandpass filter. Thus, for NIR-I FLI, all tumor and control murine models as well as resected brain were excited at 785 nm and the resultant fluorescence emission was collected from 810–830 nm. The scan time was 500 ms and the resolution was set at 170 μm. | |
2.6 Ex vivo biodistribution NIR-I fluorescence imaging analysis
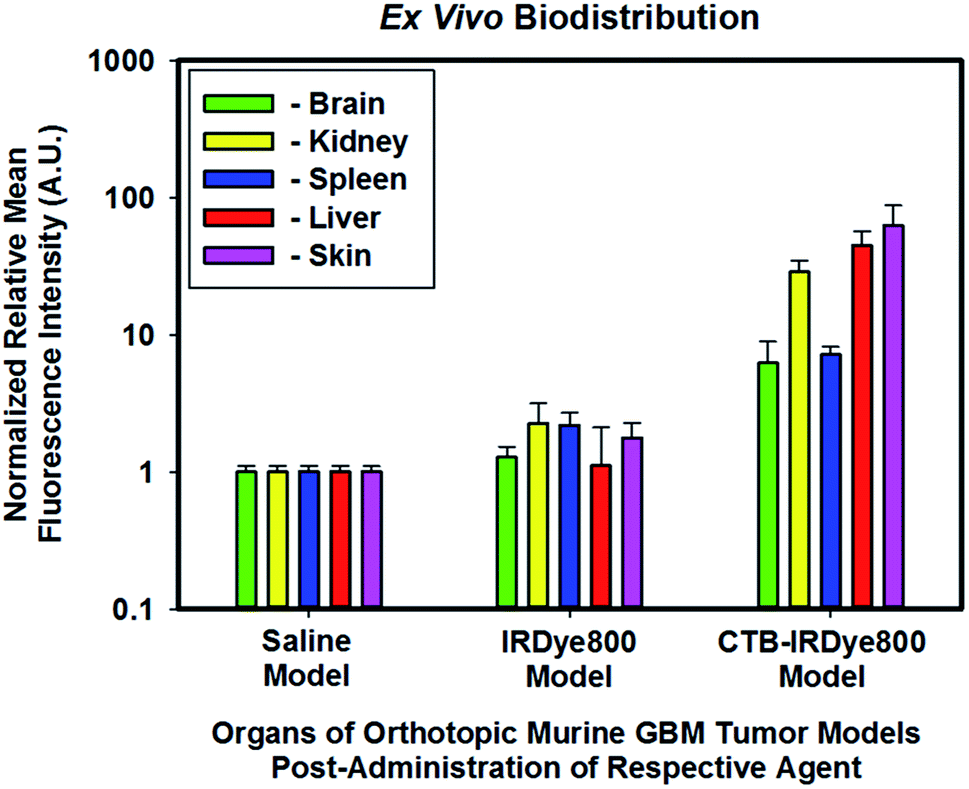
The mid-total flux cohort of orthotopic murine GBM39 tumor models that were administered unconjugated IRDye800 appeared to demonstrate a 1.27-, 2.26-, 2.56-, 1.11-, and 1.77-fold increase in the brain, kidney, spleen, liver, and skin, respectively relative to those of the high-total flux cohort of orthotopic murine GBM39 tumor models which were administered saline (Fig. 6). We anticipated these results simply because molecular fluorophores can engage in low-level nonspecific binding throughout the body, which appeared to have marginally done so. Interestingly, the cohort that was administered IRDye800 appeared to be primarily cleared by the renal system. Small molecules that are less than the kidney glomuleri pore size diameter of ∼5.5–8 nm, such as IRDye800, can readily pass through such pores and be filtered out by the renal system. The low-total flux cohort of orthotopic murine GBM39 tumor models which were administered cetuximab–IRDye800 appeared to demonstrate a 6.21-, 28.67-, 7.16-, 44.86-, and 62.83-fold increase in the brain, kidney, spleen, liver, and skin, respectively. As anticipated, these results align with the fact that hepatic clearance is the primary route for removal of biomolecules, such as immunoconjugates, due to their size, which is considerably larger than the kidney glomuleri pore diameter size of ∼5.5–8 nm. Endosomal post-processing of the immunoconjugate upon EGFR and/or EGFRvIII receptor binding, uptake, and turnover can account for the modest renal clearance, which is not unexpected. As the skin is fraught with EGFR and EGFRvIII receptors, the finding that the skin had the highest relative level of immunoconjugate was also anticipated.
