
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGut microbiota-mediated tributyltin-induced metabolic disorder in rats†
Ge-hui Yuan‡
a,
Zhan Zhang‡ab,
Xing-su Gaoa,
Jun Zhua,
Wen-hui Guoab,
Li Wangab,
Ping Dingc,
Ping Jiang *ab and
Lei Li*ab
*ab and
Lei Li*ab
aCenter for Global Health, School of Public Health, Nanjing Medical University, 101 Longmian Avenue, Nanjing 211166, China. E-mail: lilei@njmu.edu.cn; jiangping@njmu.edu.cn; Fax: +86-25-8686-8499; Fax: +86-25-8686-8402; Tel: +86-25-8686-8404 Tel: +86-25-8686-8402
bKey Lab of Modern Toxicology of Ministry of Education, School of Public Health, Nanjing Medical University, 101 Longmian Avenue, Nanjing 211166, P. R. China
cXiang Ya School of Public Health, Central South University, Changsha 410078, P. R. China
First published on 8th December 2020
Abstract
Tributyltin (TBT), an environmental pollutant widely used in antifouling coatings, can cause multiple-organ toxicity and gut microbiome dysbiosis in organisms, and can even cause changes in the host metabolomic profiles. However, little is known about the underlying effects and links of TBT-induced metabolic changes and gut microbiome dysbiosis. In this study, rats were exposed to TBT at a dose of 100 μg kg−1 body weight (BW) for 38 days, followed by multi-omics analysis, including microbiome, metabolomics, and metallomics. Results showed that TBT exposure reduced rat weight gain and decreased the serum triglyceride (TG) level. Metabolic analysis revealed that TBT fluctuated linoleic acid metabolism and glycerophospholipid metabolism in the liver; the tricarboxylic acid cycle (TCA cycle), nicotinate and nicotinamide metabolism, and arachidonic acid metabolism in serum; glycine, serine, and threonine metabolism, the one carbon pool by folate, nicotinate, and nicotinamide metabolism; and tryptophan metabolism in feces. Furthermore, TBT treatment dictated liver inflammation due to enhancing COX-2 expression by activating protein kinase R-like ER kinase (PERK) and C/EBP homologous protein (CHOP) to induce endoplasmic reticulum (ER) stress instead of stimulating arachidonic acid metabolism. Meanwhile, alteration of the intestinal flora [Acetivibrio]_ethanolgignens_group, Acetatifactor, Eisenbergiella, Lachnospiraceae_UCG-010, Enterococcus, Anaerovorax, and Bilophila under TBT exposure were found to be involved in further mediating liver inflammation, causing lipid metabolism abnormalities, such as TG, linoleic acid, and glycerophospholipids, and interfering with the energy supply process. Among these, [Acetivibrio]_ethanolgignens_group, Enterococcus, and Bilophila could be considered as potential biomarkers for TBT exposure based on receiver operator characteristic (ROC) curve analysis.
1. Introduction
Tributyltin (TBT), an organotin pollutant, is wildly distributed in marine ecosystems mainly as an antifouling component.1,2 Numerous studies have reported that TBT exposure induces neurotoxicity, immunotoxicity, reproductive toxicity, and multiple-organ toxicity.3–6 Additionally, human health could be affected due to the use of TBT-based plastics, wood catalysts, and the consumption of TBT-contaminated seafood.5 Nowadays, the main exposure route of TBT in mammals, including humans, is the intake of seafood.7 Studies have shown that female rats exposed to TBT (0.1–0.5 μg kg−1) showed obesity with increased body weight and fat mass.8–10 In addition, TBT could induce hepatic oxidative stress and histological changes.6,11,12 Short-term studies on adult rats have certified that TBT (100, 1000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg kg−1) can cause slight weight loss, while the number of cells with damaged DNA increased with the dosage of TBT.11,12 Although TBT at a dose of 100 μg kg−1 did not cause obvious oxidative stress like high-dose TBT (1000 and 10000 μg kg−1), it could induce hepatotoxicity as manifested by an elevated reactive oxygen species (ROS) level and malondialdehyde production, eventually leading DNA breakage in the liver13–15 or inducing liver necrosis with an inflammatory response.12,16 As an organ for the detoxification and metabolism of various cyclic endogenous and exogenous compounds, the liver can be attacked by TBT, and numerous experiments have shown that no matter whether low dose or high dose, or even the daily dose that a person can tolerate, TBT could still cause hepatotoxicity.17–19 Caspase-3-dependent apoptosis via increasing mitogen-activated protein kinase has also been reported to be an important mechanism of hepatotoxicity induced by TBT.13 Although the above-mentioned mechanisms can explain TBT-induced liver toxicity from some perspectives, the links and metabolic changes are still unclear. Metabolomics, as a systemic analysis of small molecule metabolites, can detect global metabolic changes in response to various stimuli in an unbiased manner by providing insights into the metabolic regulation of different compounds. Here, we investigated the metabolic changes induced by TBT at a dose of 100 μg kg−1 based on metabolomics to gain further knowledge of the etiological agent of TBT-induced hepatotoxicity.
000 μg kg−1) can cause slight weight loss, while the number of cells with damaged DNA increased with the dosage of TBT.11,12 Although TBT at a dose of 100 μg kg−1 did not cause obvious oxidative stress like high-dose TBT (1000 and 10000 μg kg−1), it could induce hepatotoxicity as manifested by an elevated reactive oxygen species (ROS) level and malondialdehyde production, eventually leading DNA breakage in the liver13–15 or inducing liver necrosis with an inflammatory response.12,16 As an organ for the detoxification and metabolism of various cyclic endogenous and exogenous compounds, the liver can be attacked by TBT, and numerous experiments have shown that no matter whether low dose or high dose, or even the daily dose that a person can tolerate, TBT could still cause hepatotoxicity.17–19 Caspase-3-dependent apoptosis via increasing mitogen-activated protein kinase has also been reported to be an important mechanism of hepatotoxicity induced by TBT.13 Although the above-mentioned mechanisms can explain TBT-induced liver toxicity from some perspectives, the links and metabolic changes are still unclear. Metabolomics, as a systemic analysis of small molecule metabolites, can detect global metabolic changes in response to various stimuli in an unbiased manner by providing insights into the metabolic regulation of different compounds. Here, we investigated the metabolic changes induced by TBT at a dose of 100 μg kg−1 based on metabolomics to gain further knowledge of the etiological agent of TBT-induced hepatotoxicity.
Gut microbiota have been reported to participate in nutritional provision and metabolism regulation in mammals,20 so we supposed that there might be a link between the metabolic effects and gut microbiome under TBT treatment. Furthermore, studies have confirmed that the gut microbiome play a critical role in producing numerous metabolites and consequently influence the host homeostasis.21 Meanwhile, intestinal microbial communities act as mediators for regulating the host physiological processes, such as immune homeostasis,22 energy and metabolic homeostasis,23 the stress response,24 and inflammation.25 For example, it was estimated that increased Clostridia and Proteobacteria are associated with inflammatory infiltration.26,27 In addition, an increasing number of literature reports have certified that the pathophysiological mechanisms of inflammation and the composition of the gut microbiota have an association with the regulation of metal elements in different disorders,28,29 such as copper (Cu), iron (Fe), and calcium (Ca), which are necessary respiratory nutrients for bacterial proliferation, while the absorption systems of Cu and Fe have been reported to promote the pathogenicity of many pathogenic bacteria.30,31 In addition, gut flora could affect the hepato-intestinal circulation of metabolites29 by transporting liver metabolites from dietary, endogenous, or xenobiotic substances (e.g., FFAs, choline metabolites, ethanol metabolites) to the intestines through the capillary system.32 Although gut microbiome dysbiosis has been reported to be caused by TBT,33 the regulation of intestinal bacteria on the metabolic profiles and metal elements needs to be extended, and the links among them under TBT exposure need clarification.
