
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable access to fully biobased epoxidized vegetable oil thermoset materials prepared by thermal or UV-cationic processes†
Samuel Malburet *a,
Chiara Di Maurob,
Camilla Noèc,
Alice Mijab,
Marco Sangermanoc and
Alain Graillota
*a,
Chiara Di Maurob,
Camilla Noèc,
Alice Mijab,
Marco Sangermanoc and
Alain Graillota
aSpecific Polymers, 150 Avenue des Cocardières, 34160, Castries, France. E-mail: samuel.malburet@specificpolymers.fr
bUniversité Côte d’Azur, Institut de Chimie de Nice, UMR 7272 CNRS, 28 Avenue Valrose, 06108 Nice Cedex 02, France
cPolitecnico di Torino, Dipartimento di Scienza Applicata e Tecnologia, C.so Duca degli Abruzzi 24, 10129, Torino, Italy
First published on 18th November 2020
Abstract
Beyond the need to find a non-toxic alternative to DiGlycidyl Ether of Bisphenol-A (DGEBA), the serious subject of non-epichlorohydrin epoxy resins production remains a crucial challenge that must be solved for the next epoxy resin generations. In this context, this study focuses on the valorization of vegetable oils (VOs) into thermoset materials by using (i) epoxidation of the VOs through the “double bonds to epoxy” synthetic route and (ii) synthesis of crosslinked homopolymers by UV or hardener-free thermal curing processes. A thorough identification, selection and physico-chemical characterization of non-edible or non-valuated natural vegetable oils were performed. Selected VOs, characterized by a large range of double bond contents, were then chemically modified into epoxides thanks to an optimized, robust and sustainable method based on the use of acetic acid, hydrogen peroxide and Amberlite® IR-120 at 55 °C in toluene or cyclopentyl methyl ether (CMPE) as a non-hazardous and green alternative solvent. The developed environmentally friendly epoxidation process allows reaching almost complete double bond conversion with an epoxy selectivity above 94% for the 12 studied VOs. Finally, obtained epoxidized vegetable oils (EVOs), characterized by an epoxy index from 2.77 to 6.77 meq. g−1 were cured using either UV or hardener-free thermal curing. Both methods enable the synthesis of 100% biobased EVO thermoset materials whose thermomechanical performances were proved to linearly increase with the EVOs' epoxy content. This paper highlights that tunable thermomechanical performances (Tα from −19 to 50 °C and Tg from −34 to 36 °C) of EVO based thermoset materials can be reached by well selecting the starting VO raw materials.
Introduction
For many decades, the declining reserves of petroleum-based raw materials and the growing environmental concerns have made renewable feedstock valorisation one of the major stakes of scientific and industrial research. With the continuous population growth, this demand will become increasingly critical and efforts must be maintained.As a large end-user of chemicals, polymer industry plays a significant role in the fossil resources consumption. Among polymeric materials, thermosets and especially epoxy thermoset received a great deal of attention. First, due to their crosslinked network, epoxy thermosets materials developed so far cannot be recycled. Significant research efforts are thus geared towards the development of innovative solutions such as chemical1 or enzymatic2 degradative pathways. Secondly, epoxy production is mostly carried out by using epichlorohydrin as main reactant, enabling to convert either acids, phenols, amines or hydroxyls into corresponding epoxides (glycidyl). Although promising results were obtained to date by valorizing biomass derivatives such as furfuryl3–5 and vanillin derivatives6–8 as well as isosorbide,9 the epoxidation routes based on epichlorohydrin reactant raise several issues. As a matter of fact, this synthetic strategy implies various side-reactions which often lead to an increase of the molecular weight and thus a decrease of the epoxy content. More, the main drawback of epichlorohydrin is linked to its high toxicity and its classification by several international health research agencies.10 Taking this into account, the synthesis of epoxides through epichlorohydrin based strategies is likely to be restricted in a near future and relevant alternatives must be considered to develop the next epoxy resin generations.
One alternative to the use of epichlorohydrin or other toxic reagents is to convert naturally occurring double bonds into epoxides. Even if several natural products exhibit natural double bonds such as limonene and eugenol for instance, their conversion generally leads to monofunctional epoxy11,12 which is not consistent with thermoset applications. For this reason, vegetable oils (VOs) appear as one of the most promising feedstocks due to their high double bond content. Moreover, they are abundant, low-cost, non-toxic and non-depletable. VOs are generally composed by triglycerides and minor components:13
• triglycerides constitute from 95 to 99% of the oil. They are comprised of three fatty acids joined at a glycerol junction. Conventional fatty acid varies from 8 to 24 carbons in length, with 0 to 3 double bonds and may even contain functional groups. A general structure of VOs' triglyceride and the most conventional fatty acids are shown in Fig. 1. The chemical nature of the fatty acids and more specifically the corresponding double bonds content have an influence on the reactivity, the stability and the physical and physiological properties of vegetable oils. Double bonds are mainly positioned on the third, sixth and ninth chain position and the related fatty acids are commonly known as omega-3, omega-6 and omega-9 respectively.
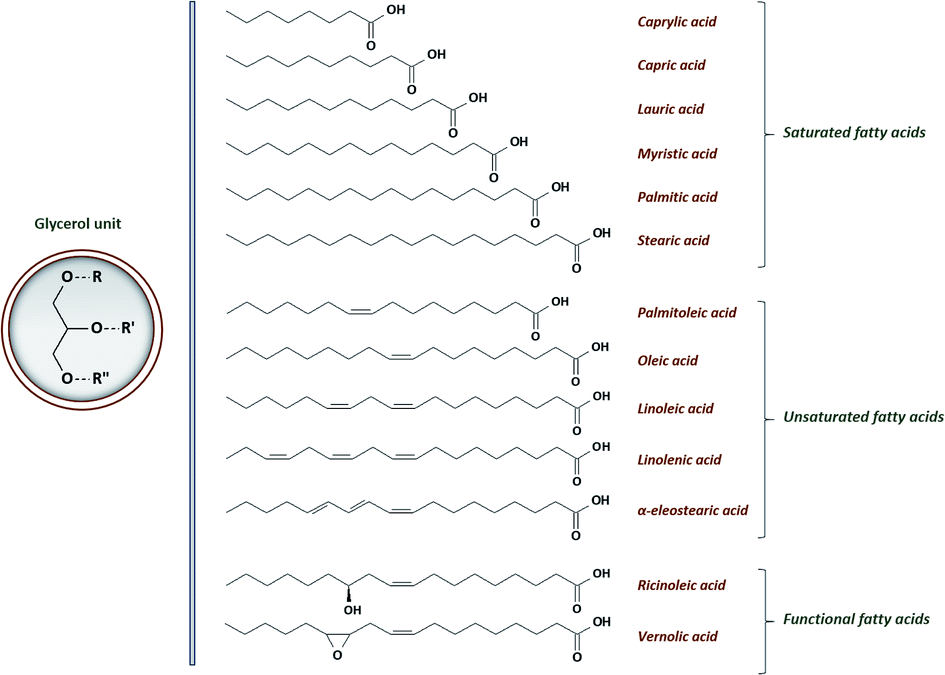 | ||
| Fig. 1 General structure of triglyceride with the most conventional fatty acids. | ||
• Minor components, representing from 1 to 5% of the oil, are phospholipids, complex lipids and non-glyceride compounds such as tocopherol, phytosterols or carotenoids.
Several EVOs are available at industrial scales such as soybean oil (Lankroflex® E2307 – Valtris Specialty Chemicals), linseed oil (Vikoflex® 7190 – Arkema) and castor oil (ERISYS® GE-35 – Emerald Performance Materials). Due to their aliphatic structures, the thermo-mechanical properties of EVOs materials are generally limited. However, recent work highlighted that the thermo-mechanical properties can be tailored through an adapted choice of the curing agents.14–17 So far, EVOs find applications mostly as plasticizers, stabilizers or acid scavengers for PVC as well as component into epoxy formulated mixtures to impart flexibility, impact resistance and/or thermal shock resistance.
Structure, composition and functionality vary across vegetable oils nature. These aspects make vegetable oils a noteworthy biomass resource to be valorized and considered in material science. In this work, our goal was to bring to light that the oils composition diversity is a major asset to synthesize EVOs based crosslinked materials exhibiting tunable thermo-mechanical properties.
To do so, both UV-cationic and hardener-free thermal processes were applied to obtain fully biobased thermoset materials. Beside EVOs, neither additives nor hardeners (except photoinitiator for UV-curing) were used in the thermoset material formulations in order to avoid any influence on the final thermo-mechanical properties and thus (i) highlight the real performances resulting from the EVOs and (ii) easily draw relationships between the chemical composition of the selected VOs/synthesized EVOs and the final properties of corresponding thermoset materials. While thermal curing process is very important for thermosets production with structural applications, in the field of polymeric coatings, the UV-curing technique shows many interesting advantages over the thermal method. It is worth mentioning the reduced energy consumption, high cure speed even at room temperature and absence of VOC emission.18–20 More, investigating and comparing both hardener-free thermal and photo-induced curing processes allow evaluating the influence of the crosslinking mechanisms on the thermo-mechanical properties of EVOs based thermoset materials.
First, criteria applied for the selection of the vegetable oils are highlighted in this paper as well as the nature and composition of these oils. Then, the selection of the epoxidation conditions is described. Following the presentation of several EVOs results, thermoset materials properties obtained by both thermal and UV curing will be finally displayed and confronted. To the best of our knowledges, it is the first time that a such study is achieved by keeping and applying a sustainable approach at each step of the value chain.