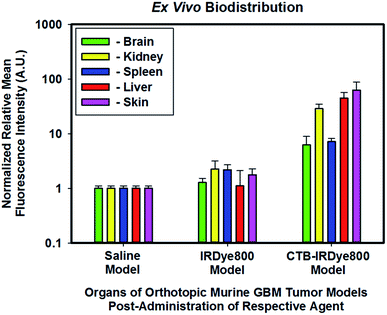 |
| | Fig. 6 Normalized relative averaged NIR-I fluorescence intensity of biodistribution of the immunoconjugate ex vivo. All organs from all three cohorts were normalized to the respective organs of the high-total flux cohort of orthotopic murine GBM tumor models which were administered saline. The excitation source was a 785 nm solid-state laser diode. Fluorescence emission in the NIR-I spectral region was collected at 820 ± 10 nm using a bandpass filter. Thus, for NIR-I FLI, all tumor and control murine models as well as resected brain were excited at 785 nm and the resultant fluorescence emission was collected from 810–830 nm. The scan time was 500 ms and the resolution was set at 170 μm. | |
3. Conclusion
GBM is a primary malignant tumor of the CNS that is the most common type of brain tumor as well as the most aggressive form of primary brain tumor types. As such, the current median survival rate for those afflicted with GBM is ∼15 months despite treatment. As it remains extraordinarily difficult to visually discriminate between anatomical structures as well as between diseased and healthy tissue under WLI, FGS has been implemented to enhance the capability of the operating surgeon to visually discriminate between such. Here, we established the feasibility of utilizing an EGFR-targeting fluorescent construct in the form of an immunoconjugate (cetuxmimab-IRDye800) to obtain visualization between diseased tissue (i) arising from a low-passage patient-derived GBM39 cell line and (ii) healthy tissue, via utilizing intracranial orthotopic murine GBM39 tumor models, in in vivo and ex vivo settings. We examined the in vivo BLI and NIR-I fluorescence emission of GBM39 proliferation in such models compared to controls, both of which progressed as would be anticipated. We next determined the NIR-I fluorescence emission of GBM39 tissue growth using resected intact and sliced whole brain ex vivo, wherein we obtained an average 6.2-fold enhancement in the cohort of GBM39 tumor models that were administered the immunoconjugate similarly relative to its controls. Ex vivo biodistribution studies revealed the immunoconjugate is primarily cleared by the liver, wherein any endosomal postprocessing affords entities that can be cleared by the kidney. In all, we established proof-of-concept of utilizing an immunoconjugate coupled with FLI to visualize tumor tissue in intracranial orthotopic murine patient-derived GBM39 tumor models. We look forward to correlating the ex vivo results with histological staining of the tumor tissue at the microscopic level in future studies.