In the present study, we integrated 16S rRNA sequencing, metabolomics, and metalomics to investigate the effects and the metabolism of TBT exposure at 100 μg kg−1 in rats. The results showed that TBT caused weight loss, TG reduction, liver inflammation, and metabolic abnormalities, including in the linoleic acid metabolism, glycerophospholipid metabolism in liver, TCA cycle, nicotinate and nicotinamide metabolism, arachidonic acid metabolism in serum, and glycine, serine, and threonine metabolism, one carbon pool by folate, nicotinate, and nicotinamide metabolism, and tryptophan metabolism in feces. Besides, TBT-induced gut microbiome dysbiosis with [Acetivibrio]_ethanolgignens_group, Acetatifactor, Eisenbergiella, Lachnospiraceae_UCG-010, Enterococcus, Anaerovorax, and Bilophila alteration is associated with inflammation and metabolic disorder.
2. Materials and methods
2.1. Animal treatment
Seven-week-old female Sprague-Dawley rats were purchased from Qinglong Mountain Animal Breeding Farm (Jiangning, Nanjing, China). All the rats were maintained at 22 °C on a 12/12 h light/dark cycle with 55% humidity. The rats were placed in plastic Macrolon cages with stainless steel lids and wood shaving T two rats per cage. Standard rat chow and filtered tap water were provided ad libitum. Rats were weighed and randomly divided into a control group and TBT group (n = 12). The control group was treated with 0.4% ethanol and the TBT group was treated daily with 100 μg kg−1 BW TBT by gavage for 38 days. On the last day of the experiment, fresh feces were collected from each cage and stored at −80 °C. At the end of the experiment, the rats were fasted for 12 h, and then sacrificed under anesthesia. Blood was collected and centrifuged at 4000 rpm for 15 min at 4 °C for the collection of serum, which was kept at −80 °C until use. The liver and intestine were excised immediately and then divided, with some sections of liver and intestine fixed and sliced for further histological and immunohistochemical analysis and the rest stored at −80 °C until use. The care and use of animals all followed the Animal Welfare Guidelines. All the protocols were approved by the Animal Care and Welfare Committee of Nanjing Medical University (IACUC-1812027).2.2. Biochemical parameters analysis of serum
Serum biochemical index TG in serum was detected by using an automatic biochemical analyzer (Hitachi 7100).2.3. Histopathology
The abdominal adipose and liver tissue of the mice were fixed in 4% paraformaldehyde for 24 h. Paraffin-embedded 5 μm sections were cut and mounted on slides. Adipose tissue and liver tissue sections were stained with hematoxylin and eosin for morphological analysis. Immunohistochemical staining of the liver tissue sections were immersed in boiling citric acid antigen retrieval buffer for antigen retrieval. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in distilled water. The slides were incubated in COX-2 monoclonal antibody (Servicebio, China; dilution 1:1000), followed by HRP-labeled goat anti-rabbit secondary antibody (Servicebio, China; dilution 1:200). The final DAB visualized positive expression was brownish yellow, and the hematoxylin-stained nucleus was blue. The stained slides were scanned by a Pannoramic SCAN system (3DHISTECH, Hungary) to obtain representative images.
2.4. Western blotting for protein expression
Total protein was extracted from the liver tissue and the concentration detected using the BCA (Beyotime Biotech, Shanghai, China) method. After mixing with 5× sodium dodecyl sulfate (SDS) loading buffer and denatured at 100 °C for 8 min, the appropriate amount of each protein mixture was then separately loaded on a 10% SDS–polyacrylamide gel. Then, the total protein was transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, MA, USA), and blocked with skim milk for 1 h at room temperature. After binding with primary antibodies of PERK, CHOP, and GAPDH (Cell Signaling Technology, MA, USA) at 4 °C for 12–16 h, the membrane was further washed with TBST (0.1% Tween-20 with 20× Tris–HCl buffered saline: 200 mmol L−1 Tris, 3 mol L−1 NaCl, pH 7.5) 5 times (5 min × 2, 15 min × 3). Then, the membrane was put in the corresponding secondary antibody (KPL, Gaitherburg, USA) for 1 h and washed with TBST 6 times (5 min × 2, 15 min × 4). Finally, the samples were coated with enhanced chemiluminescence reagent (Yeasen Biotech, Shanghai, China), and the signal was detected by using a chemiluminescence imaging system (Bio-Rad, California, USA).2.5. Metabolites analysis based on Q Exactive UPLC-MS/MS
000 rpm, 4 °C, and then 150 μL of supernatant was transferred into the injection bottle for detection.:isopropanol:water = 3:3:2; v/v/v) were homogenized with stainless steel beads for 5 min at 30 Hz on a tissuelyser, then cooled for 30 s and this process was repeated three times. The mixture was vortexed for 15 s and centrifuged at 13000 rpm for 15 min at 4 °C, and finally 200 μL of supernatant was prepared for detection.000 rpm for 15 min at 4 °C and the supernatant was collected. Next, 1 mL ice methanol was added to the remaining precipitation to homogenize at 30 Hz for 1 min, then cool for 30 s and this was repeated three times. Then, the mixture was centrifuged at 12000 rpm for 15 min at 4 °C and the supernatant was collected. Finally, 500 μL of each supernatant was mixed and centrifuged at 12000 rpm for 15 min at 4 °C, and the final supernatant was collected for detection.All the non-instrument operations were performed chilled on ice to suppress enzymatic activity. The metabolites of all the samples were detected by using a Q Exactive UPLC-MS/MS system (Thermo Scientific, Wilmington, USA) based on non-targeted metabolomics according to our previous study.34