Results and discussion
Benchmark analysis of vegetable oils sustainable raw materials
The VOs advantages reside in their composition diversity which is notably dependent on the nature of the oil, its geographical origin and its extraction process. Instead of involving intensive chemical modifications to modify the vegetable oils, they were selected accordingly to their fatty acid composition and thus their double content to reach a large range of epoxy index. In this scope, 12 vegetable oils differing mostly by their theoretical fatty acid composition were identified and used (Table 1). The main criteria considered for VOs' selection are given below:| Vegetable oil | Karanja | St John's wort | Peanut | Rapeseedd | Soybean | Rose hip | Safflower | Camelina | Hemp | Rose hip | Linseed | Perilla |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Vegetable oils are divided in 3 categories: the non-edible oils (NE), the edible oils (E) and the non-valuated oils (NV).b Evaluated by 1H NMR.c Determined by 1H qNMR.d Compared to canola oil (edible), rapeseed oil (non-edible oil) contains higher amount of erucic fatty acid. | ||||||||||||
| Label | VO 1 | VO 2 | VO 3 | VO 4 | VO 5 | VO 6 | VO 7 | VO 8 | VO 9 | VO 10 | VO 11 | VO 12 |
| Statusa | NE | NE | E | NE | E | NE | NV | NV | NV | NE | E | NV |
| Saturated fatty acids (%)b | 22 | 21 | 20 | 7 | 16 | 17 | 12 | 10 | 10 | 5 | 10 | 3 |
| Mono-unsaturated fatty acids (%)b | 56 | 63 | 53 | 62 | 26 | 20 | 18 | 30 | 12 | 16 | 19 | 21 |
| Di-unsaturated fatty acids (%)b | 21 | 16 | 27 | 25 | 49 | 57 | 70 | 28 | 65 | 46 | 13 | 18 |
| Tri-unsaturated fatty acids (%)b | 1 | — | — | 7 | 8 | 5 | — | 32 | 13 | 36 | 58 | 58 |
| Double bonds content (meq. g−1)c | 2.94 | 3.13 | 3.47 | 4.13 | 4.73 | 4.85 | 5.15 | 5.38 | 5.89 | 6.43 | 7.02 | 7.13 |
| Iodine value (IV)c | 74.7 | 79.3 | 88.0 | 104.7 | 120.0 | 123.0 | 130.8 | 136.5 | 149.6 | 163.1 | 178.1 | 180.9 |
• first, all selected VOs are constituted predominantly by the same conventional fatty acids but in different proportion. This criterion allows to set aside the influence of structure or functionality of the fatty acids. Therefore, mainly stearic, oleic, linoleic and linolenic acids constitute the selected vegetable oils.
• Secondly, these oils were selected in order to demonstrate the influence of the epoxy content on the final material properties. Hence, the double bonds index of each VO was measured and considered to reach a wide range of unsaturation.
• Finally, a signification attention has been devoted to the non-edible or non-valuated aspects of the selected oils. Non-valuated oils correspond to VOs which are non-healthy or not commonly used in the food sector. Apart from peanut, soybean and linseed oils, all selected vegetable oils are non-edible (Karanja, St John's wort, rapeseed and rose hip oils) or non-valuated (safflower, camelina, hemp and perilla oils).
Physico-chemical analysis of vegetable oils
Commonly, fatty acid composition in vegetable oils is determined by gas chromatography (GC). This latter is the AOAC Official method from American Oil Chemists' Society for the determination of fatty acids' content.21 Nevertheless, this analytical tool is destructive, time- and chemical-consuming since it implies (i) triglyceride hydrolysis, (ii) esterification by methanol or by direct transesterification followed by (iii) separation, identification and quantification by GC.Hence, 1H NMR was used in this work since this technique is largely approved as a robust and effective technic for lipid analysis. An example of 1H NMR spectrum of hemp oil is given in Fig. 2. 1H NMR spectrum of the other studied oils are displayed in Section S2 (see ESI).†
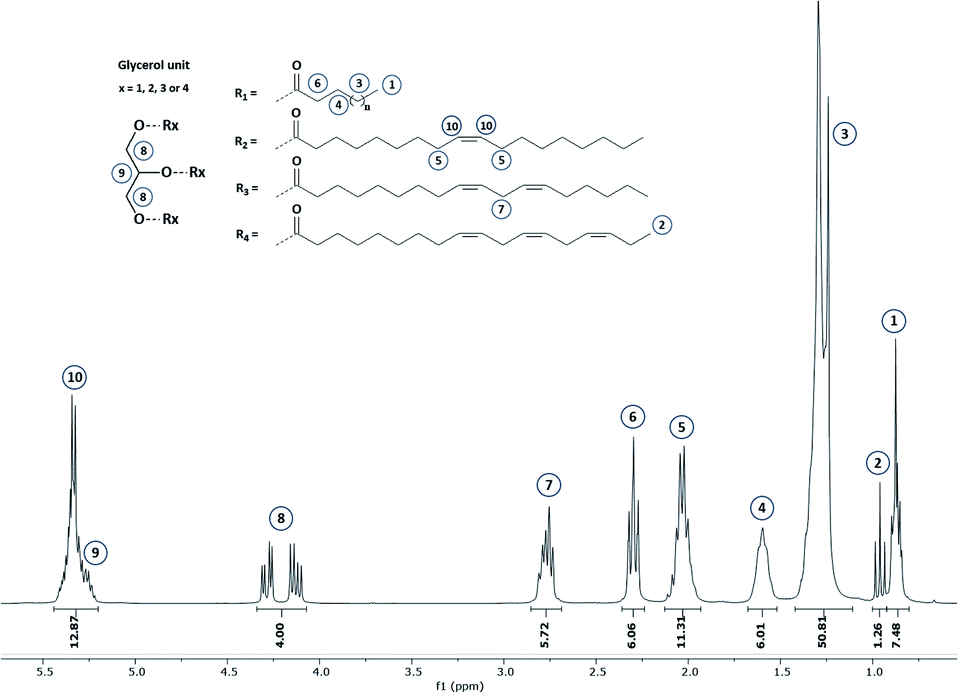 | ||
| Fig. 2 1H NMR spectrum of hemp oil – NMR 300 MHz – CDCl3. | ||
Since all fatty acid chains are linked to a common glycerol moiety, a simple methodology based on 1H NMR analysis has been set up (see ESI Section S1†). Based on this method, the proportion of each fatty acids comprised within the selected VOs has been determined and gathered in Table 1. In the same time, double bond equivalent (DBE) and subsequent iodine value (IV) were determined by 1H quantitative NMR (1H qNMR) in order to adapt the parameters that will be used during the epoxidation process. Interestingly, results obtained from this analysis confirm the possibility and interest in using several VOs as raw materials to reach a large DBE from 2.94 to 7.13 meq. g−1 and an associated IV from 74.7 to 180.9. Additional 13C NMR were also performed on representative VOs and are displayed in Section S3 (ESI).†
Epoxidation of VOs
(1) in situ epoxidation with peracids in the presence of inorganic or supported catalyst respectively with H2SO4 (bi-phasic system) or acidic ion exchange resin (AIER) (tri-phasic system).22
(2) Epoxidation with organic or inorganic peroxides using transition metals23–25 or enzymatic species26,27 as catalysts.
(3) Epoxidation via halohydrins formation.28
(4) Epoxidation with molecular oxygen.28
According to environmental reasons, epoxidation through halohydrins formation (3) cannot be considered. On one hand, this harsh method uses an excess of reactants and leads to by-products such as dihalides, halogens, ethers and salts which are highly environmentally unfriendly.
On other hand, it is noteworthy that this route implies the use of soda or other strong bases to ensure the epoxide ring formation. Therefore, this step would lead also to the formation of undesired byproducts deriving from partial or total triglyceride hydrolysis.
Although molecular oxygen (4) could be of high interest since it is one of the cheaper and greener method, this oxidant finds limitation since is only effective on simple molecules such as ethylene29 even with appropriated silver catalyst. Additionally, EVOs synthesis with molecular oxygen leads to the cleavage and degradation of the VOs leading to undesired smaller by-products, such as aldehydes and ketones, as well as short dicarboxylic acids chains.30 Therefore, it is not an efficient method for VOs epoxidation.
Overall, it can be thus established that both the first and second method are relevant for vegetable oils epoxidation. The latter exhibits several advantages in terms of energy and time consumption, as well as safety and epoxide yield. However, this technique suffers of significant drawbacks.
On the one hand, the chemoenzymatic epoxidation involves lipase as catalyst which is temperature sensitive and an expensive method. Its catalytic activity is also lower in the case of EVOs due to steric hindrance that does not occur in the process with free fatty acids.31 Furthermore, an important feature is that lipases demonstrate ability to transesterification and hydrolysis side-reactions which result in a mixture of epoxidized mono-, di- and triglycerides, epoxidized free fatty acids and glycerol.32
On the other hand, the epoxidation catalyzed by transition metal complexes is characterized by (i) expensive noble metals,23 (ii) high temperature33 and/or (iii) high catalyst loading24 to afford great epoxide yield. It must be anyhow noted that a versatile promising manganese complex was developed34 very recently and enables efficient epoxidation. Indeed, low catalyst loading (0.1 wt% with respect to the oil), mild reaction conditions (room temperature) and short reaction time (2 hours) were reported and allow reaching epoxy conversion yields and selectivities generally higher than 90%. Acting as homogeneous catalyst, its removal and recyclability remain subjects to discuss. Nevertheless, this recent approach was not considered in the scope of this work.
