
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue light-induced low mechanical stability of ruthenium-based coordination bonds: an AFM-based single-molecule force spectroscopy study†
Mohd. Muddassir *
*
Department of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia. E-mail: mmohammadarshad@ksu.edu.sa; muddassirchem@gmail.com
First published on 6th November 2020
Abstract
Metal complexes containing coordination bonds play a prominent role in many essential biological systems in living organisms. Examples of such complexes include hemoglobin containing iron, chlorophyll containing magnesium, and vitamin B12 containing cobalt. Although the thermodynamic and other collective properties of metal complexes are well established, their mechanical stability remains minimally explored. Single-molecule force spectroscopy has been used to determine the structural and mechanical properties of chemical bonds; however, it has been minimally utilized in the field of coordination chemistry. Thus, here, we select a unique molecule of interest, HA–RuII, {HA = hyaluronan and RuII = [(bpy)2Ru(4-pyNH2)2](PF6)2} and subject it to single-molecule force spectroscopy analysis to directly study its bond-rupture process. The molecule is excited by blue-light irradiation, and surprisingly, this whole process could be reversed without applying any external energy, such as heat or solvent exposure. Our results demonstrate the reversibility of the luminescent RuII complex to its original state, a phenomenon that can be further applied to other coordination compounds.
Introduction
Since Werner published his seminal theory of coordination complexes in 1893, for which he was awarded the Nobel Prize in chemistry in 1913, coordination chemistry has garnered significant research interest.1,2 The immense information on coordination complexes is of paramount importance in general chemistry, fundamental chemistry, metalloproteins, and biochemistry, as well as for highly versatile applications in various fields, such as biomedicine, industrial catalysis, and superconductivity. Conventionally, a coordination complex contains a central metal atom, which is bonded to a close selection of molecules known as ligands. Dative or coordinative bonds are a distinctive class of chemical covalent bonds, which result in strong dipolar character and complex electronic interactions between the central metal atom and ligands. The determination of the exact mechanical nature or strength of the coordinative bond is still significantly challenging. Therefore, not surprisingly, extensive investigations on metal–ligand interactions have constituted one of the central themes in coordination chemistry.Coordinative bonds are considerably weaker than covalent bonds but stronger than hydrogen bonding, van der Waal interactions, and hydrophobic interactions, and they are widely found in natural organisms.3–6 They provide sufficient mechanical stability while acting as a sacrificial motif to dissipate external force to protect covalent linkages from fracture.7–10 These peculiar properties of coordination bonds endow natural organisms with both high strength and toughness for load-bearing processes and energy consumption, and have inspired the development of many artificial materials.11–17 Moreover, most coordination bonds are reversible. They can quickly reform when an external force is applied, which endows the material with “self-healing” property and increases the creep resistance of thermosets. Recently, via metal–ligand interactions, several materials with dynamic exchangeable crosslinks incorporated into the covalent framework were designed to increase the material plasticity and strength.18–20 Based on the high exchange rate of the dynamic bonds, Nishimura et al. utilized silyl ether as a robust and thermally stable motif to prepare a malleable material.21 This material displayed tunable mechanical property coupled with both malleability and processability. Exploiting the single-molecule force spectroscopy (SMFS) technique, Li et al. found that the catechol–Fe3+ complex has different mechanical stabilities for mono-, bis-, and tris-complexes and that the stoichiometry of the complex can be triggered by varying the pH and Fe3+ concentration.22 Although there are numerous environmentally responsive metal–ligand motifs, it is not practical to modulate the mechanical properties of a material by changing the environmental conditions, such as pH and ion concentration, or by adjusting the material components. However, light is a different trigger of choice, offering the possibility of altering the mechanical properties of a material by implanting photochromic molecules.23 Ru(bpy3)2+ and its derivatives are well characterized in the fields of transition-metal photocatalysis and solar energy conversion, as well as in biomedical applications involving blue light, because of their extraordinary photophysical properties and chemical stability.24,25 Ru(II) complexes with polypyridyl ligands function as molecular light switches, because of their combination of easily constructed rigid chiral structures spanning all three spatial dimensions and rich photophysical repertoire.26–29
[Ru(bpy)3]2+ and its derivatives have been studied for their potential utility in the construction of molecule-based photoresponsive machines or to mimic the functions performed by natural systems, such as solar energy conversion and storage, due to their relatively long-lived triplet metal-to-ligand charge transfer (3MLCT) state. However, their mechanical strength remains minimally explored.30–32 A profound understanding of the mechanical properties of [Ru(bpy)3]2+ and its derivatives will stimulate the engineering and tailoring of the mechanical performance of this important class of materials. It will be interesting to reveal the mechanical strength of Ru(bpy3)2+-based complexes and elucidate the process of tuning their mechanical stability using light irradiation.