2.6. Metal element analysis
First, 100 μL of serum from each sample was diluted to 5 mL with 0.05% HNO3 + 0.05% Triton and mixed for detection. All the samples were measured by ICP-MS (Thermo Scientific, Wilmington, USA) according to our previous studies.352.7. 16S rRNA gene sequencing
Microbial DNA from the fecal samples of rats was extracted using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to the manufacturer's protocols. The final DNA concentration and quality were checked by using a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA) and 1% agarose gel electrophoresis. The V3–V4 hypervariable regions of the bacteria 16S rRNA gene were amplified with the primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) using a thermocycler PCR system (GeneAmp 9700, ABI, USA). PCR reactions were performed in triplicate with 20 μL mixtures containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. The following is the PCR reaction program: 3 min of denaturation at 95 °C, 27 cycles of 30 s at 95 °C, 30 s for annealing at 55 °C, and 45 s for elongation at 72 °C, and a final extension at 72 °C for 10 min. The PCR products were obtained for further purification using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluor™-ST (Promega, USA) according to the manufacturer's protocol. Purified amplicons were pooled in equimolar and paired-end sequences (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). Raw fastq files were quality-filtered by trimmomatic and merged by FLASH with the following criteria: (i) the reads were truncated at any site receiving an average quality score <20 over a 50 bp sliding window; (ii) sequences whose overlap were longer than 10 bp were merged according to their overlap with a mismatch of no more than 2 bp; (iii) sequences of each sample were separated according to the barcodes (exactly matching) and primers (allowing 2 nucleotide mismatching), and the reads containing ambiguous bases were removed. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE version 7 with a novel ‘greedy’ algorithm that performed chimera filtering and OTU clustering simultaneously. The taxonomy of each 16S rRNA gene sequence was analyzed by the RDP classifier algorithm against the Silva (SSU123) 16S rRNA database at a confidence threshold of 70%. The data were analyzed using the standard protocols from Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).2.8. Data analysis
Microbiome analysis was performed on the i-sanger platform. The screening of metal elements and metabolites was done using SIMCA 14.1. The relative contribution of the variables identified was obtained by considering ‘the variable importance in the projection’ (VIP) values in the PLS-DA model. The elements and metabolites that showed VIP > 1.0 and p < 0.05 were considered statistically significant. The name of the metabolite was identified in the HMDB library. The pheatmap in R3.5.4 was used to draw the heatmap. Metabolic pathways analysis and visualization were performed using the open source network tool MetaboAnalyst 3.0 and the KEGG metabolic pathway database. All the data are presented as the mean ± standard error of mean (SEM). The significance tests not described in this article were all performed using Student's t test. Correlation between variables was achieved using Pearson correlation analysis. *P < 0.05, **P < 0.01, ***P < 0.001.3. Results
3.1. TBT induced lipid disorder and liver inflammation
Compared with that of the control group, the weight gain of the rats treated with TBT was lower, and showed a statistically significant decrease at the 38th day (Fig. 1A). Consistent with this phenotype, the food consumption and utilization rate of the rats exposed to TBT during the same period were also lower than those of the control group (Fig. S1†), suggesting that a lower food consumption was related to weight gain. Besides, the TG level in the serum of the TBT group was obviously inhibited relative to the control group (Fig. 1B). To further explore whether TBT treatment could affect the area of abdominal adipose, histopathological and quantitative analysis were performed and the results showed that adipose tissue cells were smaller and per unit area significantly decreased in the TBT group compared to the control group (Fig. 1C and S2†). These results indicated that TBT might cause lipid disorders. As illustrated in Fig. 1D and E, TBT exposure induced inflammatory cell infiltration in the liver, while the immunohistochemistry of COX-2 further showed that TBT increased the positive expression of COX-2 relative to the control group. All the data indicated that TBT induced lipid disorder and hepatitic inflammation with the upregulation of COX-2. | ||
| Fig. 1 TBT-induced lipid disorder and liver inflammation in rats. (A) Effect of animal weight gain. (B) The level of TG in serum. (C) Histology of abdominal adipose tissue. (D) Histology of liver tissue with H&E staining. Black arrows point to inflammatory cell infiltration. (E) Immunohistochemistry for COX-2. The images were taken under the same size magnification. Scale bar: 100 μm. | ||
3.2. Metabolic changes, ER stress, and metal disturbances induced by TBT