In the scope of this paper, we finally selected the in situ epoxidation with peracid since it is a softer, environmentally friendly and more versatile technique for the epoxidation of a large range of vegetable oils especially when tri-phasic system is used instead of the bi-phasic one. Indeed, the main difference between tri-phasic and bi-phasic process is the use of a solid catalyst in the first case rather than an inorganic acid (H2SO4, H3PO4, etc.) catalyst for the second one. It considerably increases the selectivity but first and foremost, this catalyst can be straightforwardly separated and regenerated at least 4 times without losing significant activity.35 More interestingly, it had been demonstrated that regeneration of the solid catalyst is not required between each batch.36
In this work, acetic acid was preferred than formic acid even if this latter is still largely employed40,41 due to its higher reactivity and thus enables faster double bonds conversion. However, this acid is highly corrosive and promotes the cleavage of the oxirane rings30 making acetic acid more relevant here. In any event, the amount of acetic acid with respect to the double bonds remains an important factor. A low acetic acid concentration leads to an insufficient transfer of oxygen to the oil while a too large amount of acid rapidly introduces side-reactions. The optimum value was well studied and fixed at 0.5 molar equivalent with respect to the VOs double bonds content.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.42,43
:1.42,43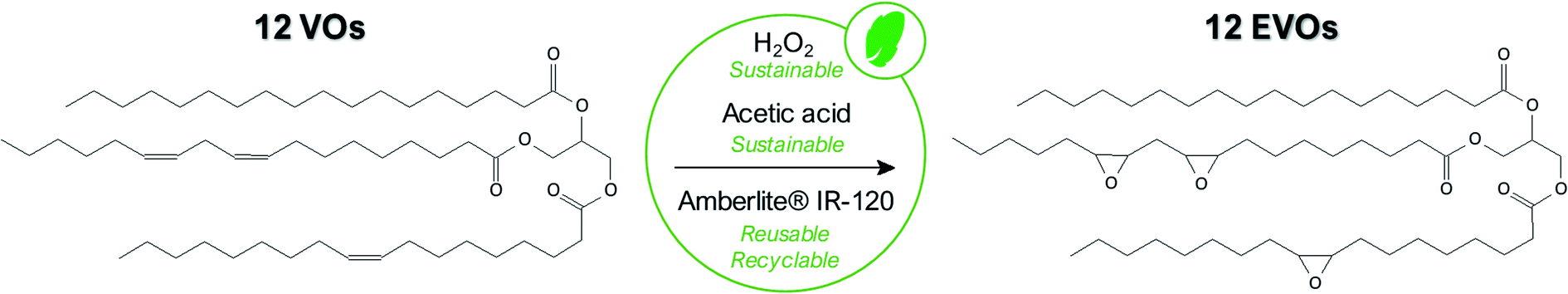 | ||
| Fig. 3 Sustainable synthesis pathway applied for VOs epoxidation. | ||
The reaction progress was monitored by means of 1H NMR (Section S4 ESI†) and finally stopped once double bonds conversion exceeds 97%. A representative 1H NMR spectrum corresponding to the epoxidized hemp oil is given in Fig. 4.
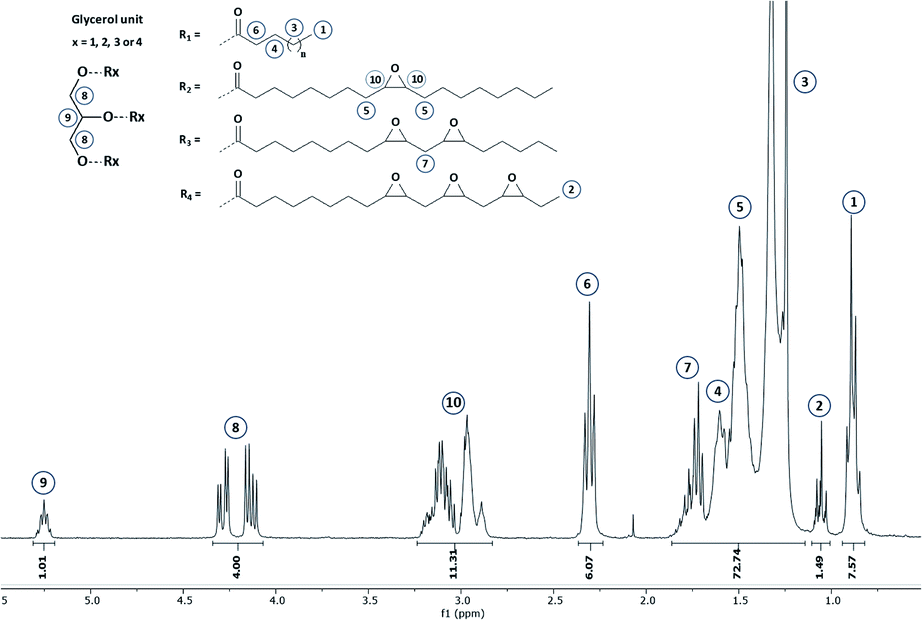 | ||
| Fig. 4 1H NMR spectrum of epoxidized hemp oil (EVO 9) – NMR 300 MHz – CDCl3. | ||
Overall results obtained after epoxidation of the whole range of selected VOs are gathered in Table 2. All the VOs exhibit a great response to the optimized epoxidation parameters since double bonds conversion is comprised from 97 to 100% according to 1H NMR analyses. It was observed that the reaction time vary from 18 to 26 h to reach such conversion rates. However, most of previous works22,35,40 put forward that this long reaction time usually promotes side-reactions and notably epoxy ring opening which reduces the final oxirane number effectively reached. Indeed, during the epoxidation process, several side reactions may occur and are mainly focused and detrimental to the epoxide ring (Fig. 5).
| EVO | Karanja | St John's wort | Peanut | Rapeseed | Soybean | Rose hip | Safflower | Camelina | Hemp | Rose hip | Linseed | Perilla | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The epoxidation of EVO 13 was performed in cylopentyl methyl ether (CPME) instead of toluene.b Equivalent to overall reaction time. Addition time of hydrogen peroxide was performed over approximately 1 h.c Determined by 1H NMR analyses.d Determined by 1H qNMR. | |||||||||||||
| Label | EVO 1 | EVO 2 | EVO 3 | EVO 4 | EVO 5 | EVO 6 | EVO 7 | EVO 8 | EVO 9 | EVO 10 | EVO 11 | EVO 12 | EVO 13a |
| Reaction time (h)b | 26 | 18 | 24 | 22 | 23 | 22 | 24 | 24 | 20 | 23 | 26 | 20 | 24 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conversion (%)c C conversion (%)c |
97 | 100 | 100 | 100 | 100 | 100 | 100 | >99 | 100 | >99 | 99 | 100 | 99 |
| Selectivity (%)c | 97 | 95 | 96 | 96 | 96 | 97 | 97 | 99 | 95 | 99 | 95 | 95 | 94 |
| EI (meq. g−1)d | 2.77 | 2.97 | 3.33 | 3.96 | 4.54 | 4.7 | 5.0 | 5.27 | 5.6 | 6.3 | 6.6 | 6.77 | 6.64 |
| EEW (g eq.−1)d | 361 | 337 | 300 | 253 | 220 | 213 | 200 | 190 | 179 | 159 | 152 | 148 | 151 |
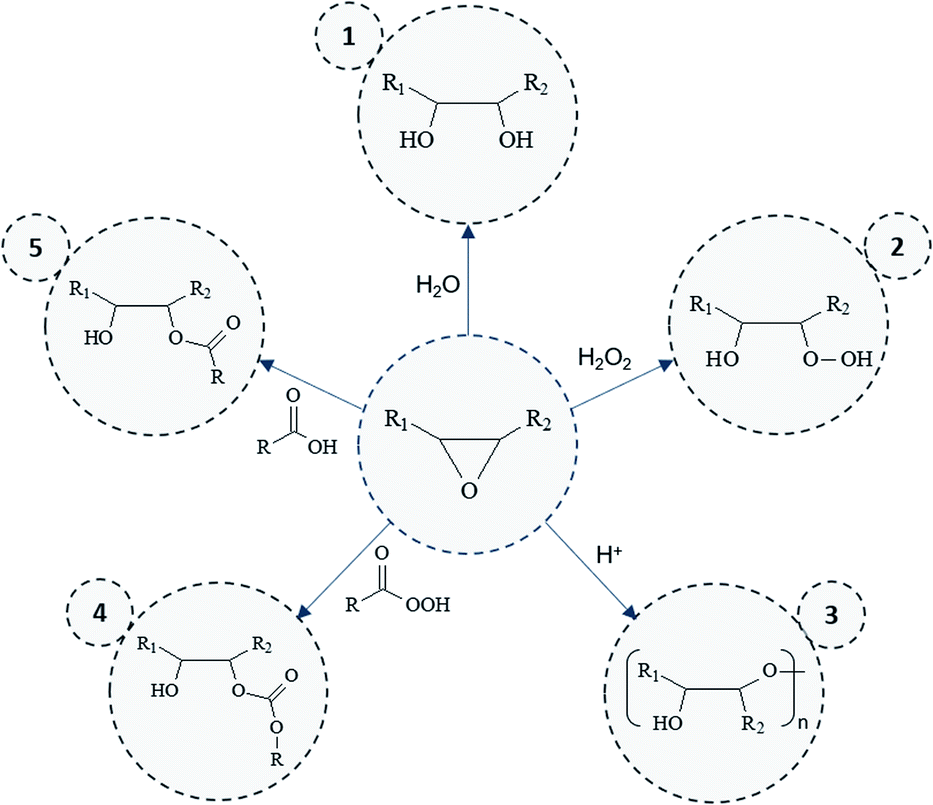 | ||
| Fig. 5 Eventual side-reactions and related by-products obtained during epoxidation process. | ||
By-products 1. and 2. (Fig. 5) could be obtained by the action of water or hydrogen peroxide. However, these reactions are limited here since vegetable oils are diluted in solvent during the epoxidation process. Moreover, hydrogen peroxide can react faster when acetic acid is used as oxidizing agent. Even though the latter reaction can be responsible of side-products 5. or 4. (Fig. 5) (in its peracetic form), the use of acetic acid rather than formic acid prevents risks of side reactions. Free fatty acids could also participate in the production of these by-products but only low amount might be observed initially.
All side-reactions lead to the formation of hydroxyl groups which may react further with another epoxy groups to give internal ethers or oligomeric ethers (dimers) as displayed in Fig. 5 (by-product 3.). Polymerization reactions are especially detrimental since they considerably impact the product viscosity which can be an issue for some implementation processes. However, by means of 1H NMR, neither hydroxyl nor ether functions were identified in the final products, which should be usually observed in the range of 3–4 ppm. This result was also confirmed by means of Size Exclusion Chromatography (SEC) analysis (Section S5 ESI†) which proves the presence of only one dominant signal attributed to the EVO.
