
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando soft X-ray absorption spectroscopic study on microporous carbon-supported sulfur cathodes†
Yao Xiaoa,
Kentaro Yamamoto *a,
Yukiko Matsuib,
Toshiki Watanabea,
Koji Nakanishia,
Tomoki Uchiyamaa,
Shoso Shingubarac,
Masashi Ishikawab,
Masayoshi Watanabed and
Yoshiharu Uchimotoa
*a,
Yukiko Matsuib,
Toshiki Watanabea,
Koji Nakanishia,
Tomoki Uchiyamaa,
Shoso Shingubarac,
Masashi Ishikawab,
Masayoshi Watanabed and
Yoshiharu Uchimotoa
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida-nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yamamoto.kentaro.4e@kyoto-u.ac.jp
bDepartment of Chemistry and Materials Engineering, Kansai University, 3-3-35 Yamate-cho, Suita, Osaka 564-8680, Japan
cDepartment of Mechanical Engineering, Kansai University, 3-3-35 Yamate-cho, Suita, Osaka 564-8680, Japan
dInstitute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
First published on 2nd November 2020
Abstract
Sulfur is a promising material for next-generation cathodes, owing to its high energy and low cost. However, sulfur cathodes have the disadvantage of serious cyclability issues due to the dissolution of polysulfides that form as intermediate products during discharge/charge cycling. Filling sulfur into the micropores of porous carbon is an effective method to suppress its dissolution. Although microporous carbon-supported sulfur cathodes show an electrochemical behavior different from that of the conventional sulfur ones, the corresponding reaction mechanism is not clearly understood. In this study, we focused on clarifying the reaction mechanism of microporous carbon-supported sulfur cathodes by operando soft X-ray absorption spectroscopy. In the microporous carbon support, sulfur was present as smaller fragments compared to conventional sulfur. During the first discharge process, the sulfur species in the microporous carbon were initially reduced to S62− and S22− and then to Li2S. The S62− and S22− species were observed first, with S22− being the main polysulfide species during the discharge process, while Li2S was produced in the final discharge process. The narrow pores of microporous carbon prevent the dissolution of polysulfides and influence the reaction mechanism of sulfur cathodes.
1. Introduction
With the rapid development of electronic equipment and energy-storage devices, the demand for high-energy storage systems has become increasingly urgent and important. Sulfur cathodes have received much attention because of their high theoretical specific capacity (1672 mA h g−1), low cost, high availability, and environment-friendliness.1–4 Therefore, sulfur cathodes are considered as promising components of next-generation energy-storage systems.5–8 However, current sulfur cathodes possess many disadvantages that prevent their rapid development. One of the most serious disadvantages is the high solubility of lithium polysulfides (Li2Sn, n = 4–8) formed during the charging and discharging processes.9–11 The dissolution of polysulfides leads to a low coulombic efficiency and rapid capacity degradation of batteries.12–14Filling sulfur into microporous carbon is a promising method to overcome the dissolution problem caused by the high solubility of the polysulfide species in liquid electrolytes.15–20 In the microporous carbon-supported sulfur cathode, the dissolution of polysulfides is suppressed because the solvent does not come into contact with sulfur.21–23 Although this approach is effective in suppressing the polysulfide dissolution, the microporous carbon-supported sulfur cathodes show an electrochemical behavior different from that of their conventional sulfur counterparts, especially in the first discharge process.15,17 The conventional sulfur cathode shows two plateaus at approximately 2.4 and 2.1 V vs. Li/Li+ during the first discharge process.11,16 During the first discharge plateau, the cyclic S8 molecules are reduced to long-chain lithium polysulfides (Li2Sn, 4 ≤ n < 8) through a disproportionation reaction, and the latter species are further reduced to short-chain lithium polysulfides (Li2Sn, 1 < n < 4) and Li2S during the second longer discharge plateau.24–27 In contrast to the conventional sulfur cathode, the microporous carbon-supported sulfur cathode shows a single discharge plateau (∼1.8 V vs. Li/Li+) during the first discharge process.15,18 To design a microporous carbon-supported sulfur cathode with high electrochemical performance, it is necessary to understand the reaction mechanism of the corresponding electrodes.
