
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origin of the enhanced photocatalytic activity of (Ni, Se, and B) mono- and co-doped anatase TiO2 materials under visible light: a hybrid DFT study†
Hanan H. Ibrahim ,
Adel A. Mohamed and
Ismail A. M. Ibrahim*
,
Adel A. Mohamed and
Ismail A. M. Ibrahim*
Department of Chemistry, Faculty of Science, Helwan University, 11795 Cairo, Egypt. E-mail: ismail.ibrahim@science.helwan.edu.eg
First published on 26th November 2020
Abstract
The characteristic properties of TiO2 (anatase) make doping necessary to enhance its photocatalytic activity. Herein, a density functional theory (DFT) study using the Heyd–Scuseria–Ernzerhof (HSE) hybrid functional was performed to precisely investigate the effect of mono- and co-doping (Ni, Se and B) on the structural, electronic and optical properties of anatase TiO2. Notably, the origin of the enhanced photocatalytic activity of the modified systems was determined. The response to visible light was enhanced for all the mono- and co-doped materials except for Bint, and the highest absorption coefficient was observed for Se4+ mono-doping and Se/Bint+sub and Ni/Bsub co-doping. The decrease in bandgap is associated with a red shift in the absorption edges with the smallest bandgap calculated for Ni/Bsub (2.49 eV). Additionally, the Ni, Se4+ and Se2− mono-doped systems and Ni/Se4+ co-doped systems are proposed as promising photocatalysts for water splitting applications and further experimental validation. Moreover, the Ni/Bint+sub and Se/Bint+sub co-doped materials can also be valuable photocatalysts for other energy applications due to their enhanced visible light activity and the prolonged lifetime of their produced charge carriers.
1. Introduction
In recent years, clean energy technology has attracted great interest for a sustainable economy and reduction of environmental pollution.1,2 Accordingly, photocatalytic materials have been extensively investigated, especially semiconductor photocatalysts. The light absorbed by a semiconductor is characterized by its electronic bandgap. Therefore, TiO2 is an ideal photocatalyst due to its characteristic properties such as low cost, low toxicity, and high thermal stability. However, due to the wide bandgap (3.2 and 3.00 eV for anatase and rutile, respectively) of pure TiO2, the absorption of light is limited to only UV light, which represents ∼5% of solar energy. Therefore, a low quantum yield of the photo-generated oxidative species is obtained. Thus, bandgap engineering of TiO2-based materials is of major interest for solar energy applications such as water splitting for H2 production.3–6 Doping with ions such as transition metals, rare earth metals, alkali metals, noble metals and non-metals was found to be an effective method for controlling the electronic structure and extending the light response to the visible light region.7 Various experimental and theoretical studies have been carried out on TiO2 materials, but nearly all the theoretical investigations were demonstrated based on ab initio DFT calculations.8–11Nickel doping was largely exhibited since the ionic radius of Ni2+ ions is slightly higher than that of Ti4+ ions.9 Several experimental methods have been employed for the synthesis of Ni-doped TiO2 such as the electrospinning technique,12 co-precipitation method,13 hydrothermal method,14 and sol–gel method.15 The visible light response and enhanced photocatalytic performance of Ni-doped anatase have been extensively reported.14,16 Based on diffuse reflectance spectroscopy (DRS) analysis, bandgap narrowing increases as a function of the dopant concentration.14,16 Quantum mechanical methods based on density functional theory were used to explain the optical response and changes in the electronic properties of TiO2 upon Ni doping.17 On the other hand, several reports on selenium doping were recently published, suggesting that it exhibits several oxidation states, including Se4+, Se0 and Se2−.18 Although, cationic Se4+ and Se0 have been significantly assigned experimentally, Se2− was only clarified theoretically in the previous work by Harb.10 Xie et al. attributed the narrowed gap (2.19 eV) and the suppressed electron/hole recombination rate upon Se4+ doping to the energy states appearing in the gap region upon the incorporation of Se.11 A DFT-based study with HSE level showed the effect of Se doping at different valence states with different concentrations on water splitting.10 The results showed that substitutional cationic and anionic Se can induce the best band positions for the water splitting reaction. Nonetheless, substitutional Se2− within the same crystal provides localized gap states capable of limiting the hole mobility and enlarging the recombination rate.10
For boron-doped TiO2, it was found that B doping has photochemical19,20 and electrochemical applications,21 such as degradation of atrazine as an organic pollutant on light absorption and high performance anode for sodium ion batteries. Based on previous experimental and theoretical studies, B can occupy different sites in the crystal lattice of anatase TiO2, either interstitial or substitutional sites, and at high concentration it can occupy both sites.20 For interstitial B, no visible light activity was observed; however, a response to visible light was detected for substitutional B due to the electronic transitions from the deep states.22,23 At high concentration, both interstitial and substitutional B were found to be stable in bulk anatase or co-doped with other elements.20,23 B was co-doped with Ni by Masae et al., who claimed the improvement of photocatalytic activity upon co-doping, but the origin of this enhancement was not clear.24
To the best of our knowledge, neither Ni/Se co-doping nor Se/B co-doping has been addressed experimentally and theoretically to date. Therefore, this study proposes new co-doped TiO2 photocatalysts for further experimental validation. In the current work, we attempted to study the effect of mono-doping and co-doping with different metals and non-metals at the HSE hybrid functional level on the photocatalytic activity of anatase TiO2. In our calculations, the structural, electronic, and optical properties were addressed and compared to the available experimental data. In addition, a systematic description of the induced levels on defect formation and the interaction between these states on co-doping were exhibited. Besides, the formation energy was also calculated, revealing the stability upon the introduction of the dopant to the anatase lattice. We found that most dopants reduced the bandgap, with a significant improvement for co-doped materials. The visible light response and the value of the absorption edge were determined from UV-Vis absorption spectra calculated at the HSE hybrid functional level together with the frequency-dependent dielectric function. The photocatalytic reaction of TiO2-based materials was investigated by determining the band edge position of modified materials with respect to the normal hydrogen electrode (NHE) potential for water splitting.