Finally, our optimized, robust and environmentally friendly procedure enables to reach and maintain a high epoxy selectivity (defined as the total amount of epoxy obtained compared to the double bonds effectively converted) from 94 to 99% which is notably superior to results commonly obtained in the literature.23,32,36,41 To the best of our knowledge, an epoxidation process exhibiting an efficiency (conversion rate and selectivity) and a reproducibility for such variety of VOs was never reported in the literature. It must be noted that, in comparison with EVO 12, the EVO 13 was synthesized using the same epoxidation route and VO raw material (perilla oil) but by substituting toluene with cyclopentyl methyl ether (CPME), a sustainable and eco-friendly solvent which can be derived from biomass.45 All obtained results allow validating that this green solvent is a noteworthy alternative to toluene. Finally, thanks to a correct selection of vegetable oils and epoxidation process parameters, a set of 12 epoxidized vegetable oils was prepared and 1H qNMR enable determining epoxy index (EI) from 2.77 meq. g−1 (EVO 1) to 6.77 meq. g−1 (EVO 12) or epoxy equivalent weight (EEW) from 148 to 361 g eq.−1.
EVOs thermoset materials
Synthesized EVOs were then used to prepare thermoset materials either by UV-curing or thermal processes.The reactivity of the synthesized EVOs towards cationic photopolymerization was evaluated by FT-IR analysis (Section S6 ESI†). A representative example corresponding to the UV-curing of epoxidated hemp oil is given in Fig. 6.
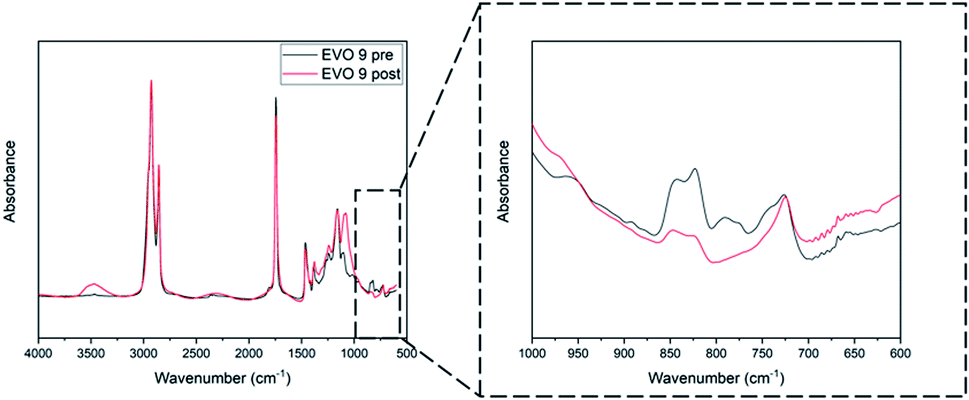 | ||
| Fig. 6 FT-IR spectrum of EVO 9 (epoxidized hemp oil) before (dark line) and after (red line) irradiation. | ||
Epoxy group conversion was measured, upon UV-irradiation, by following the decrease of the absorption peaks at 822 and 833 cm−1 which can be attributed to the epoxy groups vibrations,49,50 after one passage (equivalent to few seconds) under fusion lamp (at a conveyor belt speed of 6 m min−1 and a light intensity of 225 mW cm−2). Apart from EVO 1 (epoxidized Karanja oil), all the investigated EVOs showed an epoxy group conversion after irradiation above 90% (Table 3). This lack of reactivity observed for EVO 1 might be attributed to the presence of natural flavonoids including karanjin and pongamol which have strong photoabsorptive properties.51
| EVOs label | FT-IR conversion (%) | Tg DSC (°C) | Tα DMA (°C) | E′ at glassy state (MPa) | E′ at rubbery plateau (MPa) | ν (mmol cm−3) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Results of UV-cured EVOs materials are highlighted in brown. | ||||||||||||
| EVO 1 | — | 98 | — | −34/−9 | — | — | — | — | — | — | — | — |
| EVO 2 | 97 | 87 | −34 | −34/−10 | −18 | −19 | 418 | 1400 | 3.71 | 0.17 | 0.49 | 0.09 |
| EVO 3 | 90 | 99 | −33 | −33/−8 | −15 | −16 | 642 | 1650 | 7.67 | 0.9 | 1.00 | 0.11 |
| EVO 4 | 91 | 99 | −27 | −35/−9 | −5 | −15 | 345 | 4000 | 7.13 | 0.52 | 0.90 | 0.07 |
| EVO 5 | 91 | 83 | −25 | −35/−12 | 0 | −16 | 606 | 1100 | 9.34 | 0.55 | 1.16 | 0.07 |
| EVO 6 | 96 | 92 | −8 | −32/−10 | 3 | −15 | 541 | 600 | 11.70 | 0.38 | 1.44 | 0.06 |
| EVO 7 | 93 | 96 | −20 | −26 | 6 | −14 | 558 | 1200 | 10.90 | 0.25 | 1.33 | 0.05 |
| EVO 8 | 99 | 91 | −15 | −27/−4 | 23 | −12 | 1154 | 1000 | 43.08 | 1.27 | 4.99 | 0.19 |
| EVO 9 | 92 | 97 | 0 | −21 | 25 | −11 | 658 | 1700 | 23.75 | 1.7 | 2.74 | 0.24 |
| EVO 10 | 92 | 96 | 8 | −17 | 25 | — | 492 | — | 26.63 | — | 3.07 | — |
| EVO 11 | 90 | 85 | 13 | −12 | 42 | −10 | 1018 | 900 | 35.30 | 1.8 | 3.88 | 0.23 |
| EVO 12 | 94 | 92 | 36 | −6 | 50 | 1 | 490 | 1350 | 16.72 | 2.4 | 1.80 | 0.35 |
While DSC analysis gives information about the thermal behaviour, DMA allows the evaluation of the elastic and viscous component of the modulus of the material in a very large temperature interval. In the Tg region the tanδ curve (tanδ = E′′/E′: ratio loss modulus/storage modulus) shows maximum which is assumed to be the Tg of the cured films.49 Therefore, by combining these techniques, a good characterization of the thermal and viscoelastic properties of the crosslinked EVOs was obtained. The Tg measured by DMTA are higher than the one determined by DSC due to a well-known frequency effect.52
The tanδ peaks recorded for the UV-cured films are collected in Fig. 8. It is possible to observe the shifting of the maximum of tanδ towards higher temperature, by increasing the epoxy index in the EVOs starting monomers.
Taking into account that very similar epoxy group conversion was achieved for all the EVOs by UV-curing, with an almost complete consumption of the reactive rings (always above 90%), the epoxy content parameter is the one governing the final crosslinking density upon curing and therefore the final Tg and thermo-mechanical properties of corresponding thermoset materials.
Moreover, in the DMA thermogram (Fig. S7.2, ESI†), the loss tangent (tanδ) peaks intensity are lower and broaden with the increase of EVOs' epoxy content. This suggests an increase of the crosslinked network heterogeneity and a broad distribution of chain segment mobility.53,54 Obtained results (Fig. S7.3†) also demonstrate that the storage modulus E′ of the cured products gradually increase with increasing epoxy content. Both the E′ and the crosslink densities (Table 3), are consistent with the values reported for epoxidized vegetable oils thermally cured.23,55–57
From this analysis it is clear that it is possible to modulate the final Tg of the crosslinked material in a very large temperature interval from −18 °C up to 50 °C when the epoxy index varies from 2.97 (EVO 2) to 6.77 meq. g−1 (EVO 12). In a previous article,58 we reported the thermal properties of epoxidized castor oil cured by UV-cationic process. Interestingly this material exhibit Tg at 10 °C measured by DSC or 20 °C measured by DMA, with an epoxy index evaluated at 2.85 meq. g−1 in the starting photocurable monomer. Here, we reached these values with EVOs with a relatively higher epoxy content (from 5.27 to 5.6 meq. g−1). ECO is mainly constituted with ricinoleic acid which displays a hydroxyl function alongside the alkyl chain. Comparison of the results in between ECO and EVO 2 indicates that the hydroxyl groups which characterize ECO has a strong influence on the UV-cured thermoset material thermo-mechanical properties. Indeed, since ECO and EVO 2 exhibit a similar epoxy content, it can be inferred that ECO hydroxyl groups lead to an increase of Tg value by 23%. The presence of the hydroxyl groups is likely to strengthen the crosslinked network either through the formation of hydrogen bonds (supramolecular effects) or by chemical reaction of the cationic species that characterize the photopolymerization mechanism with the hydroxyl groups. Finally, all gathered data highlight that the thermomechanical performances of the EVO based UV-cured thermosets are driven by (i) the epoxy index of the EVO and (ii) the presence of additional functionalities such as hydroxyl moieties for ECO. It clearly appears that the selection of VOs raw materials and the accurate determination of their chemical composition is crucial to reach a given range of mechanical properties.
Herein, the EVOs' thermal autocrosslinking without initiators and hardeners were studied to test the ability of EVOs to generate crosslinked homopolymers and further to correlate the thermomechanical properties of EVOs thermosets with their initial epoxy content. The epoxidized linseed oil (EVO 11) was selected as the reference system due to the already known anionic or cationic crosslinking reactivities and properties of materials generated by this monomer.15–17,60
The selection of EVOs thermal curing parameters were obtained by combining ThermoGravimetric Analysis (TGA) with dynamic Differential Scanning Calorimetry (DSC). While the first one allows determining the thermal stability of starting monomers, the second one gives the thermodynamic parameters on the crosslinking of EVOs. Comparison of both information (Section S8, ESI†) allows to define the proper thermal curing conditions to prepare EVOs thermoset materials. TGA data are collected in Table S8.1.† The results from the TGA analysis (Fig. S8.1†) allow to observe that the EVOs monomers have a high thermal stability. The T5% values are in the range from 271 (EVO 9) to 305 °C for (EVO 1 and 7) which is similar to the value commonly obtained with other epoxy derivatives such as DGEBA.61 All the EVOs underwent a complex multistep thermal degradation in air atmosphere, with an onset temperature higher than 250 °C.
