
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired NHC-organocatalyzed Stetter reaction in aqueous conditions†
Mégane Debiaisab,
Aladin Hamoudab,
Reihana Drainab,
Philippe Barthélémy ab and
Valérie Desvergnes*ab
ab and
Valérie Desvergnes*ab
aUniv. Bordeaux, CNRS, INSERM, ARNA, UMR 5320, U1212, F-33000 Bordeaux, France. E-mail: valerie.desvergnes@u-bordeaux.fr; Tel: +33 557571176
bChemBioPharm Team, 146 Rue Leo Saignat, UFR Pharmacie, 3ième Tranche, 4ième étage, 33076 Bordeaux Cedex, France
First published on 9th November 2020
Abstract
The first bio-inspired N-Heterocyclic Carbene (NHC)-catalyzed Stetter reaction in aqueous medium is reported with benzaldehyde and chalcone as model substrates. A screening of azolium salts as precatalysts revealed the remarkable efficiency of synthetic thiazolium salt 8 (up to 90% conversion in pure water at 75 °C). The reaction was successfully extended to various simple aldehyde substrates. The effect of temperature was also investigated in order to extend the reaction to lower temperature allowing a potential application to sensitive biomolecules. This study highlighted the influence of both solvent and temperature on the 1,4-diketone 3/benzoin 4 ratio. New precatalysts 26 and 27 were designed and synthesized to explore a possible compartmentalization of the reaction in aqueous conditions. Owing to the use of inexpensive metal-free N-Heterocyclic Carbene (NHC) as a bioinspired catalyst, we anticipate that this green strategy in aqueous conditions will be attractive for bioconjugation of many biomolecule-type aldehydes and enone derivatives.
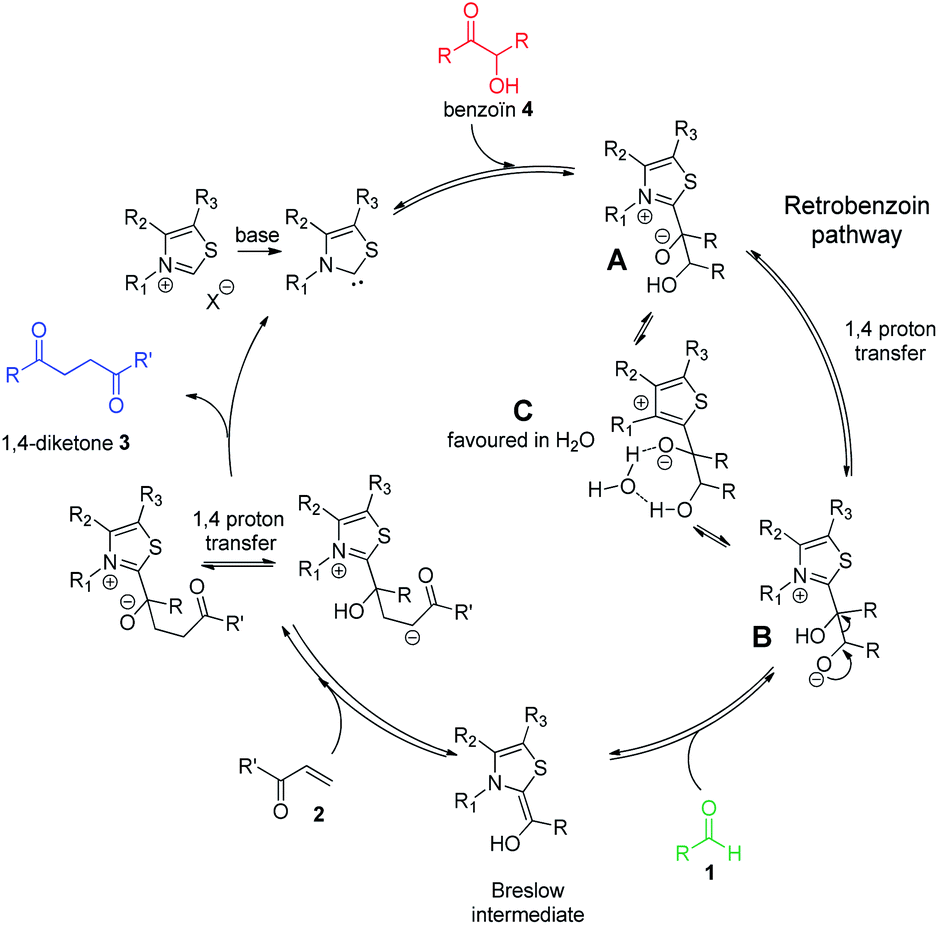
Enzyme catalysis involving cofactors is a source of inspiration for creating synthetic catalysts.1 Moreover, chemical reactivity in an aqueous medium is at the very origin of life and is an important link between chemistry and biology. Over the past decades, now-called bio-inspired synthesis has become a new way of thinking about chemistry, as it is the case for green chemistry. Organocatalysis is greener than metallocatalyzed processes2 and organocatalyzed reactions in aqueous media3 have attracted considerable interest because water is considered as the greenest solvent thanks to its nontoxicity, safety, etc. N-Heterocyclic Carbenes (NHCs) are biomimetic species, analogous to the natural cofactor thiamine diphosphate and known to address wide challenges in the field of organocatalysis4 as non-biological catalysts. NHCs were early described by Breslow as intermediate (thiamin diphosphate cofactor, vitamin B1) in biochemical pathways occurring in aqueous media.5 NHC are highly reactive neutral nucleophiles, which apparently prohibit their use in aqueous solution. Indeed, free carbenes, especially imidazolylidinenes, are reported to be moisture sensitive, leading to rapid protonation or hydrolytic cleavage of the azolium salt ring6 even if a relative stability in water has been described very recently.7 Several studies reported that pKas of various azoles are concurring with the low concentration of free carbenes in water.8 In line with the Breslow's studies,9 NHC organocatalyzed benzoin condensation was the first reaction successfully investigated in water.10 Other reactions in aqueous conditions using various electrophiles were also reported.11–16. In the context of bio-inspired organocatalysis in aqueous media, NHC-catalyzed reactions in aqueous medium represent an attractive eco-friendly approach. A related Umpolung reaction to benzoin condensation is the NHC-catalyzed Stetter reaction17 involving the conjugate addition of an aldehyde to a Michael acceptor. The Stetter reaction and the benzoin condensation have similar mechanisms both going through the formation of the widely described Breslow intermediate. For the Stetter reaction, the 1,4-diketone is obtained through a non-reversible later step following the addition of Breslow intermediate to the α,β-unsaturated compound. Benzoin condensation can be observed as a side reaction arising from the reversible addition of Breslow intermediate to the aldehyde (Scheme 1). The formation of C–C bonds through reverse polarity via Stetter reaction has proved to be of major interest as key step for total synthesis.18 As a bio-inspired reaction, it would be highly desirable extend such an organocatalyzed reaction's green chemistry perspectives.19 Herein, on the basis of our previous work,20 the Stetter reaction was investigated first in presence of water then in pure water. Benzaldehyde and chalcone were chosen as model substrates for their robustness in aqueous media (no polymerization, hydratation…) and good reactivity in the Stetter reaction.