Significant changes in the small molecular metabolites in the liver and serum were detected in the TBT group compared to the control. Based on a comparison with the control group and identification in the HMDB library, 121 and 46 differential metabolites were separately obtained from the liver and serum (ESI Tables S1 and S2†). PLS-DA analysis showed that the metabolisms in the liver (Fig. 2A) and serum (Fig. 2B) were different between the control group and TBT group, which indicated that TBT exposure could disrupt metabolic homeostasis. Furthermore, the metabolic pathways of the above-mentioned differentiated metabolites are summarized and finally presented as a bubble diagram in Fig. 2C and D. In the liver, linoleic acid metabolism, glycerophospholipid metabolism, pantothenate and CoA biosynthesis, and glyoxylate and dicarboxylate metabolism were mainly perturbed under TBT exposure (Fig. 2C). In serum, TBT treatment influenced the TCA cycle, nicotinate and nicotinamide metabolism, arachidonic acid metabolism, and retinol metabolism (Fig. 2D). The above results illustrated that TBT could induce liver and serum metabolic disorder. Among these, both linoleic acid and 2-arachidonylglycerol of linoleic acid metabolism were significantly decreased in the liver (Fig. 2E). Since linoleic acid is a precursor of arachidonic acid, we speculated that the arachidonic acid metabolism was affected by TBT exposure from the source. Moreover, two synthetic raw materials of glycerophospholipids were altered, among which phosphatidylcholine decreased and dCDP increased (Fig. 2E). Other than those, LysoPCs(15:0, 17:0, 16:1(9Z)/0:0, 16:0, 22:4(7Z,10Z,13Z,16Z), 20:3(5Z,8Z,11Z), 20:5(5Z,8Z,11Z,14Z,17Z), 18:2(9Z,12Z), 18:3(6Z,9Z,12Z)), PC(22:5(4Z,7Z,10Z,13Z,16Z)/14:1(9Z)), and PE(14:0/22:5(4Z,7Z,10Z,13Z,16Z)), which are hydrolysates of phospholipids, were reduced under TBT treatment (Fig. 2E). FADH, FAD, and cis-aconitic acid of the TCA cycle were found to be elevated in the liver (Fig. 2E), whereas in serum, oxalosuccinic acid was increased and oxoglutaric acid decreased in the TCA cycle (Fig. 2F). Niacinamide of the nicotinate and nicotinamide metabolism was decreased in TBT (Fig. 2F). Besides, prostaglandin E1 and prostaglandin A2 in the arachidonic acid metabolism were also decreased due to TBT intervention (Fig. 2F). In addition, western blotting showed that the expression of the proteins PERK and CHOP in the TBT group were significantly increased compared to in the control group, which suggested that TBT exposure might activate ER stress and further cause apoptosis (Fig. 2G and H). Due to the metal elements participation in biological activities, including ER stress, inflammation, and other reactions, the content of metal elements in serum were measured, as shown in Fig. 2I–K. Among them, Ca, Cu, and Fe are the basic elements, and we found Ca and Cu remarkably were increased and Fe decreased in the TBT group compared to the control group (Fig. 2I–K). The above results indicated that TBT not only led to abnormal lipid metabolism, interfered with energy metabolism process, induced ER stress, affected the level of Ca, Cu, and Fe, but also stimulated the expression of COX-2 to cause hepatic inflammation by ER stress instead of arachidonic acid metabolism.
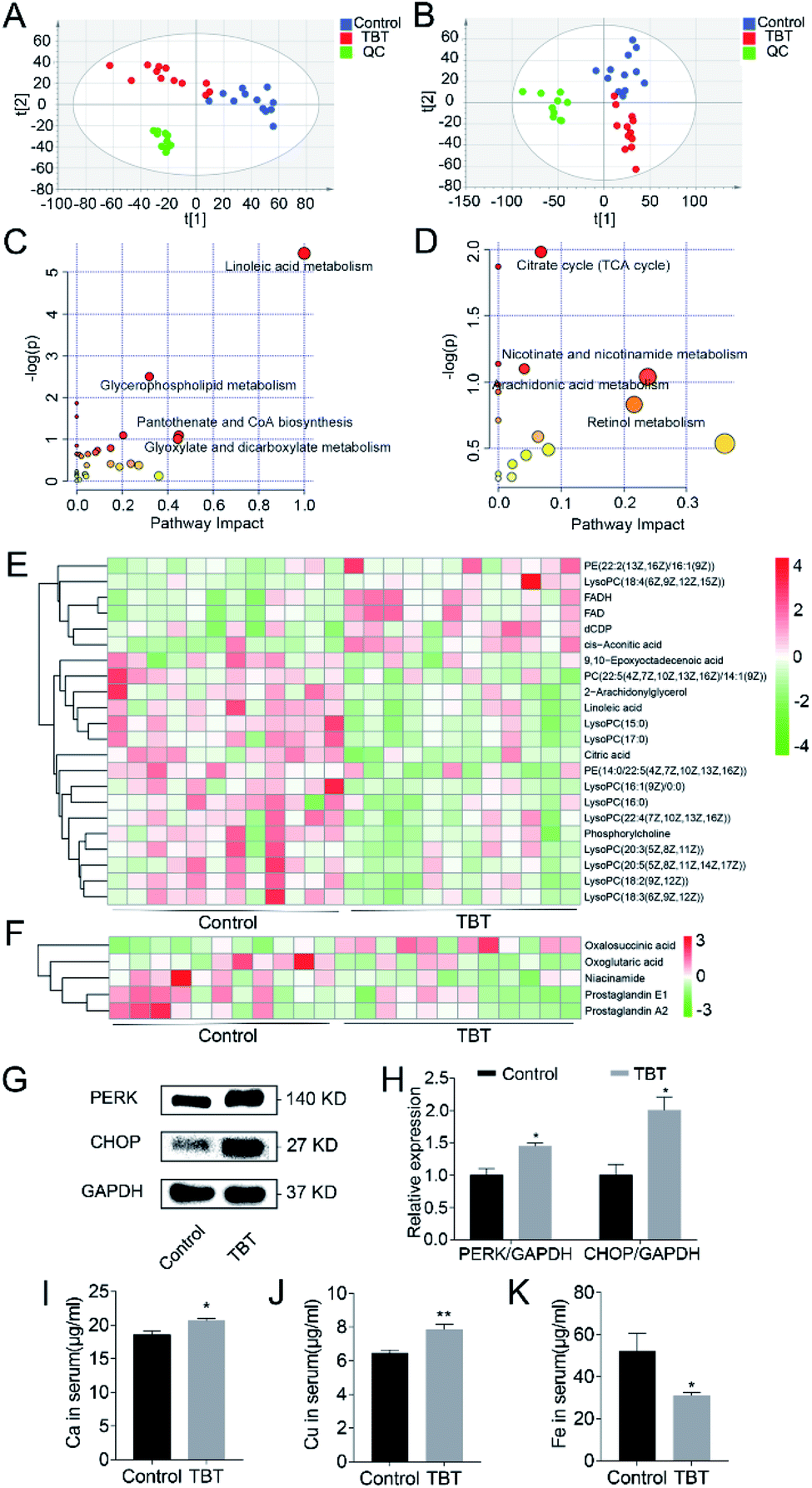 | ||
| Fig. 2 TBT induced metabolic changes, ER stress, and metal disturbances. Partial least squares discrimination analysis (PLS-DA) of the metabolites in rat liver (A) and serum (B) were performed and quality control was simplified to QC. Metabolic pathways affected by TBT treatment in the liver (C) and serum (D) are presented as a bubble diagram based on KEGG metabolic pathway database. Larger bubbles and darker colors indicate that the pathways were more affected. Metabolites that had significant changes in liver (E) and serum (F) due to TBT intervention are presented in the heatmap. (G) Western blotting of protein kinase R-like ER kinase (PERK) and C/EBP homologous protein (CHOP) in the endoplasmic reticulum (ER) stress pathway. (H) The relative expression of protein PERK and CHOP. Metal elements Ca (I), Cu (J), and Fe (K) in serum were determined by ICP-MS. | ||
3.3. TBT altered the fecal microbial communities in rats
The profile of the microbiota composition in fresh rat feces was analyzed by 16S rRNA sequencing technology and found to be shifted significantly under TBT treatment. Principal coordinates analysis (PCoA) showed that the control group and TBT group had partially discrete results (Fig. 3A), suggesting that TBT played a role in disturbing the gut microbiota. To track alterations in the phylum level, a community bar plot was used to show (Fig. 3B) that both Verrucomicrobia (control: 0.40%, TBT: 1.78%) and Bacteroidetes (control: 11.77%, TBT: 20.07%) were increased compared with in the control group, but the composition of Firmicutes (control: 82.73%, TBT: 73.85%) was decreased in the TBT group, which further indicated that TBT exposure could affect the composition of the intestinal bacteria. Diving to the genus level, TBT exposure led to [Acetivibrio]_ethanolgignens_group, Acetatifactor, Eisenbergiella, Lachnospiraceae_UCG-010 of Lachnospiraceae family, Anaerovorax, and Bilophila being significantly increased, while Enterococcus was significantly reduced compared to in the control group (Fig. 3C). As shown in Fig. 3D, KEGG functional annotation pathways predicted that the altered gut microbiome in the TBT group might take part in RNA transport, glycerolipid metabolism, glycolysis/gluconeogenesis, and the phosphotransferase system (PTS). These results suggested that TBT could induce a disturbance of the gut microbiota in rats and was involved in TBT-induced metabolic disorder.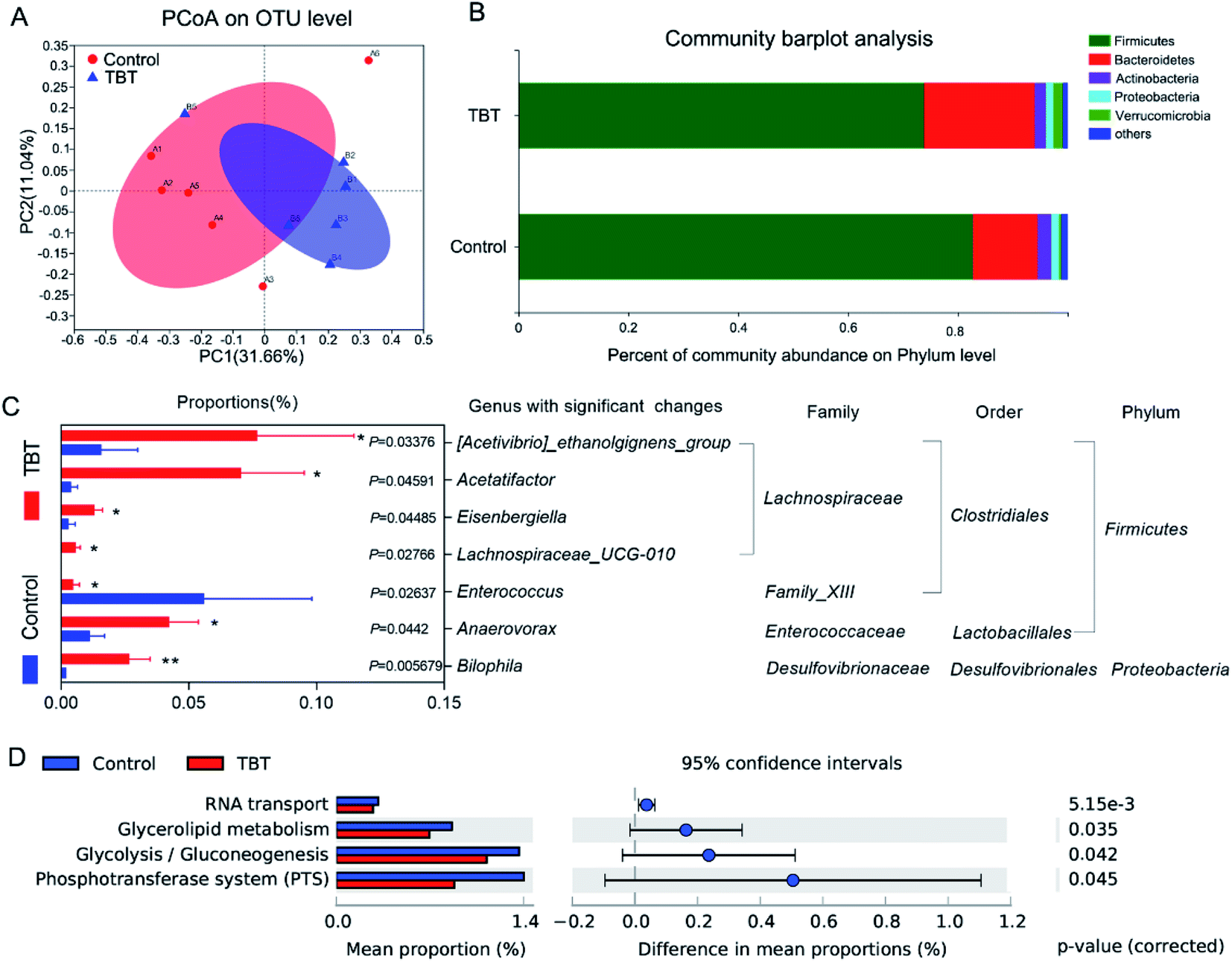 | ||
| Fig. 3 TBT changed the fecal microbiota composition in rats. (A) Principal coordinate analysis (PCoA) plots of the fecal microbiota on OTU level based on binary-Pearson distance metrics. (B) Community bar plot analysis. The average percent of community abundance of the phylum level in each group is presented as a bar plot. (C) Comparison and classification of gut microbiota at the genus level. Wilcoxon rank-sum test were performed between the control group and TBT group, *P < 0.05, **P < 0.01. D. KEGG functional annotation predicted from 16S rRNA sequencing. Predicted metabolic pathways were analyzed by Tax4Fun on the i-sanger platform using STAMP software. Differences between groups were determined using the Welsh's t-test. P < 0.05 was considered to be significant. | ||