Various spectroscopic techniques such as X-ray photoemission spectroscopy (XPS)28 and X-ray absorption spectroscopy29 (XAS) have been employed to investigate the reaction mechanism of microporous carbon-supported sulfur cathodes. Helen et al. showed that on the surface and subsurface for the microporous carbon-supported sulfur cathode during first discharge by XPS, long-chain polysulfide could not be detected and only Li2S2 and Li2S could be detected.28 They proposed a direct transformation from sulfur to Li2S2/Li2S during first discharge in the system.28 On the other hand, Dominko et al. observed that a mixture of long chain polysulfide (S82− and S62−) and a mixture of short chain polysulfides (S42− and S22−) by operando XAS and suggested the transformation from the long-chain polysulfide to short-chain polysulfides and Li2S.29 However, thus far, no quantitative studies of the reaction mechanism have been reported. In this study, the reaction pathway of the microporous-supported sulfur cathodes was examined by the operando XAS analysis, which is a powerful technique to quantitatively evaluate the sulfur species formed during discharge/charge processes.30,31
2. Experimental
2.1 Materials
A microporous carbon-supported cathode was prepared using the procedure reported previously.32 After mixing sulfur (Wako Co.) and microporous carbon (Toyobo Co.), the mixture was heated to 155 °C for approximately 5 h to allow sulfur to diffuse into the microporous carbon. Finally, the temperature was increased to 300 °C and maintained for 2 h to sublime the extra sulfur on the outer surface of the microporous carbon. Sulfur, Li2S6, Na2S4, Na2S2 and Li2S were used for the references of XAS. Li2S6 was synthesized through the previously reported procedure.26 Stoichiometric ratio of sulfur and 1 M lithium triethylborohydride/tetrahydrofuran (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol%) was stirred for 1 hour and then the solvent was removed under vacuum inside Ar-filled glovebox. Na2S4 and Na2S2 were purchased from Dojindo Molecular Technologies Inc. Sulfur and Li2S were purchased from Aldrich Co.
:1 mol%) was stirred for 1 hour and then the solvent was removed under vacuum inside Ar-filled glovebox. Na2S4 and Na2S2 were purchased from Dojindo Molecular Technologies Inc. Sulfur and Li2S were purchased from Aldrich Co.
2.2 Characterizations
The particle morphology of the microporous carbon was observed by transmission electron microscopy (TEM, JEM-2100F, JEOL) and scanning electron microscopy (SEM, SU-1500, Hitachi). The amount of sulfur in the microporous carbon was determined by thermogravimetric analysis (TGA) (DTG-60AH, Shimadzu) under an Ar atmosphere at a heating rate of 5 °C min−1. N2 adsorption and desorption isotherms were measured at −196 °C using Autosorbμ-iQ (Quantachrome).2.3 Electrochemical testing
The electrochemical performance of the microporous carbon-supported sulfur cathode was examined using a two-electrode cell. A composite cathode was prepared by mixing the microporous carbon-supported sulfur cathode, acetylene black, and alginic acid binder in 90:5:5 weight ratio, supported by carbon paper. Li foil was used as the counter electrode. 1 M lithium bis(trifluorosulfonyl)amide (LiTFSA)/fluoroethylene carbonate (FEC):1,1,2,2-tetrafluoroethyl-2,2,3,3 tetrafluoropropyl ether (hydrofluoroether, HFE), in which the ratio of FEC to HFE was 1:1 vol% and the quantity of the electrolyte was 700 μL, was used as the electrolyte in a glass fiber membrane separator. The two-electrode cells were assembled in a glovebox filled with Ar. Galvanostatic charge/discharge measurements were carried out at a current density of 0.1 C (1 C = 1672 mA h g−1) with cut-off voltages of 1.0 V for the discharge and 3.0 V for the charge process at 25 °C.