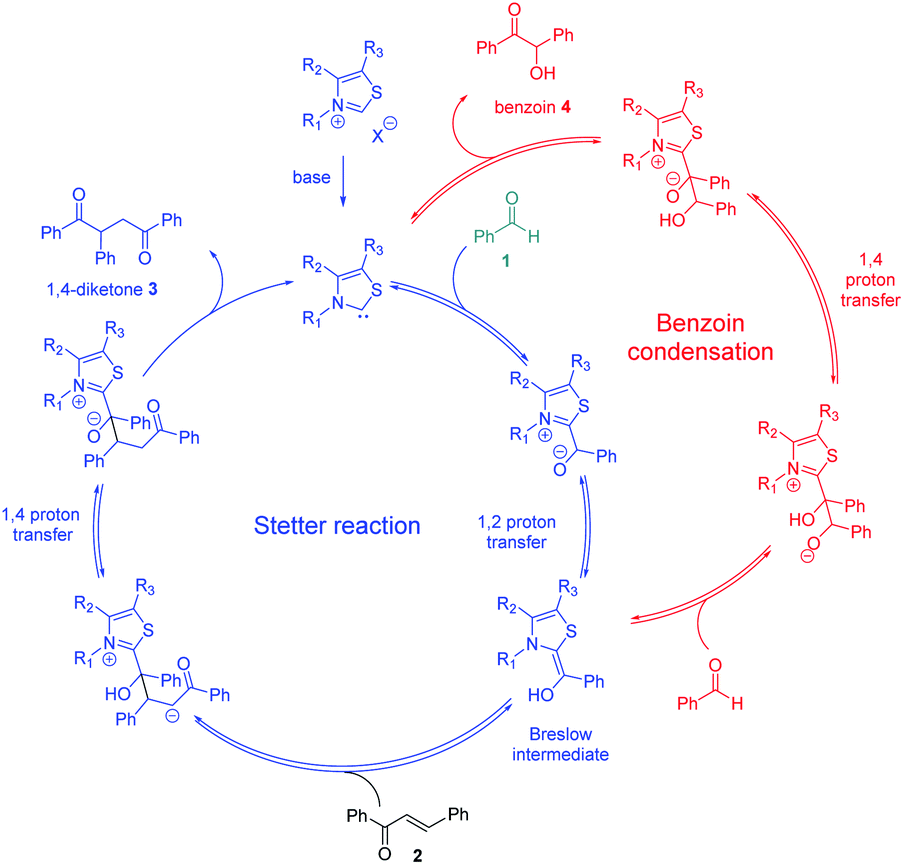 | ||
| Scheme 1 Catalytic cycles of Stetter (blue pathway)/benzoin reaction (red pathway) between benzaldehyde 1 (green) and chalcone 2. | ||
Results and discussion
Preliminary investigations involved the introduction of 10% of water in Stetter reactions using classical thiazolium salts (Fig. 1)21 as thiamine cofactor analogues and carbene precursors to compare with the same reaction under classical conditions in dry THF.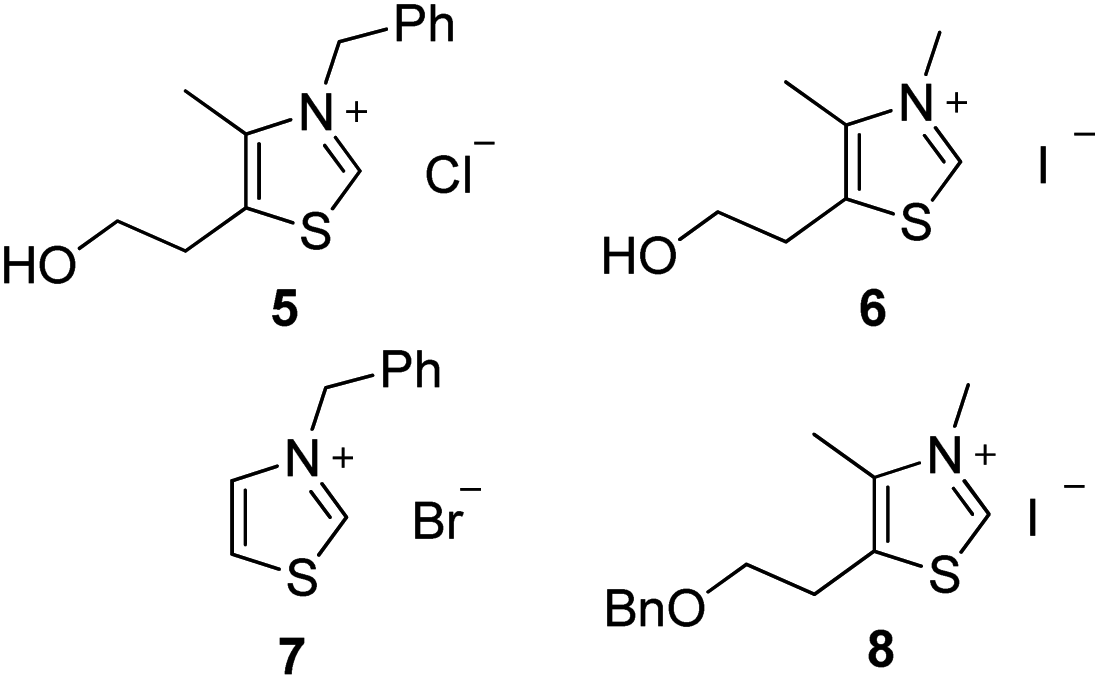 | ||
| Fig. 1 Structure of the different azolium salts used. | ||
We were pleased to observe that thiazolium salts 5–7, simple analogues of thiamine diphosphate, were able to provide carbene species and react in the presence of 10% of water. Results are reported in Table 1. Noteworthy, commercially available thiazolium salt 5, previously reported by Scheidt to catalyze conjugate addition of α-keto-carboxylates to α-β-unsaturated 2-acyl-imidazoles under neutral aqueous conditions,22 provided moderate conversions both in THF and in presence of water (Table 1 entries 1 and 2). These results encouraged us to test other simple azolium salts 6 and 7. Compound 6 turned out to be highly efficient in THF but much less in the presence of 10% of water. Compound 7 led only to benzoin formation in aqueous media without any conversion of the chalcone starting material (Table 1, entries 5 and 6). Overall, the presence of water led to a higher quantity of the side product benzoin compared to the same reaction performed in anhydrous THF23 (entry 2 vs. 1, entry 4 vs. 3 and entry 8 vs. 7).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Reaction conditionsa | Conversionb | Yield 3 | Yield 4 |
| a 1.5 equiv. of benzaldehyde in solvent (C = 1.6 M) were added to 30 mol% of azolium salt,25 then 30 mol% of DBU was added and lastly 1 equiv. of chalcone. The reaction was heated at 75 °C for 20 h.b Based on quantity of remaining of chalcone limiting reagent.c Some degradation was also observed. | |||||
| 1 | 5 | THF | 50% | 33% | 24% |
| 2 | 5 | THF/H2O (9/1) | 38% | 24% | 76% |
| 3 | 6 | THF | >90% | 86% | 0% |
| 4 | 6 | THF/H2O (9/1) | 45% | 42% | 31% |
| 5c | 7 | THF | 23% | 13% | 10% |
| 6 | 7 | THF/H2O (9/1) | 0% | 0% | 61% |
| 7 | 8 | THF | >95% | 90% | 0% |
| 8 | 8 | THF/H2O (9/1) | 76% | 62% | 13% |
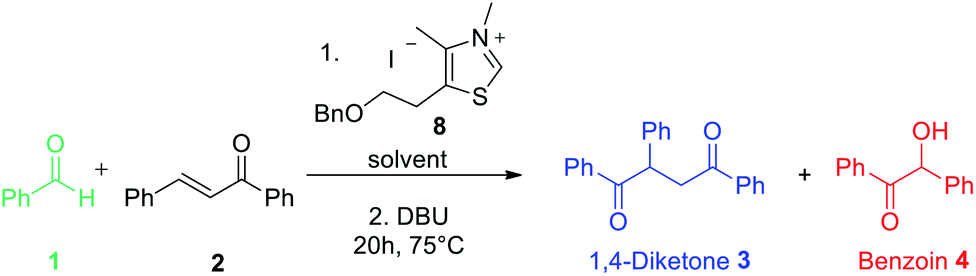
Following the encouraging results obtained with the commercially available precatalysts 5, 6, 7, we synthesized and tested thiazolium 8,10c more hydrophobic than 6 and previously reported in literature as resistant to hydrolysis under basic aqueous conditions.24 Actually, this N-methyl-5-(2′-benzyloxyethyl)-4-methylthiazolium iodide salt 8 has proved to be the most efficient to provide mainly the 1,4-diketone compound under anhydrous as well as non anhydrous conditions (Table 1 entries 7 and 8).
Thiazolium 8 was thus the best candidate to perform attempts introducing gradually water in the reaction medium. We were pleased to see that, at 75 °C, introduction of water from 10 to 100% did not drastically impact nor the conversion nor the reaction selectivity (Table 2). Nevertheless, using a 50/50 mixture of THF and water resulted unexpectedly in the lowest conversion and yield. The introduction of water resulted in formation of benzoin compound (Table 2). It is noticeable that even if the organic reactants were insoluble as described for so-called “on-water” reactions26 no substantial rate acceleration was observed in aqueous medium at 75 °C in this study.
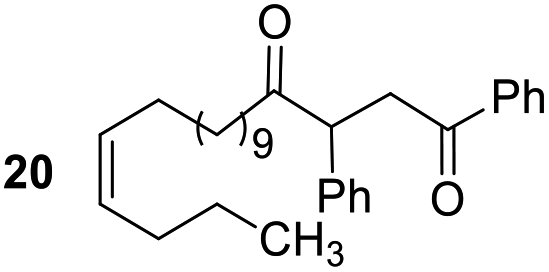
Having some optimized conditions in hands, the scope of the reaction was then extended to various simple aldehyde precursors (Table 3). Under the model conditions (20 h at 75 °C in water as solvent), the reaction took place with moderate to good yields when aliphatic or aromatic aldehydes were used as biomolecules analogs. 9, 10 and 11 lipidic aldehydes were tested as substrates and the longest carbon chain substrate led to the lowest yield. The use of hemiacetal 12 as a masked aldehyde and a very simple sugar surrogate proved also successful leading to the 1,4-diketone in a satisfactory 70% yield. Amino aldehyde-type substrates were also submitted to reaction, extending the scope of the reaction to three families of biomolecules. Reaction on chiral substrates 13 and 14, lead to the expected products without any significant stereoselectivity. Aromatic aldehydes 15 and 16 were also used. Pyrrole carboxaldehyde 16 provided the expected diketone 25 in 68% yield when the reaction was carried on for 60 hours (25% if performed at 75 °C during 20 h). The results obtained at 75 °C for 20 hours allowed to compare the reactivity with model substrate benzaldehyde 1. The last example illustrated the fact that more complex substrates require an optimization of the reaction time to reach a better conversion when performed in water. These promising results clearly extend the scope of the Stetter reaction under aqueous conditions and demonstrate that it can be efficiently applied to more complex substrates than the benzaldehyde and chalcone models.