3.4. Gut microbiome participate in TBT-induced metabolic changes
To explore the effect of TBT on the metabolism of intestinal bacteria, fecal metabolites were extracted and assessed. As shown in the ESI Table S3,† 32 differential metabolites in feces were obtained based on a comparison with the control group and identification in the HMDB library. PLS-DA analysis indicated that TBT intervention resulted in a separation from the control cluster, indicating TBT had an impact on fecal metabolism (Fig. 4A). Further pathway analysis revealed that TBT mainly affected glycine, serine, and threonine metabolism, the one carbon pool by folate, nicotinate and nicotinamide metabolism, and tryptophan metabolism (Fig. 4B). Compared with the control group, TBT exposure significantly increased phosphohydroxypyruvic acid, 5,10-methylene-THF and N-undecanoylglycine of glycine, serine and threonine metabolism (Fig. 4C). Besides, 5,10-methylene-THF was increased under TBT exposure, which also participated in the one carbon pool by folate (Fig. 4C). Moreover, quinolinic acid of nicotinate and nicotinamide metabolism was decreased in the TBT group (Fig. 4C). Additionally, as a precursor of citric acid, beta-citryl-L-glutamic acid was decreased in feces after TBT exposure (Fig. 4C). Furthermore, we explored the relationship between gut microbiome and metabolites through correlation analysis and found that Bilophila, Lachnospiraceae_UCG-010, and Anaerovorax were positively related to 5,10-methylene-THF; while Bilophila, Acetatifactor and Anaerovorax were positively and Enterococcus was negatively related to N-undecanoylglycine; and Eisenbergiella, Bilophila and Anaerovorax were negatively related to beta-citryl-L-glutamic acid (Fig. 4D). The above results suggested that the TBT-induced metabolic changes were associated with gut microbiome mediation.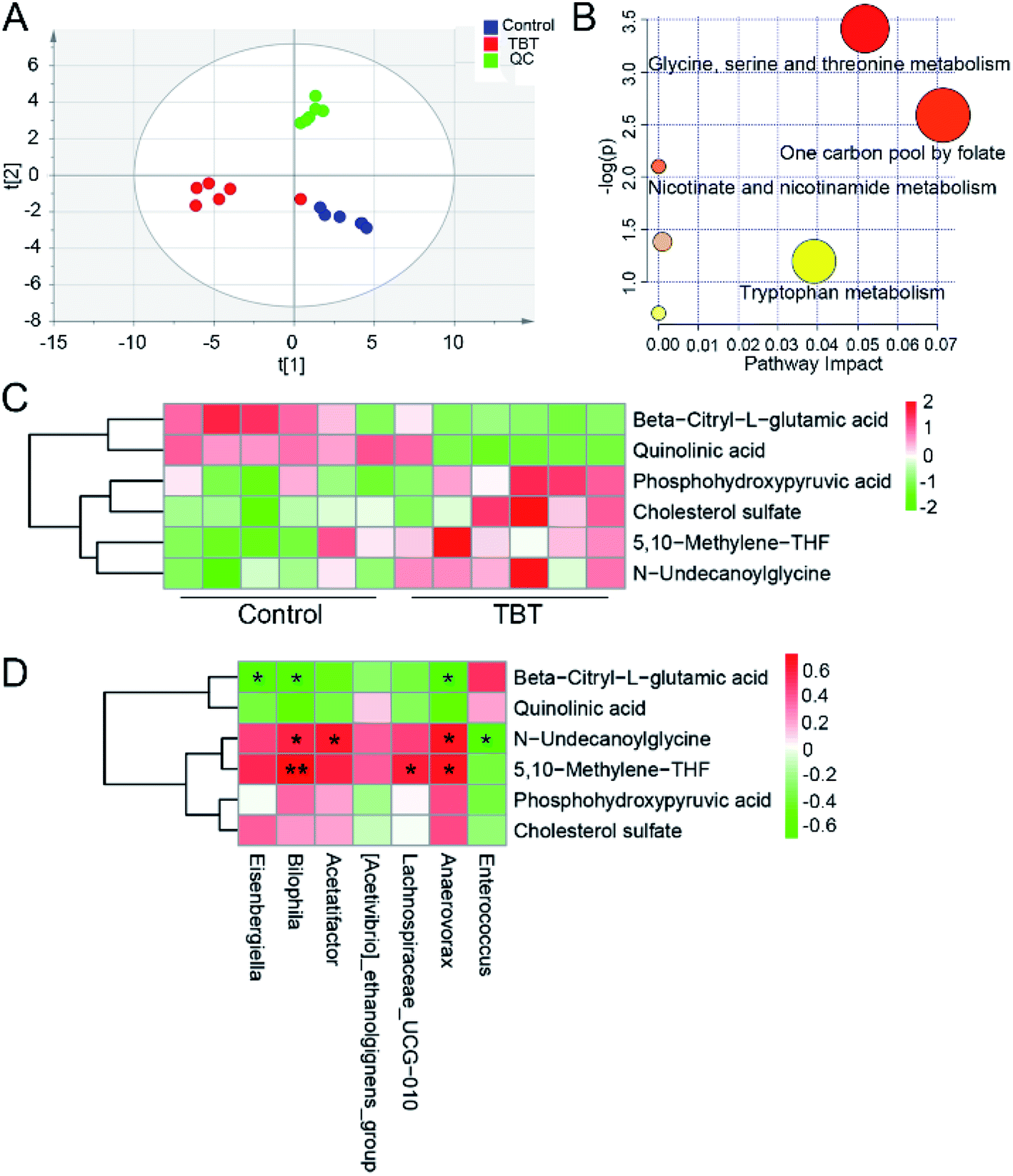 | ||
| Fig. 4 Gut microbiome participate in TBT-induced metabolic changes. (A) Partial Least Squares Discrimination Analysis (PLS-DA) of the metabolites in rat feces. (B) Metabolic pathways affected by TBT treatment in feces are presented as a bubble diagram based on the KEGG metabolic pathway database. (C) Metabolites with statistical significance under TBT exposure. (D) Correlation analysis between differential intestinal bacteria and metabolites, with significant differences marked with *. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.5. ROC curve analysis of gut microbiome under TBT exposure
In order to explore the diagnostic value of intestinal bacteria, ROC analysis was applied to assess the potential value of the differential genera as exposure biomarkers for TBT. The results showed that Bilophila (AUC = 0.9722, P = 0.0065) showed high accuracy; while Enterococcus (AUC = 0.8889, P = 0.0250), [Acetivibrio]_ethanolgignens_group (AUC = 0.875, P = 0.0308) also showed good accuracy with statistical significance; and although the AUC of Anaerovorax (AUC = 0.8611, P = 0.0374), Acetatifactor (AUC = 0.8472, P = 0.0453), Eisenbergiella (AUC = 0.8333, P = 0.0547), and Lachnospiraceae_UCG-010 (AUC = 0.8333, P = 0.0547) suggested good accuracy, they had no statistical significance. These results suggested that the altered intestinal flora caused by TBT, especially Bilophila, Enterococcus, and [Acetivibrio]_ethanolgignens_group, had the potential to be used as biomarkers for TBT exposure.4. Discussion
In this study, we found that TBT (100 μg kg−1 BW) led to a significant decrease in body weight gain and serum TG in rats accompanied with changes in liver and serum metabolism (Fig. 1 and 2). Among these changes, linoleic acid metabolism and glycerophospholipid metabolism were mainly affected by TBT exposure in the liver (Fig. 2C). According to previous studies, linoleic acid can be esterified to form neutral and polar lipids, such as phospholipids, TG, and cholesterol esters.36 In our study, we found that TBT exposure significantly inhibited linoleic acid and the downstream metabolite 2-arachidonylglycerol in the liver and prostaglandin E1 and prostaglandin A2 in serum (Fig. 2E and F), which might explain one reason why TBT suppressed TG in serum. Besides, previous studies have confirmed that increasing Cu inhibits the storage and secretion of TG,37 and so we believed that the change of elemental Cu was key evidence for the decreasing TG in the TBT group (Fig. 1B and 2J). On the other hand, Cu has been reported to be an important molecule in metabolic processes, such as inflammation and lipid peroxidation.38 As this study showed that TBT decreased Cu in serum (Fig. 2J) and induced the aggregation of inflammatory cells in liver tissue through increasing COX-2 expression in the liver (Fig. 1D and E), thus we speculated that Cu might regulate TBT-induced inflammation. Generally, linoleic acid is reported to prolong and desaturate arachidonic acid, and COX-2 is a key enzyme to catalyze arachidonic acid to produce pro-inflammatory factor prostaglandins, which is the first rate-limiting step in prostaglandin biosynthesis.39,40 Obviously, the reduction of linoleic acid, 2-arachidonylglycerol, prostaglandin E1, and prostaglandin A2 in the TBT group (Fig. 2) suggested that the activation of COX-2 in TBT group was not through the arachidonic acid metabolism. COX-2, as a key enzyme located on the membrane of ER, was reported to be N-glycosylated in the ER in the absence of arachidonic acid, and then transported to the Golgi apparatus for modification, before finally being transported