4. Experimental
4.1 Cell culture
A patient-derived GBM cell line (GBM39) was used for our studies. We acknowledge the GBM39 cell line as a gift from Dr. Sanjiv Sam Gambhir who obtained them from Dr. Paul Mischel (Ludwig Institute for Cancer Research, University of California at San Diego). The GBM39 cells had been transfected with a lentiviral vector that expresses firefly luciferase such that having done enables BLI. The GBM39 cells were grown in a defined, serum-free media of a 1:1 mixture of Neurobasal-A Medium (1×)/DMEM/F12 (1×) that also contained HEPES buffer solution (10 mM), MEM sodium pyruvate solution (1 mM), MEM non-essential amino acids solution (10 mM, 1×), GlutaMAX-I supplement (1×) and antibiotic–antimycotic (1×). All solutions are from Invitrogen/Life Technologies Inc. The full working media also contained h-EGF (20 ng mL−1), h-FGF-basic-154 (20 ng mL−1), h-PDGF-AA (10 ng mL−1), h-PDGF-BB (10 ng mL−1) and heparin solution, 0.2% (2 μg mL−1) as growth factors (all from Shenandoah, Inc.) and B-27 (Invitrogen/Life Technologies) as supplements. All cells were propagated at 37 °C in a humidified atmosphere containing 5% CO2. The GBM39 cells were cultured as floating cellular spheres. Cells were sub-cultured when they reached 80–90% confluency. The cell layer was rinsed with PBS (Gibco #10010049) and 5 mL of 0.25% (w/v) Trypsin + 0.5 mM EDTA (GIBCO #25200056) solution was added to the attached cells until cell layer was dispersed (usually within 5 minutes). The reaction was terminated with 5 mL complete growth medium and cells were collected by gently pipetting. Cells were either used accordingly or were split 1:5 by adding about 5 × 106 cells per 75 cm2 flask in 15 mL of growth medium for further propagation.
4.2 Live subject ethical statement
All maintenance, handling, monitoring, and experimental procedures were performed in accordance to a protocol that was approved by The Administrative Panel on Laboratory Animal Care (APLAC) of Stanford University, wherein such approved protocol and committee comply with all federal and state regulations governing the humane care and use of laboratory animals.
4.3 In vivo tumor models
All animals were anesthetized with inhaled 2–3% isoflurane for surgical and imaging procedures, recovered, and fed ad liditum via having free access to food (containing alfalfa) and water. Eye lubricant and a heating pad were used during anesthetization. For intracranial xenograft GBM39 tumor models, GBM39 cells that were transfected with GFP and luciferase were injected (0.5 × 106 cells; 150 μL of serum-free media) stereotactically into the striatum of anesthetized female Nu/nu mice (aged 17–18 weeks; Charles River Laboratories) using the following coordinates: 2 mm posterior to the bregma, 2 mm lateral to the midline, and 3–4 mm deep with respect to the surface of the skull. The tumors were allowed to grow for 5–6 weeks prior to the models being used. Tumor growth was monitored via firefly luciferase BLI. Animals were scored for tumor formation by quantifying the peak radiance from BLI assays. Once tumor masses were detected via utilizing BLI assays, the mice were stratified into three cohorts based on total flux prior to administration of a particular agent as follows: high-, mid-, and low-total flux cohorts. We assigned the administration of saline (200 μL, 0.9% NaCl, USP, sterile grade) was assigned to the high-total flux cohort. We assigned the administration of IRDye800CW (LI-COR®, Lincoln, NE) to the mid-total flux cohort (200 μL, 27.44 μM in PBS, 6.40 μg, 5.49 nanomoles in unconjugated form). We assigned the administration of the conjugated form of cetuximab–IRDye800 (immunoconjugate) to the low-total flux cohort (200 μL, 2 mg mL−1, 400 μg, 5.49 nanomoles of conjugated IRDye800). The ratio of the unconjugated (free) IRDye800 form to the immunoconjugate form was 1:1.
4.4 Bioluminescence imaging
BLI was performed for ∼4 weeks post-inoculation on an IVIS Spectrum (Caliper Life Science). Luciferase activity was visualized by injecting mice intraperitoneally with a prepared (15 mg mL−1 in PBS) D-luciferin (firefly) potassium salt (Biosynth International, Inc.) solution (150 μL). The bioluminescence was monitored after 10 min. After placing the mice under nose-cone anesthesia immediately prior to imaging using an exposure time of 0.3 s. The emitted total flux (photons per second) was measured by imaging the mice until peak radiance was achieved, wherein the bioluminescence emission was collected from 430–650 nm. Peak radiance was quantified with Living Image 4.0 software (PerkinElmer; Waltham, MA). BLI was repeated daily immediately prior to and after upon administration of either saline, IRDye800, or cetuximab–IRDye800.