2. Computational details
In our work, spin-polarized DFT calculations were performed on pure and defected anatase using projector augmented plane wave methods for treating the valence electrons and their corresponding ionic cores25 as implemented in the VASP code.26,27 A grid cutoff energy of 560 eV and 2 × 2 × 2 Monkhorst–Pack k-point meshes were converged for the plane wave basis set and for sampling the first Brillouin zone, respectively.28 Gaussian smearing with a value of 0.1 eV was used for the Brillouin zone integrations during the total energy calculations. The doped systems were constructed from 3 × 3 × 1 anatase supercells corresponding to 108 atoms. The valence electron configurations in the PAW potentials of the considered elements were Ni 3d8 4s2, Se 4s2 4p4, B 2s2 2p1, Ti 3d3 4s1 and O 2s2 2p4. For the geometry optimization and formation energy calculations, the generalized gradient approximation (GGA) together with the Perdew, Burke, and Ernzerhof (PBE)29 exchange-correlation functional was employed. However, the GGA functional fails to produce accurate results, especially when addressing band characteristics such as bandgap values for the strongly correlated d and f electrons. Thus, to overcome this deficiency, a hybrid functional was used by introducing the Hartree–Fock exchange in the DFT calculations such as the screened hybrid HSE06 functional, which was applied to reproduce more effective band gap values comparable to the experimental values. Therefore, the Heyd–Scuseria–Ernzerhof (HSE) hybrid functional was used to evaluate the crystal structure and electronic properties.30–32 The screening parameter of 0.207 Å−1 was used as applied in the standard HSE06 functional.32 However, the exact exchange energy was optimized by fitting the calculated band gap to the experimental value of pure anatase by checking several values (25%, 22%, 20%, 18%, 17% and 15%), as performed in our recent work.33 Hereinafter, we refer to this functional as the HSE hybrid functional.3. Results and discussion
3.1. Dopant effect on the structural and electronic properties
For pure anatase TiO2, the calculated lattice parameters are a = 3.834 Å and c = 9.624 Å, which agree with the experimental lattice parameters of a = 3.784 Å and c = 9.502 Å.34 Upon the introduction of impurity into the anatase lattice, volume distortions were observed since the doped impurity has a different configuration and ionic radius from the replaced ions of the host lattice. The lattice parameters and cell volumes of the modified systems are listed in Table 1, and the relaxed structures corresponding to the mono-doped materials are shown in Fig. 1. For all the systems, the lattice parameters of the relaxed structures increased due to the incorporation of an impurity. The bandgap value of pure anatase calculated with the HSE hybrid functional was about 3.26 eV, which is consistent with the experimental value (3.2 eV) and the previous HSE hybrid functional calculations.35,36 The DOS and PDOS calculations for pure, mono-doped, and co-doped anatase TiO2 are shown in Fig. 2. The analysis of PDOS for pure anatase shows hybridization between the O 2p and Ti 3d states, indicating the covalent character of the formed Ti–O bond, as shown in Fig. 2a.33 In our work, the bandgap represents the energy difference between the valence band maximum (VBM) to the conduction band minimum (CBM). In contrast, the energy gap signifies the energy difference between the highest occupied state and the lowest unoccupied state. The electronic structure also changes upon TiO2 doping since new states appear in the bandgap region. The position of these gap states may affect the visible light response and the charge separation ability of modified TiO2. If these states are shallow, they act as trapping centers for charge carrier pairs, endowing TiO2 with longer life photogenerated charges. However, if the impurity states move high in the bandgap, they are deep states and can act as recombination centers for the electron/hole pairs, suppressing the photoactivity.37,38| System | Lattice parameter | Cell volume (Å3) | ΔV (Å3) | |
|---|---|---|---|---|
| a (Å) | c (Å) | |||
| Pure TiO2 | 3.835 (3.784) | 9.624 (9.502) | 1273.91 | — |
| Ni–TiO2 | 3.838 (3.789) | 9.631 (9.715) | 1276.78 | 2.78 |
| Se4+–TiO2 | 3.839 (3.760) | 9.633 (9.490) | 1277.62 | 3.71 |
| Se2−–TiO2 | 3.851 | 9.665 | 1290.13 | 16.22 |
| Bsub–TiO2 | 3.842 | 9.642 | 1280.83 | 6.92 |
| Bint–TiO2 | 3.846 | 9.652 | 1285.14 | 11.23 |
| Bint+sub–TiO2 | 3.851 | 9.664 | 1289.90 | 15.99 |
| Ni/B–TiO2 | 3.840 | 9.637 | 1278.91 | 5.00 |
| Ni/Bint+sub–TiO2 | 3.851 | 9.665 | 1290.33 | 16.42 |
| Se4+/B–TiO2 | 3.849 | 9.659 | 1287.71 | 13.80 |
| Se4+/Bint+sub–TiO2 | 3.864 | 9.699 | 1303.85 | 29.94 |