|
|||
|---|---|---|---|
| Entry | Aldehyde | Product | Yielda |
| a Yield was also calculated based on remaining starting material (chalcone).b Reaction conditions were 60 h at 75 °C. | |||
| 1 | 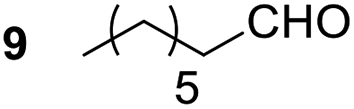 |
 |
89% |
| 2 | 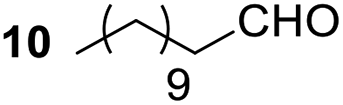 |
 |
65% |
| 3 | 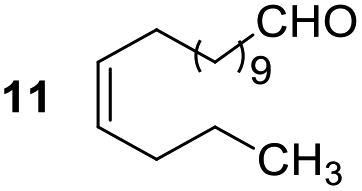 |
 |
32%, (60% brsm) |
| 4 | 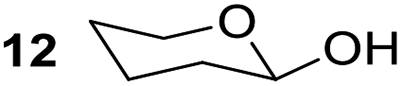 |
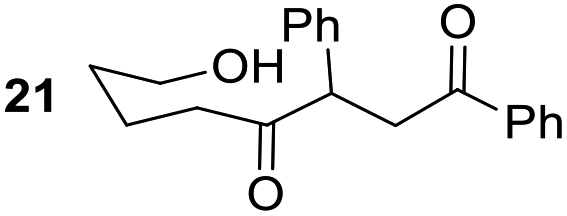 |
70%, (87% brsm) |
| 5 | 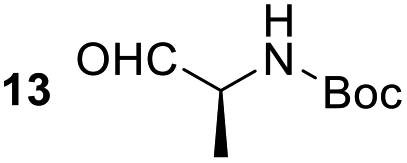 |
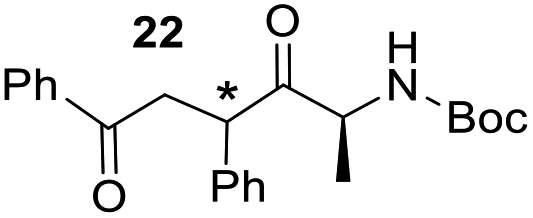 |
67%, (89% brsm), d.r. 52/48 |
| 6 | 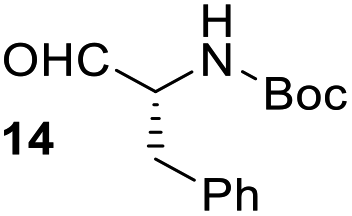 |
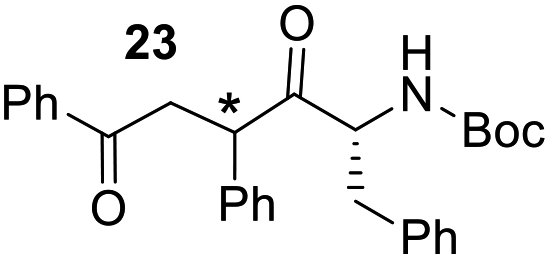 |
37%, (74% brsm), d.r. 59/41 |
| 7 | 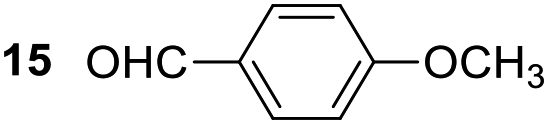 |
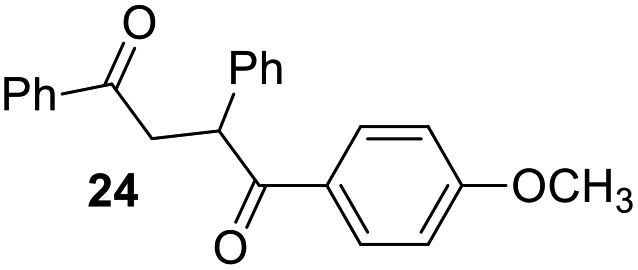 |
30%, (85% brsm), 62%b, (94% brsm) |
| 8b | 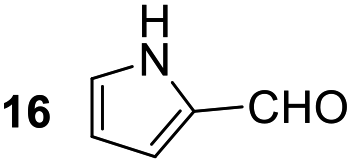 |
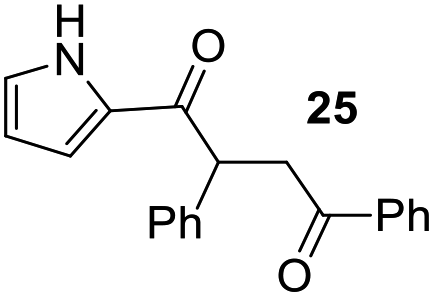 |
68%, (80% brsm) |
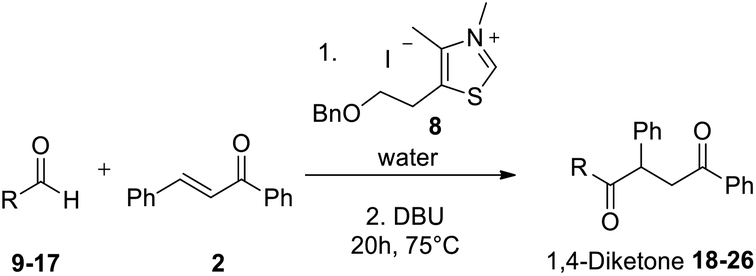
The effect of temperature onto the reaction rate and selectivity was also investigated. The reaction has been studied at 40 °C, a more compatible temperature for extension to biomolecules substrates. The ratios of aldehyde, Stetter adduct and benzoin were thoroughly monitored by 1H NMR at 95 °C and 40 °C in water by carrying out different experiments which were stopped every 2 hours until completion (Fig. 2).
 | ||
| Fig. 2 Temperature effect onto the compounds ratio and the reaction rate of Stetter reaction in water. | ||
As expected, the reaction was quicker at higher temperature (completion after 7 h at 95 °C). At both temperatures we could observe a very fast decrease of the aldehyde concentration along with an increase of the benzoin adduct. Interestingly the diketone increase (blue line) seemed closely connected to benzoin decrease (red line). It is noticeable that if the Stetter 1,4-diketone product is the major product from the very beginning at 95 °C (15 min), it becomes major after more than 50 hours when the reaction is run at 40 °C.