back to the ER.39,41 Furthermore, it has been reported to catalyze the biosynthesis of eicosanoid lipids, affecting the expression of eicosanoid-lysolipids in ER.42–45 In addition, PERK is an ER transmembrane protein response to ER stress and could promote apoptosis through activating the downstream pro-apoptotic transcription factor CHOP.46 It has been verified that the action of PERK could affect VLDL receptors that are regulators of liver TG and fatty degeneration.47–49 As shown in Fig. 2G and H, the expression of PERK and CHOP increased remarkably under TBT exposure, and thus we hypothesized that TBT-induced ER stress might be due to the enhancement COX-2 by activating the PERK and CHOP responses. In addition, a previous study certified that ER stress can activate the release of Ca ions (Ca2+), and then lead to programmed cell death.50,51 Usually, following the apoptosis of the cell, the integrity of the plasma membrane is destroyed, leading to the release of certain cellular components, such as ATP, K+, uric acid, and other metabolic substances, and the occurrence of an inflammatory response.52 Correspondingly, TBT exposure induced the increase in Ca in serum (Fig. 2I), suggesting that TBT-induced ER stress might cause apoptosis in the liver accompanied by the release of Ca to serum. A previous study reported that the loss of saturated lysophosphatidylcholine (LysoPC) to phosphatidylcholine (PC) conversion activity can enhance ER stress.53 In this study, the reduction of LysoPCs and PC in the TBT group (Fig. 2E) suggested that TBT-induced ER stress could also attributed be to abnormal metabolism. Additionally, the TCA cycle is a central part of the carbon and energy metabolism, also connecting to glycolysis, amino acid, and lipid metabolism.54 Besides, TCA cycle activity and ER homeostasis have been shown to be metabolically related.55 As in our study, TBT increased the TCA cycle intermediates cis-aconitic acid, FAD, and FADH (Fig. 2E), which illustrated that TBT could enhance the energy supply for maintaining the function of ER and metabolic progress.Previous studies have indicated that gut microbiota are important mediators in related physiological process, like host energy homeostasis and systemic inflammation, etc.25 Our bacterial sequencing results also showed that the composition of intestinal flora was shifted under TBT exposure, including [Acetivibrio]_ethanolgignens_group, Acetatifactor, Eisenbergiella, Lachnospiraceae_UCG-010, Enterococcus, Anaerovorax, and Bilophila (Fig. 4C). Among these, Bilophila, [Acetivibrio]_ethanolgignens_group, and Enterococcus showed high AUC, indicating those genera could be potential markers for TBT exposure (Fig. 5A, E and G). In line with our results, previous studies have reported that Bilophila was considered to stimulate systemic inflammation and reduce liver TG,56,57 while Acetatifactor of Lachnospiraceae family has been reported to be correlated with a reduction in body weight, energy efficiency, liver fatty acids, and serum cholesterol.58,59 Besides, the Lanchnospiraceae family is involved in the CoA metabolism of the TCA cycle, which could be damaged by overloading with Cu and this would then disrupt the supply of energy to cellular respiration by the mitochondria.60,61 Thus, we suggest that TBT exposure could cause weight gain and TG in serum would be significantly decreased with enhanced inflammation by increasing the Lachnospiraceae family and Bilophila. Moreover, it was reported that Enterococcus can catabolize glycerol,62 which might be the reason why TBT induced the reduction of TG (Fig. 1B). Based on the prediction of the flora in this study, the flora affected by TBT participated in glycerolipid metabolism, glycolysis/gluconeogenesis, and PTS (Fig. 3D), suggesting that the gut microbiome played an important role in metabolic disorders caused by TBT. Meanwhile, such a conclusion is also supported by previous studies, in which the bacterial glycerol metabolic pathway was reported to be a dehydrogenation and phosphorylation pathway,63 while the metabolism of glucose/glycolysis and the metabolism of glycerol have been reported to generate nicotinamide adenine dinucleotide molecules by Enterococcus, although in different ways.64 As our research found, TBT reduced Enterococcus and affected nicotinate and nicotinamide metabolism in serum and feces, which was related to tryptophan metabolism (Fig. 3C and 4B), allowing surmising that the above-mentioned metabolism was the main contributor to those phenomena. In addition, a previous study reported that the addition of niacin tended to increase the abundance of Acetivibrio.65 So we speculated that the decreased niacinamide in serum (Fig. 2F) and quinolinic acid in feces (Fig. 4C) caused by TBT was due to the consumption of the [Acetivibrio]_ethanolgignens_group. Moreover, niacinamide and quinolinic acid in nicotinate and nicotinamide metabolism are related to the TCA cycle, where beta-citryl-L-glutamic acid was reported to play a role as an Fe-carrier for mitochondrial aconitase.66 Besides, 5,10-methylene-THF can be formed by the decomposition of glycine catalyzed by glycine lyase in bacteria, and also, glycine can positively regulate metabolic flux to the TCA cycle.67,68 All these studies supported that beta-citryl-L-glutamic acid was found to be negatively related to Eisenbergiella, Bilophila, and Anaerovorax (Fig. 4D), 5,10-methylene-THF, and N-undecanoylglycine of glycine, while serine and threonine metabolism in feces were positively related with Bilophila and Anaerovorax in this study (Fig. 4D). Therefore, we speculated that the above-mentioned intestinal bacteria affect the intermediate steps of the TCA cycle to affect energy metabolism under TBT exposure.