| Ni/Se4+–TiO2 | 3.838 | 9.631 | 1276.69 | 2.78 |
| Ni/Se2−–TiO2 | 3.851 | 9.663 | 1289.55 | 15.64 |
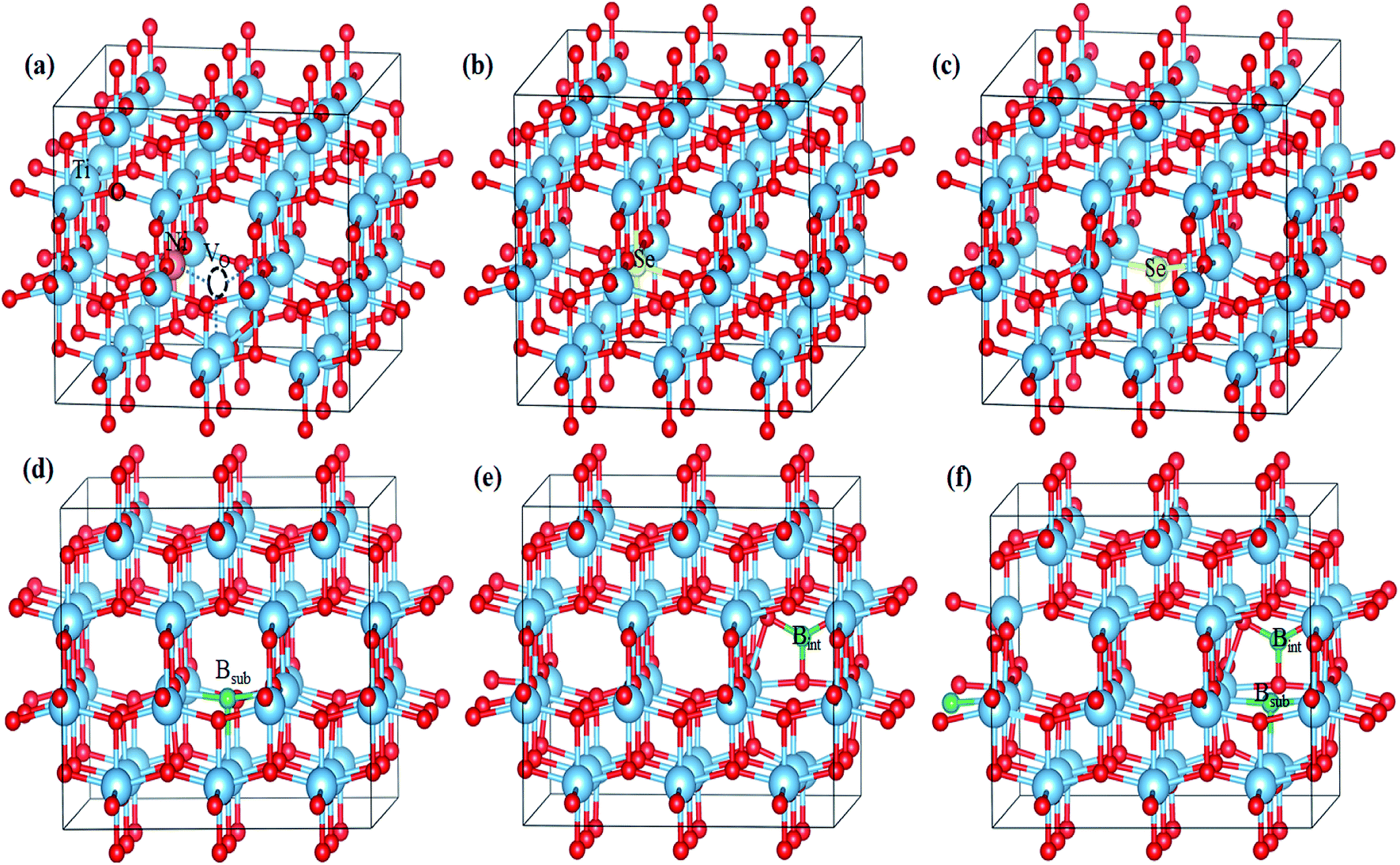 | ||
| Fig. 1 Optimized geometries of (a) Ni-doped, (b) Se4+-doped, (c) Se2−-doped, (d) Bsub-doped, (e) Bint-doped, and (f) Bint+sub-doped anatase TiO2 determined using the HSE hybrid functional level. | ||
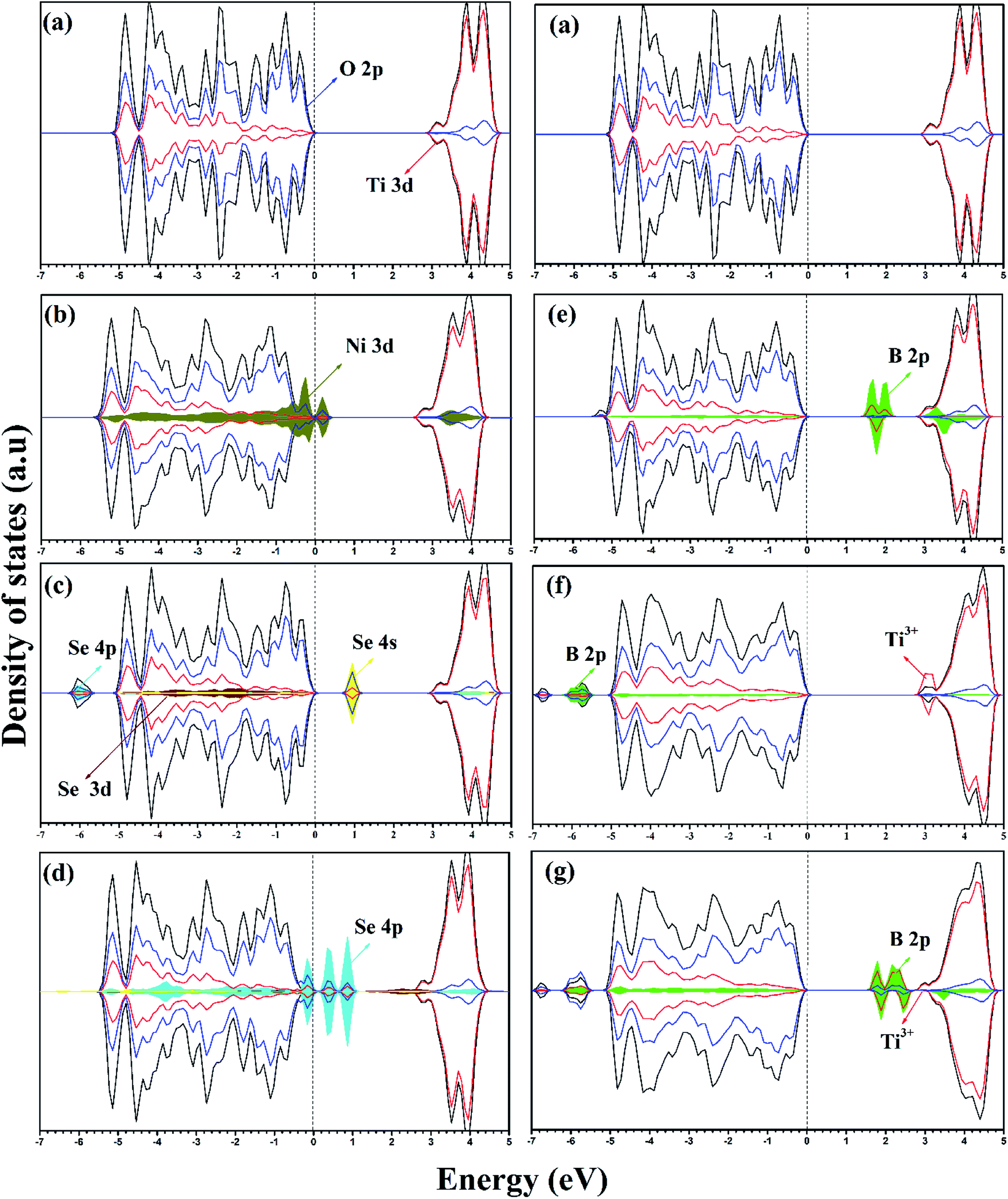 | ||
| Fig. 2 DOS and PDOS of (a) pure anatase TiO2, (b) Ni-doped, (c) Se4+-doped, (d) Se2−-doped, (e) Bsub-doped, (f) Bint-doped, and (g) Bint+sub-doped anatase TiO2 calculated using the HSE hybrid functional level. The valence band maximum is set at 0 eV. | ||
In the case of the mono-doped materials, the replacement of Ti4+ by Ni2+ leads to the formation of oxygen vacancy to preserve the charge neutrality, showing a stoichiometry of NixTi1−xO2−δ where (x = 0.028 and δ = 0.028), and the relaxed geometry is shown in Fig. 1a. The calculated Ni–O bond length is 2.06 Å, which is slightly longer than the reference Ti–O bond due to the ionic radii differences. The calculated PDOS of the Ni mono-doped anatase (Fig. 2b) shows hybridization between the Ni 3d and O 2p states, which confirms the existence of an Ni–O–Ti bond. The incorporation of Ni into the anatase TiO2 lattice preserves the diamagnetism of the system with a singlet spin state and a magnetic moment of 0 μB.39 Moreover, it can be observed that the Ni states are completely overlapped with the VBM, with some states located at 2.53 eV from the CBM. Therefore, the main bandgap is reduced to 2.89 eV with an energy gap of 2.53 eV, which is consistent with the previous results by Blanco-Vega et al.15 for Ni doped at small concentrations. Thus, based on the bandgap reduction, enhanced photoactivity towards visible light can be observed upon Ni mono-doping.
For Se-modified anatase, substitutional Se is doped at two different sites with two different stoichiometries.18 In one structure, Se4+ is substituted at the Ti4+ site with the formula of SexTi1−xO2, whereas Se2− at the oxygen site leads normally to TiO2−δSeδ. For the configuration with cationic Se, the obtained Se–O bond has a length of 2.00 Å, whereas the Ti–Se bond formed upon anionic Se doping of 2.30 Å is much longer. For both cases, a closed-shell singlet spin state was observed, corresponding to the diamagnetic properties of the formed structures. The DOS and PDOS for the cationic Se depicted in Fig. 2c shows a slight increase (0.03 eV) in the main bandgap value on the formation of the Se4+ defect; however, the energy gap originating from the incorporation of Se states in the bandgap region is about 2.18 eV. The energy gap calculated for Se4+ mono-doping is consistent with the previously observed value.11,40 The slight increase in the calculated main band gap was reported using the standard HSE06 functional.10 The calculated main bandgap value for two Se4+ doped anatase within the standard HSE06 functional was about 3.4 eV, whereas the calculated bandgap value of pure anatase was 3.3 eV.10 However, as shown in Table 2, Se4+ doping decreases the band gap of TiO2 experimentally.18 This can be attributed to the anatase to rutile phase transition on Se doping, where the anatase percentage was 50% at the calcination temperature of 650 °C.18 The deep states formed in the bandgap region is mainly due to the Se 4s states with a small contribution from the Se 4p states to the valence band (VB).41 However, the Se 4p contribution to the conduction band (CB) is noticeable. Furthermore, the Se 4s states are hybridized with the O 2p states, which gives an indication of the strength of the formed Se–O bond. The doped Se impacts the diamagnetic properties of the modified TiO2, which is associated with the closed shell singlet state, resulting in a 0 μB net magnetic moment. On the other hand, the anionic Se2−-modified anatase structure formed through oxygen substitution reveals different DOS and PDOS from the cationic doped material (Fig. 2d). The main bandgap is reduced to 3.18 eV, while the energy gap is narrowed to 1.88 eV by the Se2− impurity states in the forbidden region.10 The gap states are mainly due to the Se 4p states, which are separated by 2.34 and 1.88 eV from the CBM.41 Some of these states are overlapped with the VBM, while the other states are hybridized with the O 2p states. For both Se mono-doped cases, the electron transition is allowed from the induced bandgap states to the CB. Consequently, the photocatalytic activity of these materials under visible light is enhanced. Nevertheless, the charge separation efficiency may be affected by these deep states, and consequently the photocatalytic activity. Accordingly, it is worthwhile to co-dope these materials with another elements capable of improving the charge separation efficiency.