The EVOs reactivity in function of the temperature was thus analysed by dynamic DSC (Fig. S8.2†) in the temperature range from 25 to 300 °C (temperature ramp: 10 °C min−1). All the data are collected in Table S8.1.† The DSC study shows that all the EVOs exhibit the ability to homopolymerize under heating which is consistent with results already highlighted by Lee et al.59 or by Levchik et al.62 Overall, the selected monomers show a decreasing temperature of homopolymerization with the epoxy content (from 197 for EVO 1 to 156 °C for EVO 9 or 11), while the Tpeak value is very close for all the monomers, around 220 °C (Table S8.1†). In a study reported by Scala et al.63 it was put in evidence that epoxidation kinetic is notably dictated by 2 major parameters, the electronic and the steric effects. The electronic effect was determined by comparing fatty acid methyl ester from oleic, linoleic and linolenic acids. By increasing the degree of unsaturation and thus the electronic density, it was observed that the rate constant of epoxidation can be increased up to a factor 3. In the second case, reactivity of triglycerides with high oleic, linoleic and linolenic fatty acids against epoxidation were compared. Although, reactivity of double bonds of oleic and linoleic acids are similar, that of linolenic acid was evaluated to be 2 times higher. Located at the end of the fatty acid chain, the double bond of linolenic acid is less sterically hindered. Herein, we can assume that the difference on the crosslinking temperature onset could be assigned to the reactivity of the oxirane group alongside the alkyl chains which may be directly linked to their difference of reactivity during epoxidation process. Indeed, results presented in Table 1 clearly exhibit that the increase of the double bond and thus the epoxy content is, in most of the cases, correlated with the increase of linolenic chains for which the epoxy located at the chains' ends is more reactive. Thus, it can be assumed that the thermal initiated self-curing reaction starts on this position and then activates the polymerization of other, less reactive, epoxy groups.
Finally, based on the results obtained by TGA and DSC, all the EVOs were thermo-cured at 180 °C over 24 h. Such curing temperature, above or very close to the crosslinking onset temperature, would allow the thermal self-curing of the EVOs. It was measured by FT-IR analysis (Section S9†, Table 3) that the conversion of oxirane rings is ranged between 83% for EVO 5 and 99% in the case of EVO 3 and 4. A representative FT-IR analysis corresponding to the cured EVO 9 is displayed in Fig. 7. Moreover, after immersion in several organic solvents (Section S10 ESI†) it was confirmed that EVOs materials exhibit high crosslinking rate. Interestingly, it also affords to prove EVOs recyclability in soda solution due to presence of hydrolysable bonds.
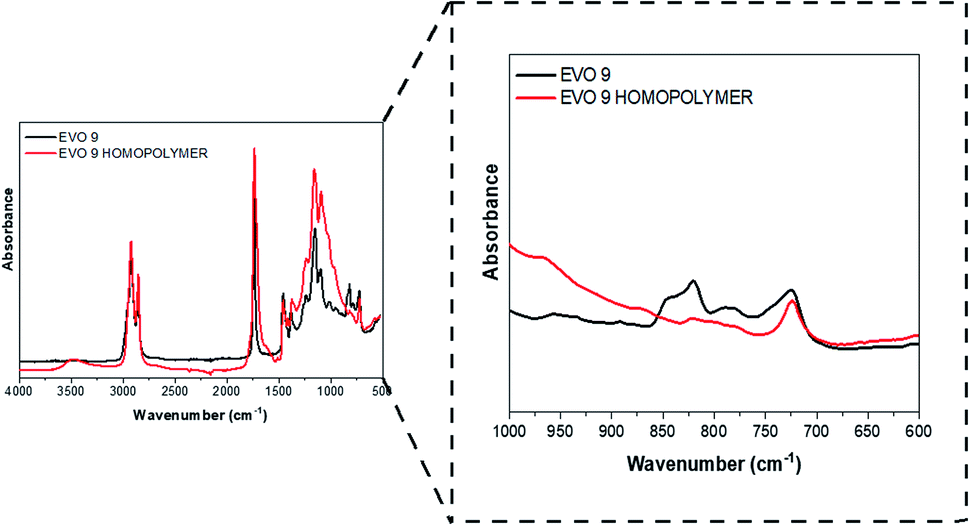 | ||
| Fig. 7 FT-IR spectrum of EVO 9 (epoxidized hemp oil) before (dark line) and after (red line) thermal curing. | ||
δ values are depending on the epoxy content on the EVO's building-blocks. It can be observed that an increase of the epoxy content lead to an increase of crosslinking density and therefore impacting the thermomechanical properties.Comprised in between −34 (EVO 2) and 36 °C (EVO 12) (DSC – Fig. 8a) or −18 (EVO 2) and 50 °C (EVO 12) (DMA – Fig. 8b) for UV-cured EVOs, the glass transition temperatures of thermo-cured EVOs reach only −35 (EVO 5) to −6 °C (EVO 12) (DSC – Fig. 8a) and −19 (EVO 2) to 1 °C (EVO 12) (DMA – Fig. 8b). These results highlight that increasing the epoxy content from 2.77 to 6.77 meq. g−1 implies an increase of the Tg value of 20 °C for the thermal curing while an increase of 70 °C was demonstrated in the case of the UV-curing process.
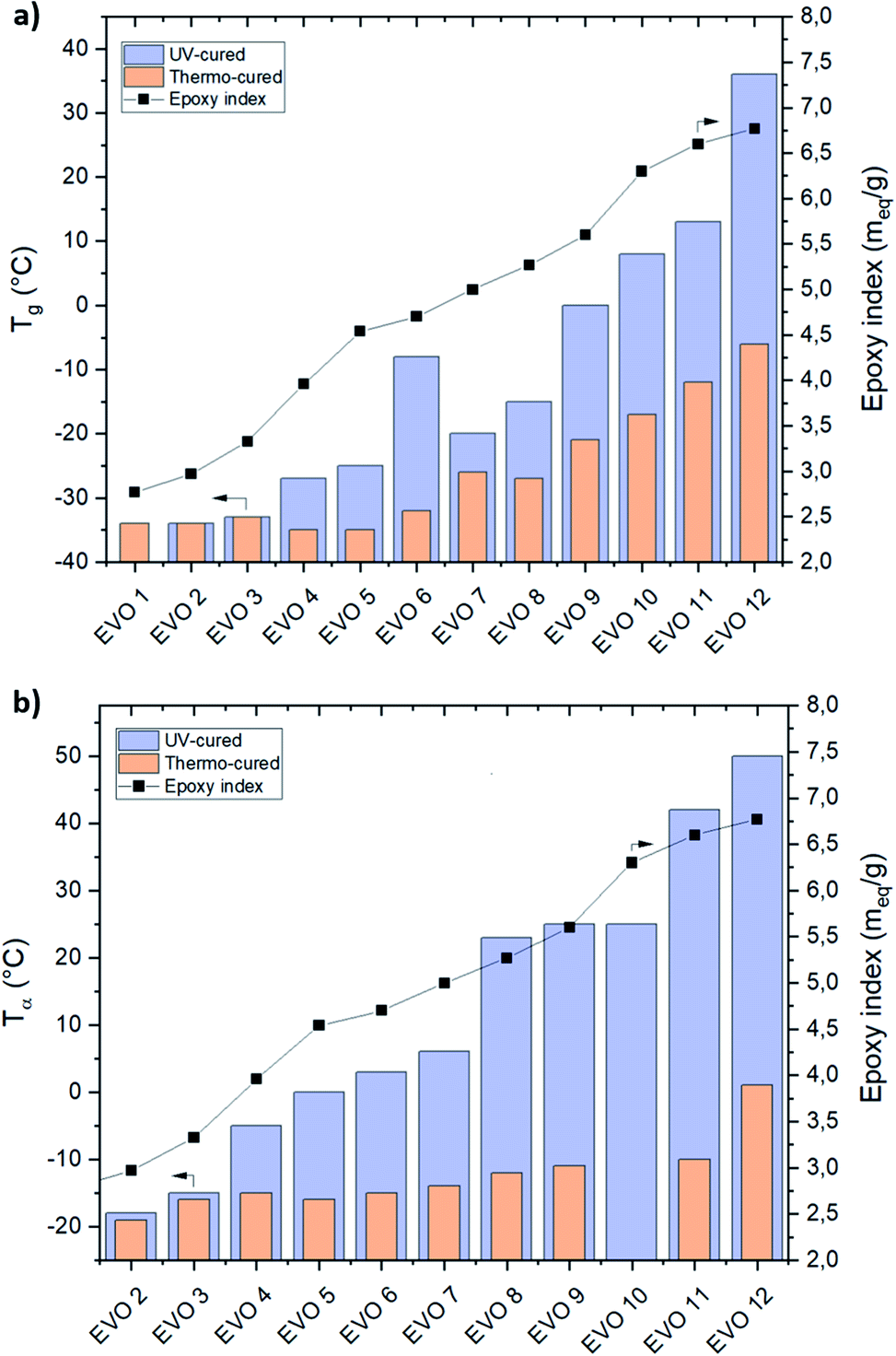 | ||
| Fig. 8 Correlation between Tg from DSC (a) or Tα from DMA (b) and epoxy index of thermal- (orange column) and UV-cured (purple column) EVOs thermoset materials. | ||
Moreover, viscoelastic properties of thermo-cured EVOs materials are in general much higher (up to 12 times higher) at the glassy state than those of UV-cured EVOs materials (Table 3). Indeed, thermo-cured materials exhibit viscoelastic properties from 600 (EVO 6) to 4000 MPa (EVO 4). It must anyhow be noted that such performances collapse to 0.25 (EVO 7) to 2.4 MPa (EVO 12) at rubbery plateau. The thermo-cured homopolymers exhibit characteristics of low crosslinked elastomers and for the high epoxy content EVOs, similar with natural rubber. In comparison, these mechanical properties are around 3.7 (EVO 2) to 43.0 MPa (EVO 8) for UV-cured materials and these properties do not fall at the rubbery plateau.