4.5 Cetuximab-IRDye800
The cetuximab–IRDye800 was produced under cGMP conditions at the University of Alabama (UAB) Vector Production Facility before shipment to Stanford University Hospital Pharmacy, which is nearly identical to that found in the literature.30 In brief, cetuximab (ImClone LLC, Eli Lilly and Company) was provided as a 2 mg mL−1 solution, concentrated, and the pH was adjusted by buffer exchange to a 10 mg mL−1 solution in 50 mmol L−1 potassium phosphate, pH 8.5. IRDye® 800CW NHS ester (LI-COR® Biosciences, Lincoln, NE) was mixed with cetuximab for 2 hours at 20 °C in the dark to enable its conjugation to such, which provided an average molar ratio of 1:2.3 cetuximab:IRDye800. After column filtration to remove unconjugated dye and exchanging buffer to phosphate-buffered saline, pH: 7, the final protein concentration adjusted to 2 mg mL−1, the product was sterilized by filtration and placed into single-use vials. After conjugation, the agent was shipped to Stanford University Hospital Pharmacy. During shipping of the immunoconjugate, the temperature was stable at 4 °C. The immunoconjugate was stored at 4 °C until used upon its receipt. The same lot was used for the cohort of animals in the treatment group.
4.6 NIR-I fluorescence imaging
All brightfield and NIR-I fluorescence images pre- and post-injection of saline, IRDye800, or the immunoconjugate were captured using a Pearl® Trilogy Small Animal Imaging System (LI-COR® Biosciences, Lincoln, NE), wherein a thermoelectrically cooled Si-based charged coupled device (CCD) camera was pre-installed by the manufacturer. The excitation source was a 785 nm solid-state laser diode. Fluorescence emission in the NIR-I spectral region was collected at 820 ± 10 nm using a bandpass filter. Thus, for all NIR-I FLI, all tumor and control murine models as well as resected brain were excited at 785 nm and the resultant fluorescence emission was collected from 810–830 nm. The scan time was 500 ms and the resolution was set at 170 μm.
4.7 Statistical analysis
Unless otherwise noted, data were expressed as the mean ± standard deviation of the mean and analyzed using one-way analysis of variance, followed by Tukey post hoc tests or unpaired t-test with Welch's correction from GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Author contributions
Project designed and managed by KSH; Imaging performed by and data acquired by NTT and RE; data processed and analyzed by KSH and NTT; all renditions of the manuscript including the final manuscript draft were written by KSH; the final manuscript was reviewed by all parties.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
We thank Dr Eben L. Rosenthal at Stanford University for providing us with cetuximab-IRDye800 immunoconjugate and allowing us to use his LI-COR® Pearl Trilogy Small Animal Imaging System for NIR-I FLI. FTC's contribution to this work was supported by the Department of Energy (DOE DE-SC0008397), the Ben and Catherine Ivy Foundation, and the National Institute of Health (NIH/NCI R21 CA205564). KSH's contribution to this work was supported, in part, by NIH/NCI NRSA fellowship (F32 CA213620).
References
- D. M. Polikarpov, D. H. Campbell, L. S. McRobb, J. Wu, M. E. Lund, Y. Lu, S. M. Deyev, A. S. Davidson, B. J. Walsh, A. V. Zvyagin and D. A. Gillatt, Near-Infrared Molecular Imaging of Glioblastoma by Miltuximab®-IRDye800CW as a Potential Tool for Fluorescence-Guided Surgery, Cancers, 2020, 12(4), 984, DOI:10.3390/cancers12040984.
- J. Yang, Z. Shi, R. Liu, Y. Wu and X. Zhang, Combined-Therapeutic Strategies Synergistically Potentiate Glioblastoma Multiforme Treatment via Nanotechnology, Theranostics, 2020, 10(7), 3223–3239, DOI:10.7150/thno.40298.