| System | Bandgap (eV) | Exp. bandgap | Energy gap (eV) | Formation energy (Ef) | |
|---|---|---|---|---|---|
| Ti-rich | O-rich | ||||
| Pure TiO2 | 3.26 | 3.20 | 3.26 | — | — |
| Ni–TiO2 | 2.89 | 2.72 | 2.53 | 4.579 | 0.118 |
| Se4+–TiO2 | 3.29 | 3.05 | 2.18 | 7.911 | −1.011 |
| Se2−–TiO2 | 3.18 | 1.88 | 1.155 | 5.616 | |
| Bsub–TiO2 | 3.21 | 1.04 | 3.873 | 8.334 | |
| Bint–TiO2 | 3.52 | 0.23 | 0.635 | 0.635 | |
| Bint+sub–TiO2 | 3.40 | 0.23 | −3.568 | 7.276 | |
| Ni/Bsub–TiO2 | 2.49 | 1.10 | 5.921 | 5.291 | |
| Ni/Bint+sub–TiO2 | 2.78 | 0.31 | 5.331 | 5.330 | |
| Se4+/Bsub–TiO2 | 2.98 | 1.23 | 10.088 | 5.627 | |
| Se4+/Bint+sub–TiO2 | 3.31 | 0.26 | 8.512 | 4.050 | |
| Ni/Se4+–TiO2 | 2.87 | 2.50 | 11.680 | −1.703 | |
| Ni/Se2−–TiO2 | 3.11 | 1.55 | 3.893 | 3.893 | |
The B-doped TiO2 was simulated by replacing B2− at the O2− site with the formula of TiO2−δBδ, and inserting B into the anatase lattice, leading to TiO2Bδ. The mixed interstitial-substitutional B system was also modeled through substitution of O2− by B from the anatase supercell with the incorporation of an additional B into the crystal system as TiO2−δB2δ. In the anatase lattice, Bsub is bound to three neighboring Ti ions with three bonds, i.e. two short (2.12 Å) and one long (2.39 Å) bonds (Fig. 1d).22 Boron added to the lattice has an odd number of valence electrons, indicating the paramagnetic properties of the neutral defect. In our study, we considered the doublet spin state, and thus the third electron is shared with a single Ti rather than bonding, explaining the longer bond length. The electrons occupy the hybrid B 2p–Ti 3d states localized deep in the bandgap region at about 1.34 and 1.04 eV for the spin up states and 1.22 eV for the spin down state below the conduction band minimum (CBM). The electronic transitions from these states can occur on visible light absorption; however, these deep states can suppress the carrier separation. Moreover, the main bandgap is slightly red shifted from the anatase bandgap, in agreement with the previous experimental observations.38,42 For interstitial B (Bint), it was reported that Bint may be tricoordinate [BO3] or tetracoordinate [BO4].22 However, the electronic characteristics of interstitial boron were rather independent of the site where the boron atom is incorporated.22 Therefore, we only considered a [BO3] structure in our work, as shown in Fig. 1e. The interstitial B atom is bonded to the three vicinal oxygen ions, where two bonds are similar (1.4 Å) and the third is shorter (1.376 Å). In this case, the boron exhibits a trigonal planer coordination.26 According to the PDOS, as shown in Fig. 2f, it is observed that no states due to boron contribution are formed in the bandgap, while the B 2p states are located below the valence band.22 The states incorporated and overlapped with the CBM are mainly due to the Ti3+ ions, which can trap the photo-generated carriers and enlarge the lifetime of these carries.43 This result indicates that the B electrons are partially delocalized and consequently donated to the lattice forming B3+.22 Despite the Ti3+ states, the main bandgap is blue shifted upon the introduction of interstitial boron, and no visible light response is detected for that system.23,44 The blue-shifted bandgap is attributed to the existence of BOx, which has a high bandgap compared to pure anatase.22,45 The situation for Bint+sub is different since the concentration of B in the lattice is increased by the introduction of two B atoms, i.e. one at an interstitial position and the other at the substitutional position, replacing an oxygen ion from the lattice, as shown in Fig. 1f. With this coupling, the bond lengths change from the mono-doped case. The three bonds surrounding the interstitial B are equal (1.379 Å), whereas the bonds around the substitutional B are still two short and one long bond (2.07 and 2.12 Å), respectively. The PDOS of Bint+sub (Fig. 2g) shows that the gap states move high in the bandgap towards the CB. Moreover, two of the shallow levels seen in Bint almost disappear in Bint+sub by the interaction between Bint and Bsub with overlap between their states. However, one shallow state due to Ti3+ is still observed below the CB, which may be associated with enhanced electron trapping. The main bandgap is slightly red shifted from Bint, but still higher than the anatase bandgap.
On co-doping, the Ni/Bsub, Ni/Bint+sub, Se4+/Bsub, Se4+/Bint+sub, Ni/Se2−, and Ni/Se4+ co-doped systems are associated with NixTi1−xO2−2δBδ, NixTi1−xO2−2δB2δ, SexTi1−xO2−δBδ, SexTi1−xO2−δB2δ, NixTi1−xO2−2δSeδ, and NixSexTi1−2xO2−δ, respectively. For all the co-doped materials, x = 0.028 and δ = 0.028 if they exist. For the co-doping models, different configurations were considered by distributing the dopants (adjacent and separated) in the supercell. It was found that the formation of adjacent dopants is more energetically favorable with respect to other configurations.46 Therefore, the lowest energy structures were selected to examine their electronic and optical properties and their catalytic activity (see ESI† for fractional coordinates). Ni/Bsub co-doping exhibits different ionic features from the mono-doped materials. The PDOS for Ni/Bsub shows an interaction between the two dopants, and overlapping is observed between the Ni 3d and B 2p states due to the bond formed between Ni and B. Also, the Ni 3d states are completely overlapped with the VBM, giving rise to bandgap narrowing of (2.49 eV); however, the B states appear deep in the bandgap region and separated by 1.10 eV from the CBM. Upon geometrical optimization of the Ni/BSub co-doped material, the local magnetic moment over the Ni ion becomes 0.843 μB; however, the magnetic moment over the whole lattice is 1.02 μB. In the Ni/Bint+sub co-doped system, an interaction exists between Ni and boron at the VBM and the total magnetic moment is 1.886 μB, while the local magnetic moment on Ni ion is 1.182 μB. Herein, the Ni 3d states are still overlapped with the VBM; however, most of the B states are found deep in the bandgap (Fig. 3b). Moreover, there are states with shallow character, corresponding to the formation of Ti3+. Due to these impurity states, many transitions can be observed upon visible light illumination and the charge carrier separation can be enhanced by the existing shallow states, in addition to the red shift in the calculated main bandgap (2.78 eV).
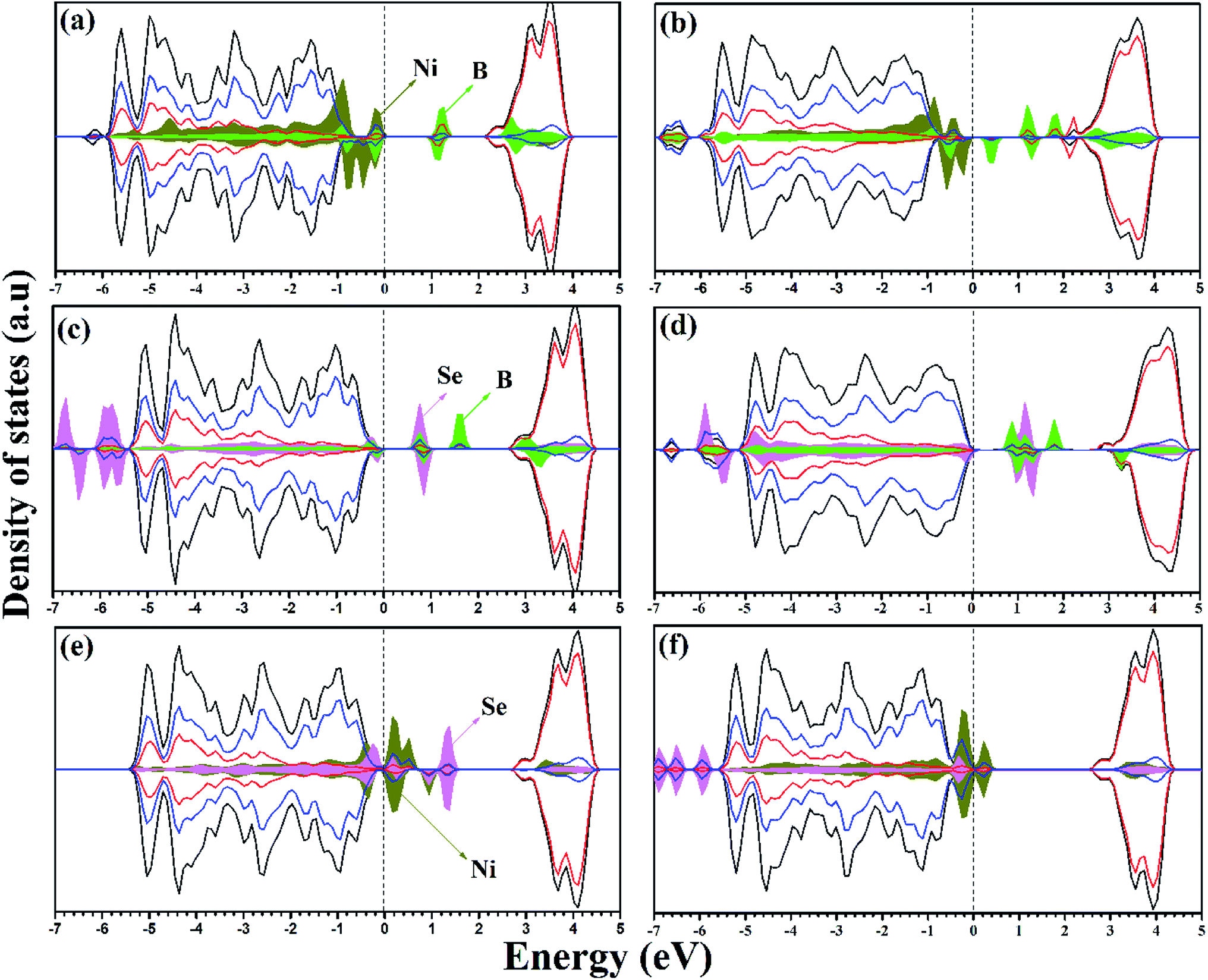 | ||
| Fig. 3 DOS and PDOS of (a) Ni/Bsub co-doped, (b) Ni/Bint+sub co-doped, (c) Se4+/Bsub co-doped, (d) Se4+/Bint+sub co-doped, (e) Ni/Se2− co-doped, and (f) Ni/Se4+ co-doped anatase TiO2 calculated using the HSE hybrid functional level. The valence band maximum is set at 0 eV. | ||