2.4 XAFS measurements
S K-edge operando XAS spectra of the microporous carbon-supported sulfur cathode were observed in the partial fluorescence yield (PFY) mode at the BL27SU beamline of the SPring-8 synchrotron radiation facility at Hyogo, Japan. A homemade cell33 was used for the operando XAS. The microporous carbon-supported sulfur cathode composite material was mounted on a polyimide film to serve as the working electrode. LiTFSA/FEC:HFE was used in a glass fiber membrane as the electrolyte solution, and Li foil was used as the counter electrode. The operando cell was assembled in an argon-filled glovebox and transferred into a chamber for the XAS measurements under ultrahigh vacuum. The operando XAS measurements were performed under galvanostatic discharge at 0.1 C.3. Results and discussion
The SEM and TEM images of microporous carbon are shown in Fig. 1a and b, respectively. The SEM image showed the particle size was 1–2 μm and the TEM image showed that the microporous carbon has amorphous structure with pore of ∼1 nm. The polar size distribution of the microporous carbon support was also confirmed by N2 adsorption measurements, as shown in Fig. S1,† which showed a pore size of ∼1 nm. The TGA (Fig. S2†) showed a weight loss of approximately 24% at ∼450 °C for the microporous carbon-supported sulfur, indicating that the sulfur content was ∼24%. | ||
| Fig. 1 (a) SEM and (b) TEM images of microporous carbon. | ||
The microporous carbon-supported sulfur cathode showed a discharge capacity of 2380 mA h g−1, with a long single plateau at a voltage of ∼1.4 V in the first discharge process and a charge capacity of 900 mA h g−1 (Fig. 2a). After the first discharge/charge cycle, the microporous carbon-supported sulfur cathode showed a different discharge curve, with a reversible capacity of 900 mA h g−1 and a high coulombic efficiency (Fig. 2b). A large irreversible discharge capacity, exceeding the theoretical capacity of sulfur (1672 mA h g−1), was also observed in previous reports.17,21,28,32,34 This phenomenon is due to the formation of a solid electrolyte interphase (SEI) layer on the electrode composite.23,35
 | ||
| Fig. 2 Charge/discharge profile (a) and cycle performance (b) of the microporous carbon-supported sulfur cathode. | ||
To examine the electronic structure of the pristine microporous-supported sulfur cathode, we performed ex situ S K-edge XAS measurements for the microporous carbon-supported sulfur and ring-like sulfur cathodes. The S K-edge XANES and EXAFS results are presented in Fig. 3a and b, respectively. The two sulfur samples showed an absorption feature at 2471.8 eV, which was attributed to the S 1s to S–S π*36 state transition of elemental sulfur (Fig. 3a). However, the absorption peak intensity of the microporous carbon-supported sulfur cathode was lower than that of conventional sulfur, indicating that the transition from S 1s to S–S π* was reduced in the former sample. In the Fourier-transformed EXAFS spectra (Fig. 3b), the intensity of the peak at 1.6 Å, attributed to the S–S bonds, was lower for the microporous carbon-supported sulfur cathode than that for the conventional sulfur sample. This result indicates a decrease in the coordination number of S–S bonds in the microporous carbon-supported sulfur sample compared to that in conventional sulfur. Since sulfur in microporous carbons has been reported by X-ray photoemission spectroscopy17 and electron energy loss spectroscopy21 to be present in a chain rather than a ring structure, the present XAS results imply that sulfur changed from a ring to a chain structure after melting into the microporous carbon.
 | ||
| Fig. 3 (a) S K-edge XANES profiles and (b) S K-edge Fourier-transformed EXAFS profiles obtained from XAS measurements for sulfur and microporous carbon-supported sulfur cathodes. | ||
Operando XAS measurements were conducted to examine the electrochemical structural changes of the microporous carbon-supported sulfur cathode (Fig. 4a). Before the discharge process, absorption peaks were observed at 2472 and 2480 eV. The peak at 2472 eV is attributed to sulfur, as observed in Fig. 3, whereas that at 2480 eV corresponds to the sulfur component of the LiTFSA compound in the electrolyte.25,37 During the discharge process, an absorption peak attributed to linear polysulfides appeared at 2470 eV;36 then, two new peaks attributed to Li2S were observed at 2472.8 and 2475.2 eV.