- G. Reifenberger, H.-G. Wirsching, C. B. Knobbe-Thomsen and M. Weller, Advances in the Molecular Genetics of Gliomas — Implications for Classification and Therapy, Nat. Rev. Clin. Oncol., 2017, 14(7), 434–452, DOI:10.1038/nrclinonc.2016.204.
- H. Zong, L. F. Parada and S. J. Baker, Cell of Origin for Malignant Gliomas and Its Implication in Therapeutic Development, Cold Spring Harbor Perspect. Biol., 2015, 7(5) DOI:10.1101/cshperspect.a020610.
- Q. T. Ostrom, H. Gittleman, J. Fulop, M. Liu, R. Blanda, C. Kromer, Y. Wolinsky, C. Kruchko and J. S. Barnholtz-Sloan, CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012, Neuro-Oncology, 2015, 17(Suppl 4), iv1–iv62, DOI:10.1093/neuonc/nov189.
- A. Shergalis, A. Bankhead, U. Luesakul, N. Muangsin and N. Neamati, Current Challenges and Opportunities in Treating Glioblastoma, Pharmacol. Rev., 2018, 70(3), 412–445, DOI:10.1124/pr.117.014944.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1, Adv. Drug Delivery Rev., 2001, 46(1), 3–26, DOI:10.1016/S0169-409X(00)00129-0.
- D. Wang, C. Wang, L. Wang and Y. Chen, A Comprehensive Review in Improving Delivery of Small-Molecule Chemotherapeutic Agents Overcoming the Blood-Brain/Brain Tumor Barriers for Glioblastoma Treatment, Drug Deliv., 2019, 26(1), 551–565, DOI:10.1080/10717544.2019.1616235.
- Y. J. Y. Zuchero, X. Chen, N. Bien-Ly, D. Bumbaca, R. K. Tong, X. Gao, S. Zhang, K. Hoyte, W. Luk, M. A. Huntley, L. Phu, C. Tan, D. Kallop, R. M. Weimer, Y. Lu, D. S. Kirkpatrick, J. A. Ernst, B. Chih, M. S. Dennis and R. J. Watts, Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies, Neuron, 2016, 89(1), 70–82, DOI:10.1016/j.neuron.2015.11.024.
- E. Chang, C. Pohling, A. Natarajan, T. H. Witney, J. Kaur, L. Xu, G. Gowrishankar, A. L. D'Souza, S. Murty, S. Schick, L. Chen, N. Wu, P. Khaw, P. Mischel, T. Abbasi, S. Usmani, P. Mallick and S. S. Gambhir, AshwaMAX and Withaferin A Inhibits Gliomas in Cellular and Murine Orthotopic Models, J. Neuro Oncol., 2016, 126(2), 253–264, DOI:10.1007/s11060-015-1972-1.
- Z. An, O. Aksoy, T. Zheng, Q.-W. Fan and W. A. Weiss, Epidermal Growth Factor Receptor (EGFR) and EGFRvIII in Glioblastoma (GBM): Signaling Pathways and Targeted Therapies, Oncogene, 2018, 37(12), 1561–1575, DOI:10.1038/s41388-017-0045-7.