In the Se/Bsub co-doped anatase lattice, the PDOS and DOS is illustrated in Fig. 3c. The Se-occupied states can be observed at about 0.91 eV above the VBM, which overlap with the B states due to Se–B bond formation, while a separated B state is introduced in the bandgap at about 1.23 eV from CBM, indicating the allowed electron transitions on visible light irradiation. Moreover, the band gap is narrowed to 2.98 eV with a total magnetic moment of 0.67 μB for the whole system. The deep B states provide centers from which electrons can be excited on light absorption. Therefore, the charge carrier population increases; however, they may act as recombination centers for these carriers. Thus, the photocatalytic activity of Se4+/B is enhanced by the response to visible light, although the lifetime of the photo-produced carriers may be affected. Se/Bint+sub displays different electronic structures, and its calculated DOS and PDOS are illustrated in Fig. 3d. Similar to Ni/Bint+sub, the interaction between Se and B increases upon the incorporation of interstitial B. Moreover, the deep states due to substitutional B observed on Se/Bsub co-doping are placed in the forbidden gap at about 1.41 eV from the CBM on Se/Bint+sub co-doping. In addition, a shallow Ti3+ state appears, which is overlapped with the CBM. The calculated DOS analysis indicates that the bandgap is slightly blue shifted (3.31 eV); however, the visible light response can be enhanced by the transitions from the previously mentioned bandgap states. In addition, the shallow state can prolong the lifetime of the carriers. Thus, Se/Bint+sub-modified anatase TiO2 can be suggested as an enhanced photocatalyst.
Upon Ni/Se2− co-doping, the calculated bandgap is about 3.11 eV with the impurity states overlapping with the VBM in addition to the bandgap states separated by about 1.55 eV from the CBM. The PDOS with this co-doping shows a higher contribution of Ni 3d and Se states to the VB, as shown in Fig. 3e. According to the DOS calculations, it can be confirmed that the Se states are overlapped with the top of the VB, and the deep states exited in the bandgap are mainly due to the Se 4p states. Therefore, the Se2−, states which are either overlapped with Ni or isolated at about 1.56 eV from the VBM in Ni/Se2−, are due to the contribution of the Se 4p states. In the second case, Ni/Se4+ is co-doped as dual metal doping by the substitution of two close Ti ions. According to the DOS and PDOS shown in Fig. 3f, a state is observed at 2.50 eV from the CBM, indicating that more carriers are generated on visible light absorption. The calculated band gap is about 2.87 eV, which is higher than the case of Ni/Se2− co-doping, but still lower than pure TiO2, revealing a better response to visible light. For both materials (Ni/Se4+ and Ni/Se2−), the photocatalytic activity under visible light is improved due to the bandgap reduction and higher number of photogenerated carriers, but the deep states may affect the stability of the photogenerated carriers.
3.2. Formation energy calculations
To reveal the stability and possibility of defect doping in the anatase lattice, the defect formation energy (Ef) of the modified systems was calculated using the following equation:| Ef = Edoped − Eundoped − (nMμM + nN−MμN−M) + (nOμO + nTiμTi) | (1) |
The lower the formation energy of defects, the more likely they are formed. Under O-rich conditions, the formation energy of metal mono-doping is lower than that of non-metal mono-doping. On the other hand, Se2− or B doping shows higher stability under Ti-rich conditions. Under both conditions, the formation energy is higher for Bsub than Bint and Bint+sub, which can verify the previously mentioned discussion of the metastable character of substitutional boron.22,33 The Bint+sub defect formation energy is the lowest value (−3.568 eV), suggesting that B at high concentration tends to occupy both the interstitial and substitutional sites.
The calculated formation energy for the co-doped defects demonstrates that defect co-doping is more facilitated under O-rich than Ti-rich conditions. However, the values for all the systems are more positive than the mono-doped cases except for Ni/Se4+, which exhibits a higher negative formation energy than Ni or Se4+ mono-doping. Moreover, Ni/Bsub, Ni/Bint+sub and Ni/Se2− co-doping and Bint mono-doping show the same value of formation energy under both conditions, indicating the independence of formation from the growth conditions.
3.3. Dopant effect on the optical properties
The linear optical properties can be obtained depending on the frequency-dependent dielectric function: ε(ω) = ε1(ω) + iε2(ω), where ε1and ε2 represent the real and imaginary parts of the dielectric function, respectively, and ω is the photon frequency. The imaginary part ε2(ω) can be calculated from the momentum matrix element between the VB and CB wavefunctions, whereas the real part ε1(ω) can be obtained from the Kramers–Krönig relationship.47 The UV-Vis optical absorption can be calculated by determining the optical absorption coefficient α(ω) for each material (in cm−1) as a function of the wavelength of the incident light according to the following equation:48
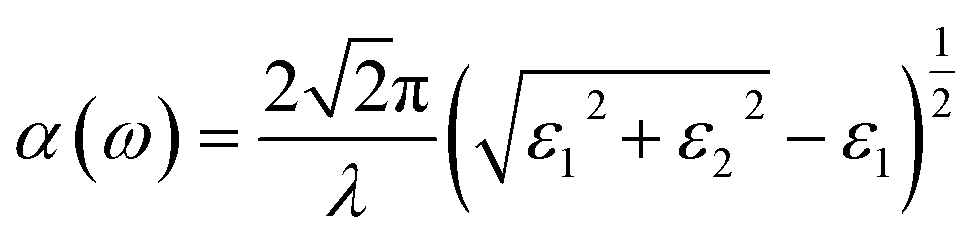 | (2) |
The UV-Vis optical absorption spectrum of anatase TiO2 is illustrated in Fig. 4a. The calculated bandgap for pristine anatase TiO2 is about 3.26 eV, which is about 0.06 eV higher compared to the experimental value.33 Therefore, all the anatase TiO2-based applications are limited to UV light, which contributes about ∼5% to solar irradiation. The spectra of the mono-doped and co-doped materials over pure anatase TiO2 are depicted in Fig. 4a and b, respectively. For all the materials, the absorption edge is red shifted considering the reference spectrum of pure TiO2 except for the Bint-doped materials. For the Ni mono-doped system, the light response extends up to about 2.5 eV, whereas the absorption edges are red shifted to about 1.6 and 2.2 eV for Se2− and Se4+ mono-doping, respectively. In B mono-doping, three different spectra for the three configurations Bsub, Bint and Bint+sub are observed. For Bsub, the absorption edge is red shifted to about 1.04 eV with a high absorption coefficient in the visible light region besides the absorption due to the main bandgap transition shown at about 3.18 eV. Bint and Bint+sub show different trends, where the absorption edge for both is blue shifted with absorption coefficients in the IR region, which are consistent with the states observed below the CB in the DOS.