Atomic force microscopy (AFM)-based SMFS (AFM-SMFS) has become a powerful tool for detecting protein folding, metal–ligand bond strength, surface adhesion, and many other biomechanical interactions,33–38 which cannot be uncovered by other traditional thermodynamics methods. Here, we exploit AFM-SMFS to study the mechanical strength of a Ru–pyridine complex, using synthesized single-chain polymeric nanoparticles as pyridine cores, which are ubiquitous in natural products and widely used for the construction of supramolecular compounds. In this approach, pyridine was modified on a hyaluronic acid chain, and the free Ru(bpy2)2+ units were buried inside the polymer chain; this is analogous to metalloprotein, but with simplified chemical environments. When the AFM cantilever stretches the nanoparticle, the folded chain is unraveled, resulting in the stepwise rupture of the Ru–N(pyridine) bonds. Thus, we can quantitatively measure the bond strength. This method can significantly increase the efficiency of obtaining sufficient high-quality single-molecule data, and the synthetic procedure is straightforward. By introducing external blue light, we can detect the changes in the mechanical properties, if any.23
In this study, we demonstrate that the Ru–N(pyridine) bond ruptures at 50–150 pN at a pulling speed of 1000 nm s−1. Thus, this bond is considerably weaker than typical covalent bonds (∼1–3 nN),39,40 stronger than hydrogen bonding (∼150 pN)41,42 and hydrophobic interactions (∼30–60 pN),43,44 and comparable to other coordinative bonds reported in the literature.22,45 Moreover, the Ru–N(pyridine) bond is considerably weakened under exposure to blue light (450 nm). Using dynamic pulling experiments based on Bell's model,46 we extract the kinetic parameters of the Ru–N bond. Our results indicate that the Ru–N motif could be an ideal candidate for use as a load-bearing material with tunable mechanical properties, and the SMFS results could guide the bottom-up design of new materials.
Experimental section
Chemicals and reagents
cis-[Ru-(bpy)2Cl2]·2H2O (97%) from Sigma-Aldrich, 4-aminopyridine (99%), NH4PF6 (99.9%, trace metals basis), KBr (99%, trace metals basis), EDC (98%) from Sigma-Aldrich, HA (MW: 150 kDa; polydispersity: 1.4, Freda Biopharm, Shandong, China) and were used as received.Physical measurements
All the synthetic procedures were carried out using Schlenk techniques and vacuum-line systems under a dry argon atmosphere, unless otherwise specified. The infrared (IR) spectra were recorded with a VECTOR 22 spectrometer using KBr pellets in the 400–4000 cm−1 region. Elemental analyses of C, H, and N were performed on a PerkinElmer 240C elemental analyzer. Electronic spectra were recorded on the UV-1700 PharmaSpec UV-vis spectrophotometer (550 Jasco). The data were reported in λmax/nm. The 1H NMR spectra were recorded using a Bruker DRX-400 spectrometer. The chemicals and solvents were purchased from commercial sources.[(bpy)2Ru(4-pyNH2)2](PF6)2 was synthesized as reported earlier.47 Briefly, the precursor complex, cis-[Ru-(bpy)2Cl2]·2H2O (228 mg, 5 mmol), and 4-aminopyridine (941 mg, 10 mmol) were dissolved in a water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol solution (1:1, 50 mL), and the mixture was heated to reflux for 12 h, at which point the color of the solution changed from dark red to cherry red. The cherry-red solution was filtered hot to remove any impurity, and the solvent was removed under vacuum. Finally, NH4PF6 (244 mg, 15 mmol) was added to obtain a red solid precipitate. Single crystals suitable for X-ray diffraction measurements were obtained after one week of slow evaporation of an acetonitrile solution at room temperature. The X-ray crystal structure of the complex with the atomic numbering scheme is shown in Fig. S1.†
:ethanol solution (1:1, 50 mL), and the mixture was heated to reflux for 12 h, at which point the color of the solution changed from dark red to cherry red. The cherry-red solution was filtered hot to remove any impurity, and the solvent was removed under vacuum. Finally, NH4PF6 (244 mg, 15 mmol) was added to obtain a red solid precipitate. Single crystals suitable for X-ray diffraction measurements were obtained after one week of slow evaporation of an acetonitrile solution at room temperature. The X-ray crystal structure of the complex with the atomic numbering scheme is shown in Fig. S1.†
Synthesis of HA–RuII complex
Briefly, 60.0 mg of HA (MW: 150 kDa) was dissolved in deionized water in an open round-bottom flask. Thereafter, 46.5 mg of EDC was added to the HA solution and stirred for 0.5 h to convert the carboxyl groups of HA to amine-reactive acylisourea intermediates. Next, 178.4 mg (0.200 mmol) of the RuII complex pre-dissolved in 5 mL of acetonitrile was transferred into the EDC/HA solution to conjugate the RuII complex to HA. The reaction was conducted for 24 h at room temperature under magnetic stirring. The conjugate was purified by dialysis against excess Milli-Q water using dialysis tubing with the molecular weight cutoff of 3.5 kDa. The final product was lyophilized and stored at −20 °C for subsequent single-molecule AFM experiments. The conjugation rate was estimated to be ∼22% based on the NMR spectrum of the HA–Ru complex (Fig. S2†).X-ray diffraction studies
Single crystal X-ray diffraction data collection was carried out at 100 K using a Bruker SMART APEX CCD diffractometer, equipped with a graphite mono-chromated Mo-Kα (λ = 0.71073 Å) radiation source. Data reduction and unit cell refinement for complex [(bpy)2Ru(4-pyNH2)2](PF6)2 were performed using SAINT-Plus.48 Using OLEX2,49 the SHELXL-2014/7 (ref. 50) software was used to solve the structure of complex [(bpy)2Ru(4-pyNH2)2](PF6)2 by the direct method; the refinement was carried out by full-matrix least-squares, based on F2 values against all reflections. Hydrogen atoms were fixed at the calculated positions with isotropic thermal parameters. Details of the crystallographic data collection, structural determination, and refinement are summarized in Table S1,† and the selected bond distances and angles for the complexes are listed in Table S2.† CCDC number: 1416079.AFM force spectroscopy experiments
AFM force spectroscopy experiments were carried out on a commercial atomic force microscope (NanoWizard II, JPK Instruments, Berlin, Germany) mounted on the top of an Axiovert 200 inverted microscope (Carl Zeiss, Jena, Germany). Light from a 452 nm blue laser (Melles-Griot, Carlsbad, CA) was focused on an ×10 objective to excite the HA–RuII complex at 1000 mW cm−2. Only force–distance curves containing at least three consecutive rupture events were selected for analysis. The force–distance curves were recorded by a commercial software from JPK and analyzed by custom-written procedures in Igor Pro 6.3 (Wavemetrics, Inc.).First, the glass coverslips (Fisher Scientific) were placed in a warm chromium acid solution for 3 h to remove residual organic matter; afterward, the coverslips were rinsed with Milli-Q water and dried under a stream of nitrogen. Next, several drops (∼50–100 μL) of the HA–RuII complex solution (1 mg mL−1) were added onto a clean glass coverslip. The complex was allowed to adsorb onto the coverslip for ∼30 min before the AFM experiment, followed by the addition of a Tris buffer (∼1500 μL, pH 7.4, 100 mM) into the fluid chamber. The AFM experiments were conducted after allowing the system to equilibrate for 30 min. Silicon nitride cantilevers (type MLCT, from Bruker) were used in all operations without any covalent attachment or modification on the substrate. The spring constants of the tips, calibrated by the thermal fluctuation method, were in the range of 0.031–0.055 N m−1. All the AFM measurements were carried out at 25 ± 1 °C.