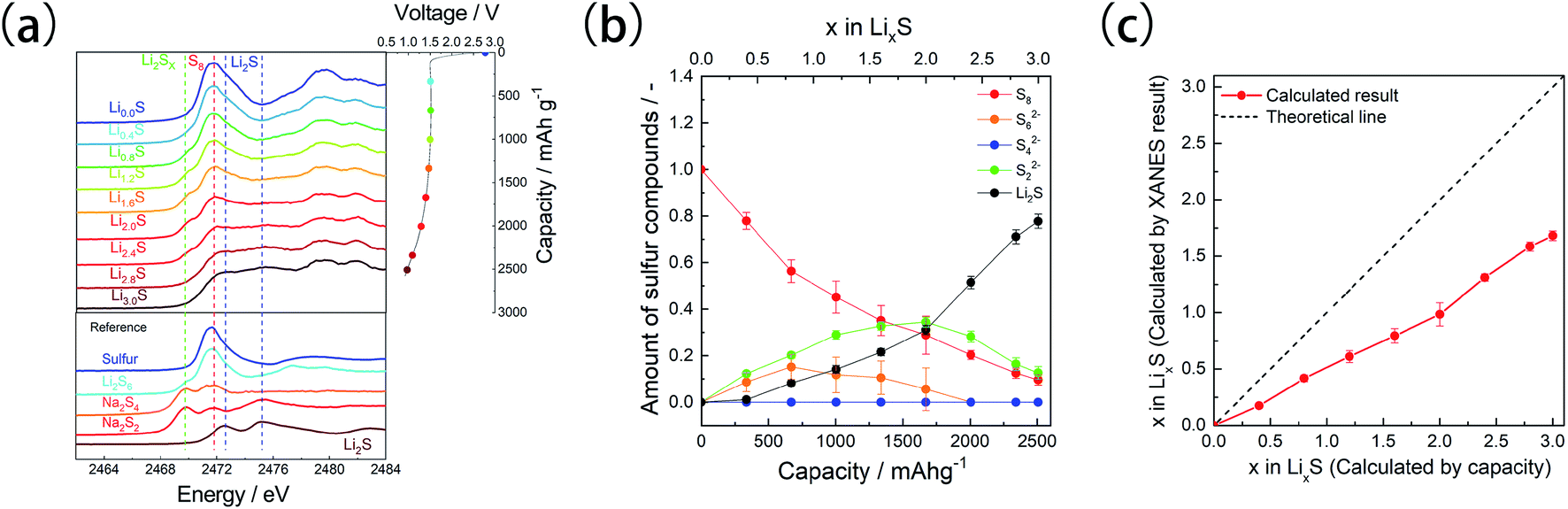 | ||
| Fig. 4 (a) Sulfur K-edge XANES spectra obtained from the operando XAS measurements of the microporous carbon-supported sulfur cathode. (b) Sulfur compound ratio of the microporous carbon-supported sulfur cathode during the first discharge, calculated by the linear combination fitting (LCF) analysis of XANES spectra. (c) Lithium compositions calculated from the LCF analysis and electrochemical capacity data. | ||
Linear combination fitting (LCF)26 was performed to estimate the ratio of the sulfur components during the discharge process (Fig. 4b). The spectra of pristine microporous carbon-supported sulfur S8, long-chain polysulfides S62−, medium/long-chain lithium polysulfides S42−, short-chain polysulfides S22−, and Li2S were used as references for the LCF analysis. The obtained fitting results were shown in Fig. S3.† Although these references were not exactly corresponding to polysulfide species forming during discharge process, they are useful to discuss the ratio change of polysulfide species.25,26 The polysulfide species S62− and S22− were formed in the initial stage, with a corresponding decrease in the amount of S8 species, whereas in the intermediate stage, the amounts of S22− and Li2S increased and those of S8 and S62− decreased. In the final stage, the analysis showed an increase in the Li2S amount and a decrease in the amounts of S8 and S22−. Unreacted S8 and S22− species were present even in the discharge state, that is, above the theoretical capacity of sulfur (1672 mA h g−1).