- C. W. Brennan, R. G. W. Verhaak, A. McKenna, B. Campos, H. Noushmehr, S. R. Salama, S. Zheng, D. Chakravarty, J. Z. Sanborn, S. H. Berman, R. Beroukhim, B. Bernard, C.-J. Wu, G. Genovese, I. Shmulevich, J. Barnholtz-Sloan, L. Zou, R. Vegesna, S. A. Shukla, G. Ciriello, W. K. Yung, W. Zhang, C. Sougnez, T. Mikkelsen, K. Aldape, D. D. Bigner, E. G. Van Meir, M. Prados, A. Sloan, K. L. Black, J. Eschbacher, G. Finocchiaro, W. Friedman, D. W. Andrews, A. Guha, M. Iacocca, B. P. O'Neill, G. Foltz, J. Myers, D. J. Weisenberger, R. Penny, R. Kucherlapati, C. M. Perou, D. N. Hayes, R. Gibbs, M. Marra, G. B. Mills, E. Lander, P. Spellman, R. Wilson, C. Sander, J. Weinstein, M. Meyerson, S. Gabriel, P. W. Laird, D. Haussler, G. Getz, L. Chin, C. Benz, J. Barnholtz-Sloan, W. Barrett, Q. Ostrom, Y. Wolinsky, K. L. Black, B. Bose, P. T. Boulos, M. Boulos, J. Brown, C. Czerinski, M. Eppley, M. Iacocca, T. Kempista, T. Kitko, Y. Koyfman, B. Rabeno, P. Rastogi, M. Sugarman, P. Swanson, K. Yalamanchii, I. P. Otey, Y. S. Liu, Y. Xiao, J. T. Auman, P.-C. Chen, A. Hadjipanayis, E. Lee, S. Lee, P. J. Park, J. Seidman, L. Yang, R. Kucherlapati, S. Kalkanis, T. Mikkelsen, L. M. Poisson, A. Raghunathan, L. Scarpace, B. Bernard, R. Bressler, A. Eakin, L. Iype, R. B. Kreisberg, K. Leinonen, S. Reynolds, H. Rovira, V. Thorsson, I. Shmulevich, M. J. Annala, R. Penny, J. Paulauskis, E. Curley, M. Hatfield, D. Mallery, S. Morris, T. Shelton, C. Shelton, M. Sherman, P. Yena, L. Cuppini, F. DiMeco, M. Eoli, G. Finocchiaro, E. Maderna, B. Pollo, M. Saini, S. Balu, K. A. Hoadley, L. Li, C. R. Miller, Y. Shi, M. D. Topal, J. Wu, G. Dunn, C. Giannini, B. P. O'Neill, B. A. Aksoy, Y. Antipin, L. Borsu, S. H. Berman, C. W. Brennan, E. Cerami, D. Chakravarty, G. Ciriello, J. Gao, B. Gross, A. Jacobsen, M. Ladanyi, A. Lash, Y. Liang, B. Reva, C. Sander, N. Schultz, R. Shen, N. D. Socci, A. Viale, M. L. Ferguson, Q.-R. Chen, J. A. Demchok, L. A. L. Dillon, K. R. M. Shaw, M. Sheth, R. Tarnuzzer, Z. Wang, L. Yang, T. Davidsen, M. S. Guyer, B. A. Ozenberger, H. J. Sofia, J. Bergsten, J. Eckman, J. Harr, J. Myers, C. Smith, K. Tucker, C. Winemiller, L. A. Zach, J. Y. Ljubimova, G. Eley, B. Ayala, M. A. Jensen, A. Kahn, T. D. Pihl, D. A. Pot, Y. Wan, J. Eschbacher, G. Foltz, N. Hansen, P. Hothi, B. Lin, N. Shah, J. Yoon, C. Lau, M. Berens, K. Ardlie, R. Beroukhim, S. L. Carter, A. D. Cherniack, M. Noble, J. Cho, K. Cibulskis, D. DiCara, S. Frazer, S. B. Gabriel, N. Gehlenborg, J. Gentry, D. Heiman, J. Kim, R. Jing, E. S. Lander, M. Lawrence, P. Lin, W. Mallard, M. Meyerson, R. C. Onofrio, G. Saksena, S. Schumacher, C. Sougnez, P. Stojanov, B. Tabak, D. Voet, H. Zhang, L. Zou, G. Getz, N. N. Dees, L. Ding, L. L. Fulton, R. S. Fulton, K.