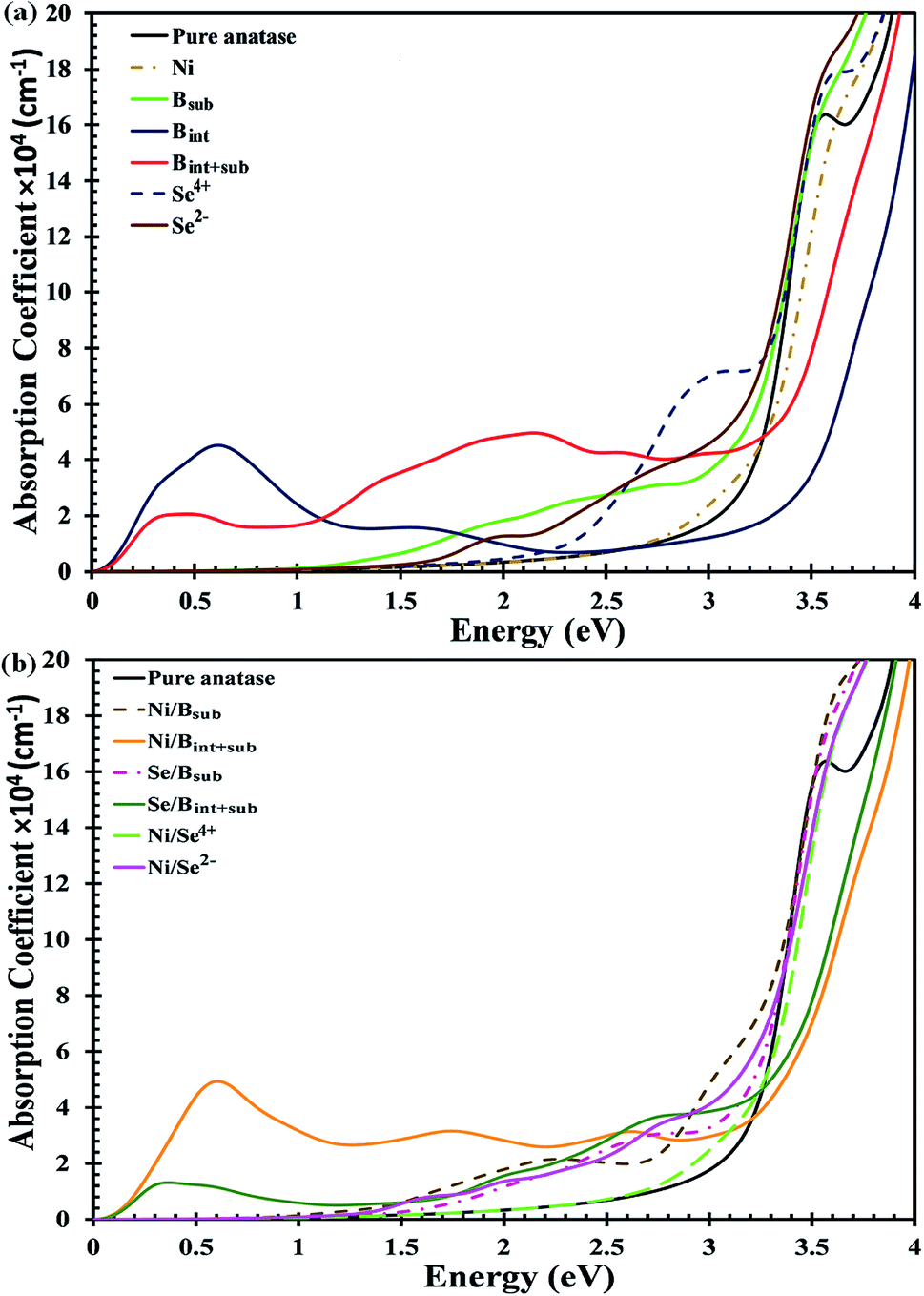 | ||
| Fig. 4 Calculated absorption coefficients with HSE hybrid functional for (a) mono-doped, and (b) co-doped anatase TiO2 materials. | ||
Upon co-doping, all the co-doped materials show an extended visible light response, indicating improved photoactivity over mono-doped materials. Ni/Bint+sub co-doping exhibits high absorption efficiency, indicating a high population of photogenerated charge carriers in addition to the red-shifted absorption due to the main bandgap excitations. Moreover, intense peaks are detected in the IR region, which confirm the transition from the shallow states located directly below the CB. The main absorption peak for Ni/Bsub co-doping is formed at about 2.5 eV. In addition, several peaks appear in the visible light region of the spectrum, but with a higher absorption coefficient than that of Ni/Bint+sub. Upon Se/Bsub co-doping, several absorptions can be observed in the visible light region with no absorption in the IR region as a result of the transitions from the deep states to the CB. Se/Bint+sub co-doping also shows visible light absorption, but with higher absorption coefficients than that of Se/Bsub co-doping. Additionally, a high intensity peak appears in the IR region, confirming the shallow state electron excitation expected from the DOS results. For Ni/Se co-doped anatase, the absorption corresponding to the bandgap excitations is red shifted to visible light for both the Ni/Se4+ and Ni/Se2− co-doped materials with energy values of about 2.9 and 3.1 eV, respectively. In addition, various peaks are shown for Ni/Se2− as a result of the electron transitions from the forbidden gap states to the CB.
Consequently, the visible light response is enhanced for all the materials except Bint-modified anatase. However, the absorption efficiency is better for the Se4+ mono-doped and Ni/Bsub and Se/Bint+sub co-doped systems, revealing the strength of these materials as effective photocatalysts under visible light.
3.4. Relative band position for water splitting
Thermodynamically, the photocatalytic reaction of adsorbed species is governed by the position of the edges of the valence band (VBE) and conduction band (CBE) of semiconducting photocatalysts. For water splitting and fuel production or the degradation of organic materials, there are standard conditions that the relevant potential of the acceptor species should lie below the CBE of the photocatalyst (more positive), whereas the potential level of the donor species should be located above the VBE (more negative).50Accordingly, the photocatalytic activity of the modified anatase TiO2 was evaluated for water splitting as an application for H2 production. The values of the VBE and CBE positions of pure anatase were calculated with respect to the normal hydrogen electrode (NHE) potential.5,6,51 For the doped system, the positions of edges of the valence and conduction bands were detected from the DOS analysis depending on their relative positions compared to pure anatase TiO2.10 The calculations for pure anatase TiO2 shows that the VBE is located at 2.94 eV, while the CBE position is 0.32 eV more negative than the reduction potential of H+/H2, which agrees with the previous experimental values.51 As shown in Fig. 5, the CBE position of the Se2− mono-doped system is almost similar to that of TiO2, whereas the VBE is shifted up by about 1.39 eV over TiO2 (about 0.32 eV more positive than the O2/H2O potential). This reveals that the reduction ability of H+ is maintained for Se2− mono-doped TiO2 with an increased tendency to release oxygen. In contrast, the CBE positions for the Ni and Se4+ mono-doped and Ni/Se4+ co-doped systems are shifted up by about 0.02, 0.08, and 0.08 eV over TiO2, respectively indicating that the reduction ability of H+ remains possible. In addition, their VBE positions are about 0.96, 0.55 and 0.87 eV more positive than the oxidation potential of O2/H2O, respectively, revealing that the ability to release oxygen is strong. Therefore, all the modified anatase materials, as shown in Fig. 5, can be candidates for water splitting and hydrogen production except for the Bsub mono-doped and NiSe2− co-doped systems. In the case of Bsub and Ni/Se2−, the CBE is about 0.30 and 0.34 eV more negative than the reduction potential of H+/H2; however, the VBE is about 0.49 and 0.02 eV more negative than the oxidation potential of O2/H2O, respectively. This means that the reduction ability of H+ ions is improved for Bsub and remains possible for Ni/Se2−, while the VBE position is unsuitable for the oxygen evolution reaction.