Computational methods
The geometry of [(bpy)2Ru(4-pyNH2)2]2+ was initially optimized at the B3LYP51 level of density functional theory (DFT) using the Gaussian 09 program.52 Ru was described with the Stuttgart RECPs and associated basis sets (SDDALL),53 and the 6-31G(d) basis sets were used for all other atoms. Subsequently, vertical electronic excitations at the geometry of the ground-state were calculated using time-dependent density functional theory (TD-DFT). Natural transformation orbital (NTO) and natural bonding orbital (NBO) analyses were also performed to characterize the excited states.Results
HA–RuII polymer synthesis for SMFS
We first synthesized a HA polymer grafted with [(bpy)2Ru(4-pyNH2)2]2+ for SMFS measurement (Fig. 1A). It is difficult to apply force on a multiligand metal-coordination complex using traditional techniques, and only a few attempts have been made using the AFM-SMFS technique.22,54–56 In this study, we used a polymer composed of RuII–bipyridyl with a HA backbone to mimic the folded metalloprotein (Fig. 1B). By stretching the folded polymer, the coordination bonds ruptured and released the bond length sequestered by the metal ligands. The sequential rupture of the coordination bond gives rise to the sawtooth-like profile in the force-extension curves. Conversely, in the absence of the RuII complex, stretching the polymer would only yield a nonlinear rupture event in the force-extension curve. This approach could significantly increase the sampling efficiency while avoiding the nonspecific interactions between the AF microscope tip and the substrate surface.54,57 Fig. 1A shows the structure of the synthetic polymer composed of the HA backbone. The carboxyl groups of HA were first converted to amine-reactive O-acylisourea intermediates with EDC. Next, [(bpy)2Ru(4-pyNH2)2](PF6)2 was added to the HA intermediate solution to conjugate to the carboxyl group on HA at a random position. The reacted mixtures were dialyzed against excessive pure water to remove the unconjugated [(bpy)2Ru(4-pyNH2)2] and EDC molecules. Confirmed by 1HNMR (Fig. S2†), the grafted ratio of successfully conjugated HA–RuII(bpy)2(4-iminePy)2 was ∼22%. IR spectrum analysis was also exploited to further characterize the final conjugates (Fig. S3†). The absorption in the range of 1620–1641 cm−1 can be assigned to the shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, which indicates the formation of an amide bond. The absorption bands due to the –CH2 vibrations of the cyclohexyl ring were observed at 2857–2948 cm−1. The other medium-intensity bands at 742–758 cm−1 were attributed to the aromatic ring vibration. The UV-vis spectrum of the HA–RuII complex exhibits low energy bands around 483 nm, which can be assigned to the MLCT of Ru (dπ) → ligand (π*) transitions, typical for polypyridyl RuII complexes. There are four maxima in the UV region (ca. 342, 292, 243, and 197 nm), which have been assigned to the π → π* transitions in the complex (Fig. S4†). The control complex of [(bpy)2Ru(4-pyNH2)2]2+ exhibits an MLCT band at 453 nm. When the [(bpy)2Ru(4-pyNH2)2]2+ and HA–RuII complexes were excited at their MLCT wavelengths, they showed weak emission spectra with maximum emission wavelengths at 625 and 613 nm, respectively (Fig. S5†). Based on NMR studies and photophysical property observation, we conclude that the local structure of the RuII metal complex is not disturbed during the reaction with HA.
O stretching, which indicates the formation of an amide bond. The absorption bands due to the –CH2 vibrations of the cyclohexyl ring were observed at 2857–2948 cm−1. The other medium-intensity bands at 742–758 cm−1 were attributed to the aromatic ring vibration. The UV-vis spectrum of the HA–RuII complex exhibits low energy bands around 483 nm, which can be assigned to the MLCT of Ru (dπ) → ligand (π*) transitions, typical for polypyridyl RuII complexes. There are four maxima in the UV region (ca. 342, 292, 243, and 197 nm), which have been assigned to the π → π* transitions in the complex (Fig. S4†). The control complex of [(bpy)2Ru(4-pyNH2)2]2+ exhibits an MLCT band at 453 nm. When the [(bpy)2Ru(4-pyNH2)2]2+ and HA–RuII complexes were excited at their MLCT wavelengths, they showed weak emission spectra with maximum emission wavelengths at 625 and 613 nm, respectively (Fig. S5†). Based on NMR studies and photophysical property observation, we conclude that the local structure of the RuII metal complex is not disturbed during the reaction with HA.