-L. Kanchi, E. R. Mardis, R. K. Wilson, S. B. Baylin, D. W. Andrews, L. Harshyne, M. L. Cohen, K. Devine, A. E. Sloan, S. R. VandenBerg, M. S. Berger, M. Prados, D. Carlin, B. Craft, K. Ellrott, M. Goldman, T. Goldstein, M. Grifford, D. Haussler, S. Ma, S. Ng, S. R. Salama, J. Z. Sanborn, J. Stuart, T. Swatloski, P. Waltman, J. Zhu, R. Foss, B. Frentzen, W. Friedman, R. McTiernan, A. Yachnis, D. N. Hayes, C. M. Perou, S. Zheng, R. Vegesna, Y. Mao, R. Akbani, K. Aldape, O. Bogler, G. N. Fuller, W. Liu, Y. Liu, Y. Lu, G. Mills, A. Protopopov, X. Ren, Y. Sun, C.-J. Wu, W. K. A. Yung, W. Zhang, J. Zhang, K. Chen, J. N. Weinstein, L. Chin, R. G. W. Verhaak, H. Noushmehr, D. J. Weisenberger, M. S. Bootwalla, P. H. Lai, T. J. Triche, D. J. Van Den Berg, P. W. Laird, D. H. Gutmann, N. L. Lehman, E. G. VanMeir, D. Brat, J. J. Olson, G. M. Mastrogianakis, N. S. Devi, Z. Zhang, D. Bigner, E. Lipp and R. McLendon, The Somatic Genomic Landscape of Glioblastoma, Cell, 2013, 155(2), 462–477, DOI:10.1016/j.cell.2013.09.034.
- S. Jawhari, M.-H. Ratinaud and M. Verdier, Glioblastoma, Hypoxia and Autophagy: A Survival-Prone ‘Ménage-à-Trois, Cell Death Dis., 2016, 7(10), e2434, DOI:10.1038/cddis.2016.318.
- M. Fakih and R. Wong, Efficacy of the Monoclonal Antibody EGFR Inhibitors for the Treatment of Metastatic Colorectal Cancer, Curr. Oncol., 2010, 17(Suppl. 1), S3–S17 Search PubMed.
- K. Shitara, T. Yokota and S. Utsunomiya, Cetuximab for Patients With Colon Cancer and Hepatic Metastasis Complicated by Liver Dysfunction and Icterus, Gastrointest. Cancer
Res., 2009, 3(4), 171 Search PubMed.
- W. Ping Li, L. A. Meyer, D. A. Capretto, C. D. Sherman and C. J. Anderson, Receptor-Binding, Biodistribution, and Metabolism Studies of 64Cu-DOTA-Cetuximab, a PET-Imaging Agent for Epidermal Growth-Factor Receptor-Positive Tumors, Canc. Biother. Rad., 2008, 23(2), 158–171, DOI:10.1089/cbr.2007.0444.
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma, N. Engl. J. Med., 2005, 352(10), 987–996, DOI:10.1056/NEJMoa043330.
- Y. Zheng, H. Yang, H. Wang, K. Kang, W. Zhang, G. Ma and S. Du, Fluorescence-Guided Surgery in Cancer Treatment: Current Status and Future Perspectives, Ann. Transl. Med., 2019, 7(Suppl. 1), S6, DOI:10.21037/atm.2019.01.26.
- Q. T. Nguyen and R. Y. Tsien, Fluorescence-Guided Surgery with Live Molecular Navigation — a New Cutting Edge, Nat. Rev. Cancer, 2013, 13(9), 653–662, DOI:10.1038/nrc3566.
- T. Nagaya, Y. A. Nakamura, P. L. Choyke and H. Kobayashi, Fluorescence-Guided Surgery, Front. Oncol., 2017, 7, 314, DOI:10.3389/fonc.2017.00314.
- J. C. DeLong, R. M. Hoffman and M. Bouvet, Current Status and Future Perspectives of Fluorescence-Guided Surgery for Cancer, Expet Rev. Anticancer Ther., 2016, 16(1), 71–81, DOI:10.1586/14737140.2016.1121109.
- L. E. Kelderhouse, V. Chelvam, C. Wayua, S. Mahalingam, S. Poh, S. A. Kularatne and P. S. Low, Development of Tumor-Targeted Near Infrared Probes for Fluorescence Guided Surgery, Bioconjugate Chem., 2013, 24(6), 1075–1080, DOI:10.1021/bc400131a.