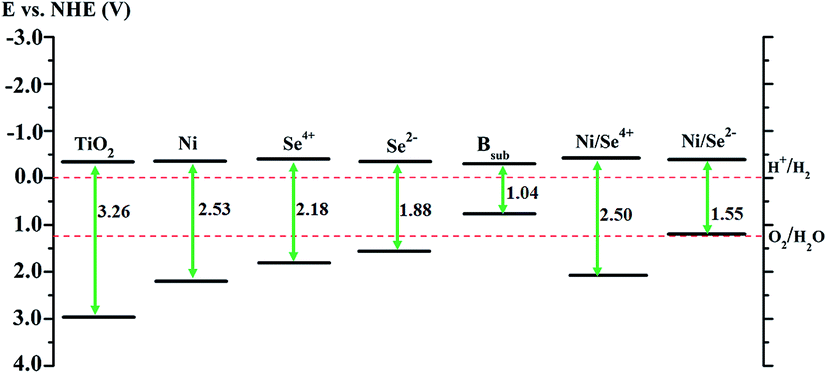 | ||
| Fig. 5 Calculated band positions of mono- and co-doped TiO2 anatase materials. The values are with respect to the NHE potential in (V). | ||
4. Conclusion
In summary, the screened coulomb hybrid HSE functional was used to study the structural, electronic and optical properties of modified anatase TiO2. It was found that the bandgaps are narrowed for all the systems except for the Bint, Bint+sub, and Se4+ mono-doped and Se/Bint+sub co-doped materials with the smallest bandgap calculated for Ni/Bsub (2.49 eV). For all the mono- and co-doped materials, the calculated DOS showed new states in the forbidden gap by the incorporation of the dopant into the lattice. The electrons occupying these states are the reason for the enhanced photoactivity of modified anatase under visible light irradiation. The reduction in the bandgaps is associated with the red shift observed for the absorption edge on the Ni, Se2−, Bsub mono-doped and Ni/Bsub, Ni/Bint+sub, Se/Bsub, Ni/Se4+, Ni/Se2− co-doped systems. The optical response was explored from the absorption coefficient calculations, which indicated a red-shifted absorption edge for all the modified anatase materials. The relative band position determination showed that the Ni, Se4+ and Se2− mono-doped in addition to Ni/Se4+ co-doped materials are expected to be candidates for water splitting and hydrogen production. In contrast, the Ni/Bint+sub and Se/Bint+sub co-doped photocatalysts can be used for other energy applications with a visible light response and prolonged lifetime of photogenerated carriers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The calculations were performed using computational resources provided by the Bibliotheca Alexandrina on its High-Performance Computing (HPC) infrastructure.References
- K. Li, B. Peng and T. Peng, Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels, ACS Catal, 2016, 6(11), 7485–7527, DOI:10.1021/acscatal.6b02089.
- C. Byrne, G. Subramanian and S. C. Pillai, Recent Advances in Photocatalysis for Environmental Applications, J. Environ. Chem. Eng., 2018, 6(3), 3531–3555, DOI:10.1016/j.jece.2017.07.080.
- A. Miyoshi, S. Nishioka and K. Maeda, Water Splitting on Rutile TiO2-Based Photocatalysts, Chem.–Eur. J., 2018, 24, 18204–18219, DOI:10.1002/chem.2018007990.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, One-Dimensional TiO2 Nanotube Photocatalysts for Solar Water Splitting, Adv. Sci., 2017, 4(1), 1–31, DOI:10.1002/advs.201600152.
- M. M. Fadlallah and U. Eckern, Cation Mono- and Co-Doped Anatase TiO2 Nanotubes: An Ab Initio Investigation of Electronic and Optical Properties, Phys. Status Solidi Basic Res., 2020,(1), 257, DOI:10.1002/pssb.201900217.
- M. Fadlallah and U. Eckern, Electronic and Optical Properties of Metal-Doped TiO2 Nanotubes: Spintronic and Photocatalytic Applications, New J. Phys, 2020, 22, 093028, DOI:10.1088/1367-2630/abae87.
- J. Liu, M. Weng, S. Li, X. Chen, J. Cen, J. Jie, W. Xiao, J. Zheng and F. Pan, High-Throughput HSE Study on the Doping Effect in Anatase TiO2, Phys. Chem. Chem. Phys., 2020, 22(1), 39–53, 10.1039/c9cp04591k.
- D. K. Božanić, G. A. Garcia, L. Nahon, D. Sredojević, V. Lazić, I. Vukoje, S. P. Ahrenkiel, V. Djoković, Ž. Šljivančanin and J. M. Nedeljković, Interfacial Charge Transfer Transitions in Colloidal TiO2 Nanoparticles Functionalized with Salicylic Acid and 5-Aminosalicylic Acid: A Comparative Photoelectron Spectroscopy and DFT Study, J. Phys. Chem. C, 2019, 123(47), 29057–29066, DOI:10.1021/acs.jpcc.9b10110.
- I. Ganesh, A. K. Gupta, P. P. Kumar, P. S. C. Sekhar, K. Radha, G. Padmanabham and G. Sundararajan, Preparation and Characterization of Ni-Doped Materials for Photocurrent and Photocatalytic Applications, Sci. World J., 2012, 2012, 1–16, DOI:10.1100/2012/127326.
- M. Harb, New Insights into the Origin of Visible-Light Photocatalytic Activity in Se-Modified Anatase TiO2 from Screened Coulomb Hybrid DFT Calculations, J. Phys. Chem. C, 2013, 117(48), 25229–25235, DOI:10.1021/jp406714e.
- W. Xie, R. Li and Q. Xu, Enhanced Photocatalytic Activity of Se-Doped TiO2 under Visible Light Irradiation, Sci. Rep., 2018, 8(1), 1–10, DOI:10.1038/s41598-018-27135-4.
- C. Amritha, S. Kurian, T. Raguram and K. S. Rajni, Structural, Optical, Functional, Morphological and Compositional Analysis of Ni-Doped TiO2 Nanofibers Prepared by Electrospinning Technique, IOP Conf. Ser. Mater. Sci. Eng., 2019, 577(1), 012073, DOI:10.1088/1757-899x/577/1/012073.
- L. B. Patle, V. R. Huse and A. L. Chaudhari, Band Edge Movement and Structural Modifications in Transition Metal Doped TiO2 Nanocrystals for the Application of DSSC, Mater. Res. Express, 2017, 4(10), 105045, DOI:10.1088/2053-1591/aa919a.
- M. Shaban, A. M. Ahmed, N. Shehata, M. A. Betiha and A. M. Rabie, Ni-Doped and Ni/Cr Co-Doped TiO2 Nanotubes for Enhancement of Photocatalytic Degradation of Methylene Blue, J. Colloid Interface Sci., 2019, 555, 31–41, DOI:10.1016/j.jcis.2019.07.070.
- M. P. Blanco-Vega, J. L. Guzmán-Mar, M. Villanueva-Rodríguez, L. Maya-Treviño, L. L. Garza-Tovar, A. Hernández-Ramírez and L. Hinojosa-Reyes, Photocatalytic Elimination of Bisphenol A under Visible Light Using Ni-Doped TiO2 Synthesized by Microwave Assisted Sol-Gel Method, Mater. Sci. Semicond. Process., 2017, 71, 275–282, DOI:10.1016/j.mssp.2017.08.013.
- L.-L. Lai, W. Wen and J.-M. Wu, Ni-Doped Rutile TiO2 Nanoflowers: Low-Temperature Solution Synthesis and Enhanced Photocatalytic Efficiency, RSC Adv, 2016, 6(30), 25511–25518, 10.1039/c6ra01752e.
- M. R. Elahifard, S. Ahmadvand and A. Mirzanejad, Effects of Ni-Doping on the Photo-Catalytic Activity of TiO2 Anatase and Rutile: Simulation and Experiment, Mater. Sci. Semicond. Process., 2018, 84, 10–16, DOI:10.1016/j.mssp.2018.05.001.
- S. Mathew, P. Ganguly, V. Kumaravel, J. Harrison, S. J. Hinder, J. Bartlett and S. C. Pillai, Effect of Chalcogens (S, Se, and Te) on the Anatase Phase Stability and Photocatalytic Antimicrobial Activity of TiO2, Mater. Today Proc., 2020, 1–7, DOI:10.1016/j.matpr.2020.01.336.
- W.-K. Wang, J.-J. Chen, M. Gao, Y.-X. Huang, X. Zhang and H.-Q. Yu, Photocatalytic Degradation of Atrazine by Boron-Doped TiO2 with a Tunable Rutile/Anatase Ratio, Appl. Catal. B Environ., 2016, 195, 69–76, DOI:10.1016/j.apcatb.2016.05.009.