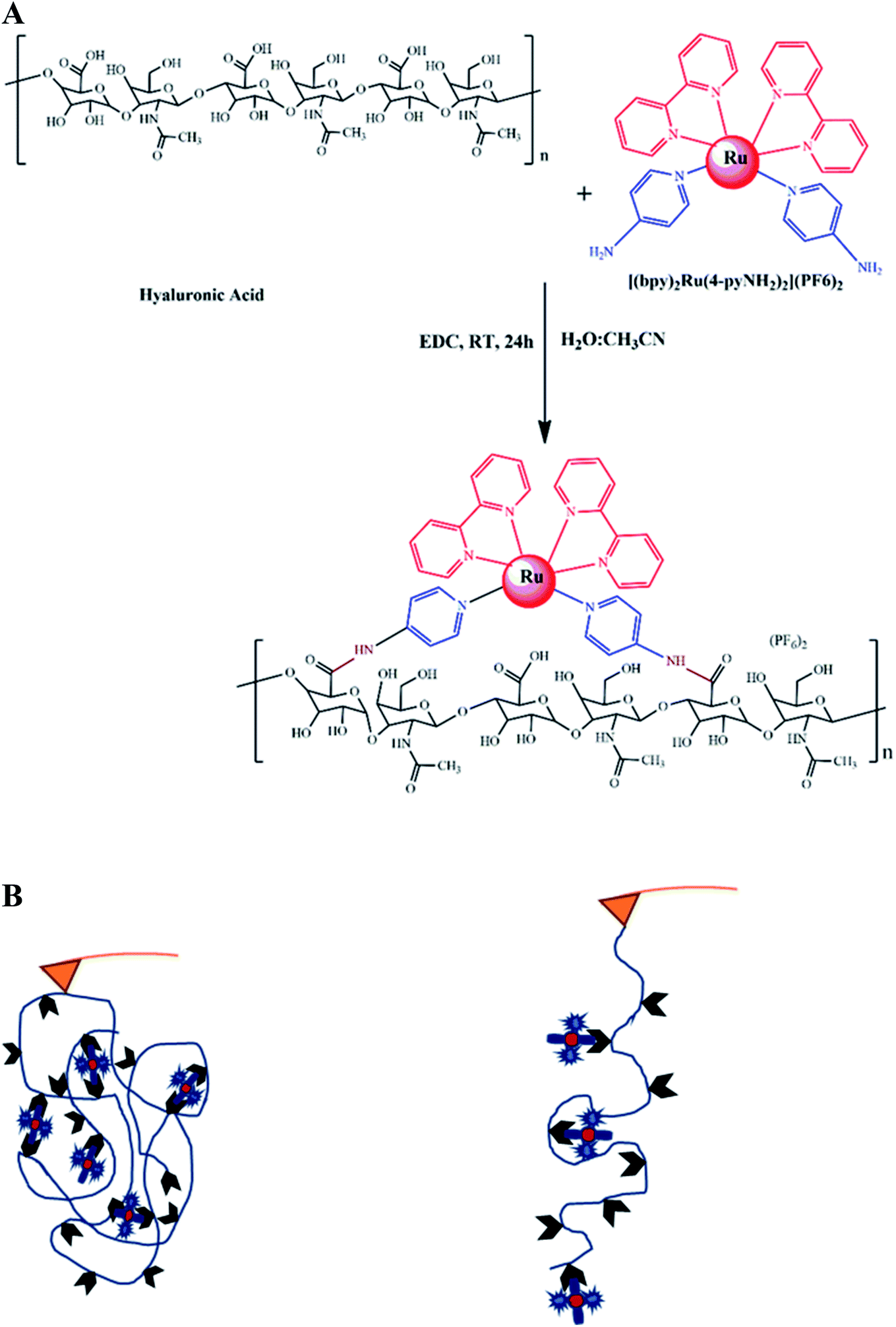 | ||
| Fig. 1 (A) The synthetic scheme of the HA–RuII complex. (B) Schematic representations of the HA–RuII complex, the scheme of single-molecule AFM experiments using the “multi-fishhook” approach. The RuII complex was conjugated to a hyaluronan polymer through amide bonds. In a typical simple experiment, the cantilever tip was brought into contact with the polymeric molecule, resulting in stretching, indicated by force-extension curves with sawtooth-like appearance. The individual force peaks in the curves correspond to the mechanical unfolding of individual domains, and the last peak corresponds to the stretching of the fully unfolded polymer chain and its subsequent detachment from either the AFM tip or glass substrate. Therefore, the last peak of each trace was excluded in the data analysis. | ||
SMFS analysis of the HA–RuII nanoparticles
Next, we used AFM to directly measure the mechanical stability of the RuII–N coordination bond (Fig. 1A). HA–RuII was first prepared as a stock solution at a concentration of 1 mg mL−1. Thereafter, several drops of HA–RuII were deposited on the glass slide surface to make the nanoparticles attach to the surface nonspecifically. During the force spectroscopy measurement, a silicon nitride AFM cantilever approaches the HA–RuII grafted substrate at a constant speed of 1000 nm s−1 and holds on the surface at 4–6 nN for 2 s. We relied on nonspecific interactions to anchor the polymer between the AFM tip and the substrate at two random positions. Subsequently, the cantilever was retracted at the same speed. The experimental scheme is depicted in Fig. 1B. As the distance between the tip and the substrate surface increased, the behavior of the polymer under extension was monitored by the deflection of the AFM tip. Two representative force-extension curves are depicted in Fig. 2A. A characteristic and reproducible sawtooth-like force-extension profile was observed, corresponding to the mechanical unfolding of the individual RuII–N bonds. The rupture-force values of the peaks are in the range of 100–200 pN. To avoid the interference of nonspecific interactions, the curves with more than three rupture peaks were further analyzed. The last peak of the force-extension curve corresponds to the polymer detachment either from the AFM tip or the substrate surface. Noteworthily, the mechanical strength of the nonspecific interactions between the HA molecule and the cantilever tip (or the substrate surface) can reach up to 300 pN, according to previous studies,22,54 which is much higher than that of the Ru–N bonds. We use the worm-like chain (WLC) model, an entropic elastic model that predicts the relationship between the extension of the polymer and its entropic restoring force, to identify the stretched polymer based on its mechanical response under external force (red lines on each curve). All the peaks in the same trace can be fitted using a fixed persistence length of ∼0.4 nm (Fig. S8A†), which is consistent with the persistence length of HA reported in the literature.58 This further confirmed that only a single HA polymer was picked up during each force measurement. The curves with abnormal persistence lengths were excluded in our data analysis. As the RuII complex was grafted on the HA polymer at a random position, the distance between individual peaks on the force-extension curve varied widely. To further confirm that the sawtooth peaks resulted from the rupture of the HA–RuII bonds, we performed control experiments using unmodified HA and HA–4-iminopyridine, and similar sawtooth-like patterns were not observed (Fig. S6 and S7†). We maintained the pickup rate at ∼1% by adjusting the amount of HA–RuII on the surface to avoid multiple chain-stretching events. | ||