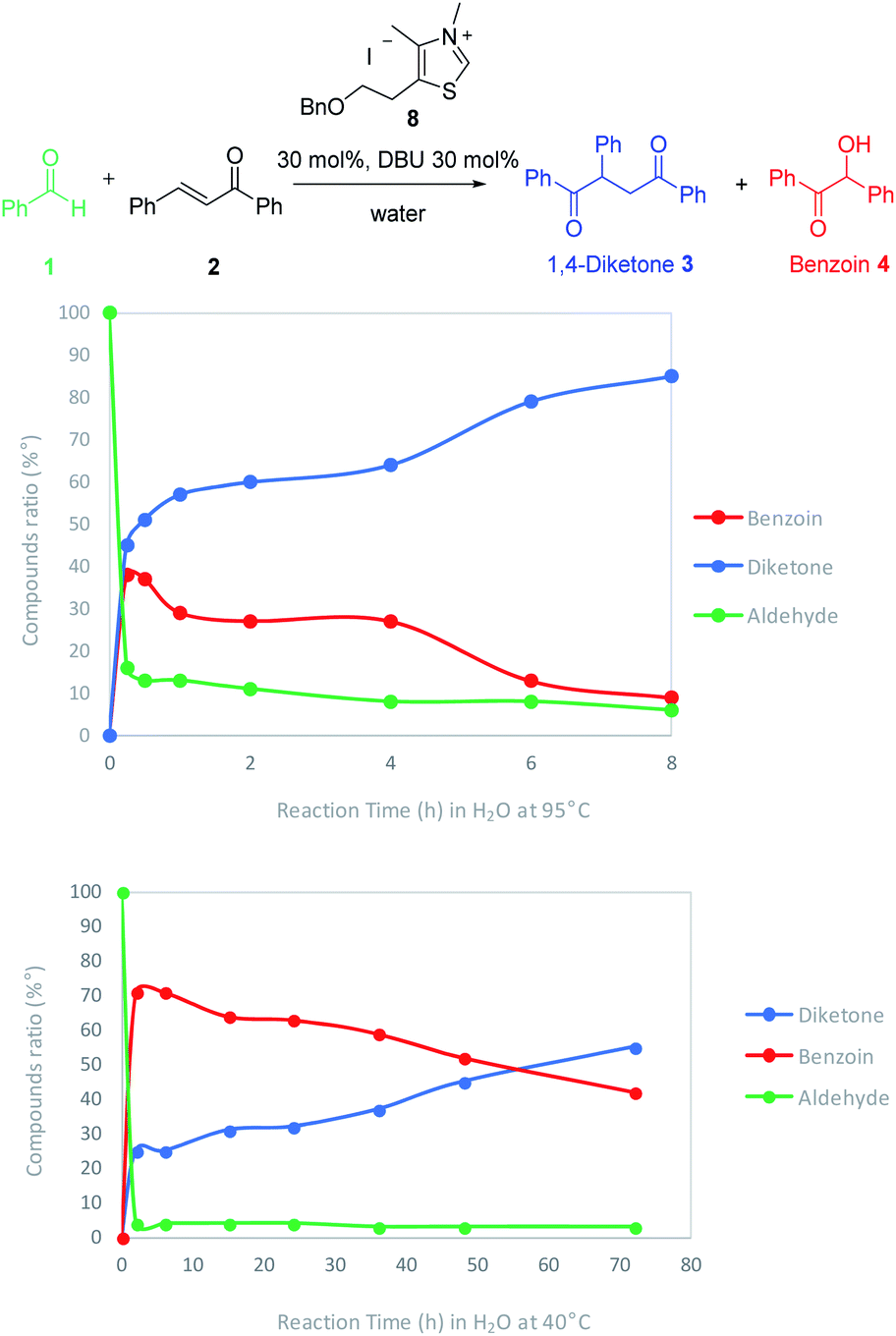
Considering the two connected catalytic pathways (Scheme 1), several points should then be pointed out. Water may be seen as a proton shuttle10g then the 1,2-proton transfer step in the catalytic cycle is certainly no more the limiting step. The generation of Breslow intermediate being then faster than in THF when water was the solvent, two hypotheses could explain the overall slower reaction when water replaced THF. Either the kinetics of the 1,4-addition onto the enone is slower in the presence of water or the generation of the Breslow intermediate from the retrobenzoin reaction (Scheme 1 red pathway) is slower. In order to solve this question and ultimately improve the reaction efficiency at low temperatures, we decided to perform the reaction starting from benzoin as masked aldehyde by analogy with the elegant use of α′-hydroxyenones as surrogates for α,β-unsaturated aldehydes in NHC catalyzed reactions.27 Benzoin was then used as starting material in our optimized reaction conditions both in THF and with water as solvent. The reaction was performed at 95 °C to be fast enough to allow NMR monitoring after 2 h, 6 h, 15 h and 24 h (Fig. 3).28
 | ||
| Fig. 3 Monitoring of Stetter reaction with benzoin as starting material in water and in THF. | ||
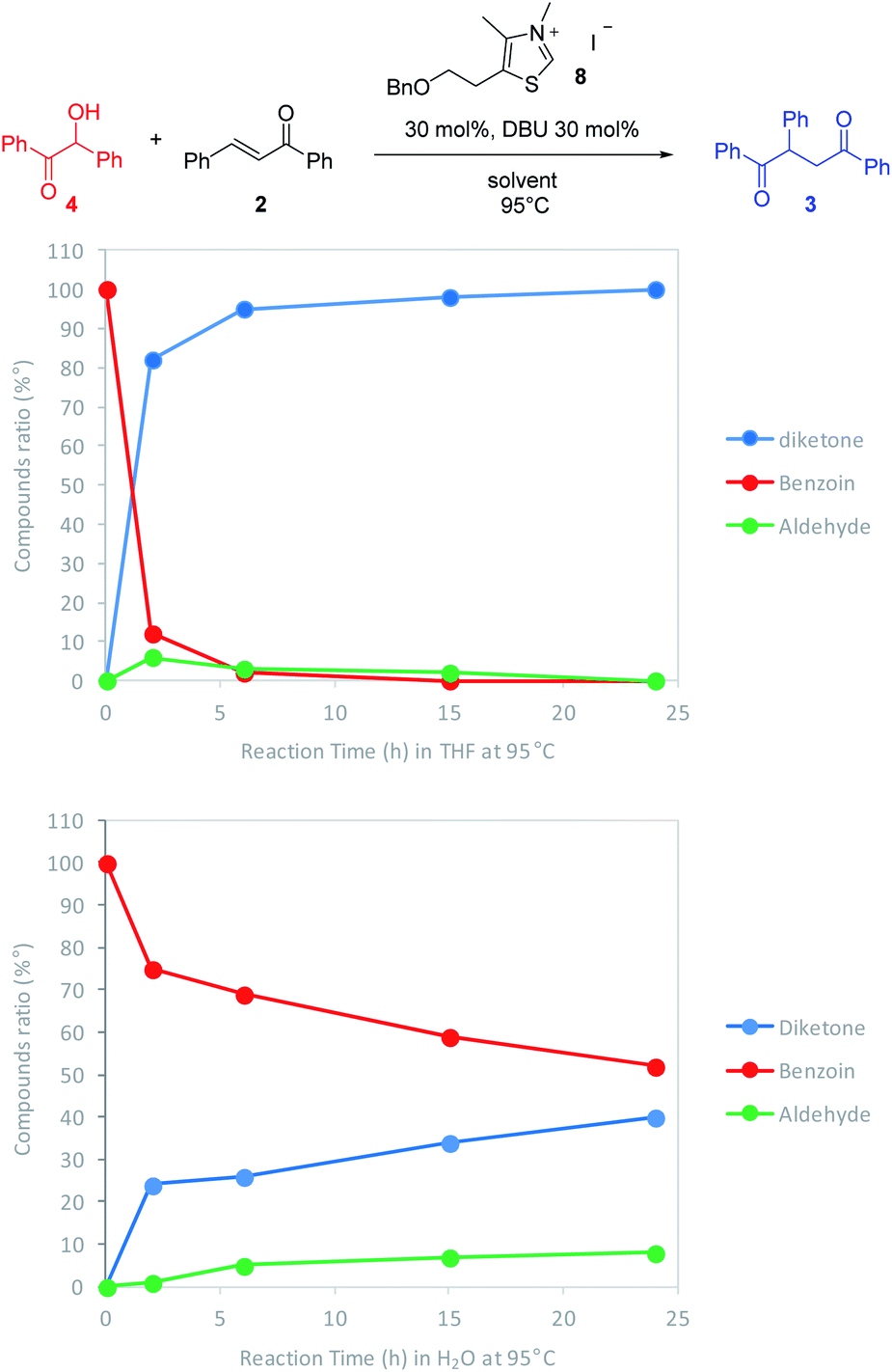
As expected, the nature of the solvent affected considerably the reaction kinetics. In both solvents, the diketone formation (blue line) is closely related to the benzoin decrease (red line). Benzaldehyde (green line) is consumed as it is released. Nevertheless, the benzoin decrease (i.e. pathway including retrobenzoin reaction) is very slow and disfavored in aqueous medium. The retrobenzoin step, allowing the formation of the Breslow intermediate, therefore appears to be the rate-limiting step in aqueous medium. This could be explained by the presence of H-bonded stabilized transient solvated species C, impacting drastically B concentration and then Breslow intermediate formation rate (Scheme 2).
 | ||
| Scheme 2 Catalytic cycle of Stetter reaction starting from benzoin 4 and involving retrobenzoin pathway. | ||
Unlike a reaction “in water” where reactants are dissolved, macroscopic observation of the reaction medium shows the non-solubility of reactants. We can observe a highly heterogeneous mixture and an emulsion appears when insoluble reactants are stirred. By analogy with transition state proposed in literature for “on water” catalytic reactions,29 it can be assumed that organic reagents (azolium salts, enone, aldehyde) form hydrophobic droplets inside water (Fig. 4, see picture in ESI†).
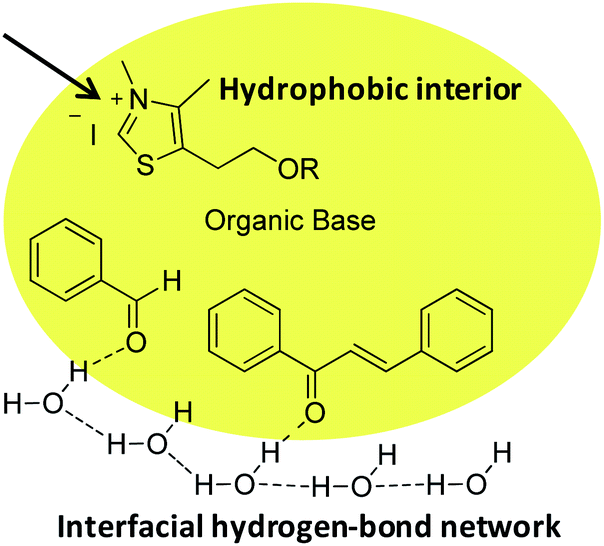 | ||
| Fig. 4 Proposed molecular arrangement for “on water” NHC-catalyzed Stetter reaction between benzaldehyde and chalcone. | ||
Hence, potential activation of aldehyde, by dangling OH groups on water surface, would make it even more electrophilic. An “on water” type chemistry process would then explain the increase in benzoin formation. Moreover, azolium 8, more lipophilic than its hydroxyl analogue 6, would then be inside the organic lipophilic phase, protected from hydrolysis and solvatation, which would explain its better performance. This lipophilic phase of the reaction medium constitutes a non polar interior that could be seen as a surrogate of hydrophobic enzyme pocket in enzyme/coenzyme natural systems.