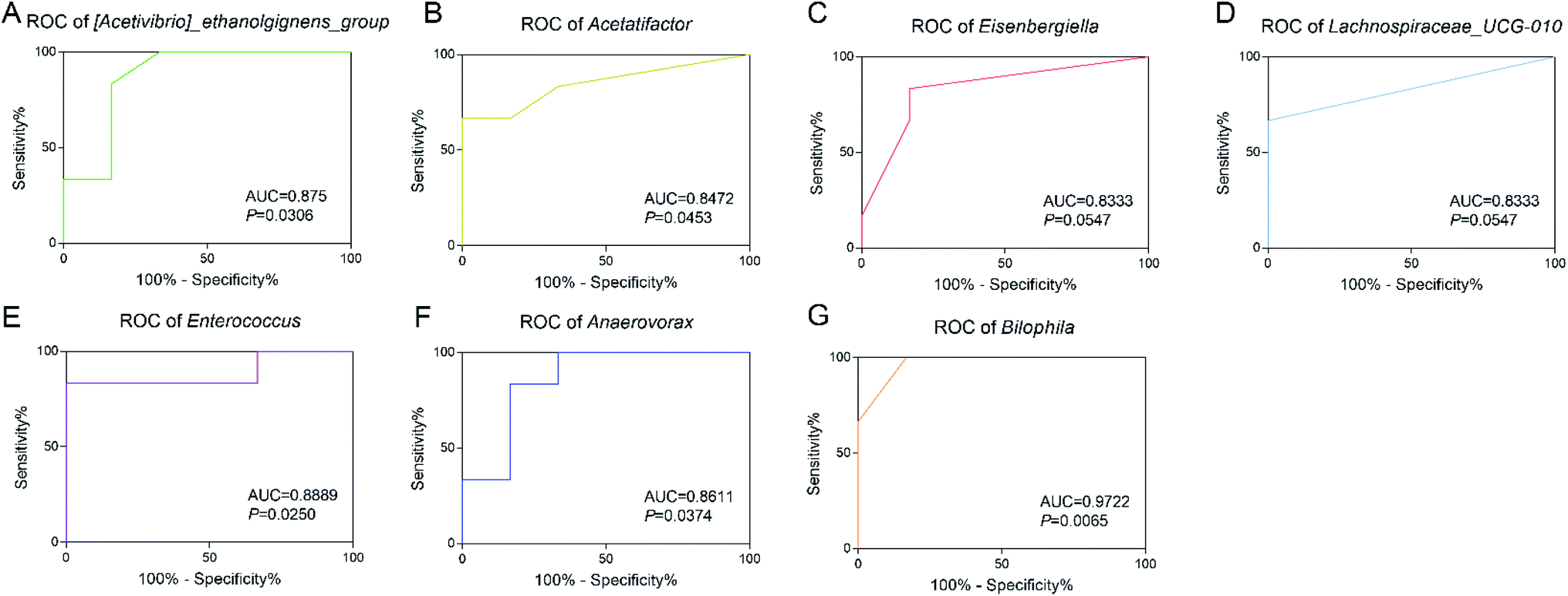 | ||
| Fig. 5 Receiver operator characteristic (ROC) curve analysis of gut microbiome under TBT exposure. ROC analysis of (A) [Acetivibrio]_ethanolgignens, (B) Acetatifactor, (C) Eisenbergiella, (D) Lachnospiraceae_UCG-010, (E) Enterococcus, (F) Anaerovorax, and (G) Bilophila was performed using GraphPad Prism 7 software. The area under the curve (AUC) indicated the accuracy of the prediction and P < 0.0332 was considered statistically significant. | ||
5. Conclusions
Our study demonstrated that gut microbiome might be involved in TBT-induced inflammation and lipid metabolic disorder. TBT could trigger liver inflammation due to enhancing COX-2 expression by activating PERK and CHOP in ER stress instead of stimulating the arachidonic acid metabolism. Besides, Ca and Cu were found to respond to TBT-caused inflammation and lipid disorder. Intestinal flora [Acetivibrio]_ethanolgignens_group, Acetatifactor, Eisenbergiella, Lachnospiraceae_UCG-010, Anaerovorax, and Bilophila were altered under TBT exposure and were involved in mediating inflammation and lipid metabolism abnormalities, such as TG, linoleic acid, and glycerophospholipids, and also interfered with the energy supply process of the TCA cycle. Among these, Bilophila (AUC = 0.9722, P = 0.0065), Enterococcus (AUC = 0.8889, P = 0.0250), and [Acetivibrio]_ethanolgignens_group (AUC = 0.875, P = 0.0308) have a high diagnostic value for TBT exposure risk.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81673228, 81973096 and 82003504), The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (19KJB330004), Science and Technology Development Fund of Nanjing Medical University (NMUB2019012).References
- A. K. Leonardi and C. K. Ober, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 241–264 CrossRef.
- K. A. Dafforn, J. A. Lewis and E. L. Johnston, Mar. Pollut. Bull., 2011, 62, 453–465 CrossRef CAS.
- I. B. Castro, J. Iannacone, S. Santos and G. Fillmann, Chemosphere, 2018, 205, 253–259 CrossRef CAS.
- H. Tian, P. Wu, W. Wang and S. Ru, Aquat. Toxicol., 2015, 162, 117–125 CrossRef CAS.
- B. Antizar-Ladislao, Environ. Int., 2008, 34, 292–308 CrossRef CAS.
- S. Mitra, R. Gera, V. Singh and S. Khandelwal, Food Chem. Toxicol., 2014, 64, 335–343 CrossRef CAS.
- Y. Mattos, W. B. Stotz, M. S. Romero, M. Bravo, G. Fillmann and I. B. Castro, Sci. Total Environ., 2017, 595, 209–217 CrossRef CAS.
- K. He, J. Zhang and Z. Chen, Endokrynol. Pol., 2014, 65, 485–490 CrossRef CAS.
- C. S. da Costa, L. Miranda-Alves, M. A. La Merrill, I. V. Silva and J. B. Graceli, Toxicol. Lett., 2019, 307, 59–71 CrossRef CAS.
- J. F. P. de Araujo, P. L. Podratz, G. C. Sena, E. Merlo, L. C. Freitas-Lima, J. G. M. Ayub, A. F. Z. Pereira, A. P. Santos-Silva, L. Miranda-Alves, I. V. Silva and J. B. Graceli, Toxicol. Lett., 2018, 295, 99–114 CrossRef CAS.
- X. Wu, J. Liu, Y. Duan, S. Gao, Y. Lu, X. Li, Q. Zhu, X. Chen, J. Lin, L. Ye and R. S. Ge, Front. Pharmacol., 2017, 8, 704 CrossRef.
- H. G. Liu, Y. Wang, L. Lian and L. H. Xu, Environ. Toxicol., 2006, 21, 166–171 CrossRef CAS.
- Y. Zhang, Y. Chen, L. Sun, J. Liang, Z. Guo and L. Xu, Environ. Toxicol., 2014, 29, 234–242 CrossRef CAS.
- Z. H. Li, P. Li and Z. C. Shi, Environ. Toxicol., 2016, 31, 937–944 CrossRef CAS.
- H. Liu, Z. Guo, L. Xu and S. Hsu, Environ. Toxicol., 2008, 23, 77–83 CrossRef CAS.
- H. C. da Silva de Assis, A. Sanchez-Chardi, R. C. Dos Reis, L. Nicaretta, C. Mencinauski, S. C. Jakobi, P. H. da Silva, A. R. Zampronio, E. Pelletier and C. A. de Oliveira Ribeiro, Environ. Toxicol. Pharmacol., 2005, 19, 113–120 CrossRef CAS.
- M. Zhou, M. Feng, L. L. Fu, L. D. Ji, J. S. Zhao and J. Xu, Food Chem. Toxicol., 2016, 97, 316–326 CrossRef CAS.
- H. Yan, H. Guo, D. Cheng, R. Kou, C. Zhang and J. Si, Environ. Toxicol., 2018, 33, 752–758 CrossRef CAS.
- B. D. Bertuloso, P. L. Podratz, E. Merlo, J. F. de Araujo, L. C. Lima, E. C. de Miguel, L. N. de Souza, A. L. Gava, M. de Oliveira, L. Miranda-Alves, M. T. Carneiro, C. R. Nogueira and J. B. Graceli, Toxicol. Lett., 2015, 235, 45–59 CrossRef CAS.
- R. X. Ding, W. R. Goh, R. N. Wu, X. Q. Yue, X. Luo, W. W. T. Khine, J. R. Wu and Y. K. Lee, J. Food Drug Anal., 2019, 27, 623–631 CrossRef CAS.
- Y. Gu, X. Wang, J. Li, Y. Zhang, H. Zhong, R. Liu, D. Zhang, Q. Feng, X. Xie, J. Hong, H. Ren, W. Liu, J. Ma, Q. Su, H. Zhang, J. Yang, X. Wang, X. Zhao, W. Gu, Y. Bi, Y. Peng, X. Xu, H. Xia, F. Li, X. Xu, H. Yang, G. Xu, L. Madsen, K. Kristiansen, G. Ning and W. Wang, Nat. Commun., 2017, 8, 1785 CrossRef.
- M. Yi, S. Yu, S. Qin, Q. Liu, H. Xu, W. Zhao, Q. Chu and K. Wu, J. Hematol. Oncol., 2018, 11, 47 CrossRef.
- P. D. Cani and N. M. Delzenne, Curr. Opin. Clin. Nutr. Metab. Care, 2007, 10, 729–734 CrossRef.
- S. A. Bassett, W. Young, K. Fraser, J. E. Dalziel, J. Webster, L. Ryan, P. Fitzgerald, C. Stanton, T. G. Dinan, J. F. Cryan, G. Clarke, N. Hyland and N. C. Roy, Sci. Rep., 2019, 9, 14026 CrossRef.
- P. C. Barko, M. A. McMichael, K. S. Swanson and D. A. Williams, J. Vet. Intern. Med., 2018, 32, 9–25 CrossRef CAS.