| Fig. 2 (A and C) Two representative sawtooth-like force-extension traces at a pulling speed of 1000 nm s−1 for the HA–RuII complex without (in black) and with continuous blue-light illumination (in blue), respectively, in a Tris buffer (containing 100 mM Tris, 50 mM NaCl, pH 7.2). The height of these peaks directly corresponds to the rupture force. Each peak corresponds to a rupture event between RuII and 4-iminopyridine. Red lines correspond to worm-like chain (WLC) fitting to the rupture events using the same persistence length of ∼0.4 nm, indicating single-molecule pulling events. The last peak corresponds to the stretching of the fully unfolded polymer chain and its subsequent detachment from either the AFM tip or the glass substrate. (B and D) The rupture-force histogram obtained at a pulling speed of 1000 nm s−1 without (in black, n = 649) and with continuous blue-light illumination (in blue, n = 457), respectively. | ||
Oxidation state of the central metal ion plays an important role in overall stability of the complex. Xian Hao and co-worker elegantly shown the great potential of oxidation state through single-molecule force analysis by combining several interdisciplinary tools, particularly including ligand design and synthesis, surface self-assembly chemistry, single-crystal electrochemistry, STM imaging, and high-resolution electrochemical AFM force imaging and spectroscopy on Os–terpy systems (here Os is osmium(II) and terpy is 2,2′:6′,2′′-terpyridine), which displays that the Os–terpy bonds are ruptured at 100 ± 30 pN at open circuit potentials, but at 130 ± 60 pN in the oxidized state of the central metal and at the lower value of 80 ± 30 pN in the reduced state thus truly shows the effect of redox state on the coordinative bonding, which is further proved by DFT.59 Nonetheless, experiments involving similar types of complexes using more distortion, variable oxidation state under different conditions, different coordination spheres, geometries, and ligand fields are in progress in our laboratories to get fascinating results.
Mechanical strength of the RuII–N bonds revealed by SMFS
Subsequently, we analyzed the rupture-force distribution of the RuII–N bonds. Since 4-aminopyridine molecules were directly conjugated to the HA backbone covalently and coordinated to RuII, the only weak bonds among the system are the coordination bonds of Ru–N. This coordination bond ruptures before the polymer detaches from the cantilever (or substrate) and releases the sequestered length. As shown in Fig. 2B, the average rupture force is indicated by only one peak located at ∼121 ± 8 pN, at the pulling speed of 1000 nm s−1. This value is comparable to that of the RuII–bis(terpyridine) complex (95 pN).60It is well known that [Ru(bpy)3]2+ and its derivatives exhibit remarkable photophysical properties and stability. Therefore, we expect to observe the different mechanical responses of the RuII–N bond with and without continuous blue-light illumination (∼450 nm) during the force measurement, under the same experimental condition. The force-extension curves show a sawtooth-like profile with blue-light illumination. Two representative force-extension curves are depicted in Fig. 2C. The control experiments showed that, in this experimental setup, the photoexcitation effects on the HA molecules or HA–iminopyridine were negligible. The rupture force of the RuII–N bonds was reduced to 92 ± 8 pN on average (Fig. 2D), which indicates that the photoexcitation lowered the mechanical stability of the bonds by ∼25%.
Interestingly, a similar photo-weakening phenomenon was also observed in other biological systems. Wouter D. Hoff et al. found that blue-light illumination could reduce the mechanical stability of a photoactive yellow protein by ∼30%.61 The distribution of the fitted persistence length for all peaks under this condition is also centered at ∼0.4 nm (Fig. S8B†).