There is thus a significant gap of performances in between the materials obtained by both crosslinking techniques which might be attributed to, at least, 3 major factors. First, even if the thermal homopolymerization in systems with multi-epoxide functions was already reported, the corresponding mechanism still remains under debate. Several reactions could occur during the heating of the epoxides. The most probable ones are the epoxides isomerizations to generate unsaturated alcohols59,64 and the etherification reactions by intermolecular additions. An ionic mechanism was spotlighted to explain the curing of thermo-cured epoxy homopolymers.46 It can moreover be assumed that, in parallel to this ring opening polymerization initiated by the heating, a competitive mechanism corresponding to the reaction in between epoxies with some hydroxyl functions can occurs. Finally, another conceivable mechanism is the transesterification reaction by action of the forming hydroxyl on the ester bond of the triglyceride,65 which is likely to have an impact on the structure of the thermo-cured crosslinked networks and finally on the thermomechanical performances.
Finally, thermal- or UV-curing lead to close epoxy conversion in a range between 83 to 99% or 90 to 99% respectively. These results demonstrate that main deviations in between both crosslinking technics are induced by mechanistic phenomena. Within this scope, further studies will be dedicated to the understanding of these curing behaviors.
Conclusions
A list of 12 VOs mostly non-edible or non-valuated and differing by their fatty acid composition was built up in order to naturally reach a wide range of double bonds content. Thanks to a thorough selection of the epoxidation process parameters, high double bonds conversion (from 97 to 100%) with high selectivity (from 94 to 99%) have been obtained by using a versatile, green and robust one-step process. To further reduce the environmental impact of this process, toluene was replaced by cyclopentyl methyl ether as green solvent without incidence on the final quality of the related EVO. Finally, synthesized EVOs exhibit an epoxy index from 2.77 to 6.77 meq. g−1. UV-cationic and hardener-free thermal processes were applied to obtain fully biobased thermoset materials based on synthesized EVOs and the influence of the epoxy index on the thermo-mechanical properties of the related thermoset materials was evaluated through DSC and DMA analyses.The synthesized EVOs monomers showed a very high reactivity towards cationic photopolymerization with a very high epoxy group conversion upon irradiation (always above 90%). Moreover, EVOs have shown an outstanding ability of forming thermoset materials just by heating and without initiator or hardener since epoxy conversion reached from 83 to 99%. Hence, either by UV or thermal curing, EVOs reactivity allowed to prepare fully biobased epoxy thermosets.
A linear increase of the Tg values by increasing the EVOs' epoxy content was observed for both thermal and UV processes. Thus, it was demonstrated that, by simply acting on the selection of the starting VO, it was possible to modulate the epoxy content and therefore the final thermomechanical performances of the corresponding crosslinked material in a very high temperature interval (from −19 to 50 °C).
Due to their low viscosity, stability and flexibility, this platform of EVOs would pave the way towards the preparation of new coatings or biocomposites as well as new raw materials finding application in 3D-printing, as sustainable polyurethanes (biobased polyols – epoxide ring opening), non-isocyanate polyurethane (cyclic carbonate – epoxide carbonatation) or vinyl ester resins (epoxide ring opening with (meth)acrylic acid) for instance.
Experimental section
Materials
Amberlite® IR-120 hydrogen form, anhydrous sodium sulfate Na2SO4 (99%) and all solvents (>95%) used were purchased from Sigma Aldrich. Hydrogen peroxide aqueous solution (35%, w/w) was purchased from Alfa Aesar. Acetic acid (99%) was purchased from abcr GmbH. Vegetable oils were obtained from various suppliers. The cationic photoinitiator triarylsulfonium hexafluoroantimonate salt, mixed 50 wt% in propylene carbonate were purchased from Sigma Aldrich (Milan, Italy). All reagents, reactants and solvents were used as received without further purification.Characterization techniques
The double bond equivalent (DBE) and Epoxide Index (EI) was determined according to 1H NMR titration method. The method consists in solubilizing a known mass of the product and of an internal standard. The number of moles of epoxide or double bonds functions per gram of product (meq. g−1) was measured by comparing the standardized integration of the standard with the standardized integration of the oxirane rings or the double bonds.
The equivalent epoxy weight (EEW) was determined thanks to the EI (eq. g−1) previously mentioned using the following eqn (1):
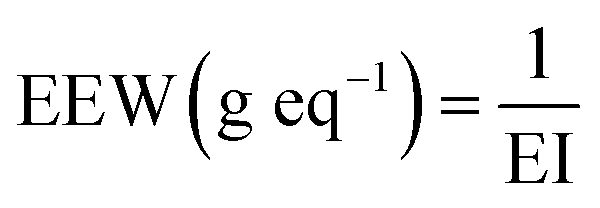 | (1) |
The iodine value (IV) was determined thanks to the DBE (eq. g−1) previously mentioned using the following eqn (2):
| IV = DBE × 100 × Mw (I2) | (2) |
Double bonds conversion was determined by comparison of the olefinic hydrogens value at 5.2–5.5 ppm.
The conversion of double bonds was determined as follows eqn (3):
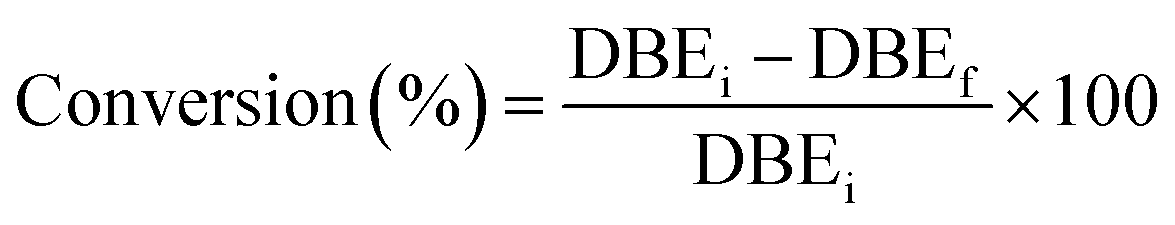 | (3) |
Selectivity was determined by comparison of the oxirane hydrogens value at 2.7–3.3 ppm with the double bonds effectively converted at 5.2–5.5 ppm.
The selectivity was determined as follows eqn (4):
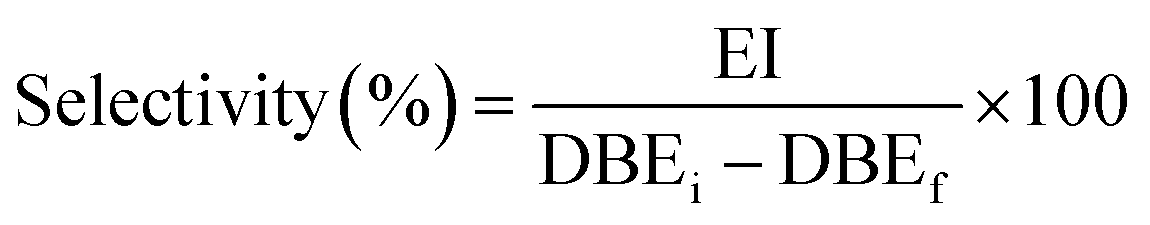 | (4) |
000000 Da) columns connected in series and a refractive index (RI) detector.The EVO's thermal homopolymerization reactions and products were studied on a Mettler-Toledo DSC 3 instrument equipped with STAR software in a temperature interval from 25 to 300 °C, at 10 °C min−1, under air atmosphere. About 5–10 mg of sample were placed in 40 μL aluminium crucibles. A second scan run (from −80 to 180 °C) at the scanning rate of 10 °C min−1 was performed on thermoset materials to obtain the glass transition temperature (Tg).
δ) was recorded as a function of temperature.Dynamic Mechanical Analysis (DMA) on thermally cured homopolymers were carried out on Mettler-Toledo DMA 1 instrument, equipped with STAR© software for curves analysis. The analysed samples had rectangular dimensions of 30 × 7 × 2 mm (length × width × thickness). Each formulation was analysed 3 times and the values averaged. Storage modulus (E′) and damping factor (tanδ) were collected at 3 °C min−1 heating rate from −80 to 80 °C and 1.0 Hz frequency. The DMA was operated using tension method for the determination of the modulus (E′). The glass transition temperature was assigned at the maximum of damping factor (tanδ = E′′/E′).
Crosslinking density was calculated by the eqn (5), according to the rubber elasticity theory:
| ν = E′/3RT | (5) |
Epoxidation of vegetable oils (EVOs)
Quantities were adjusted regarding the unsaturation content (evaluated by 1H NMR titration) of the VOs. Typically, the epoxidation is carried out under the following molar ratio conditions CC/acetic acid/H2O2 (35% w/w); 1/0.5/1.5 eq. with Amberlite® IR-120 loaded at 10 wt% with respect to (w.r.t) acetic acid and hydrogen peroxide weight.
Once completion of reaction, the reaction mixture was filtrated to remove Amberlite® IR-120 and 600 mL of diethyl ether was added. The organic layer was washed three times with water (3 × 100 mL), dried on anhydrous Na2SO4 and then filtered. Solvents were removed under reduced pressure at 60 °C using a rotary evaporator. A clear red liquid epoxidized Karanja oil (EVO 1) was obtained (94.9 g) with a yield of 92%. A sample was submitted to 1H NMR and 1H qNMR.
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.38–4.03 (m, 4H), 3.28–2.73 (m, 6H), 2.3 (t, 6H), 1.89–0.97 (m, 86H), 0.86 (m, 9H).