The lithium content calculated by the LCF analysis (Fig. 4c) increased in the x range of 0–3.0, but its value was lesser than that estimated by electrochemical measurements, and it was lesser than 2.0 even in the final discharge state (x = 3.0). These results indicate the decomposition of the electrolyte and the presence of unreacted sulfur species. It has been reported that the FEC solvent undergoes decomposition at 1.5 V (ref. 35) on the surface of the microporous carbon. In the microporous carbon-supported sulfur cathode, lithium-ion transport to the sulfur is sluggish compared to conventional sulfur cathode because the electrolyte cannot enter the micropores.17 Therefore, lithium-ion conduction becomes the rate-limiting step, and the sulfur species located deep inside the micropores remain unreacted. A similar reaction distribution was observed in all-solid-state battery systems,38 in which the reaction proceeds from the solid electrolyte side because ionic conduction is slower than electronic conduction.
The reaction mechanism of the microporous carbon-supported sulfur cathode during discharge is illustrated in Fig. 5. In the initial stage, sulfur forms chain structures inside the micropores. In the intermediate stage of discharge, S62− and S22− polysulfides are formed because the solvent cannot enter the micropores, and these polysulfides do not dissolve in the liquid electrolyte. In the final discharge process, Li2S is the main sulfide species, with residual amounts of S22− and long-chain S8 species inside the micropores.
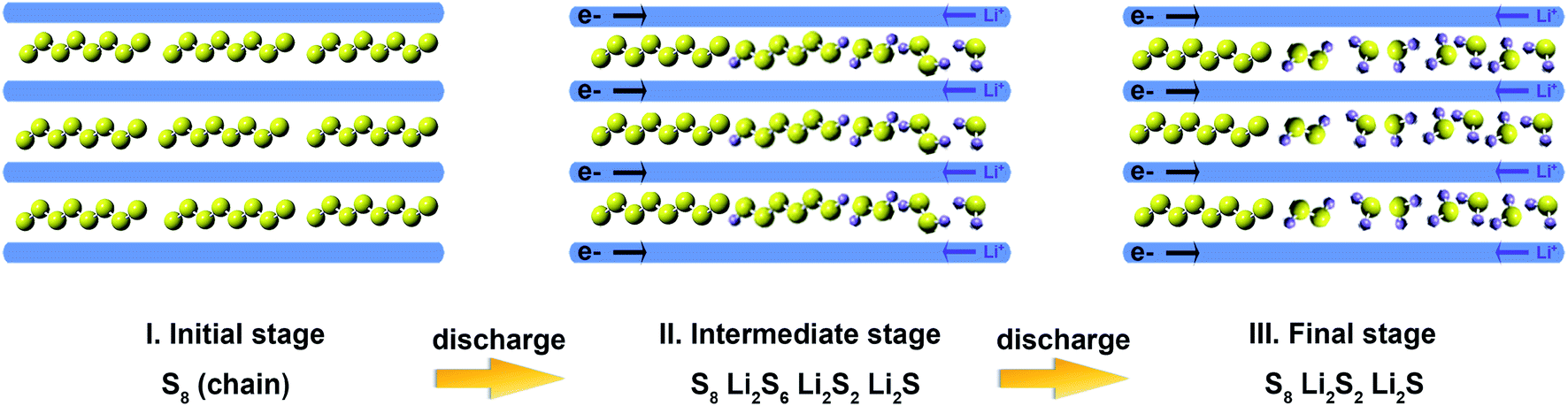 | ||
| Fig. 5 Reaction model for the microporous carbon-supported sulfur cathodes. | ||
The microporous carbon can prevent the dissolution of polysulfides in the liquid electrolyte, resulting in a change in the sulfur cathode reaction pathway. This change leads to a single plateau at approximately 1.4 V during the first discharge, as observed in the solid electrolyte.