Cyclic illumination experiment to demonstrate the reversibility of the RuII–N interaction
Next, we investigated the reversibility of this photo-sensitive system. We performed three consecutive series of force spectroscopy experiments on the HA–RuII complex with and without blue-light illumination, without changing the cantilever or the sample (Fig. 3A–C). We first conducted SMFS without blue-light illumination in a Tris buffer (containing 100 mM Tris, 50 mM NaCl, pH 7.2), and the rupture force of the RuII–N bond was determined as 118 ± 8 pN on average (n = 387), which is consistent with our previous result. Thereafter, we introduced the blue light and continued the force measurements. In this case, the average rupture force of the RuII–N bonds was reduced to 90 ± 8 pN (n = 152, Fig. 3B). The average rupture force was restored after the blue light was turned off. Fig. 3C shows that the average rupture force is 113 ± 8 pN (n = 326), indicating the reversibility of the HA–RuII complex without requiring any external energy source, such as heat or solvent exposure. The possible explanation for this reversibility lies in its fluorescence property. The HA–RuII complex exhibits weak fluorescence despite its large structure, suggesting that a considerable portion of absorbed light is converted to heat. The reversibility examined here is based on the fact that this energy could be reversed after the completion of the series B experiment (illumination of blue light), resulting in an increase in the rupture force to the original value (Fig. 3C). | ||
| Fig. 3 Single-molecule force spectroscopy experiments of the HA–RuII complex in the reversible condition. (A) Experimental series in the neutral condition. (B) Series during continuous blue-light illumination. (C) Series after turning off the blue light and proceeding with the experiment. All the experiments were performed with the same tip and sample traces at a pulling speed of 1000 nm s−1 in a Tris buffer (containing 100 mM Tris, 50 mM NaCl, pH 7.2). | ||
Free-energy landscape for the RuII–N bonds with and without blue-light illumination
The question of interest here is what change in the energy landscape can explain the difference in mechanical stability (average unfolding forces) between the two pulling geometries? There are two possibilities. (1) The pulling geometry does not change the transition state, but simply alters the relative position of the transition state along the reaction coordinate; (2) the pulling geometry alters the structure of the transition state, which, in turn, affects both the energy and position of the rate-limiting barrier.62To further investigate the molecular mechanism and free-energy landscape of the RuII–N dynamic bond, we performed dynamic loading experiments. We changed the loading rate by changing the pulling speed. As shown in Fig. 4(A), the rupture force of the RuII–N bond is loading-rate dependent. The higher the loading rate exerted on the bond, the higher the rupture force. Exploiting the widely used Bell–Evans model,63,64 we extracted the kinetic parameters, free-energy barrier (ΔG), and distance to the transition state (Δx) of the RuII–N bond using
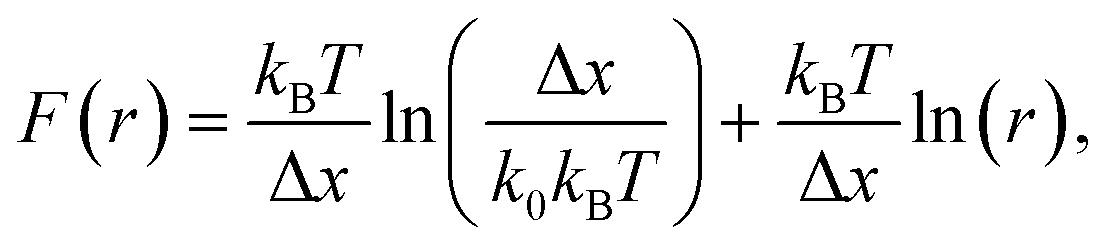 | (1) |
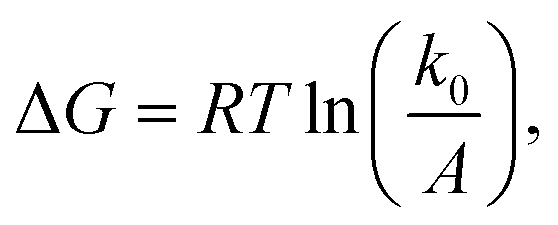 | (2) |
 | ||
| Fig. 4 (A) Plot of the rupture forces at different loading rates for the HA–RuII complex without (black) and with continuous blue-light illumination (blue), respectively, in a Tris buffer (containing 100 mM Tris, 50 mM NaCl, pH 7.2). The lines correspond to the fitting of the Bell–Evans model to the experimental data. The R2 values (rupture force vs. logarithmic loading rate) for the HA–RuII complex are 0.932 and 0.922 without and with continuous blue-light illumination, respectively. | ||
The linear nature of the plot is consistent with a single pathway model in which the force continuously tilts the free-energy landscape, thereby lowering the height of the barrier.66
First-principles calculation
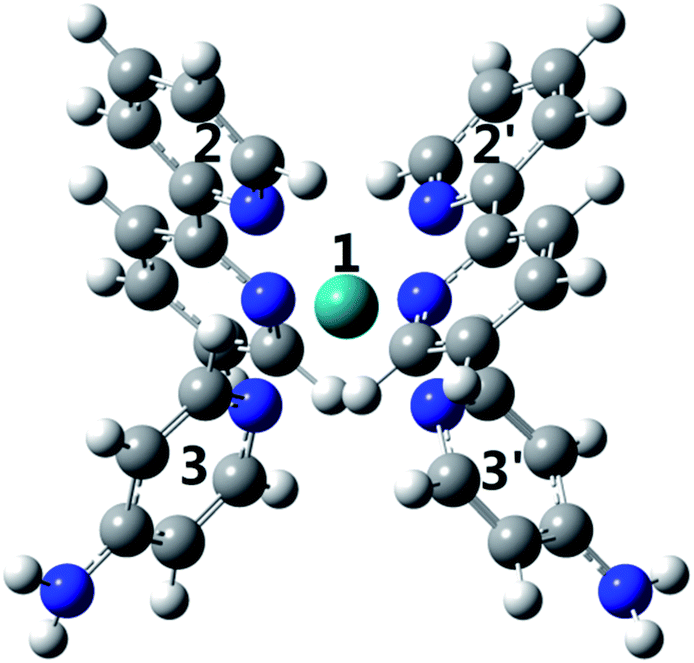
To investigate why the blue-light induced excitation in [(bpy)2Ru(4-pyNH2)2]2+ can speed up its coordination bond rupture, we perform a first-principles study of the electronic structure of the ground state and the electronically excited states of [(bpy)2Ru(4-pyNH2)2]2+ using DFT. As shown in Fig. 5, the optimized ground-state structure of [(bpy)2Ru(4-pyNH2)2]2+ has six nitrogen atoms around RuII, exhibiting an octahedron-like conformation. For the electronic excitation, the main absorption peak at 468.09 nm (S5) near the blue ray is found in our calculations, in good agreements with experiments. To see how the electron clouds evolve upon photoexcitation, we plot the NTO analysis of S5, as shown in Fig. 6. The figure shows that electrons mainly transfer from fragment 3 to fragment 2 (or 3′ to 2′) and that the symmetric units follow the same process. Due to the significantly decreased electron population at fragments 3 and 3′, the interactions between the units of RuII and two 4-pyNH2 can be expected to be largely weakened after the optical excitation by blue light. To provide a quantitative description of the inter-fragment interaction, the pair interaction potential is calculated by the following equation:
 | (3) |
 | ||
| Fig. 5 The optimized geometry of the ground-state [Ru(4-Ampy)2]2+. | ||
 | ||
| Fig. 6 Natural transition orbitals for S0 → S5 of [Ru(4-Ampy)2]2+. | ||
[Ru(bpy)3]2+ and its derivatives are essential for mimicking the functions performed by natural systems, such as solar energy conversion and storage. Therefore, it is required to understand the mechanical properties of [Ru(bpy)3]2+ and its derivatives to tune the mechanical performance of this important class of materials, as their coordination bonds are very important.