- Y. Vazquez-Baeza, E. R. Hyde, J. S. Suchodolski and R. Knight, Nat. Microbiol., 2016, 1, 16177 CrossRef CAS.
- Y. Minamoto, C. C. Otoni, S. M. Steelman, O. Buyukleblebici, J. M. Steiner, A. E. Jergens and J. S. Suchodolski, Gut Microbes, 2015, 6, 33–47 CrossRef CAS.
- P. M. Spritzer, S. B. Lecke, V. C. Fabris, P. K. Ziegelmann and L. Amaral, Biol. Trace Elem. Res., 2017, 175, 254–262 CrossRef CAS.
- Q. Zhai, S. Cen, J. Jiang, J. Zhao, H. Zhang and W. Chen, Environ. Res., 2019, 171, 501–509 CrossRef CAS.
- M. Y. Zeng, N. Inohara and G. Nunez, Mucosal Immunol., 2017, 10, 18–26 CrossRef CAS.
- G. Porcheron, A. Garenaux, J. Proulx, M. Sabri and C. M. Dozois, Front. Cell. Infect. Microbiol., 2013, 3, 90 Search PubMed.
- A. Tripathi, J. Debelius, D. A. Brenner, M. Karin, R. Loomba, B. Schnabl and R. Knight, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 397–411 CrossRef CAS.
- H. Guo, H. Yan, D. Cheng, X. Wei, R. Kou and J. Si, Environ. Toxicol. Pharmacol., 2018, 60, 202–208 CrossRef CAS.
- J. Zhu, C. Wang, X. Gao, J. Zhu, L. Wang, S. Cao, Q. Wu, S. Qiao, Z. Zhang and L. Li, RSC Adv., 2019, 9, 10766–10775 RSC.
- S. Zhao, S. Cao, L. Luo, Z. Zhang, G. Yuan, Y. Zhang, Y. Yang, W. Guo, L. Wang, F. Chen, Q. Wu and L. Li, Clin. Chim. Acta, 2018, 485, 323–332 CrossRef CAS.
- J. Whelan and K. Fritsche, Adv. Nutr., 2013, 4, 311–312 CrossRef.
- S. Guttmann, O. Nadzemova, I. Grunewald, M. Lenders, E. Brand, A. Zibert and H. H. Schmidt, PLoS One, 2020, 15, e0230025 CrossRef CAS.
- A. Ayala, M. F. Munoz and S. Arguelles, Oxid. Med. Cell. Longevity, 2014, 2014, 360438 CrossRef.
- X. Liao, W. Wang, C. Fan, N. Yang, J. Zhao, Y. Zhang, R. Gao, G. Shen, S. Xia and G. Li, Int. J. Mol. Med., 2017, 40, 75–82 CrossRef CAS.
- E. Saadi, S. Tal and L. Barki-Harrington, Biochem. J., 2018, 475, 3141–3151 CrossRef CAS.
- M. Y. Yang, C. H. Wu, T. W. Hung and C. J. Wang, Antioxidants, 2019, 9, 26 CrossRef.
- X. Liu, S. H. Moon, C. M. Jenkins, H. F. Sims and R. W. Gross, Cell Chem. Biol., 2016, 23, 1217–1227 CrossRef.
- S. J. Desai, B. Prickril and A. Rasooly, Nutr. Cancer, 2018, 70, 350–375 CrossRef CAS.
- L. J. Marnett and R. N. DuBois, Annu. Rev. Pharmacol. Toxicol., 2002, 42, 55–80 CrossRef CAS.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS.
- H. Hu, M. Tian, C. Ding and S. Yu, Front. Immunol., 2018, 9, 3083 CrossRef CAS.
- X. Wu, N. Xu, M. Li, Q. Huang, J. Wu, Y. Gan, L. Chen, H. Luo, Y. Li, X. Huang, Z. Su and Y. Liu, Front. Pharmacol., 2019, 10, 1134 CrossRef CAS.
- C. Hetz and F. R. Papa, Mol. Cell, 2018, 69, 169–181 CrossRef CAS.
- Y. Hu, J. Liu, Y. Yuan, J. Chen, S. Cheng, H. Wang and Y. Xu, Environ. Toxicol. Pharmacol., 2018, 64, 112–121 CrossRef CAS.
- P. A. Lindahl and M. J. Moore, Biochemistry, 2016, 55, 4140–4153 CrossRef CAS.
- S. Lee and K. T. Min, Mol. Cells, 2018, 41, 1000–1007 CAS.
- P. I. Merksamer and F. R. Papa, J. Cell Sci., 2010, 123, 1003–1006 CrossRef CAS.
- X. Rong, C. J. Albert, C. Hong, M. A. Duerr, B. T. Chamberlain, E. J. Tarling, A. Ito, J. Gao, B. Wang, P. A. Edwards, M. E. Jung, D. A. Ford and P. Tontonoz, Cell Metab., 2013, 18, 685–697 CrossRef CAS.
- R. Rathod, B. Gajera, K. Nazir, J. Wallenius and V. Velagapudi, Metabolites, 2020, 10, 103 CrossRef CAS.
- E. R. Gansemer, K. S. McCommis, M. Martino, A. Q. King-McAlpin, M. J. Potthoff, B. N. Finck, E. B. Taylor and D. T. Rutkowski, iScience, 2020, 23, 101116 CrossRef CAS.
- Z. Feng, W. Long, B. Hao, D. Ding, X. Ma, L. Zhao and X. Pang, Gut Pathog., 2017, 9, 59 CrossRef.
- J. M. Natividad, B. Lamas, H. P. Pham, M. L. Michel, D. Rainteau, C. Bridonneau, G. da Costa, J. van Hylckama Vlieg, B. Sovran, C. Chamignon, J. Planchais, M. L. Richard, P. Langella, P. Veiga and H. Sokol, Nat. Commun., 2018, 9, 2802 CrossRef.
- J. Liu, W. Hao, Z. He, E. Kwek, Y. Zhao, H. Zhu, N. Liang, K. Y. Ma, L. Lei, W. S. He and Z. Y. Chen, Food Funct., 2019, 10, 2847–2860 RSC.
- P. Pathak, C. Xie, R. G. Nichols, J. M. Ferrell, S. Boehme, K. W. Krausz, A. D. Patterson, F. J. Gonzalez and J. Y. L. Chiang, Hepatology, 2018, 68, 1574–1588 CrossRef CAS.
- C. He, A. J. Ryan, S. Murthy and A. B. Carter, J. Biol. Chem., 2013, 288, 20745–20757 CrossRef CAS.
- S. H. Duncan, A. Barcenilla, C. S. Stewart, S. E. Pryde and H. J. Flint, Appl. Environ. Microbiol., 2002, 68, 5186–5190 CrossRef CAS.
- A. Bizzini, C. Zhao, A. Budin-Verneuil, N. Sauvageot, J. C. Giard, Y. Auffray and A. Hartke, J. Bacteriol., 2010, 192, 779–785 CrossRef CAS.
- E. C. Lin, Annu. Rev. Microbiol., 1976, 30, 535–578 CrossRef CAS.
- Y. Doi, Appl. Microbiol. Biotechnol., 2018, 102, 10183–10192 CrossRef CAS.
- D. Luo, Y. Gao, Y. Lu, M. Qu, X. Xiong, L. Xu, X. Zhao, K. Pan and K. Ouyang, Anim. Nutr., 2017, 3, 180–185 CrossRef.
- M. Hamada-Kanazawa, M. Narahara, M. Takano, K. S. Min, K. Tanaka and M. Miyake, Biol. Pharm. Bull., 2011, 34, 1455–1464 CrossRef CAS.
- T. Tezuka and Y. Ohnishi, J. Bacteriol., 2014, 196, 1369–1376 CrossRef.
- Z. X. Cheng, C. Guo, Z. G. Chen, T. C. Yang, J. Y. Zhang, J. Wang, J. X. Zhu, D. Li, T. T. Zhang, H. Li, B. Peng and X. X. Peng, Nat. Commun., 2019, 10, 3325 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07502g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |