
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the electrochemical performance of a natural molybdenite/N-doped graphene composite anode for lithium-ion batteries via short-time microwave irradiation†
Shuonan Wang a,
Yun Haia,
Bin Zhoub,
Hao Liu*b and
Libing Liao*a
a,
Yun Haia,
Bin Zhoub,
Hao Liu*b and
Libing Liao*a
aBeijing Key Laboratory of Materials Utilization of Nonmetallic Minerals and Solid Wastes, National Laboratory of Mineral Materials, School of Materials Science and Technology, China University of Geosciences, Beijing, 100083, PR China. E-mail: clayl@cugb.edu.cn
bSchool of Science, China University of Geosciences, Beijing, 100083, PR China. E-mail: liuhao1398@cugb.edu.cn
First published on 26th November 2020
Abstract
In the present work, low-cost natural molybdenite was used to make a MoS2/N-doped graphene composite through coulombic attraction with the aid of (3-aminopropyl)-triethoxysilane and the electrochemical performance was greatly improved by solvent-free microwave irradiation for tens of seconds. The characterization results indicated that most (3-aminopropyl)-triethoxysilane can decompose and release N atoms to further improve the N-doping degree in NG during the microwave irradiation. In addition, the surface groups of N-doped graphene were removed and the particle size of MoS2 was greatly decreased after the microwave irradiation. As a result, the composite electrode prepared with microwave irradiation exhibited a better rate performance (1077.3 mA h g−1 at 0.1C and 638 mA h g−1 at 2C) than the sample prepared without microwave irradiation (1013.6 mA h g−1 at 0.1C and 459.1 mA h g−1 at 2C). Therefore, the present results suggest that solvent-free microwave irradiation is an effective way to improve the electrochemical properties of MoS2/N-doped graphene composite electrodes. This work also demonstrates that natural molybdenite is a promising low-cost anode material for lithium-ion batteries.
1. Introduction
Molybdenite (MoS2), as a typical transition metal sulfide, is a promising anode material for lithium-ion batteries. It has a high theoretical specific capacity of 670 mA h g−1.1 After the initial cycle, MoS2 decomposes and forms Li2S and Mo atoms, and Mo atoms used to be considered to have no capacity.2 But a lot of studies have shown that the reversible capacity of MoS2 can reach 1000 mA h g−1, and extra capacity arises from the Mo atoms accommodating six Li+ ions over the prolonged discharge process,3 so molybdenite has great potential in lithium-ion batteries. However, MoS2 nanosheets are often subject to poor cycling stability and inferior rate capability because of their irreversible restacking, volume variation and poor electronic conductivity.4–6Integrating MoS2 with N-doped graphene (NG) can combat these drawbacks.1,4,5,7–9 First, NG can form a network to alleviate the aggregation and volume expansion of nanomaterials.10,11 In addition, N-doping can improve the electrical conductivity of carbon-based materials and offer ion transportation passages, which contribute to achieving a better rate performance during cycling.12–14 Furthermore, nitrogen doping in graphene can generate defects and active sites which enable the effective trapping of lithium polysulfides produced during lithiation of MoS2, leading to improvement of the cycling performance.15–18 Previous work has shown that NG can significantly improve the electrochemical properties of MoS2 anodes.1,19
A compact interface between these two materials is essential for charge transfer during the charging/discharging processes.20,21 Because of the negative surface charge of MoS2 and graphene,22–25 chemical modifiers or cross-linkers are usually used to ensure a compact interface through coulombic attraction7,26 or cross-linking.19 These chemicals can hinder the charge transfer process and can always be removed after the formation of the composite through traditional heating methods.7,19,26 For example, Li et al.26 fabricated a 3D MoS2@C/RGO composite through a surface-modification triggered self-assembly process. In order to disperse MoS2 into GO solution and trigger the self-assembly process between MoS2 and GO, polydopamine (PDA) coating of MoS2 was carried out based on π–π stacking and hydrogen bonding interactions. A sintering process at 400 °C was applied to transform PDA into N-doped amorphous carbon. As a result, the composite had a high capacity and good rate performance.
In contrast to traditional heating methods, microwave irradiation is an energy-efficient and fast heating method for material synthesis. Solvents usually need to be microwave absorbing and thermally conductive materials.27–31 For the last decade, solvent-free microwave irradiation synthesis methods have been gradually developed32–38 and some novel uses of solvent-free microwave irradiation have been explored such as microwave carbonization39 and the synthesis of nanoparticles.36 Previous studies showed that graphene can effectively absorb microwaves and generate a lot of heat and the temperature can rapidly rise above 1000 °C in a few seconds, and the particles loaded on the graphene can transform into nanoparticles from microparticles.36 These facts inspire us to use this method to treat MoS2/graphene composites or similar configurations to remove the chemical modifiers or cross-linkers and reduce graphene in a short time to improve the electrochemical performance of composite electrodes.
On the other hand, most reported MoS2 anode materials are synthesized with a relatively high cost.40 Nevertheless, molybdenite can be formed in nature and the reserves of molybdenite ore are abundant. High purity (>98.5%) but low cost natural molybdenite ($14.50–18.20 per kg vs. ∼$140 per kg for synthesized MoS2, Aladdin Co., >98%) can be obtained by using appropriate purification processes such as exfoliation41,42 and flotation.43,44 Several reports have demonstrated that natural molybdenite anodes can exhibit excellent electrochemical properties by size-controlling45 and selecting a suitable binder.44 Because the introduction of NG to form composites has many advantages, it is worth exploring the electrochemical properties of a natural molybdenite/NG composite electrode and the influence of microwave irradiation on this type of electrode.
In this work, we synthesized a natural molybdenite/NG composite and the natural molybdenite and NG were combined through coulombic attraction with the assistance of (3-aminopropyl)-triethoxysilane (APTES). This composite was further irradiated using microwaves for tens of seconds to enhance the interfacial interaction and the electronic conductivity through the removal of APTES and surface groups on NG. Therefore, the rate performance of this type of composite was greatly improved. This work not only demonstrated that the electrochemical performance of the MoS2/NG composite or similar composites can be improved through solvent-free microwave irradiation for a short time, but also showed the potential application prospects of natural molybdenite ore as a promising low-cost anode material for lithium-ion batteries.
2. Experimental
2.1 Materials
Natural molybdenite ore was obtained from Guangzhou Haoyu stone craft firm. (3-Aminopropyl)-triethoxysilane (APTES) was purchased from Aladdin Industrial Corporation, Shanghai, China. N-doped graphene was supplied by Nanjing XFNano Materials Tech Co., Ltd.2.2 Synthesis of MoS2/NG and MoS2/NG-MW
As shown in Fig. 1, natural molybdenite was crushed and sieved (50 mesh), and the sieved powder was ball-milled for 4 h at 560 rpm in a planetary ball mill (QM-3SP2, Nanjing NanDa Instrument Plant). 0.1546 g molybdenite was dispersed in 50 ml ethanol–water mixture (volume ratio, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and the suspension was ultrasonicated for 5 min. 0.3 ml APTES was added to the molybdenite suspension under magnetic stirring for 1 h. 30 mg N-doped graphene was dispersed in 50 ml ethanol–water mixture (volume ratio, 1:1), and the suspension was ultrasonicated for 1 h. The two suspensions above were mixed under magnetic stirring for 1.5 h. The solid was obtained via centrifugation and dried at 60 °C for 12 h. Finally, the mixture was heated under argon flow to 250 °C with a heating rate of 5 °C min−1 and maintained at this temperature for 2 h. This sample was marked as MoS2/NG. Then, the MoS2/NG was put in a glass bottle and sealed with a balloon in the glove box. The bottle was put into a microwave oven (Midea, M1-L213B, 700 W) on full power for 30 s. This sample was marked as MoS2/NG-MW.
:1), and the suspension was ultrasonicated for 5 min. 0.3 ml APTES was added to the molybdenite suspension under magnetic stirring for 1 h. 30 mg N-doped graphene was dispersed in 50 ml ethanol–water mixture (volume ratio, 1:1), and the suspension was ultrasonicated for 1 h. The two suspensions above were mixed under magnetic stirring for 1.5 h. The solid was obtained via centrifugation and dried at 60 °C for 12 h. Finally, the mixture was heated under argon flow to 250 °C with a heating rate of 5 °C min−1 and maintained at this temperature for 2 h. This sample was marked as MoS2/NG. Then, the MoS2/NG was put in a glass bottle and sealed with a balloon in the glove box. The bottle was put into a microwave oven (Midea, M1-L213B, 700 W) on full power for 30 s. This sample was marked as MoS2/NG-MW.
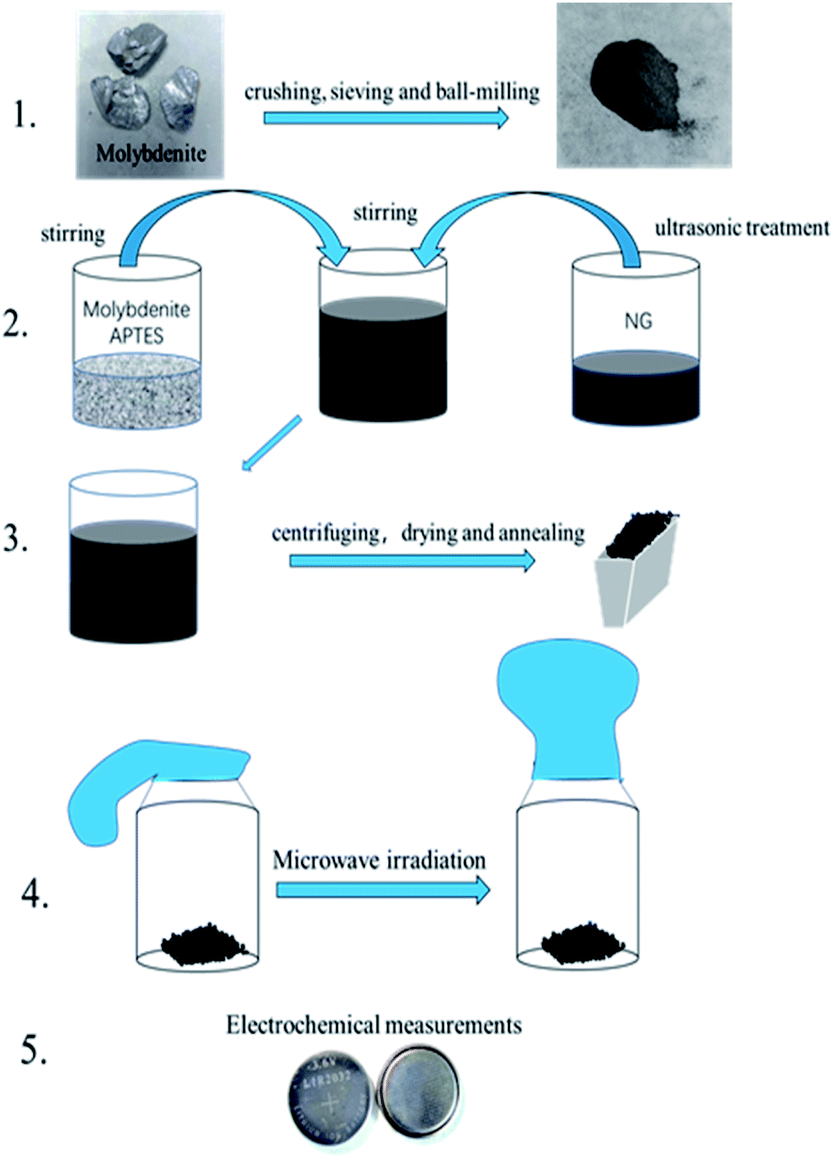 | ||
| Fig. 1 Design idea for the MoS2/NG-MW electrode material. | ||
2.3 Characterization
The structural characteristics of the as-prepared samples were investigated by X-ray diffraction (XRD, D8 Advance, Bruker, Germany) with Cu Kα radiation (λ = 0.15406 Å) at a voltage of 40 kV and a current of 100 mA, as well as X-ray photoelectron spectroscopy (XPS, K-Alpha, Thermo Scientific, America). The Raman spectra were obtained using a Laser Micro-Raman Spectrometer (Raman, Labram HR Evolution, Horiba Jobin Yvon, France) with a 532 nm laser. Fourier transform infrared spectra (FT-IR, Spectrum One, PerkinElmer, America) were obtained using the KBr pellet method. The morphology and composition information were characterized by scanning electron microscopy (FESEM, MERLIN VP Compact, ZEISS, Germany) with energy-dispersive spectroscopy (EDS), transmission electron microscopy (TEM, FEI G2 F20 S-TWIN TMP, America) and thermogravimetric analysis (TGA, TGA/DSC-1, Mettler-Toledo, Switzerland) carried out at a heating rate of 10 °C min−1 from 25 to 700 °C.2.4 Electrochemical measurements
CR2032 coin-type lithium half-cells were assembled in a glove box filled with a highly pure Ar atmosphere (O2 and H2O levels <0.5 ppm), using metallic lithium as the counter electrode with a polypropylene separator (Celgard 2000), and 1 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (volume ratio, 1:1) (provided by Nanjing Mojiesi Energy Technology Co., Ltd) as the electrolyte. The anode preparation was mixed with 90% MoS2/NG-MW and 10% binder (sodium alginate) additive, using deionized water as the solvent for blending the mixture. Then, the homogenous slurry was uniformly painted on copper foil and subsequently dried at 60 °C for 12 h in a vacuum oven before obtaining the final working electrode. The pristine MoS2 electrode was made using 70% molybdenite, 20% conductive agent (acetylene black) and 10% binder (sodium alginate) additive. The mass loading of active material (MoS2) was ∼1.0 mg cm−2. The half-cells were allowed to stand for 12 h before the electrochemical measurements. The electrochemical impedance spectroscopy (EIS) tests were performed on an electrochemical workstation (Ivium-Vertex, Ivium Technologies, Holland) in the frequency range from 0.1 Hz to 100 kHz. The cyclic voltammetry (CV) was performed on an electrochemical work station (CS150, CorrTest, China) in the voltage range 3.0–0.1 V at a scan rate of 5 mV s−1. The galvanostatic charge/discharge tests and rate performance were carried out on a battery testing system (CT-4008, Neware, China).
3. Results and discussion
The phase identification was carried out by X-ray Powder Diffraction (XRD). As shown in Fig. 2(a), there are 9 peaks at 14.4°, 32.7°, 33.5°, 35.9°, 39.5°, 44.2°, 49.8°, 58.3° and 60.1°. All the diffraction peaks can be matched to the standard card of 2H-MoS2 (JCPDS no. 37-1492), indicating that the primary component of the three samples is 2H-MoS2. Compared with pristine MoS2, the relative intensities of the diffraction peaks of the (002) planes of MoS2/NG and MoS2/NG-MW are lower, indicating that the degree of preferred orientation of MoS2 in these two samples decreased. On the other hand, the diffraction peak of NG is very weak because of the low content of NG in MoS2/NG and MoS2/NG-MW. | ||
| Fig. 2 (a) XRD patterns of pristine MoS2, MoS2/NG and MoS2/NG-MW. SEM images of (b) pristine MoS2, (c) MoS2/NG and (d) MoS2/NG-MW; EDS spectra of (e) MoS2/NG and (f) MoS2/NG-MW. | ||
The detailed morphology and layer structure of the materials were investigated by SEM. As shown in Fig. 2(b), the pristine MoS2 is an aggregate of layered MoS2 and the particle size is 2–5 μm. As shown in Fig. 2(c) and the EDS analysis of MoS2/NG, the atomic ratio of S to Mo is 2.02:1, which is consistent with the atomic ratio of MoS2. But in Fig. 2(d) and the EDS analysis of MoS2/NG-MW, the atomic ratio of S to Mo is 1.53:1, indicating that MoS2 decomposed and this caused the loss of S. As shown in Fig. 2(c) and (d), MoS2 is interspersed with NG, the composite presents a flower-like structure and the MoS2 particles in the composite material do not show agglomeration. The surface modification with APTES results in the presence of amino groups on the MoS2 surface, and the electrostatic attraction between the amino groups and graphene reduced the agglomeration.46 The preferential orientation of MoS2 is weak, which is consistent with the XRD pattern. By comparing Fig. 2(c) and (d), it can be clearly observed that after microwaving, the particle size of MoS2 decreased from 2–5 μm to 0.5–1 μm. Besides, the existence of microspheres in MoS2/NG-MW also indicates that some MoS2 decomposed and recombined.
The structural information was further revealed by TEM and the results are shown in Fig. 3. It can be clearly observed that there are MoS2 nanosheets with particle sizes ranging from several hundred nanometers to one micrometer (Fig. 3(a) and (b)). The high-resolution transmission electron microscopy (HRTEM) image of MoS2/NG-MW is displayed in Fig. 3(c) and the interplanar spacing is 0.274 nm which corresponds to the (100) plane of 2H-MoS2. Fig. 3(d) shows the elemental distributions of Mo, S, C and N elements, revealing that the MoS2 (bright parts in the STEM image) was wrapped in graphene (gray parts in the STEM image). Therefore, the above TEM results agree with the previous SEM and XRD results.
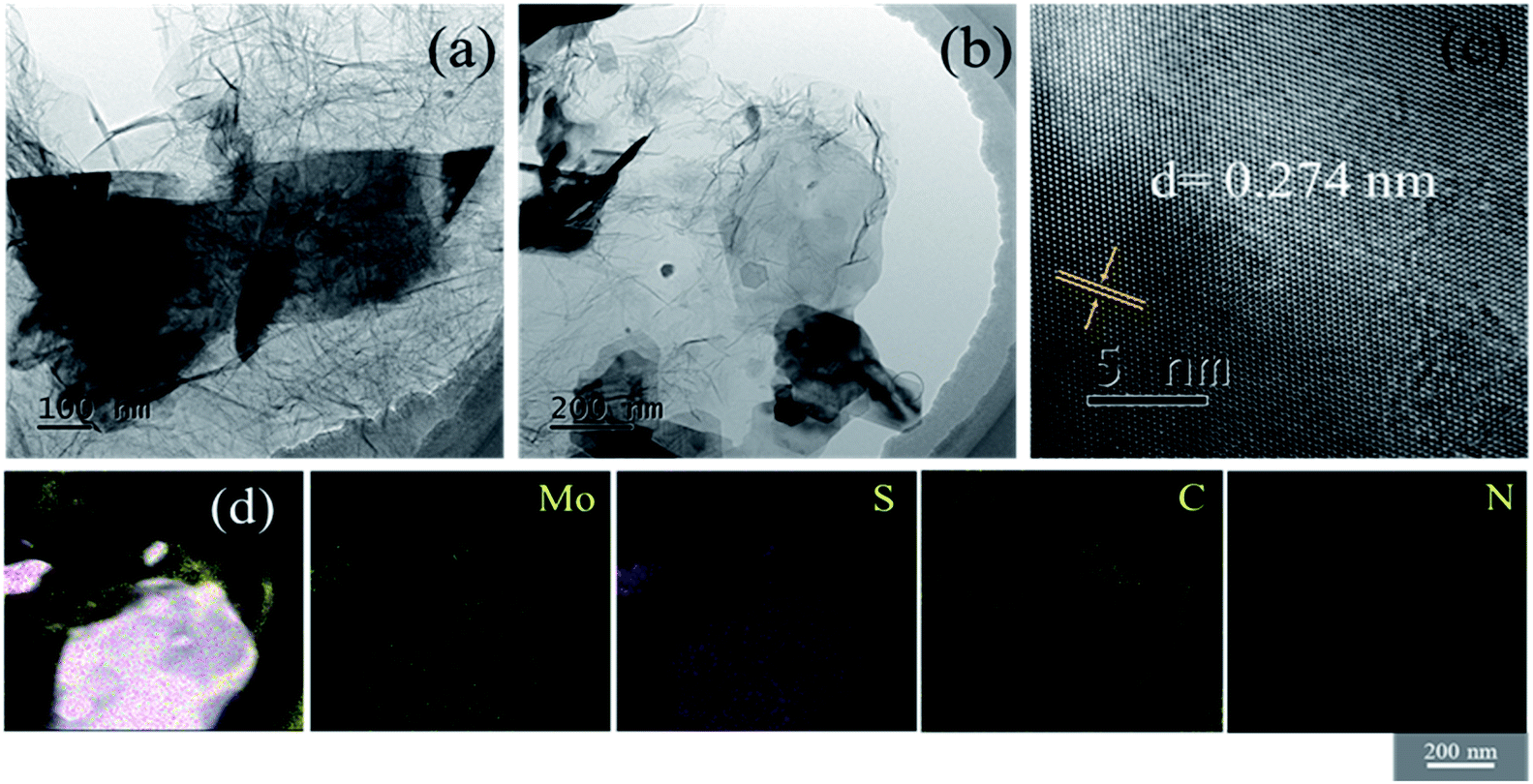 | ||
| Fig. 3 The (a) and (b) TEM images, (c) HRTEM image and (d) STEM and element mapping images of MoS2/NG-MW. | ||
Furthermore, XPS spectra were used to distinguish the elements’ oxidation states and the results are shown in Fig. 4. Mo, S, C and O are seen in the total spectra in Fig. 4(a), but the peak of N (∼399 eV) cannot be observed in the total spectra because of the presence of a nearby strong Mo 3p peak (∼395 eV). According to the Mo 3d spectra of pristine MoS2, MoS2/NG and MoS2/NG-MW in Fig. 4(b), the three peaks at 232.6, 229.4 and 226.5 eV are attributed to Mo 3d3/2, Mo 3d5/2, and S 2s, respectively, corresponding with 2H-MoS2.47 The S 2p spectra of pristine MoS2, MoS2/NG and MoS2/NG-MW are shown in Fig. 4(c), and the two peaks at 162.2 and 163.4 eV correspond to the S 2p3/2 and S 2p1/2 states of MoS2.47 But in the spectrum of MoS2/NG-MW, there are two new peaks at 164.1 and 164.9 eV which correspond to the S 2p3/2 and S 2p1/2 states of elemental S, indicating that microwave irradiation caused a high temperature so that some of the MoS2 decomposed and elemental S was produced. Due to the low content of elemental S, the diffraction peak of elemental S does not appear in the XRD pattern. The C 1s spectrum is depicted in Fig. 4(d), and there are four peaks at 284.8, 285.5, 286.6 and 288.5 eV which correspond with C–C, C–O, C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.16,48,49 The C–O (285.5 eV) peak area of MoS2/NG-MW is smaller than that of MoS2/NG, and the C–N (286.6 eV) peak area of MoS2/NG-MW is larger than that of MoS2/NG.
O, respectively.16,48,49 The C–O (285.5 eV) peak area of MoS2/NG-MW is smaller than that of MoS2/NG, and the C–N (286.6 eV) peak area of MoS2/NG-MW is larger than that of MoS2/NG.
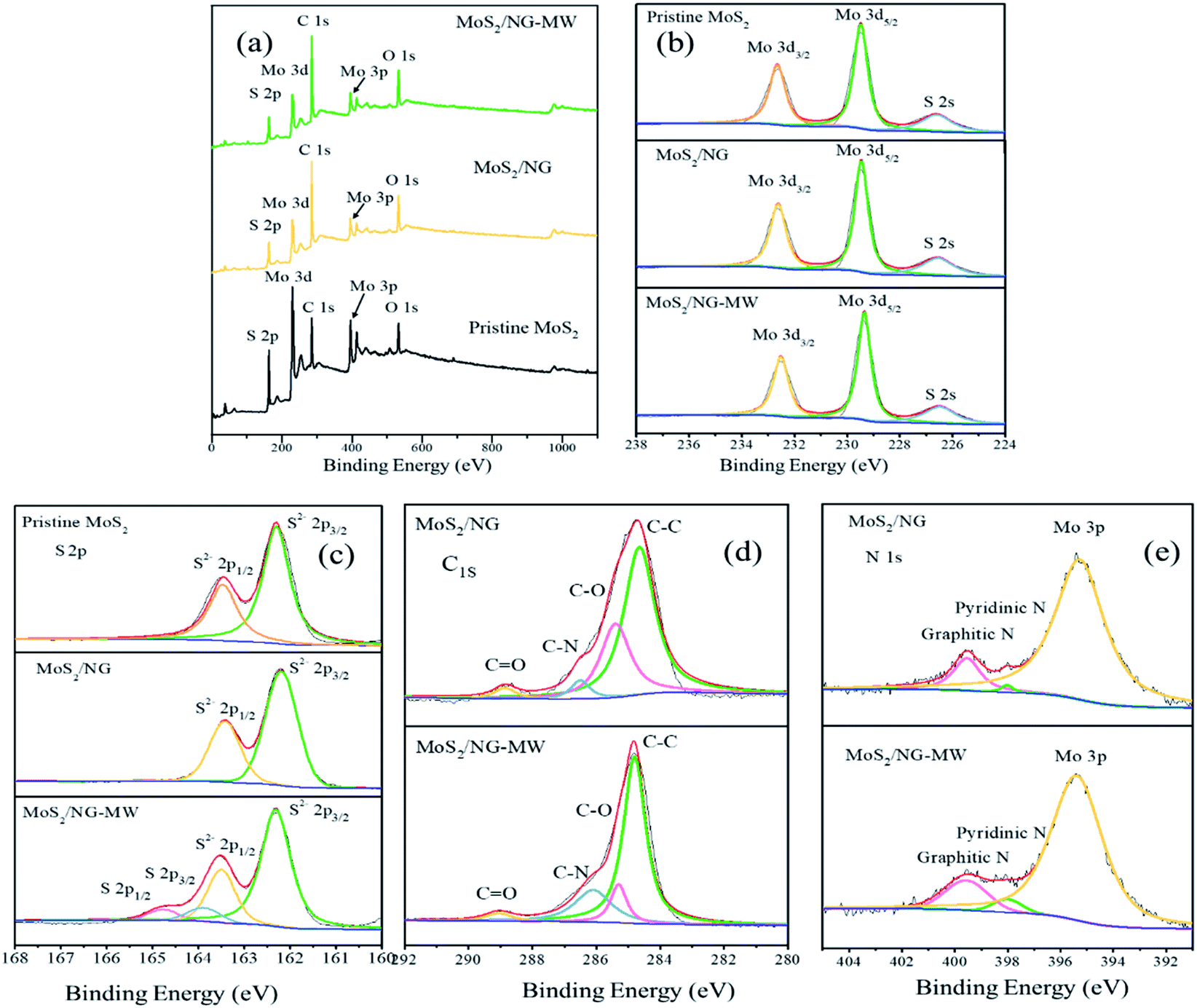 | ||
| Fig. 4 (a) Total spectra, (b) Mo 3d, (c) S 2p, (d) C 1s, (e) N 1s high-resolution XPS spectra of pristine MoS2, MoS2/NG and MoS2/NG-MW. | ||
At the same time, the peaks at 398.5 and 401.4 eV in the high-resolution N 1s spectrum (Fig. 4(e)) can be attributed to the pyridinic N and graphitic N.1,50 The area of these two peaks is larger for MoS2/NG-MW than MoS2/NG, which coincides with the change in the C–N peak area. These results suggest that the functional groups on the NG surface were consumed but the N-doping degree was enhanced during microwave irradiation. They also demonstrate that the APTES decomposed and the released N atoms entered the NG layer during microwave irradiation.
The FT-IR spectra are shown in Fig. 5(a), and the FT-IR spectrum of MoS2/NG shows a broad absorption band at 3234 cm−1, which can be assigned to the O–H stretching vibration arising from the hydroxyl groups of the NG sheets.51 This peak disappeared after microwave irradiation, indicating that NG is reduced during the process of microwave irradiation. The peaks at 3065 cm−1, 1662 cm−1 and 1039 cm−1 are associated with the N–H stretching vibrations of amino groups, N–H bond bending of amino groups and Si–O–C,47 and are ascribed to the APTES. Nevertheless, the N–H peaks almost disappear and the intensity of the Si–O–C peak greatly decreases in MoS2/NG-MW, indicating that most of the APTES decomposed during the process of microwave irradiation. The FT-IR results agree with the previous XPS results.
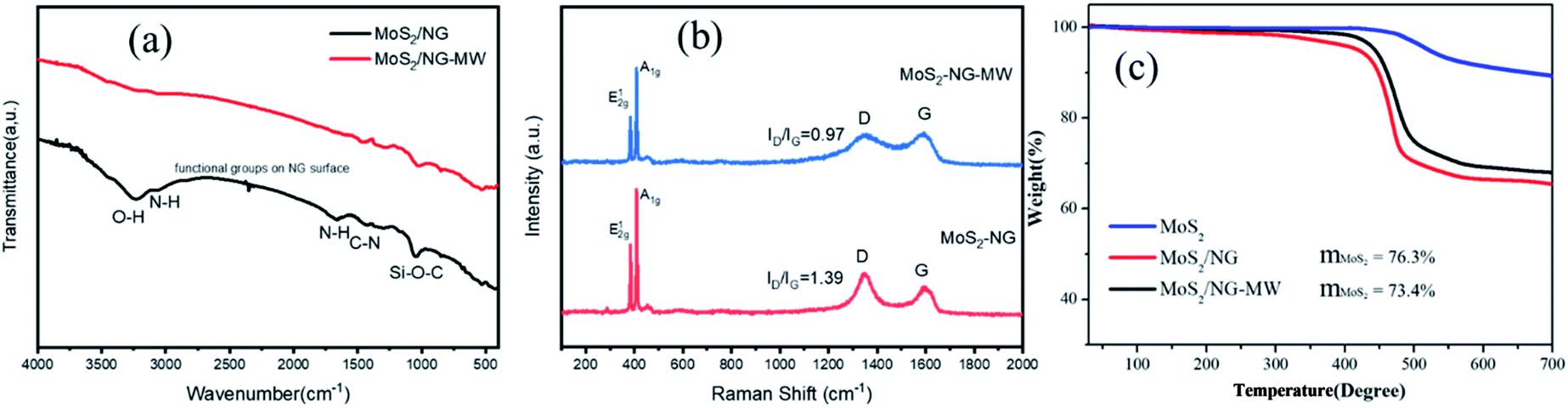 | ||
| Fig. 5 (a) FT-IR and (b) Raman spectra of MoS2/NG and MoS2/NG-MW; (c) TGA curves of pristine MoS2, MoS2/NG and MoS2/NG-MW. | ||
The structural features of the as-obtained samples were further explored through Raman spectroscopy. As shown in Fig. 5(b), the two distinct Raman peaks at 384 cm−1 and 409 cm−1 are attributed to the typical E12g and A1g vibration modes of hexagonal MoS2 crystals. There is no difference in peak separation (24.86 cm−1) but the relative peak intensity ratios of A1g and E12g provide important information on the crystallinity and degree of order of MoS2. The A1g/E12g values for MoS2/NG and MoS2/NG-MW are 1.36 and 1.25 respectively, suggesting that MoS2/NG has better crystallinity. For the D band and G band of NG, the D band is associated with defects and the G band corresponds to the stretching mode of sp2 bonded carbon.52 The ratio between the D band and G band decreased from 1.39 to 0.97 after microwave irradiation, suggesting that oxygen functional groups on the NG surface were removed during this process. This coincides with the results of FT-IR and XPS.
In order to know the amount of each component in the sample, TGA was performed and the results are shown in Fig. 5(c). The main temperature range for weight loss is 400–500 °C. The weight loss of pristine MoS2 is attributed to the oxidation of MoS2 to MoO3 and SO2. And the weight losses of MoS2/NG and MoS2/NG-MW are attributed to the oxidation of MoS2 and NG to MoO3, SO2 and CO2. The remaining weights of pristine MoS2, MoS2/NG and MoS2/NG-MW were 89.1%, 68.1% and 65.4%, respectively, after heating to 700 °C. The mass fraction of MoS2 is found to be around 76.3% and 73.4% in MoS2/NG and MoS2/NG-MW, respectively. The mass fraction of MoS2 in MoS2/NG-MW is lower than in MoS2/NG, which is associated with the decomposition of MoS2 during microwave irradiation. Therefore, the TGA results coincide with the previous EDS and XPS results.
The CV curves of pristine MoS2 and MoS2/NG-MW electrodes are shown in Fig. 6(a) and (b). For the first cyclic curve, two pronounced reductions at 0.85 V and 0.4 V are observed. The peak at 0.85 V can be ascribed to the intercalation of Li+ into MoS2 interlayers and the resulting 2H-MoS2 to 1T-MoS2 phase transition: MoS2 + xLi+ + xe− → LixMoS2.53 The peak at 0.4 V can be attributed to the reduction of LixMoS2 to Mo particles and Li2S: LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2S.1 The oxidation peak at 2.35 V can be ascribed to the decomposition of Li2S: Li2S − 2e− → 2Li+ + S.1 In the subsequent cyclic curve, there are three distinct reduction peaks at 1.9 V, 1.1 V and 0.3 V, which can be assigned to the following three reactions: 2Li+ + S + 2e− → Li2S, MoS2 + xLi+ + xe− → LixMoS2, LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2S. The charge–discharge profiles of the pristine MoS2 and MoS2/NG-MW electrodes are shown in Fig. 6(c) and (d). As shown in Fig. 6(c) and (d), the two potential plateaus at 1.0 V and 0.5 V can be attributed to the intercalation of Li+ into the MoS2 interlayers and the reduction of LixMoS2 to Mo particles and Li2S. In the subsequent discharge curves, the two potential plateaus disappeared and a new potential plateau appeared at 1.9 V, in accordance with the formation of Li2S. In the charge curves, the obvious potential plateau at about 2.3 V corresponds to the delithiation of Li2S. Compared with the pristine MoS2 electrode, the MoS2/NG-MW electrode has charge–discharge profiles with better repeatability, indicating that the MoS2/NG-MW electrode has better cycling performance. For the MoS2/NG-MW electrode, the discharge capacities are 1470, 1098 and 1056 mA h g−1 for the first three cycles. As for the pristine MoS2 electrode, the discharge capacities are only 1185, 813 and 770 mA h g−1 for the first three cycles. The capacity loss of about 30% in the first cycle was the result of an irreversible process such as the formation of an inorganic solid electrolyte interphase (SEI) film and the irreversibility of Li2S.3
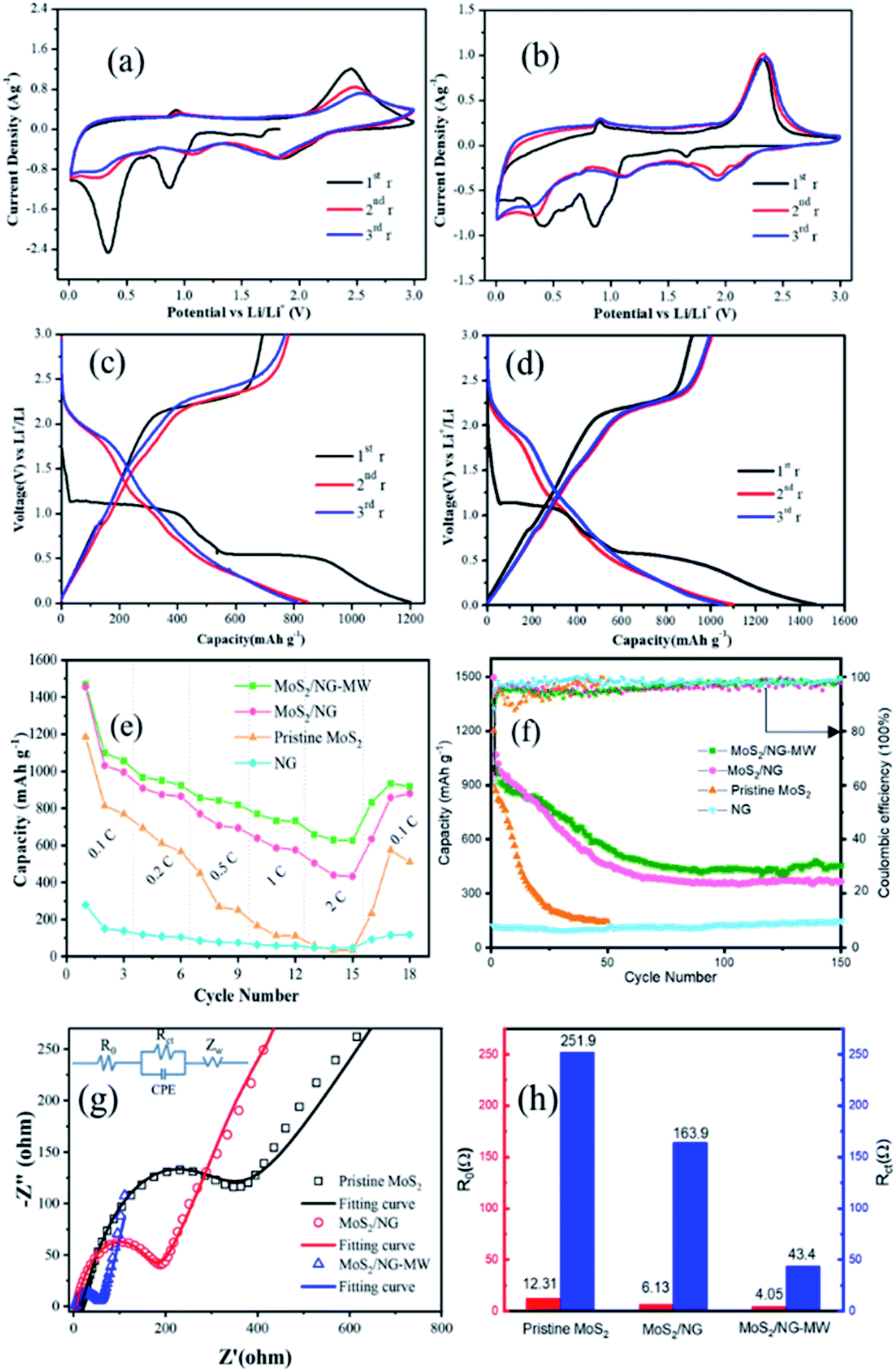 | ||
| Fig. 6 CV curves of (a) pristine MoS2 and (b) MoS2/NG-MW electrodes at a scanning rate of 0.5 mV s−1; the galvanostatic charge and discharge profiles of (c) pristine MoS2 and (d) MoS2/NG-MW electrodes at 0.1C; (e) rate performance of pristine MoS2, MoS2/NG and MoS2/NG-MW electrodes at various current densities; (f) cycling performance and coulombic effciency of MoS2, MoS2/NG and MoS2/NG-MW electrodes at 0.1C; (g) EIS curves of pristine MoS2, MoS2/NG and MoS2/NG-MW electrodes; (h) the impedance parameters calculated from the equivalent circuit model fitting. | ||
Rate performances were tested at different current densities and the results are shown in Fig. 6(e). At rates of 0.1, 0.2, 0.5, 1 and 2C, the MoS2/NG electrode exhibits average specific discharge capacities of 1013.6, 882.5, 723.8, 600.5 and 459.1 mA h g−1, while the MoS2/NG-MW electrode exhibits better average specific discharge capacities of 1077.3, 956.7, 834, 745.5 and 638 mA h g−1, respectively. In addition, the specific discharge capacities can recover to 879.5 and 918.4 mA h g−1 for the MoS2/NG and MoS2/NG-MW electrodes when the current density goes back to 0.1C. In contrast, the capacity of the pristine MoS2 electrode is only 509.4 mA h g−1 at the same rate.
It is obvious that the MoS2/NG-MW electrode has the best rate performance among these three electrodes. The cycling performances of pristine MoS2, MoS2/NG and MoS2/NG-MW electrodes were measured at a rate of 0.1C. As shown in Fig. 6(f), the capacity of the pristine MoS2 electrode decreased sharply for the first 30 cycles, and only 142.2 mA h g−1 was preserved up to the 50th cycle. For the MoS2/NG and MoS2/NG-MW electrodes, the capacities apparently decreased for the first 70 cycles, but the capacities remained stable for the next 80 cycles, which can be attributed to the fact that the flower-like structure could reduce the loss of active material. After 70 and 150 cycles, the specific discharge capacities of the MoS2/NG electrode are 387.4 and 366.6 mA h g−1, and those of the MoS2/NG-MW electrode are 459.8 and 453.7 mA h g−1, respectively. The result confirms that MoS2/NG-MW has a more compact interface between MoS2 and NG, so the loss of active material is reduced, leading to the better cycling performance of MoS2/NG-MW. The initial coulombic efficiency is about 85% and quickly increases to above 95% after several cycles.