Considering the hypothesis of the transition state proposed in Scheme 2, we assessed that it should be possible to limit C formation by further increasing lipophilicity of azolium salt.10a Such a compound would be less close to the interface with water and therefore less accessible for interfacial H bonding with water. Such conditions should consequently favor retrobenzoin step to regenerate Breslow intermediate, making it available to attack the enone and enhance subsequently diketone formation (Scheme 3).
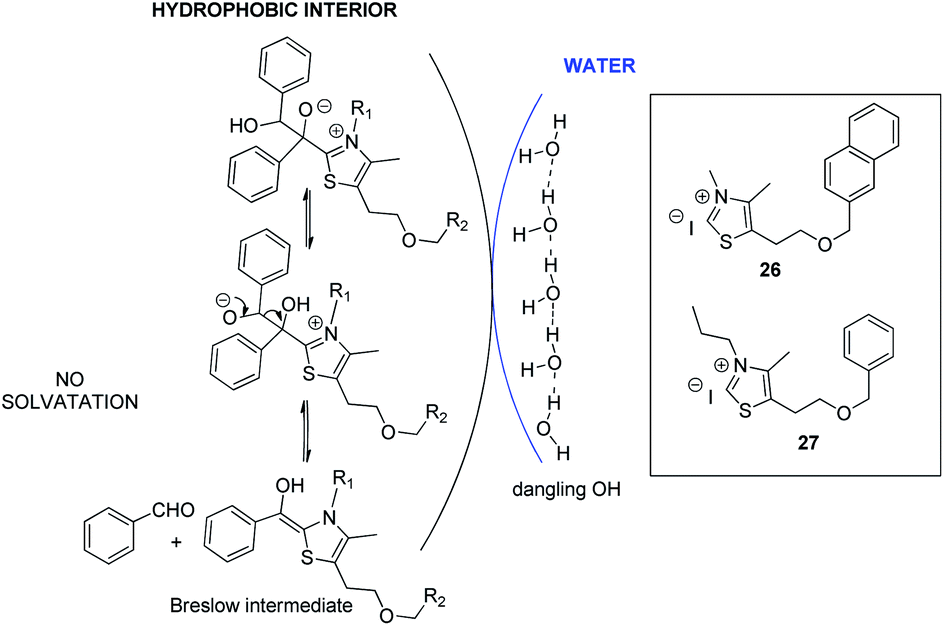 | ||
| Scheme 3 Retrobenzoin pathway in the organic droplets. | ||
Azolium 26 and azolium 27 analogues of the benzylated salt 8 were then synthesized30 and their efficiency was compared to 8 (Table 4).
| Entry | Conditionsa | Conversionb | Diketone 3/benzoin 4 ratioc |
|---|---|---|---|
| a 1.5 equiv. of benzaldehyde in water (1.6 M) were added to 30 mol% of azolium salt, then 30 mol% of DBU was added and lastly 1 equiv. of chalcone. The reaction was heated for 20 h at 75 °C.b Conversion was calculated from the quantity of recovered chalcone limiting reagent.c Based on 1H NMR crude spectra signals integrations. | |||
| 1 | 8, 75 °C, 20 h | 90% | 81/19 |
| 2 | 26, 75 °C, 20 h | 58% | 97/3 |
| 3 | 27, 75 °C, 20 h | 84% | 90/10 |
Interestingly, results highlighted that with azolium 27 (R1 = propyle vs. methyle for 8) the proportion of diketone was slightly increased. As expected, further increasing the lipophilicity of the azolium 26 in which the benzyl group in R2 is replaced by a naphthyle, made the diketone even more favored. The observed results seem to point towards a compartmentalization of the lipophilic and hydrophilic environments. However, the catalyst design still needs to be improved since the conversion with azolium 26 does not exceed 55%, probably because it is still solid at 75 °C. Moreover, the same reaction performed with inorganic bases such as NaOH or LiOH led to conversions that have never exceeded 25%. Rewardingly, the use of more lipophilic base Bu4N+OH− allowed a 45% conversion reaction in favor of diketone (44% yield). This drastic effect of the nature of the base argues for a reaction taking place in an organic droplet phase.
Conclusion
In summary, a Stetter reaction occurring in aqueous medium has been reported for the first time. Similarly to previously reported enzymatic reactions31 the results reported in this contribution allow for an eco-friendly approach to bioinspired organocatalysis. A study based on the ratio of expected 1,4-diketone compound 3 versus benzoin side product 4 supports the hypothesis that, with water as unique solvent, the rate limiting step was no longer proton transfer but rather the retrobenzoin reaction. Also, benzoin as masked aldehyde was used for the first time and our study in both organic solvent and water highlighted that retrobenzoin kinetic in water was slower than in THF. Design and synthesis of the azolium salt were carried out to verify a possible compartmentalization of the reaction in aqueous conditions. Indeed, if organocatalytic reactions in water have been well reviewed32 NHC-catalyzed Stetter reaction, reported herein for the first time in aqueous medium, needs certainly further mechanistic study to elucidate the role of water onto the Breslow intermediate reactivity. A more in-depth structure–activity study of azolium will underway to identify and tune key catalysts features such as stability, efficiency and selectivity in water. The bio-inspired Stetter reaction's approach presents many important aspects of click-chemistry, including atom economy and the wide availability of starting materials. Even if extensively used in total synthesis, very few examples involved biomolecules. The biomolecules are most of the time insoluble or unstable in organic solvents. Hence, the development of this reaction in an aqueous environment would be of great interest to make the link between chemistry and biology, expanding meaningfully the toolbox of bio-conjugation chemistry.Experimental
Synthesis of azolium salts
First step of a two-step sequence: synthesis of 5-methyl-4-[2-(phenylmethoxy)ethyl]-thiazole. To a suspension of NaH 60% (726 mg, 18.16 mmol) in mineral oil in anhydrous DMF (21 mL), at 0 °C, were added dropwise benzyl bromide (2.16 mL, 18.16 mmol) followed by a solution of 2-(4-methylthiazol-5-yl)ethanol (1.67 mL, 13.97 mmol) anhydrous DMF (10 mL). The reaction mixture was stirred during 16 h at room temperature and ethyl acetate (20 mL) was added. The organic layer was washed with sat. aq. NH4Cl (3 × 10 mL), brine (3 × 10 mL) and water (3 × 10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure to afford the benzylated product as a yellow oil (2.6 g, 80%). NMR data are consistent with literature.
Rf: 0.36 (pentane/EtOAc: 70/30).
1H NMR (300 MHz, CDCl3) δ (ppm): 8.46 (s, 1H), 7.33–7.17 (m, 5H), 4.44 (s, 2H), 3.56 (t, J = 5.4 Hz, 2H), 2.95 (t, J = 5.4 Hz, 2H), 2.34 (s, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 150.0, 149.5, 138.4, 128.7, 128.4, 127.9, 127.9, 73.3, 70.3, 27.3, 15.3. IR (ATR) ν (cm−1): 3024, 2922, 2858, 1597, 1496, 1453, 1361, 1096, 912, 737, 697.
Second step: 1,5-dimethyl-4-[2-(phenylmethoxy)ethyl]-thiazolium iodide 8. A solution of iodomethane (788 μL, 12.66 mmol) in anhydrous acetonitrile (2 mL) was added on first step resulting compound 4-methyl-5-[2-(phenylmethoxy)ethyl]-thiazole (3.11 g, 13.33 mmol). The reaction mixture was stirred at 90 °C during 24 h then at room temperature during 12 h. The solvent was then removed under reduced pressure, diethyl ether (10 mL) was added and the mixture thus obtained was triturated in order to remove impurities, affording the azolium 8 as a brown gum without any further purification (2.5 g, 53%).
Rf: 0.56 (pentane/EtOAc: 70/30). Mp: 89–91 °C.
1H NMR (300 MHz, CDCl3) δ (ppm): 10.86 (s, 1H), 7.46–7.23 (m, 5H), 4.55 (s, 2H), 4.36 (s, 3H), 3.73 (t, J = 5.4 Hz, 2H), 3.13 (t, J = 5.4 Hz, 2H), 2.52 (s, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 157.2, 142.2, 137.1, 135.1, 128.7, 128.2, 128.1, 73.6, 67.8, 41.7, 28.0, 12.8. IR (ATR) ν (cm−1): 3436, 3027, 2924, 2863, 1593, 1452, 1362, 1097, 918, 735, 700. NMR spectra for compound 8 are reported in the ESI.