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.43–4.02 (m, 4H), 3.26–2.81 (m, 6H), 2.33 (t, 6H), 1.87–1.16 (m, 77H), 1.00–0.77 (m, H).
1H NMR (300 MHz, chloroform-d) δ 5.25 (q, 1H), 4.35–4.02 (m, 4H), 3.17–2.78 (m, 6H), 2.31 (t, 6H), 1.86–0.99 (m, 82H), 0.88 (m, 9H).
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.36–4.01 (m, 4H), 3.23–2.74 (m, 8H), 2.29 (t, 6H), 1.84–1.11 (m, 78H), 1.11–0.97 (m, 1H), 0.87 (m, 9H).
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.34–4.04 (m, 4H), 3.25–2.78 (m, 9H), 2.30 (t, 6H), 1.88–1.10 (m, 67H), 1.10–1.00 (m, 1H), 0.94–0.75 (m, 8H).
1H NMR (300 MHz, chloroform-d) δ 5.25 (q, 1H), 4.43–4.02 (m, 4H), 3.28–2.80 (m, 9H), 2.33 (t, 6H), 1.87–1.16 (m, 77H), 1.00–0.77 (m, 9H).
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.37–4.04 (m, 4H), 3.18–2.81 (m, 10H), 2.30 (t, 6H), 1.87–1.11 (m, 76H), 0.88 (m, 10H).
1H NMR (300 MHz, chloroform-d) δ 5.25 (q, 1H), 4.38–4.00 (m, 4H), 3.29–2.78 (m, 13H), 2.31 (t, 7H), 1.90–1.13 (m, 85H), 1.14–0.97 (m, 4H), 0.88 (m, 7H).
1H NMR (300 MHz, chloroform-d) δ 5.27 (q, 1H), 4.40–4.05 (m, 4H), 3.29–2.79 (m, 11H), 2.33 (t, 6H), 1.93–1.15 (m, 69H), 1.15–1.00 (m, 1H), 1.00–0.73 (m, 8H).
1H NMR (300 MHz, chloroform-d) δ 5.23 (q, 1H), 4.38–4.00 (m, 4H), 3.27–2.71 (m, 14H), 2.29 (t, 7H), 1.89–1.13 (m, 73H), 1.13–0.95 (m, 3H), 0.87 (m, 7H).
1H NMR (300 MHz, chloroform-d) δ 5.24 (q, 1H), 4.35–4.00 (m, 4H), 3.22–2.75 (m, 14H), 2.28 (t, 6H), 1.88–1.10 (m, 64H), 1.10–0.95 (m, 5H), 0.84 (m, 4H).
1H NMR (300 MHz, chloroform-d) δ 5.19 (q, 1H), 4.32–3.98 (m, 4H), 3.20–2.73 (m, 13H), 2.25 (t, 6H), 1.86–1.08 (m, 63H), 1.08–0.91 (m, 5H), 0.81 (m, 4H).
1H NMR (300 MHz, chloroform-d) δ 5.27 (q, 1H), 4.39–4.05 (m, 4H), 3.33–2.81 (m, 13H), 2.34 (t, 6H), 1.94–1.15 (m, 62H), 1.15–0.98 (m, 5H), 0.97–0.77 (m, 4H).
EVOs curing by UV-cationic process
All the EVOs were mixed with 2 wt% of triarylsulfonium hexafluoroantimonate salts (cationic photoinitiator). The liquid formulations were crosslinked with a Fusion System lamp in air at a conveyor belt speed of 6 m min−1 and a light intensity of 225 mW cm−2. Samples thickness was about 100 μm.EVOs curing by thermal process
The homopolymerization reactions were carried out using the optimized curing program determined by the help of DSC studies. The EVOs thermosets were obtained by isothermal curing in an oven, at 180 °C for 24 hours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ECOXY project. ECOXY is a project funded by the European Commission. This project has received funding from the Bio Based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation program (grant agreement no. 744311).Notes and references
- A. R. de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites, Mater. Horiz., 2016, 3, 241–247 RSC.
- N. Eliaz, E. Z. Ron, M. Gozin, S. Younger, D. Biran and N. Tal, Microbial Degradation of Epoxy, Materials, 2018, 11, 2123 CrossRef.
- F. Hu, S. Kumar, Y. John, J. La Scala, J. M. Sadler and G. R. Palmese, Preparation and Characterization of Fully Furan-Based Renewable Thermosetting Epoxy-Amine Systems, Macromol. Chem. Phys., 2015, 216(13), 1381–1489 CrossRef.
- A. Marotta, V. Ambrogi, P. Cerruti and A. Mija, Green approaches in the synthesis of furan-based diepoxy monomers, RSC Adv., 2018, 8, 16330–16335 RSC.
- A. Marotta, N. Faggio, V. Ambrogi, P. Cerruti, G. Gentile and A. Mija, Curing Behavior and Properties of Sustainable Furan-Based Epoxy/Anhydride Resins, Biomacromolecules, 2019, 20(10), 3831–3841 CrossRef CAS.
- M. Shibata and T. Ohkita, Fully biobased epoxy resin systems composed of a vanillin-derived epoxy resin and renewable phenolic hardeners, Eur. Polym. J., 2017, 92, 165–173 CrossRef CAS.
- M. Fache, R. Auvergne, B. Boutevin and S. Caillol, New vanillin-derived diepoxy monomers for the synthesis of biobased thermosets, Eur. Polym. J., 2015, 67, 527–538 CrossRef CAS.
- G. H. M. de Kruijff, T. Goschler, N. Beiser, A. Stenglein, O. M. Türk and S. R. Waldvogel, Sustainable access to biobased biphenol epoxy resins by electrochemical dehydrogenative dimerization of eugenol, Green Chem., 2019, 21, 4815–4823 RSC.
- M. Chrysanthos, J. Galy and J.-P. Pascault, Preparation and properties of bio-based epoxy networks derived from isosorbide diglycidyl ether, Polymer, 2011, 52(16), 3611–3620 CrossRef CAS.
- J. W. Cherrie, M. Gorman Ng, J. Lamb, A. Shafrir, M. van Tongeren, R. Mistry, M. Sobey, C. Corden, L. Rushton and S. Hutchings, Health, socio-economic and environmental aspects of possible amendments to the EU Directive on the protection of workers from the risks related to exposure to carcinogens and mutagens at works 1-chloro-2,3-epoxypropane (epichlorohydrin), IOM Research Project: P937/25, 2011, pp. 1–63 Search PubMed.
- T. Modjinou, D.-L. Versace, S. Abbad-Andaloussi, V. Langlois and E. Renard, Antibacterial and antioxidant photoinitiated epoxy co-networks of resorcinol and eugenol derivatives, Mater. Today Commun., 2011, 12, 19–28 CrossRef.
- R. Ciriminna, F. Parrino, C. De Pasquale, L. Palmisanoc and M. Pagliaro, Photocatalytic partial oxidation of limonene to 1,2 limonene oxide, Chem. Commun., 2018, 54, 1008–1011 RSC.
- E. W. Hammond, VEGETABLE OILS/Types and Properties, Encyclopedia of Food Sciences and Nutrition, 2003, pp. 5899–5904 Search PubMed.
- X.-Y. Jian, X.-P. An, Y.-D. Li, J.-H. Chen, M. Wang and J.-B. Zeng, All Plant Oil Derived Epoxy Thermosets with Excellent Comprehensive Properties, Macromolecules, 2017, 50, 5729–5738 CrossRef CAS.
- J.-M. Pin, N. Sbirrazzuoli and A. Mija, From Epoxidized Linseed Oil to Bioresin: An Overall Approach of Epoxy/Anhydride Cross-Linking, ChemSusChem, 2015, 8, 1232–1243 CrossRef CAS.
- J.-M. Pin, N. Guigo, L. Vincent, N. Sbirrazzuoli and A. Mija, Cationic copolymerization as synergic strategy to combine epoxidized linseed oil and furfuryl alcohol: design of a fully bio-based thermoset, ChemSusChem, 2015, 8, 4149–4161 CrossRef CAS.
- G. Falco, N. Sbirrazzuoli and A. Mija, Biomass derived epoxy thermosets: from reactivity to final properties, Mater. Today Commun., 2019, 21, 100683 CrossRef CAS.
- M. Sangermano, N. Razza and J. V. Crivello, Cationic UV-Curing: Technology and Applications, Macromol. Mater. Eng., 2014, 299(7), 775–793 CrossRef CAS.
- M. Sangermano, I. Roppolo and M. Messori, UV-cured functional coatings, in Photocured Materials, ed. A. Tiwari, RSC, London, 2014, 13, pp. 121–133 Search PubMed.
- M. Sangermano, I. Roppolo and A. Chiappone, New Horizons in Cationic Photopolymerization, Polymers, 2018, 10, 136–143 CrossRef.
- American Oil Chemists' Society – AOCS, Official Methods Ce 1–62: fatty acid composition by gas chromatography, USA, 1997 Search PubMed.
- M. R. Janković, S. V. Sinadinović-Fišer and O. M. Govedarica, Kinetics of the Epoxidation of Castor Oil with Peracetic Acid Formed in situ in the Presence of an Ion-Exchange Resin, Ind. Eng. Chem. Res., 2014, 53, 9357–9364 CrossRef.
- A. E. Gerbase, J. R. Gregorio, M. Martinelli, M. C. Brasil and A. N. F. Mendes, Dynamic mechanical and thermal behavior of epoxy resins based on soybean oil, J. Am. Oil Chem. Soc., 2002, 79, 179–181 CrossRef CAS.
- J. Jiang, Y. Zhang, L. Yan and P. Jiang, Epoxidation of soybean oil catalyzed by peroxo phosphotungstic acid supported on modified halloysite nanotubes, Appl. Surf. Sci., 2012, 258(17), 6637–6642 CrossRef CAS.