In this work, we quantified the strength of the RuII–N coordination bonds using SMFS, for the first time, which could yield tremendous insights into the molecular mechanism with and without blue-light illumination.
We discovered that the rupture forces for the RuII–N coordination bonds are in the range of ∼100–125 pN at a pulling speed of 1000 nm s−1, which are comparable to those for stable mechanical metallo-proteins.56,67 The experimental approaches presented in this study could serve as a general tool to explore the complicated mechanical stabilities of different types of coordination bonds and provide quantitative information for the mechanical design of load-bearing biomaterials.
Conclusion
In summary, we synthesized and characterized the HA–RuII molecule using a simplified experimental scheme, the “multiple fishhooks” approach. Our experiments confirm that the HA–RuII molecule can be reversibly and reproducibly toggled between two different structural isomers utilizing blue fluorescent light. Our molecular system could be activated (in the dark) and slowed down (under blue-light illumination); subsequently, once the blue light was turned off, it recovered to its original state. Compared to other systems that require external energy, such as heat or solvent exposure, to reverse the photoexcitation effect, our complex maintains reproducibility without any external energy source up to a significant extent. Light, which has been overlooked in this field, has been proven as a great source for tuning the mechanical property of materials, and our results indicate that it can be very beneficial when applied as a trigger. Furthermore, the design, synthesis, and other molecular parameters (such as the identity of the metal ion and ligand) can be varied to further improve the material properties, which can lead to very exciting results. Future research in this direction will focus on elucidating the specific roles of such kinds of systems. These studies will likely provide new insights into the nature of this important class of chemical bonds. The information about single molecules extracted from these investigations will provide more in-depth insights into the fundamentals and will have a significant impact in coordination chemistry.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
I am grateful to Researchers Supporting Project number (RSP-2020/141), King Saud University, Riyadh, Saudi Arabia, for financial assistance.References
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- K.-H. Ernst, F. R. W. P. Wild, O. Blacque and H. Berke, Angew. Chem., Int. Ed., 2011, 50, 10780–10787 CrossRef CAS.
- B. Song, S. Kandapal, J. Gu, K. Zhang, A. Reese, Y. Ying, L. Wang, H. Wang, Y. Li, M. Wang, S. Lu, X.-Q. Hao, X. Li, B. Xu and X. Li, Nat. Commun., 2018, 9, 4575 CrossRef.
- S. J. Wezenberg and B. L. Feringa, Nat. Commun., 2018, 9, 1984 CrossRef.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, in Annual Review of Materials Research, ed. D. R. Clarke and P. Fratzl, 2011, vol. 41, pp. 99–132 Search PubMed.
- E. Munch, M. E. Launey, D. H. Alsem, E. Saiz, A. P. Tomsia and R. O. Ritchie, Science, 2008, 322, 1516–1520 CrossRef CAS.
- H. B. Zeng, D. S. Hwang, J. N. Israelachvili and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12850–12853 CrossRef CAS.
- W. C. Yount, D. M. Loveless and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 14488–14496 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS.
- J. Wu, P. Li, C. Dong, H. Jiang, X. Bin, X. Gao, M. Qin, W. Wang, C. Bin and Y. Cao, Nat. Commun., 2018, 9, 620 CrossRef.
- D. G. Barrett, D. E. Fullenkamp, L. He, N. Holten-Andersen, K. Y. C. Lee and P. B. Messersmith, Adv. Funct. Mater., 2013, 23, 1111–1119 CrossRef CAS.
- N. Becker, E. Oroudjev, S. Mutz, J. P. Cleveland, P. K. Hansma, C. Y. Hayashi, D. E. Makarov and H. G. Hansma, Nat. Mater., 2003, 2, 278–283 CrossRef CAS.
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, Angew. Chem., Int. Ed., 2014, 53, 12026–12044 CrossRef CAS.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- W. C. Yount, D. M. Loveless and S. L. Craig, Angew. Chem., Int. Ed., 2005, 44, 2746–2748 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS.
- Y. R. Li, J. Wen, M. Qin, Y. Cao, H. B. Ma and W. Wane, ACS Biomater. Sci. Eng., 2017, 3, 979–989 CrossRef CAS.