First step of a two-step sequence. To a suspension of NaH 60% (726 mg, 18.16 mmol) in mineral oil in anhydrous DMF (21 mL), at 0 °C, were added dropwise 2-bromo-methyl-naphthalene (4.01 g, 18.16 mmol) followed by a solution of 2-(4-methylthiazol-5-yl)ethanol (1.67 mL, 13.97 mmol) anhydrous DMF (10 mL). The reaction mixture was stirred during 16 h at room temperature and ethyl acetate (20 mL) was added. The organic layer was washed with sat. aq. NH4Cl (3 × 10 mL), brine (3 × 10 mL) and water (3 × 10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The crude was purified by flash chromatography over silica gel (EP/AcOEt: 70/30 to 60/40) to provide the intermediate compound as a yellow oil (3.715 g, 72%).
1H NMR (300 MHz, CDCl3) δ (ppm): 8.58 (s, 1H), 7.87–7.79 (m, 3H), 7.75 (s, 1H), 7.52–7.41 (m, 3H), 4.70 (s, 2H), 4.21 (s, 3H), 3.70 (t, J = 6.3 Hz, 2H), 3.08 (t, J = 6.6 Hz, 2H), 2.41 (s, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm) 149.5, 149.1, 135.4, 133.1, 132.8, 128.0, 127.9, 127.7, 127.6, 126.1, 126.0, 125.5, 72.9, 69.9, 26.9, 14.8. HRMS (ESI) calculated for C17H17NOSNa+ [M + Na]+ 306.09231, found 306.09220.
Second step. A solution of iodomethane (775 μL, 12.45 mmol) in anhydrous acetonitrile (2 mL) was added on the compound obtained from the first step (3.715 g, 13.10 mmol). The reaction mixture was stirred at 90 °C during 24 h then at room temperature during 12 h. The solvent was then removed under reduced pressure, THF (10 mL) was added and the mixture thus obtained was triturated in order to remove impurities, affording the azolium 26 as a light brown powder without any further purification (4.1 g, 74%).
Mp: 111–112 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 10.92 (bs, 1H), 7.88–7.78 (m, 3H), 7.74 (s, 1H), 7.53–7.38 (m, 3H), 4.69 (s, 2H), 4.28 (s, 3H), 3.75 (t, J = 5.4 Hz, 2H), 3.09 (t, J = 5.4 Hz, 2H), 2.43 (s, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm) 156.9, 142.1, 135.0, 134.6, 133.1, 132.9, 128.4, 127.9, 127.7, 126.8, 126.3, 126.2, 125.9, 73.5, 67.8, 41.6, 27.9, 12.7. IR (ATR) ν (cm−1): 3431, 2990, 2945, 2861, 1667, 1598, 1481, 1334, 1273, 1098, 918, 860, 752. HRMS (ESI) m/z calcd for C18H20NOS+ [M+] 298.12601, found 298.12560. NMR spectra for compound 26 are reported in the ESI.†
Second step. A solution of iodopropane (1.29 mL, 13 mmol) in anhydrous acetonitrile (2 mL) was added on the compound obtained from the first step (3.715 g, 13.10 mmol). The reaction mixture was stirred at 90 °C during 24 h then at room temperature during 12 h. The solvent was then removed under reduced pressure, THF (10 mL) was added and the mixture thus obtained was triturated in order to remove impurities, affording the azolium 27 as a light brown powder without any further purification (3.72 g, 71%).
Mp: 76–78 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 10.92 (s, 1H), 7.45–7.19 (m, 5H), 4.65 (t, J = 7.5 Hz, 2H), 4.55 (s, 2H), 3.74 (t, J = 5.4 Hz, 2H), 3.15 (t, J = 5.4 Hz, 2H), 2.53 (s, 3H), 2.10–1.86 (m, 2H), 1.05 (t, J = 7.5 Hz, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm) 157.1, 141.5, 137.1, 135.6, 128.7, 128.2, 128.0, 73.6, 67.8, 55.1, 41.7, 28.1, 23.6, 12.7, 10.8. IR (ATR) ν (cm−1): 3040, 2963, 2863, 1586, 1450, 1311, 1101, 1042, 745, 701. HRMS (ESI) m/z calcd for C16H22NOS+ [M+] 276.14221, found 276.14105. NMR spectra for compound 27 are reported in the ESI.†
Synthesis of compounds 18–25
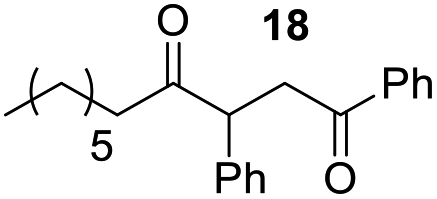
Rf: 0.69 (EP/AcOEt: 95/5) 1H NMR (300 MHz, CDCl3) δ (ppm): 7.94 (d, J = 8.4 Hz, 2H), 7.59–7.18 (m, 8H), 4.41 (dd, J = 3.6 Hz, 10.2 Hz, 1H), 4.02 (dd, J = 10.2 Hz, 18 Hz, 1H), 3.10 (dd, J = 3.6 Hz, 18 Hz, 1H), 2.68–2.37 (m, 2H), 1.64–1.42 (m, 2H), 1.31–1.08 (m, 8H), 0.87 (t, J = 6.3 Hz, 3H) 13C NMR (75.46 MHz, CDCl3) δ (ppm): 209.5, 198.3, 138.3, 136.6, 133.2, 129.1, 128.6, 128.4, 128.2, 127.6, 53.4, 42.4, 41.9, 31.7, 29.1, 29.0, 23.7, 22.7, 14.2. IR (ATR) ν (cm−1): 2925, 2856, 1684, 1598, 1580, 1494, 1451, 753, 700. HRMS (ESI) m/z calcd for C23H29O2+ [M + H]+ 337.21621, found 337.21631. NMR spectra for compound 18 are reported in the ESI.†
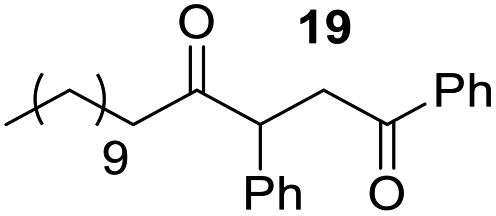
Rf: 0.55 (EP/AcOEt: 95/5) 1H NMR (300 MHz, CDCl3) δ (ppm): 7.93 (d, J = 7.5 Hz, 2H), 7.57–7.20 (m, 8H), 4.41 (dd, J = 3.3 Hz, 10.2 Hz, 1H), 4.03 (dd, J = 10.2 Hz, 18 Hz, 1H), 3.09 (dd, J = 3.3 Hz, 18 Hz, 1H), 2.71–2.37 (m, 2H), 1.71–1.44 (m, 2H), 1.42–1.09 (m, 16H), 0.87 (t, J = 6.6 Hz, 3H) 13C NMR (75.46 MHz, CDCl3) δ (ppm): 209.2, 198.0, 138.2, 136.5, 133.1, 129.0, 128.5, 128.3, 128.0, 127.5, 53.3, 42.4, 41.8, 31.9, 29.6, 29.4, 29.3, 29.0, 23.6, 22.7, 14.1. IR (ATR) ν (cm−1): 2923, 2853, 1714, 1685, 1599, 1582, 1494, 1453, 752, 698, 690. HRMS (ESI) m/z calcd for C27H37O2+ [M + H]+ 393.27881, found 393.27895. NMR spectra for compound 19 are reported in the ESI.†
Rf: 0.32 (toluene/acetone: 99/1). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.03–7.86 (m, 2H), 7.63–7.04 (m, 8H), 5.44–5.26 (m, 2H), 4.44 (dd, J = 3.6 Hz, 10.2 Hz, 1H), 4.06 (dd, J = 10.2 Hz, 18 Hz, 1H), 3.14 (dd, J = 3.6 Hz, 18 Hz, 1H), 2.71–2.37 (m, 2H), 2.12–1.88 (m, 4H), 1.69–1.42 (m, 2H), 1.40–0.98 (m, 16H), 0.96–0.80 (m, 3H). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 209.6, 198.4, 138.3, 136.7, 133.3, 130.0, 130.0, 129.2, 128.7, 128.5, 128.2, 127.6, 53.4, 42.5, 42.0 32.1, 29.6, 29.5, 29.5, 29.4, 29.1, 27.0, 22.5, 14.2. IR (ATR) ν (cm−1): 2927, 2855, 1713, 1683, 1598, 1450, 1400, 1202, 753, 690. HRMS (ESI) m/z calcd for C31H43O2+ [M + H]+ 447.32576, found 447.32574. NMR spectra for compound 20 are reported in the ESI.†
Rf: 0.43 (CH2Cl2). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.88 (d, J = 8.1 Hz, 2H), 7.58–7.06 (m, 8H), 4.36 (dd, J = 3.6 Hz, 10.5 Hz, 1H), 3.98 (dd, J = 10.5 Hz, 18 Hz, 1H), 3.44 (t, J = 6.3 Hz, 2H), 3.07 (dd, J = 3.3 Hz, 18 Hz, 1H), 2.76 (brs, 1H), 2.71–2.36 (m, 2H), 1.70–1.29 (m, 4H). 13C NMR (75.5 MHz, CDCl3) δ (ppm) 209.5, 198.2, 137.9, 136.3, 133.2, 129.0, 128.4, 128.2, 128.0, 127.5, 61.9, 53.1, 42.3, 41.2, 31.7, 19.6. IR (ATR) ν (cm−1) 3402, 2935, 2864, 1712, 1681, 1598, 1581, 1494, 1450, 1073, 754, 696. HRMS (ESI) m/z calcd for C20H21O3− [M − H+] 309.14962, found 309.14921. NMR spectra for compound 21 are reported in the ESI.†
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48) was obtained which were not separated.