To confirm the difference in the electrochemical performance of the pristine MoS2, MoS2/NG and MoS2/NG-MW electrodes, EIS tests were performed. The Nyquist plots and fitted equivalent circuit are shown in Fig. 6(g). The semicircular loop at the high–medium frequencies is related to resistance R0, electrode/electrolyte interface resistance (CPE) and charge transfer resistance (Rct), and the sloped line at low frequencies represents the Warburg impedance (Zw), which is connected to the Li+ diffusion in the electrode materials.45 The impedance parameters calculated from the equivalent circuit model fitting are shown in Fig. 6(h). The R0 and Rct of the MoS2/NG-MW electrode are 4.05 Ω and 43.4 Ω, which are much lower than those of the pristine MoS2 electrode (R0 = 12.31 Ω, Rct = 251.9 Ω) and the MoS2/NG electrode (R0 = 6.13 Ω, Rct = 163.9 Ω). This fact confirms that the electrical conductivity is improved. Nevertheless, the impedance spectra of the MoS2/NG and MoS2/NG-MW electrodes become similar after tens of cycles (Fig. S1, ESI†).
We have compared the obtained results with the literature (Table S1, ESI†) and found that the MoS2/NG-MW composite possesses a good rate performance but the cycling performance is relatively poor. In order to know the reason for the poor cycling performance, SEM was performed and the results are shown in Fig. 7. As shown in Fig. 7(a–e), it is hard to find the layered structure of MoS2 after 150 cycles, and a thick SEI film can be observed on the surface of the electrode. According to the EDS results, the content of sulfur in the MoS2/NG and MoS2/NG-MW electrodes greatly decreases after 150 cycles. According to the CV results and other reports,2 the reaction processes of MoS2-based anode materials are similar to those of Li–S batteries after the first several cycles. Therefore, the problem of polysulfide dissolution has a great impact on the cycling stability of the electrodes. After long-term cycling, the content of sulfur in the MoS2/NG and MoS2/NG-MW electrodes greatly decreases due to this problem, leading to the poor cycling performance of these two electrodes. In addition, the difference between the MoS2/NG and MoS2/NG-MW electrodes becomes small after tens of cycles, leading to similar impedance of these two electrodes. Several measures can be taken to improve the electrochemical performance of our samples. For example, other binders, such as carboxymethyl cellulose (CMC), may be more suitable than sodium alginate for a MoS2-based anode.44,45 In addition, adding some porous or surface polarization materials, which can trap the soluble polysulfide, in the composite electrode or separator may also lead to an improvement of the cycling stability.
 | ||
| Fig. 7 SEM images of (a) MoS2/NG-MW and (b) MoS2/NG electrodes after 1 cycle; SEM images of (c) and (d) MoS2/NG-MW and (e) MoS2/NG electrodes after 150 cycles and EDS results of the MoS2/NG and MoS2/NG-MW electrodes. | ||
Based on the above results, it can be concluded that microwave irradiation can greatly improve the rate performance and cycling performance of the MoS2/NG composite. Firstly, the APTES can decompose and the released N atoms enter into the NG layer during microwave irradiation. Therefore, MoS2/NG-MW has a more compact interface and the N-doping degree is enhanced, which are beneficial for the charge transfer during the charging/discharging processes. Secondly, the functional groups on the NG surface are removed during microwave irradiation and this can further improve the electronic conductivity of the composite. Thirdly, the particle size of MoS2 decreases from 2–5 μm to 0.5–1 μm and the transfer path for ions and electrons can be greatly shortened during cycling. Therefore, the rate performance of the MoS2/NG-MW electrode has been improved by microwave irradiation.
4. Conclusions
In summary, a natural molybdenite/NG composite was synthesized with the assistance of APTES, and the electrochemical performance of the composite was improved via short-time microwave irradiation. The influence of microwave irradiation was verified and the results indicated that part of the MoS2 was decomposed during microwave irradiation and its particle size decreased from 2–5 μm to 0.5–1 μm, which can greatly shorten the transfer paths for ions and electrons during cycling. Besides, most of the APTES decomposed and the released N atoms entered into the NG layer during microwave irradiation, which is beneficial for charge transfer during the charging/discharging processes. Furthermore, the functional groups on the NG surface are consumed during microwave irradiation and this can further improve the electronic conductivity of the composite. As a result, MoS2/NG-MW exhibited a good rate performance (1077.3 mA h g−1 at 0.1C and 638 mA h g−1 at 2C) and good cycling performance (459.8 and 453.7 mA h g−1 after 70 and 150 cycles at 0.1C), which are superior to those of MoS2/NG. This work not only suggested that short-time solvent-free microwave irradiation could improve the electrochemical performance of the MoS2/NG composite or similar NG composites, but also indicated that natural molybdenite ore is a promising low-cost anode material for lithium-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities (No. 2652019108) and the National Natural Science Foundation of China (No. 21875223).Notes and references
- B. Chen, Y. Meng, F. He, E. Liu, C. Shi, C. He, L. Ma, Q. Li, J. Li and N. Zhao, Nano Energy, 2017, 41, 154–163 CrossRef CAS.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- L. Wang, Q. Zhang, J. Zhu, X. Duan, Z. Xu, Y. Liu, H. Yang and B. Lu, Energy Storage Mater., 2019, 16, 37–45 CrossRef.
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839–844 CrossRef CAS.
- J. Jiao, K. Du, Y. Wang, P. Sun, H. Zhao, P. Tang, Q. Fan, H. Tian, Q. Li and Q. Xu, Mater. Chem. Phys., 2020, 240, 122169 CrossRef CAS.
- F. Tu, Y. Han, Y. Du, X. Ge, W. Weng, X. Zhou and J. Bao, ACS Appl. Mater. Interfaces, 2019, 11, 2112–2119 CrossRef CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS.
- L. Ma, J. Ye, W. Chen, D. Chen and J. Yang Lee, Nano Energy, 2014, 10, 144–152 CrossRef CAS.
- B. Yu, Y. Chen, Z. Wang, D. Chen, X. Wang, W. Zhang, J. He and W. He, J. Power Sources, 2020, 447, 227364 CrossRef CAS.
- D. Zhou, W.-L. Song, X. Li, L.-Z. Fan and Y. Deng, J. Alloys Compd., 2017, 699, 730–737 CrossRef CAS.
- N. Wu, W. Du, X. Gao, L. Zhao, G. Liu, X. Liu, H. Wu and Y. B. He, Nanoscale, 2018, 10, 11460–11466 RSC.
- X. Tang, G. Wen and Y. Song, Appl. Surf. Sci., 2018, 436, 398–404 CrossRef CAS.
- X. Ge, S. Liu, M. Qiao, Y. Du, Y. Li, J. Bao and X. Zhou, Angew. Chem., Int. Ed., 2019, 58, 14578–14583 CrossRef CAS.
- X. Zhou, L. Yu and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1600451 CrossRef.
- D. Su, M. Cortie and G. Wang, Adv. Energy Mater., 2017, 7, 1602014 CrossRef.
- X. Li, Y. Bai, M.-S. Wang, G. Wang, Y. Ma, L. Li, B. Xiao and J. Zheng, Sustainable Energy Fuels, 2019, 3, 1427–1438 RSC.
- C. Li, X.-L. Sui, Z.-B. Wang, Q. Wang and D.-M. Gu, Ceram. Int., 2018, 44, 13419–13425 CrossRef CAS.