- A. Campanella, M. A. Baltanas, M. C. Capel-Sanchez, J. M. Campos-Martin and J. L. G. Fierro, Soybean oil epoxidation with hydrogen peroxide using an amorphous Ti/SiO2 catalyst, Green Chem., 2004, 6, 330–334 RSC.
- T. Vlček and Z. S. Petrović, Optimization of the Chemoenzymatic Epoxidation of Soybean Oil, J. Am. Oil Chem. Soc., 2006, 83, 247–252 CrossRef.
- C. Bérubéa, X. Barbeaub, P. Lagüeb and N. Voyer, Revisiting the Juliá-Colonna enantioselective epoxidation: supramolecular catalysis in water, Chem. Commun., 2017, 53, 5099–5102 RSC.
- S. Guenter, R. Rieth and K. T. Rowbottom, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, 6th edn, 2003, 12, pp. 269–284 Search PubMed.
- R. A. Sheldon and J. Dakka, Heterogeneous catalytic oxidations in the manufacture of fine chemicals, Catal. Today, 1994, 19, 215–246 CrossRef CAS.
- S. Dinda, A. V. Patwardhan, V. V. Goud and N. C. Pradhan, Epoxidation of cottonseed oil by aqueous hydrogen peroxide catalysed by liquid inorganic acids, Bioresour. Technol., 2008, 99(9), 3737–3744 CrossRef CAS.
- S. M. Danov, O. A. Kazantsev, A. L. Esipovich, A. S. Belousov, A. E. Rogozhin and E. A. Kanakov, Recent advances in the field of selective epoxidation of vegetable oils and their derivatives: a review and perspective, Catal. Sci. Technol., 2017, 7, 3659–3675 RSC.
- E. Milchert, K. Malarczyk and M. Kłos, Technological Aspects of Chemoenzymatic Epoxidation of Fatty Acids, Fatty Acid Esters and Vegetable Oils: A Review, Molecules, 2015, 20, 21481–21493 CrossRef CAS.
- M. Farias, M. Martinelli and D. P. Bottega, Epoxidation of soybean oil using a homogeneous catalytic system based on a molybdenum (VI) complex, Appl. Catal., A, 2010, 384, 213–219 CrossRef CAS.
- J. Chen, M. J. de Liedekerke Beaufort, L. A. Gyurik, J. M. Dorresteijn, M. Otte and R. J. M. Klein Gebbink, Highly efficient epoxidation of vegetable oils catalyzed by a manganese complex with hydrogen peroxide and acetic acid, Green Chem., 2019, 21, 2436–2447 RSC.
- S. Sinadinović-Fišer, M. Janković and Z. S. Petrović, Kinetics of in situ epoxidation of soybean oil in bulk catalyzed by ion exchange resin, J. Am. Oil Chem. Soc., 2001, 78(7), 725–731 CrossRef.
- V. V. Goud, A. V. Patwardhan, S. Dinda and N. C. Pradhan, Epoxidation of Karanja (Pongamia glabra) oil catalyzed by acidic ion exchange resin, Eur. J. Lipid Sci. Technol., 2007, 109, 575–584 CrossRef CAS.
- H. Kawanami, Y. Himeda and G. Laurenczy, Chapter Ten - Formic Acid as a Hydrogen Carrier for Fuel Cells Toward a Sustainable Energy System, Adv. Inorg. Chem., 2017, 70, 395–427 CrossRef CAS.
- J.-S. Kim and G.-G. Choi, Pyrolysis of Lignocellulosic Biomass for Biochemical Production, Waste Biorefinery, 2018, 323–348 CAS.
- T. Ravinder, B. Ramesh, G. Seenayya and G. Reddy, Fermentative production of acetic acid from various pure and natural cellulosic materials by Clostridium lentocellum SG6, World J. Microbiol. Biotechnol., 2000, 16, 507–512 CrossRef CAS.
- E. Santacesaria, R. Tesser, M. Di Serio, R. Turco, V. Russo and D. Verde, A biphasic model describing soybean oil epoxidation with H2O2 in a fed-batch reactor, Chem. Eng. J., 2011, 173(1), 198–209 CrossRef CAS.
- L. K. Jia, L. X. Gong, W. J. Ji and C. Y. Kan, Synthesis of vegetable oil-based polyol with cottonseed oil and sorbitol derived from natural sources, Chin. Chem. Lett., 2011, 22(11), 1289–1292 CrossRef CAS.
- A. P. Chavan and P. R. Gogate, Ultrasound assisted synthesis of epoxidized sunflower oil and application as plasticizer, J. Ind. Eng. Chem., 2015, 21(25), 842–850 CrossRef CAS.
- R. Mungroo, N. C. Pradhan, V. V. Goud and A. K. Dalai, Epoxidation of Canola Oil with Hydrogen Peroxide Catalyzed by Acidic Ion Exchange Resin, J. Am. Chem. Soc., 2008, 85, 887–896 CAS.
- S. Sinadinović-Fišer, M. Janković and O. Borota, Epoxidation of castor oil with peracetic acid formed in situ in the presence of a ion exchange resin, Chem. Eng. Process., 2012, 62, 106–113 CrossRef.
- G. de Gonzalo, A. R. Alcántara and P. D. de María, Cyclopentyl-methyl ether (CPME): versatile eco-friendly solvent for applications in Biotechnology and Biorefineries, ChemSusChem, 2019, 12(10), 2083–2097 CrossRef CAS.
- J. V. Crivello, Photoinitiators for Free Radical Cationic and Anionic Photopolymerization, G. Bradley, Wiley, New York, 1998, p. 329 Search PubMed.
- M. Sangermano, Advances in Cationic Photopolymerization, Pure Appl. Chem., 2012, 84, 2089–2101 CAS.
- M. Sangermano, UV Cured nanostructured epoxy coatings, Epoxy Polymers: New Materials and Innovations, ed. J. P. Pascault and R. J. J. Williams, Wiley, Weinheim, 2010 Search PubMed.
- J. V. Crivello and R. Narayan, Epoxidized Triglycerides as Renewable Monomers in Photoinitiated Cationic Polymerization, Chem. Mater., 1992, 4, 692–699 CrossRef CAS.
- S. J. Park, F. L. Jin and J. R. Lee, Thermal and mechanical properties of tetrafunctional epoxy resin toughened with epoxidized soybean oil, Mater. Sci. Eng., A, 2004, 374, 109–114 CrossRef.
- P. A. Shenoy, S. S. Khot, M. C. Chavan, J. V. Takawale and S. Singh, Study of sunscreen activity of aqueous, methanol and acetone extracts of leaves of Pongamia pinnata (L.) Pierre, Fabaceae, IJGP, 2010, 4(4), 270–274 CrossRef.
- L. E. Nielsen, Mechanical Properties of Polymers and Composites, Marcel Dekker, New York, 1994 Search PubMed.
- A. Anila, S. Wenfang, S. Xiaofeng and N. Kangming, Physical and thermal properties of UV curable waterborne polyurethane dispersions incorporating hyperbranched aliphatic polyester of varying generation number, Polymer, 2005, 46, 11066–11078 CrossRef.
- S. G. Tan and W. S. Chow, Curing Characteristics and Thermal Properties of Epoxidized Soybean Oil Based Thermosetting Resin, J. Am. Oil Chem. Soc., 2011, 88, 915–923 CrossRef CAS.
- F. L. Jin and S. J. Park, Thermal and rheological properties of vegetable oil-based epoxy resins cured with thermally latent initiator, J. Ind. Eng. Chem., 2007, 13, 808–814 CAS.
- S. J. Park, F. L. Jin and J. R. Lee, Synthesis and thermal properties of epoxidized vegetable oil, Macromol. Rapid Commun., 2004, 25, 724–727 CrossRef CAS.
- S. G. Tan and W. S. Chow, Biobased Epoxidized Vegetable Oils and Its Greener Epoxy Blends: A Review, Polym.-Plast. Technol. Eng., 2010, 49(15), 1581–1590 CrossRef CAS.
- C. Noè, S. Malburet, A. Bouvet-Marchand, A. Graillot, C. Loubat and M. Sangermano, Cationic photopolymerization of bio-renewable epoxidized monomers, Prog. Org. Coat., 2019, 133, 131–138 CrossRef.
- H. Lee and K. Neville, Handbook of Epoxy Resins, Mc Graw- Hill, New York, 1967 Search PubMed.
- N. Supanchaiyamat, A. J. Hunt, P. S. Shuttleworth, C. Ding, J. H. Clark and A. S. Matharu, Bio-based thermoset composites from epoxidized linseed oil and expanded starch, RSC Adv., 2014, 4, 23304–23313 RSC.
- F. Ng, L. Bonnet, G. David and S. Caillol, Novel biobased and food contact epoxy coatings for glass toughening applications, Prog. Org. Coat., 2017, 109, 1–8 CrossRef CAS.
- S. V. Levchik, G. Camino, M. P. Luda, L. Costa, B. Costes, Y. Henry, E. Morel and G. Muller, Mechanistic Study of Thermal Behavior and Combustion Performance of Epoxy Resins: I Homopolymerized TGDDM, Polym. Adv. Technol., 1994, 6, 53–62 CrossRef.
- J. La Scala and R. P. Wool, Effect of FA composition on epoxidation kinetics of TAG, J. Am. Oil Chem. Soc., 2002, 79(4), 373–378 CrossRef CAS.
- R. J. Morgan, Structure-Property Relations of Epoxies Used as Composite Matrices, Adv. Polym. Sci., 1985, 72, 3–10 CrossRef.
- S. Caillol, M. Desroches, G. Boutevin, C. Loubat, R. Auvergne and B. Boutevin, Synthesis of new polyester polyols from epoxidized vegetable oils and biobased acids, Eur. J. Lipid Sci. Technol., 2012, 114, 1447–1459 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR method, 1H NMR spectra, 13C NMR spectra, SEC chromatograms, FT-IR spectra, DSC, DMA and TGA thermograms. See DOI: 10.1039/d0ra07682a |
| This journal is © The Royal Society of Chemistry 2020 |