:48) was obtained which were not separated.Rf: 0.20 (EP/AcOEt: 90/10). 1H NMR (300 MHz, CDCl3) δ (ppm) 8.01–7.91 (m, 4H, d1 + d2), 7.64–7.51 (m, 2H, d1 + d2), 7.50–7.39 (m, 4H, d1 + d2), 7.39–7.22 (m, 10H, d1 + d2), 5.54 (d, J = 7.5 Hz, 1H, d2), 5.26 (d, J = 7.5 Hz, 1H, d1), 4.75–4.55 (m, 3H, d1 + d2), 4.52–4.38 (m, 3H, d1 or d2), 4.06 (dd, J = 3 Hz, 10.2 Hz, 1H, d2), 4.01 (dd, J = 3.2 Hz, 10.2 Hz, 1H, d1), 3.32 (dd, J = 3.6 Hz, 18 Hz, 1H, d2), 3.18 (dd, J = 3.6 Hz, 18.3 Hz, 1H, d1), 1.57 (d, J = 7.2 Hz, 3H, d2), 1.49 (s, 9H, d1), 1.41 (s, 9H, d2), 1.18 (d, J = 6 Hz, 3H, d1) 13C NMR (75.5 MHz, CDCl3) δ (ppm) 208.9 d2, 207.7 d1, 198.1 d2, 197.7 d1, 155.2 d2, 154.8 d1, 133.4, 129.4, 129.1, 128.6, 128.4, 128.4, 128.2, 128.1, 127.9, 127.7, 79.6 d1 or d2, 79.4 d1 or d2, 54.7 d2, 54.0 d1, 49.8 d1, 49.4 d2, 44.1 d1, 42.5 d1, 30.3 d2, 29.7 d1, 28.4 d1, 28.4 d2, 18.6 d1, 17.4 d2. IR (ATR) ν (cm−1) 3423, 2978, 2932, 1705, 1684, 1493, 1449, 1365, 1241, 1164, 1004, 753, 690. HRMS (ESI) m/z calcd for C27H28NO4+ [M + H]+ 382.20128, found 382.20113. NMR spectra for compound 22 are reported in the ESI.†
:41) was obtained which were not separated.Rf: 0.2 (EP/AcOEt: 95/5). 1H NMR (300 MHz, CDCl3) δ (ppm) 8.02–7.87 (m, 2H, d1 + d2), 7.63–6.89 (m, 13H, d1 + d2), 5.21 (d, J = 9 Hz, 1H, d1), 5.08 (d, J = 9 Hz, 1H, d2), 4.79–4.52 (m, 2H, d1 + d2), 4.08–3.86 (m, 1H, d1 + d2), 3.44–2.98 (m, 2.4H, d1 + d2), 2.76 (dd, J = 7.5 Hz, 13.8 Hz, 0.6H, d1) 1.54–1.12 (m, 9H, d1 + d2). 13C NMR (75.5 MHz, CDCl3) δ (ppm) 207.7, 206.9 (d1 and d2), 198.1, 197.7 (d1 and d2), 155.4, 154.8 (d1 and d2), 137.3, 137.1, 136.7, 136.5, 136.4, 133.4, 133.4, 129.8, 129.5, 129.3, 129.2, 128.7, 128.5, 128.2, 127.9, 127.7, 126.8, 126.4 (d1 and d2), 79.9, 79.5 (d1 and d2), 59.2, 58.7 (d1 and d2), 50.9, 50.0 (d1 and d2), 43.1, 42.4 (d1 and d2), 38.0, 36.7 (d1 and d2), 29.8, 28.4, 28.4, 28.0 (d1 and d2). IR (ATR) ν (cm−1) 3379, 3063, 3030, 3005, 2977, 2929, 1706, 1684, 1599, 1582, 1494, 1452, 1392, 1366, 1333, 1286, 1245, 1206, 1164, 1076, 1054, 1004, 987, 946, 911, 860, 751, 732, 697, 648, 618, 558, 522. HRMS (ESI) m/z calcd for C29H32NO4+ [M + H]+ 458.23258, found 458.23274. NMR spectra for compound 23 are reported in the ESI.†
Rf: 0.4 (pentane/AcOEt: 90/10). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.12–7.95 (m, 4H), 7.63–7.16 (m, 8H), 6.90 (d, J = 8.7 Hz, 2H), 5.31 (dd, J = 3.6 Hz, 9.9 Hz, 1H), 4.22 (dd, J = 9.9 Hz, 18 Hz, 1H), 3.83 (s, 3H), 3.29 (dd, J = 3.9 Hz, 18 Hz, 1H). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 198.3, 197.5, 163.5, 139.3, 136.7, 133.3, 131.4, 129.5, 129.3, 128.7, 128.3, 127.4, 113.8, 55.5, 48.5, 43.9. IR (ATR) ν (cm−1): 3063, 2965, 2921, 2842, 1666, 1600, 1575, 1494, 1450, 1249, 998, 843, 754, 701. HRMS (ESI) m/z calcd for C23H21O3+ [M + H]+ 345.14852, found 345.14835. NMR spectra for compound 24 are reported in the ESI.†
Rf: 0.25 (pentane/AcOEt: 85/15). 1H NMR (300 MHz, CDCl3) δ (ppm) 9.48 (bs, 1H), 8.04–7.96 (m, 2H), 7.61–7.21 (m, 8H), 7.07–7.02 (m, 1H), 6.97–6.92 (m, 1H), 6.28–6.21 (m, 1H), 5.06 (dd, J = 4.2 Hz, 9.9 Hz, 1H), 4.17 (dd, J = 9.9 Hz, 18 Hz, 1H), 3.32 (dd, J = 4.2 Hz, 18 Hz, 1H). 13C NMR (75.46 MHz, CDCl3) δ (ppm) 198.1, 189.0, 139.8, 136.7, 133.3, 131.3, 129.1, 128.7, 128.3, 128.2, 127.4, 124.8, 117.4, 111.0, 48.8, 42.9. IR (ATR) ν (cm−1): 3278, 2922, 2849, 1683, 1631, 1596, 1400, 1371, 1240, 1112, 898, 745, 693. HRMS (ESI) m/z calcd for C20H18NO2+ [M + H]+ 304.13321, found 304.13307. NMR spectra for compound 25 are reported in the ESI.†
Procedure for Stetter reaction on benzoin substrate
The benzoin compound 4 (1.2 mmol) and DBU (0.3 mmol) were added to a solution of the 8 (0.3 mmol) in 600 μL of solvent. The brown solution became black. The chalcone 2 (1 mmol) was then added and the reaction was stirred at 95 °C during the corresponding time (see ESI†). After this period, the solvent was evaporated to provide the crude product. Ratios were measured onto 1H NMR spectra of the reaction crude medium of each sample.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mégane Debiais and Reihana Drain thank INSERM for Master training period fellowship. Aladin Hamoud thanks the MESRI for Ph. D grant. Dr Frédéric Robert and Dr Arnaud Gissot are warmly acknowledged for fruitful discussions. We thank CNRS, university of Bordeaux and INSERM for financial support.References
- (a) M. Fontecave and V. Artero, C. R. Chim., 2011, 14, 362 CrossRef CAS; (b) C. K. Prier and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13992 CrossRef CAS; (c) K. Tahara, L. Pan, T. Ono and Y. Hisaeda, Beilstein J. Org. Chem., 2018, 14, 2553 CrossRef CAS; (d) M. Desage-El Murr, ChemCatChem, 2020, 12, 53 CrossRef CAS; (e) For selected recent exemples, see: K. Hanauer, C. Förster and K. Heinze, Eur. J. Inorg. Chem., 2018, 3537 CrossRef CAS; M. Largeron, P. Deschamps, K. Hammad and M.-B. Fleury, Green Chem., 2020, 22, 1894 RSC; Q. Yang, Y. Zhang, W. Zeng, Z.-C. Duan, X. Sang and D. Wang, Green Chem., 2019, 21, 5683 RSC.
- For selected recent reviews, see: (a) D. Kristofikova, V. Modrocka, M. Meciarova and R. Sebesta, ChemSusChem, 2020, 13, 2828 CrossRef CAS; (b) C. Jamieson and A. J. B. Watson, Catal. Sustainability, 2016, 47 CAS; (c) V. da Gama Oliveira, M. F. do Carmo Cardoso and L. da Silva Magalhaes Forezi, Catalysts, 2018, 8, 605 CrossRef.