- R. Jaiswal, N. Patel, A. Dashora, R. Fernandes, M. Yadav, R. Edla, R. S. Varma, D. C. Kothari, B. L. Ahuja and A. Miotello, Efficient Co-B-Codoped TiO2 Photocatalyst for Degradation of Organic Water Pollutant under Visible Light, Appl. Catal. B Environ., 2016, 183, 242–253, DOI:10.1016/j.apcatb.2015.10.041.
- B. Wang, F. Zhao, G. Du, S. Porter, Y. Liu, P. Zhang, Z. Cheng, H. K. Liu and Z. Huang, Boron-Doped Anatase TiO2 as a High-Performance Anode Material for Sodium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8(25), 16009–16015, DOI:10.1021/acsami.6b03270.
- E. Finazzi, C. Di Valentin and G. Pacchioni, Boron-Doped Anatase TiO2 : Pure and Hybrid DFT Calculations, J. Phys. Chem. C, 2009, 113(1), 220–228, DOI:10.1021/jp8072238.
- C. Sun and D. J. Searles, Origin of the Visible Light Absorption of Boron/Nitrogen Co-Doped Anatase TiO2, J. Phys. Chem. C, 2013, 117(50), 26454–26459, DOI:10.1021/jp408858u.
- M. Masae, L. Sikong, K. Kooptarnond, W. Taweepreda and F. Saito, Photoactivity and Hydrophilicity of B and Ni Co-Doped TiO2 Films, Adv. Mater. Res., 2011, 148–149, 1473–1479, DOI:10.4028/www.scientific.net/amr.148-149.1473.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979, DOI:10.1103/physrevb.50.17953.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/physrevb.54.11169.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188–5192, DOI:10.1103/physrevb.13.5188.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/physrevlett.77.3865.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid Functionals Based on a Screened Coulomb Potential, J. Chem. Phys., 2003, 118(18), 8207–8215, DOI:10.1063/1.1564060.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)], J. Chem. Phys., 2006, 124(21), 219906, DOI:10.1063/1.2204597.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals, J. Chem. Phys., 2006, 125(22), 224106, DOI:10.1063/1.2404663.
- H. H. Ibrahim, A. A. Mohamed and I. A. M. Ibrahim, Electronic and Optical Properties of Mono and Co-Doped Anatase TiO2: First Principles Calculations, Mater. Chem. Phys., 2020, 252, 123285, DOI:10.1016/j.matchemphys.2020.123285.
- J. K. Burdett, T. Hughbanks, G. J. Miller, J. V. Smith and J. W. Richardson, Structural-Electronic Relationships in Inorganic Solids: Powder Neutron Diffraction Studies of the Rutile and Anatase Polymorphs of Titanium Dioxide at 15 and 295 K, J. Am. Chem. Soc., 1987, 109(12), 3639–3646, DOI:10.1021/ja00246a021.
- M. Niu, D. Cheng and D. Cao, Understanding Photoelectrochemical Properties of B–N Codoped Anatase TiO2 for Solar Energy Conversion, J. Phys. Chem. C, 2013, 117(31), 15911–15917, DOI:10.1021/jp4038792.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, Visible-Light Activation of TiO2 Photocatalysts: Advances in Theory and Experiments, J. Photochem. Photobiol. C Photochem. Rev., 2015, 25, 1–29, DOI:10.1016/j.jphotochemrev.2015.08.003.
- X. Li, J. Shi, H. Chen, R. Wan, C. Leng, S. Chen and Y. Lei, A DFT Study on the Modification Mechanism of (Cr, C) Co-Doping for the Electronic and Optical Properties of Anatase TiO2, Comput. Mater. Sci., 2017, 129, 295–303, DOI:10.1016/j.commatsci.2016.12.029.
- S. Bagwasi, B. Tian, J. Zhang and M. Nasir, Synthesis, Characterization and Application of Bismuth and Boron Co-Doped TiO2: A Visible Light Active Photocatalyst, Chem. Eng. J., 2013, 217, 108–118, DOI:10.1016/j.cej.2012.11.080.
- H. Peng, J. Li, S.-S. Li and J.-B. Xia, First-Principles Study of the Electronic Structures and Magnetic Properties of 3d Transition Metal-Doped Anatase TiO2, J. Phys. Condens. Matter, 2008, 20(12), 125207, DOI:10.1088/0953-8984/20/12/125207.
- E. M. Rockafellow, J. M. Haywood, T. Witte, R. S. Houk and W. S. Jenks, Selenium-Modified TiO2 and Its Impact on Photocatalysis, Langmuir, 2010, 26(24), 19052–19059, DOI:10.1021/la1026569.
- J. W. Zheng, A. Bhattcahrayya, P. Wu, Z. Chen, J. Highfield, Z. Dong and R. Xu, The Origin of Visible Light Absorption in Chalcogen Element (S, Se, and Te)-Doped Anatase TiO2 Photocatalysts, J. Phys. Chem. C, 2010, 114(15), 7063–7069, DOI:10.1021/jp9115035.
- W. Zhao, W. Ma, C. Chen, J. Zhao and Z. Shuai, Efficient Degradation of Toxic Organic Pollutants with Ni2O3/TiO2−xBx under Visible Irradiation, J. Am. Chem. Soc., 2004, 126(15), 4782–4783, DOI:10.1021/ja0396753.
- C. Fu, Y. Gong, Y. Wu, J. Liu, Z. Zhang, C. Li and L. Niu, Photocatalytic Enhancement of TiO2 by B and Zr Co-Doping and Modulation of Microstructure, Appl. Surf. Sci., 2016, 379, 83–90, DOI:10.1016/j.apsusc.2016.03.192.
- K. Yang, Y. Dai and B. Huang, Origin of the Photoactivity in Boron-Doped Anatase and Rutile TiO2 Calculated from First Principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76(19), 195201, DOI:10.1103/physrevb.76.195201.
- W.-K. Wang, J.-J. Chen, M. Gao, Y.-X. Huang, X. Zhang and H.-Q. Yu, Photocatalytic Degradation of Atrazine by Boron-Doped TiO2 with a Tunable Rutile/Anatase Ratio, Appl. Catal. B Environ., 2016, 195, 69–76, DOI:10.1016/j.apcatb.2016.05.009.
- R. Long and N. J. English, Synergistic Effects on Band Gap-Narrowing in Titania by Codoping from First-Principles Calculations, Chem. Mater., 2010, 22(5), 1616–1623, DOI:10.1021/cm903688z.
- M. Gajdoš, K. Hummer, G. Kresse, J. Furthmüller and F. Bechstedt, Linear Optical Properties in the Projector-Augmented Wave Methodology, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(4), 1–9, DOI:10.1103/physrevb.73.045112.
- S. Lardhi, D. Noureldine, M. Harb, A. Ziani, L. Cavallo and K. Takanabe, Determination of the Electronic, Dielectric, and Optical Properties of Sillenite Bi12TiO20 and Perovskite-like Bi4Ti3O12 Materials from Hybrid First-Principle Calculations, J. Chem. Phys., 2016, 144(13), 134702, DOI:10.1063/1.4945344.
- V. Wang, W. Xiao, D.-M. Ma, R.-J. Liu and C.-M. Yang, Structural, Electronic, and Optical Properties of GaInO3 : A Hybrid Density Functional Study, J. Appl. Phys., 2014, 115(4), 043708, DOI:10.1063/1.4863210.
- A. L. Linsebigler, G. Lu and J. T. Yates, Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results, Chem. Rev., 1995, 95(3), 735–758, DOI:10.1021/cr00035a013.
- Y. Xu and M. A. A. Schoonen, The Absolute Energy Positions of Conduction and Valence Bands of Selected Semiconducting Minerals, Am. Mineral., 2000, 85(3–4), 543–556, DOI:10.2138/am-2000-0416.
Footnote |
| † Electronic supplementary information (ESI) available: Fractional coordinates of the lowest energy co-doped structures. See DOI: 10.1039/d0ra07781j |
| This journal is © The Royal Society of Chemistry 2020 |