4. Conclusions
In this study, the reaction mechanism of the sulfur cathode in the microporous carbon during discharge was observed by operando XAS. The sulfur cathode was reduced to long-chain (S62−) and short-chain (S22−) polysulfide species during the initial discharge, while the short-chain S22− units were the main polysulfide species in the subsequent discharge; large amounts of Li2S were formed during the final discharge. The reaction mechanism of the microporous carbon-supported sulfur cathode is different from that of a conventional sulfur cathode, as the microporous carbon support prevents the dissolution of polysulfides. This study elucidated the reaction mechanism of sulfur cathodes in microporous carbon by operando soft XAS. We believe that our results can provide further insights into the deeper understanding of the behavior of sulfur in carbon-supported cathodes, and it will be helpful for designing new cathodes with high performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Japan Science and Technology Agency (JST), Advanced Low Carbon Technology Research and Development Program (ALCA), Specially Promoted Research for Innovative Next Generation Batteries (SPRING) Project (Grant Number: JPMJAL1301). Synchrotron radiation experiments were performed at beam line BL27SU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal number 2017A1025, 2017B1037, 2018A1025, 2018B1031, 2019A1021, 2019B1023, 2019B1634).Notes and references
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S.-F. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- M. Vijayakumar, N. Govind, E. Walter, S. D. Burton, A. Shukla, A. Devaraj, J. Xiao, J. Liu, C. Wang, A. Karim and S. Thevuthasan, Phys. Chem. Chem. Phys., 2014, 16, 10923–10932 RSC.
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CAS.
- Z. Lin and C. Liang, J. Mater. Chem. A, 2015, 3, 936–958 RSC.
- G. Y. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS.
- X. L. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- X. L. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325 CrossRef.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 RSC.
- S. Xin, L. Gu, N. H. Zhao, Y. X. Yin, L. J. Zhou, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS.
- Z. Li, L. X. Yuan, Z. Q. Yi, Y. M. Sun, Y. Liu, Y. Jiang, Y. Shen, Y. Xin, Z. L. Zhang and Y. H. Huang, Adv. Energy Mater., 2014, 4, 1301473 CrossRef.
- Z. Li, Y. Jiang, L. X. Yuan, Z. Q. Yi, C. Wu, Y. Liu, P. Strasser and Y. H. Huang, ACS Nano, 2014, 8, 9295–9303 CrossRef CAS.
- Z. H. Peng, W. Y. Fang, H. B. Zhao, J. H. Fang, H. W. Cheng, T. N. L. Doan, J. Q. Xu and P. Chen, J. Power Sources, 2015, 282, 70–78 CrossRef CAS.
- Y. H. Xu, Y. Wen, Y. J. Zhu, K. Gaskell, K. A. Cychosz, B. Eichhorn, K. Xu and C. S. Wang, Adv. Funct. Mater., 2015, 25, 4312–4320 CrossRef CAS.
- M. Helen, T. Diemant, S. Schindler, R. J. Behm, M. Danzer, U. Kaiser, M. Fichtner and M. A. Reddy, ACS Omega, 2018, 3, 11290–11299 CrossRef CAS.
- S. Z. Niu, G. M. Zhou, W. Lv, H. F. Shi, C. Luo, Y. B. He, B. H. Li, Q. H. Yang and F. Y. Kang, Carbon, 2016, 109, 1–6 CrossRef CAS.