- B. Adhikari, Y. Yamada, M. Yamauchi, K. Wakita, X. Lin, K. Aratsu, T. Ohba, T. Karatsu, M. J. Hollamby, N. Shimizu, H. Takagi, R. Haruki, S.-i. Adachi and S. Yagai, Nat. Commun., 2017, 8, 15254 CrossRef.
- S. S. Rozenel, C. R. Azpilcueta, M. M. Flores-Leonar, J. P. F. Rebolledo-Chávez, L. Ortiz-Frade, C. Amador-Bedolla and E. Martin, Catal. Today, 2018, 310, 2–10 CrossRef CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef.
- X.-J. He and L.-F. Tan, Inorg. Chem., 2014, 53, 11152–11159 CrossRef CAS.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS.
- L.-N. Ji, X.-H. Zou and J.-G. Liu, Coord. Chem. Rev., 2001, 216–217, 513–536 CrossRef CAS.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS.
- R. Horvath, J. Lombard, J.-C. Leprêtre, M.-N. Collomb, A. Deronzier, J. Chauvin and K. C. Gordon, Dalton Trans., 2013, 42, 16527–16537 RSC.
- H. D. Gafney and A. W. Adamson, J. Am. Chem. Soc., 1972, 94, 8238–8239 CrossRef CAS.
- R. Horvath and K. C. Gordon, Coord. Chem. Rev., 2010, 254, 2505–2518 CrossRef CAS.
- Y. Li, T. Wang, L. Xia, L. Wang, M. Qin, Y. Li, W. Wang and Y. Cao, J. Mater. Chem. B, 2017, 5, 4416–4420 RSC.
- Y. Li, H. Liu, T. Wang, M. Qin, Y. Cao and W. Wang, Chemphyschem, 2017, 18, 1466–1469 CrossRef CAS.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS.
- C. J. Liu, W. Q. Shi, S. X. Cui, Z. Q. Wang and X. Zhang, Curr. Opin. Solid State Mater. Sci., 2006, 9, 140–148 CrossRef.
- N. Hosono, A. M. Kushner, J. Chung, A. R. A. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef CAS.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS.
- M. Grandbois, M. Beyer, M. Rief, H. Clausen-Schaumann and H. E. Gaub, Science, 1999, 283, 1727 CrossRef CAS.
- S. Zou, H. Schönherr and G. J. Vancso, J. Am. Chem. Soc., 2005, 127, 11230–11231 CrossRef CAS.
- L. Dougan, A. S. R. Koti, G. Genchev, H. Lu and J. M. Fernandez, Chemphyschem, 2008, 9, 2836–2847 CrossRef CAS.
- Y. Zhang, Y. Yu, Z. Jiang, H. Xu, Z. Wang, X. Zhang, M. Oda, T. Ishizuka, D. Jiang, L. Chi and H. Fuchs, Langmuir, 2009, 25, 6627–6632 CrossRef CAS.
- Y. Zhang, C. Liu, W. Shi, Z. Wang, L. Dai and X. Zhang, Langmuir, 2007, 23, 7911–7915 CrossRef CAS.
- G. Yuan, Q. Ma, T. Wu, M. Wang, X. Li, J. Zuo and P. Zheng, Sci. Rep., 2019, 9, 10518 CrossRef.
- G. I. Bell, Science, 1978, 200, 618 CrossRef CAS.
- C. D. Ellis, L. D. Margerum, R. W. Murray and T. J. Meyer, Inorg. Chem., 1983, 22, 1283–1291 CrossRef CAS.
- V. Saint, Inc., Madison, Wisconsin, USA, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, Ö. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2014, 30, 4358–4366 CrossRef CAS.
- P. Zheng, G. M. Arantes, M. J. Field and H. B. Li, Nat. Commun., 2015, 6, 7569 CrossRef CAS.
- P. Zheng, S.-i. J. Takayama, A. G. Mauk and H. Li, J. Am. Chem. Soc., 2013, 135, 7992–8000 CrossRef CAS.
- X. T. Han, M. Qin, H. Pan, Y. Cao and W. Wang, Langmuir, 2012, 28, 10020–10025 CrossRef CAS.
- X. Han, M. Qin, H. Pan, Y. Cao and W. Wang, Langmuir, 2012, 28, 10020–10025 CrossRef CAS.
- X. Hao, N. Zhu, T. Gschneidtner, E. Ö. Jonsson, J. Zhang, K. Moth-Poulsen, H. Wang, K. S. Thygesen, K. W. Jacobsen, J. Ulstrup and Q. Chi, Nat. Commun., 2013, 4, 2121 CrossRef.
- M. Kudera, C. Eschbaumer, H. E. Gaub and U. S. Schubert, Adv. Funct. Mater., 2003, 13, 615–620 CrossRef CAS.
- J. M. Zhao, H. Lee, R. A. Nome, S. Majid, N. F. Scherer and W. D. Hoff, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11561–11566 CrossRef CAS.
- A. Yadav, S. Paul, R. Venkatramani and S. R. K. Ainavarapu, Sci. Rep., 2018, 8, 1989 CrossRef.
- G. I. Bell, Science, 1978, 200, 618–627 CrossRef CAS.
- E. Evans and K. Ritchie, Biophys. J., 1999, 76, 2439–2447 CrossRef CAS.
- F. R. Kersey, W. C. Yount and S. L. Craig, J. Am. Chem. Soc., 2006, 128, 3886–3887 CrossRef CAS.
- V. Barsegov, D. K. Klimov and D. Thirumalai, Biophys. J., 2006, 90, 3827–3841 CrossRef CAS.
- P. Zheng, G. M. Arantes, M. J. Field and H. Li, Nat. Commun., 2015, 6, 7569 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1416079. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07274e |
| This journal is © The Royal Society of Chemistry 2020 |