- S.-K. Liu, X.-B. Hong, Y.-J. Li, J. Xu, C.-M. Zheng and K. Xie, Chin. Chem. Lett., 2017, 28, 412–416 CrossRef CAS.
- S. Xia, Y. Wang, Y. Liu, C. Wu, M. Wu and H. Zhang, Chem. Eng. J., 2018, 332, 431–439 CrossRef CAS.
- X. Liu, Q. Yang, M. Mi, W. Kong, Y. Ge, J. Ma and J. Hu, J. Alloys Compd., 2019, 800, 99–106 CrossRef CAS.
- W. Zhang, S. Fang, N. Wang, J. Zhang, B. Shi, Z. Yu and J. Yang, Inorg. Chem. Front., 2020, 7, 2487–2496 RSC.
- J. H. Lee, S. H. Kwon, S. Kwon, M. Cho, K. H. Kim, T. H. Han and S. G. Lee, Nanomaterials, 2019, 9, 268 CrossRef CAS.
- M. Morant-Giner, I. Brotons-Alcázar, N. Y. Shmelev, A. L. Gushchin, L. T. Norman, A. N. Khlobystov, A. Alberola, S. Tatay, J. Canet-Ferrer, A. Forment-Aliaga and E. Coronado, Chem.–Eur. J., 2020, 26, 6670–6678 CrossRef CAS.
- C. Zhao, R. Wang, Y. Zhang, L. Chen, T. Li, X. Deng, P. Zhang and X. Lu, Electrochim. Acta, 2019, 320, 134591 CrossRef CAS.
- Y. Fang, Q. Huang, P. Liu, J. Shi and G. Xu, Colloids Surf., A, 2018, 540, 112–122 CrossRef CAS.
- S. Li, P. Liu, X. Huang, Y. Tang and H. Wang, J. Mater. Chem. A, 2019, 7, 10988–10997 RSC.
- E. Alsharaeh, F. Ahmed, Y. Aldawsari, M. Khasawneh, H. Abuhimd and M. Alshahrani, Sci. Rep., 2016, 6, 29854 CrossRef CAS.
- E.-S. M. Duraia, A. Fahami and G. W. Beall, J. Electron. Mater., 2018, 47, 7288–7295 CrossRef CAS.
- S. Shi, X. Hua and H. Guo, Ceram. Int., 2018, 44, 13495–13501 CrossRef CAS.
- Y. Aldawsari, Y. Mussa, F. Ahmed, M. Arsalan and E. Alsharaeh, Materials, 2019, 12, 2248 CrossRef CAS.
- Z. W. Lu, Y. H. Wang, Z. Dai, X. P. Li, C. Y. Zhang, G. Z. Sun, C. S. Gong, X. J. Pan, W. Lan, J. Y. Zhou and E. Q. Xie, Electrochim. Acta, 2019, 325, 134920 CrossRef CAS.
- D. Voiry, J. Yang, J. Kupferberg, R. Fullon, C. Lee, H. Y. Jeong, H. S. Shin and M. Chhowalla, Science, 2016, 353, 1413–1416 CrossRef CAS.
- B. K. Barman and K. K. Nanda, ACS Sustainable Chem. Eng., 2018, 6, 4037–4045 CrossRef CAS.
- J. Xu, R. Zhang, S. Lu, H. Liu, Z. Li, X. Zhang and S. Ding, Nanotechnology, 2018, 29, 305708 CrossRef.
- J. A. Rudd, C. E. Gowenlock, V. Gomez, E. Kazimierska, A. M. Al-Enizi, E. Andreoli and A. R. Barron, J. Mater. Sci. Technol., 2019, 35, 1121–1127 CrossRef.
- S. Xu, G. Zhong, C. Chen, M. Zhou, D. J. Kline, R. J. Jacob, H. Xie, S. He, Z. Huang, J. Dai, A. H. Brozena, R. Shahbazian-Yassar, M. R. Zachariah, S. M. Anlage and L. Hu, Matter, 2019, 1, 759–769 CrossRef.
- S. Liu, P. Yan, H. Li, X. Zhang and W. Sun, Front. Chem., 2020, 8, 104 CrossRef CAS.
- Y. Lin, D. W. Baggett, J.-W. Kim, E. J. Siochi and J. W. Connell, ACS Appl. Mater. Interfaces, 2011, 3, 1652–1664 CrossRef CAS.
- Q. Shi, D. Liu, Y. Wang, Y. Zhao, X. Yang and J. Huang, Small, 2019, 15, 1901724 CrossRef CAS.
- S. Chen, J. Gao, B. M. Srinivasan and Y.-W. Zhang, Acta Phys.-Chim. Sin., 2019, 35, 1119–1127 Search PubMed.
- W. Zhao, T. Jiang, Y. Shan, H. Ding, J. Shi, H. Chu and A. Lu, Nanomaterials, 2018, 8, 843 CrossRef.
- N. Savjani, E. A. Lewis, R. A. D. Pattrick, S. J. Haigh and P. O’Brien, RSC Adv., 2014, 4, 35609–35613 RSC.
- P. F. A. Braga, A. P. Chaves, A. B. Luz and S. C. A. França, Int. J. Miner. Process., 2014, 127, 23–27 CrossRef CAS.
- S. Li, H. Tang, P. Ge, F. Jiang, J. Zhou, C. Zhang, H. Hou, W. Sun and X. Ji, ACS Appl. Mater. Interfaces, 2018, 10, 6378–6389 CrossRef CAS.
- F. Jiang, S. Li, P. Ge, H. Tang, S. A. Khoso, C. Zhang, Y. Yang, H. Hou, Y. Hu, W. Sun and X. Ji, Front. Chem., 2018, 6, 389 CrossRef.
- W. Chen, S. Qi, L. Guan, C. Liu, S. Cui, C. Shen and L. Mi, J. Mater. Chem. A, 2017, 5, 5332–5341 RSC.
- C. Chen, Y. He, G. Xiao, Y. Xia, H. Li and Z. He, Appl. Surf. Sci., 2018, 444, 511–521 CrossRef CAS.
- S. Tao, D. Wu, S. Chen, B. Qian, W. Chu and L. Song, Chem. Commun., 2018, 54, 8379–8382 RSC.
- Z. Jing, X. Dai, X. Xian, Q. Zhang, H. Zhong and Y. Li, Materials, 2020, 13, 2529 CrossRef CAS.
- J. Liu, A. X. Wei, M. Chen and X. Xia, J. Mater. Chem. A, 2018, 6, 3857–3863 RSC.
- T. Liu, X. Zhang, B. Li, J. Ding, Y. Liu, G. Li, X. Meng, Q. Cai and J. Zhang, RSC Adv., 2014, 4, 50765–50770 RSC.
- P. Krawczyk, B. Gurzęda and A. Bachar, Appl. Surf. Sci., 2019, 481, 466–472 CrossRef CAS.
- Y. Miki, D. Nakazato, H. Ikuta, T. Uchida and M. Wakihara, J. Power Sources, 1995, 54, 508–510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07758e |
| This journal is © The Royal Society of Chemistry 2020 |