- A. Haque, Md. S. H. Faizi, J. A. Rather and M. S. Khan, Next Generation NIR Fluorophores for Tumor Imaging and Fluorescence-Guided Surgery: A Review, Bioorg. Med. Chem., 2017, 25(7), 2017–2034, DOI:10.1016/j.bmc.2017.02.061.
- P. S. Low, S. Singhal and M. Srinivasarao, Fluorescence-Guided Surgery of Cancer: Applications, Tools and Perspectives, Curr. Opin. Chem. Biol., 2018, 45, 64–72, DOI:10.1016/j.cbpa.2018.03.002.
- S. van Keulen, N. Nishio, S. Fakurnejad, A. Birkeland, B. A. Martin, G. Lu, Q. Zhou, S. U. Chirita, T. Forouzanfar, A. D. Colevas, N. S. van den Berg and E. L. Rosenthal, The Clinical Application of Fluorescence-Guided Surgery in Head and Neck Cancer, J. Nucl. Med., 2019, 60(6), 758–763, DOI:10.2967/jnumed.118.222810.
- Y. H. Quan, C. H. Oh, D. Jung, J.-Y. Lim, B. H. Choi, J. Rho, Y. Choi, K. N. Han, B.-M. Kim, C. Kim, J.-H. Park and H. K. Kim, Evaluation of Intraoperative Near-Infrared Fluorescence Visualization of the Lung Tumor Margin With Indocyanine Green Inhalation, JAMA Surg., 2020, 155(8), 732–740, DOI:10.1001/jamasurg.2020.1314.
- R. P. Judy, J. J. Keating, E. M. DeJesus, J. X. Jiang, O. T. Okusanya, S. Nie, D. E. Holt, S. P. Arlauckas, P. S. Low, E. J. Delikatny and S. Singhal, Quantification of Tumor Fluorescence during Intraoperative Optical Cancer Imaging, Sci. Rep., 2015, 5(1), 16208, DOI:10.1038/srep16208.
- J. D. Predina, A. D. Newton, J. Keating, A. Dunbar, C. Connolly, M. Baldassari, J. Mizelle, L. Xia, C. Deshpande, J. Kucharczuk, P. S. Low and S. Singhal, A Phase I Clinical Trial of Targeted Intraoperative Molecular Imaging for Pulmonary Adenocarcinomas, Ann. Thorac. Surg., 2018, 105(3), 901–908, DOI:10.1016/j.athoracsur.2017.08.062.
- J. T. Senders, I. S. Muskens, R. Schnoor, A. V. Karhade, D. J. Cote, T. R. Smith and M. L. D. Broekman, Agents for Fluorescence-Guided Glioma Surgery: A Systematic Review of Preclinical and Clinical Results, Acta Neurochir., 2017, 159(1), 151–167, DOI:10.1007/s00701-016-3028-5.
- W. S. Tummers, S. E. Miller, N. T. Teraphongphom, A. Gomez, I. Steinberg, D. M. Huland, S. Hong, S.-R. Kothapalli, A. Hasan, R. Ertsey, B. A. Bonsing, A. L. Vahrmeijer, R.-J. Swijnenburg, T. A. Longacre, G. A. Fisher, S. S. Gambhir, G. A. Poultsides and E. L. Rosenthal, Intraoperative Pancreatic Cancer Detection Using Tumor-Specific Multimodality Molecular Imaging, Ann. Surg. Oncol., 2018, 25(7), 1880–1888, DOI:10.1245/s10434-018-6453-2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07245a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Nutte Tarn Teraphongphomb,
Robert D. Ertseyb,
Eben L. Rosenthalb and
Frederick T. Chin*a
*ab,
Nutte Tarn Teraphongphomb,
Robert D. Ertseyb,
Eben L. Rosenthalb and
Frederick T. Chin*a