- E. Markevich, G. Salitra, A. Rosenman, Y. Talyosef, F. Chesneau and D. Aurbach, J. Mater. Chem. A, 2015, 3, 19873–19883 RSC.
- D. Zheng, G. Wang, D. Liu, J. Si, T. Ding, D. Qu, X. Yang and D. Qu, Adv. Mater. Technol., 2018, 3, 1700233 CrossRef.
- M. Cuisinier, P.-E. Cabelguen, S. Evers, G. He, M. Kolbeck, A. Garsuch, T. Bolin, M. Balasubramanian and L. F. Nazar, J. Phys. Chem. Lett., 2013, 4, 3227–3232 CrossRef CAS.
- M. Cuisinier, C. Hart, M. Balasubramanian, A. Garsuch and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1401801 CrossRef.
- G. R. Li, S. Wang, Y. N. Zhang, M. Li, Z. W. Chen and J. Lu, Adv. Mater., 2018, 30, 1705590 CrossRef.
- M. Helen, M. A. Reddy, T. Diemant, U. Golla-Schindler, R. J. Behm, U. Kaiser and M. Fichtner, Sci. Rep., 2015, 5, 12146 CrossRef CAS.
- R. Dominko, A. Vizintin, G. Aquilanti, L. Stievano, M. J. Helen, A. R. Munnangi, M. Fichtner and I. Arcon, J. Electrochem. Soc., 2018, 165, A5014–A5019 CrossRef CAS.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef.
- L. Zhang, T. Qian, X. Y. Zhu, Z. L. Hu, M. F. Wang, L. Y. Zhang, T. Jiang, J. H. Tian and C. L. Yan, Chem. Soc. Rev., 2019, 48, 5432–5453 RSC.
- T. Takahashi, M. Yamagata and M. Ishikawa, Prog. Nat. Sci.: Mater. Int., 2015, 25, 612–621 CrossRef CAS.
- K. Yamamoto, Y. Y. Zhou, N. Yabuuchi, K. Nakanishi, T. Yoshinari, T. Kobayashi, Y. Kobayashi, R. Yamamoto, A. Watanabe, Y. Orikasa, K. Tsuruta, J. Park, H. R. Byon, Y. Tamenori, T. Ohta and Y. Uchimoto, Chem. Mater., 2020, 32, 139–147 CrossRef CAS.
- W. H. Zhang, D. Qiao, J. X. Pan, Y. L. Cao, H. X. Yang and X. P. Ai, Electrochim. Acta, 2013, 87, 497–502 CrossRef CAS.
- E. Markevich, G. Salitra, A. Rosenman, Y. Talyosef, F. Chesneau and D. Aurbach, Electrochem. Commun., 2015, 60, 42–46 CrossRef CAS.
- X. Li, M. Banis, A. Lushington, X. F. Yang, Q. Sun, Y. Zhao, C. Q. Liu, Q. Z. Li, B. Q. Wang, W. Xiao, C. H. Wang, M. S. Li, J. W. Liang, R. Y. Li, Y. F. Hu, L. Goncharova, H. M. Zhang, T. K. Sham and X. L. Sun, Nat. Commun., 2018, 9, 4509 CrossRef.
- C. W. Lee, Q. Pang, S. Ha, L. Cheng, S. D. Han, K. R. Zavadil, K. G. Gallagher, L. F. Nazar and M. Balasubramanian, ACS Cent. Sci., 2017, 3, 605–613 CrossRef CAS.
- K. Chen, S. Shinjo, A. Sakuda, K. Yamamoto, T. Uchiyama, K. Kuratani, T. Takeuchi, Y. Orikasa, A. Hayashi, M. Tatsumisago, Y. Kimura, T. Nakamura, K. Amezawa and Y. Uchimoto, J. Phys. Chem. C, 2019, 123, 3292–3298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08299f |
| This journal is © The Royal Society of Chemistry 2020 |