- For selected reviews, see: (a) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687 RSC; (b) N. Mase and C. F. Barbas, Org. Biomol. Chem., 2010, 8, 4043 RSC; (c) S. Bhowmick, A. Mondal, A. Ghosh, B. Ayndrila and K. C. Bhowmick, Tetrahedron: Asymmetry, 2015, 26, 1215 CrossRef CAS; (d) K. C. Bhowmick and T. Chanda, Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. Z. Wei and W. Cue Berkeley, 2nd edn, 2018, pp. 291–324 Search PubMed; (e) H. Pelissier, Curr. Org. Chem., 2018, 22, 323 CrossRef; (f) M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491 CrossRef CAS.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (b) C. D. Campbell, K. B. Ling and A. D. Smith, in N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Catalysis by Metal Complexes 32, ed. C. S. J. Cazin, ©Springer Science+Business Media B.V., 2011, pp. 263–297 Search PubMed; (c) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC; (d) S. J. Ryan, L. D. Candish and W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC; (e) N-Heterocyclic Carbenes, in Organocatalysis, ed. A. T. Biju, 2019, ©Wiley-VCH Verlag GmbH & Co. KGaA Search PubMed; (f) For recent reviews on NHC catalysis, see: X.-Y. Chen, Z.-H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690 CrossRef CAS; S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425 CrossRef; C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583 CrossRef; K. J. R. Murauski, A. A. Jaworski and K. A. Scheidt, Chem. Soc. Rev., 2018, 47, 1773 RSC; X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862 CrossRef; S. Mukherjee and A. T. Biju, Chem.–Asian J., 2018, 13, 2333 CrossRef; D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef.
- (a) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS; (b) P. A. Frey in Thiamine: Catalytic Mechanisms and Role in Normal and Disease States, ed. F. Jordan and M. S. Patel, Marcel Dekker Inc., New York, 2004, pp. 1–14 Search PubMed.
- (a) R. Breslow, J. Am. Chem. Soc., 1957, 79, 1762 CrossRef CAS; (b) J. M. Duclos and P. Haake, Biochemistry, 1974, 13, 5358 CrossRef CAS; (c) M. K. Denk, J. M. Rodezno, S. Gupta and A. J. Lough, J. Organomet. Chem., 2001, 617–618, 242 CrossRef CAS; (d) O. Holloczki, P. Terleczky, D. Szieberth, G. Mourgas, D. Gudat and L. Nyulászi, J. Am. Chem. Soc., 2011, 133, 780 CrossRef CAS.
- P. Jain, V. R. Chaudhari and A. Kumar, Phys. Chem. Chem. Phys., 2019, 21, 24126 RSC.
- (a) T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366 CrossRef CAS; (b) E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alde and A. C. O'Donoghue, Chem. Commun., 2011, 47, 1559 RSC; (c) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421 CrossRef CAS.
- (a) H. Stetter, R. Y. Raemsch and H. Kuhlmann, Synthesis, 1976, 733 CrossRef CAS; (b) H. Stetter and H. Kuhlmann, Org. React., 1991, 40, 407 CAS.
- (a) K. Iwamoto, M. Hamaya, N. Hashimoto, H. Kimura, Y. Suzuki and M. Sato, Tetrahedron Lett., 2006, 47, 7175 CrossRef CAS; (b) K. Iwamoto, M. Hamaya, N. Hashimoto, H. Kimura, M. Oike and M. Sato, Org. Biomol. Chem., 2008, 6, 912 RSC; (c) H. Zhao, F. W. Jr. Foss and R. Breslow, J. Am. Chem. Soc., 2008, 130, 12590 CrossRef CAS; (d) X. Bi, L. Wu, C. Yan, X. Jing and H. Zhu, J. Chil. Chem. Soc., 2011, 56, 663 CrossRef CAS; (e) C. Yao, D. Wang, J. Lu, B. Qin, H. Zhang, T. Li and C. Yu, Tetrahedron Lett., 2011, 52, 6162 CrossRef CAS; (f) K. A. Hoop, D. C. Kennedy, T. Mishki, G. P. Lopinski and J. P. Pezacki, Can. J. Chem., 2012, 90, 262 CrossRef CAS; (g) J. Yan, R. Sun, K. Shi, K. Li, L. Yang and G. Zhong, J. Org. Chem., 2018, 83, 7547 CrossRef CAS.
- For oxidative carbonylation see: M. Yoshida, Y. Katagiri, W. Zhu and K. Shishido, Org. Biomol. Chem., 2009, 7, 4062 RSC.
- For asymmetric hydration see: H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2010, 132, 2860 CrossRef CAS.
- X. Li, K. Liu, X. Xu, L. Ma, H. Wang, D. Jiang, Q. Zhang and C. Lu, Chem. Commun., 2011, 47, 7860 RSC.
- E. G. Delany, C.-L. Fagan, S. Gundala, K. Zeitler and S. J. Connon, Chem. Commun., 2013, 49, 6513 RSC.
- W. W. Y. Leong, X. Chen and Y. R. Chi, Green Chem., 2013, 15, 1505 RSC.
- P. Suresh, S. Thamotharan and S. S. Ganesan, Catal. Commun., 2018, 111, 47 CrossRef CAS.
- (a) H. Stetter, Angew. Chem., Int. Ed., 1976, 15, 639 CrossRef; (b) S. Mizuhara and L. Handler, J. Am. Chem. Soc., 1954, 76, 571 CrossRef CAS; (c) For review see: M. Gravel and J.-L. Holmes, Comprehensive Organic Synthesis II, vol. 4, 2014, pp. 1384–1406 CrossRef CAS; S. R. Yetra, A. Patra and A. T. Biju, Synthesis, 2015, 47, 1357 CrossRef CAS.
- (a) H. Stetter and H. Kuhlmann, Synthesis, 1975, 379 CrossRef CAS; (b) B. M. Trost, C. D. Shuey, F. Dininno and S. S. Mcelvain, J. Am. Chem. Soc., 1979, 101, 1284 CrossRef CAS; (c) K. L. Baumann, D. E. Butler, C. F. Deering, K. E. Mennen, A. Millar, T. N. Nanninga, C. W. Palmer and B. D. Roth, Tetrahedron Lett., 1992, 33, 2283 CrossRef CAS; (d) P. E. Harrington and M. A. Tius, Org. Lett., 1999, 1, 649 CrossRef CAS; (e) P. E. Harrington and M. A. Tius, J. Am. Chem. Soc., 2001, 123, 8509 CrossRef CAS; (f) C. C. Galopin, Tetrahedron Lett., 2001, 42, 5589 CrossRef CAS; (g) S. Randl and S. Blechert, J. Org. Chem., 2003, 68, 8879 CrossRef CAS; (h) S. Anjaiah, S. Chandrasekhar and R. Grée, Adv. Synth. Catal., 2004, 346, 1329 CrossRef CAS; (i) K. C. Nicolaou, A. Li and D. J. Edmonds, Angew. Chem., Int. Ed., 2006, 45, 7086 CrossRef CAS; (j) K. C. Nicolaou, Y. F. Tang and J. H. Wang, Chem. Commun., 2007, 1922 RSC; (k) J. Xu, E. J. E. Caro-Diaz and E. A. Theodorakis, Org. Lett., 2010, 12, 3708 CrossRef CAS; (l) S. Sommerwerk, S. Kern, L. Heller and R. Csuk, Tetrahedron Lett., 2014, 55, 6243 CrossRef CAS.
- M. Raj and V. K. Singh, Chem. Commun., 2009, 6687 RSC.
- (a) J. Labarre-Lainé, R. Beniazza, V. Desvergnes and Y. Landais, Org. Lett., 2013, 15, 4706 CrossRef; (b) J. Labarre-Lainé, I. Perinan, V. Desvergnes and Y. Landais, Chem.–Eur. J., 2014, 20, 9336 CrossRef; (c) A. Hamoud, P. Barthélémy and V. Desvergnes, Org. Biomol. Chem., 2018, 16, 1760 RSC.
- Imidazolium and triazolium salts have been unsuccessfully tested.
- M. M. Myers, A. R. Bharadwaj, B. C. Milgram and K. A. Scheidt, J. Am. Chem. Soc., 2005, 127, 14675 CrossRef CAS.
- THF was used after being dried according the following procedure: D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351 CrossRef CAS.
- K. Yamashita, T. Osaki, H. Yokota, M. Nango and K. Tsuda, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1711 CrossRef CAS.
- A drawback of organocatalysis is the usually high catalysts loading needed compared to transition metal catalyzed processes. Reaction were also performed using 15 and 20 mol% of thiazolium salt and 30 mol% were found to provide the best yields. (see additional information in ESI†).
- (a) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS; (b) S. Narayan, J. Muldoon, M. G. Finn, V. V. Folkin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS; (c) M. C. Pirrung, Chem.–Eur. J., 2006, 12, 1312 CrossRef CAS.
- (a) P.-C. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714 CrossRef CAS; (b) P.-C. Chiang, Y. Kim and J. W. Bode, Chem. Commun., 2009, 4566 RSC.
- Compounds ratios were calculated using 1H NMR crude spectra signals integrations.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129(17), 5492 CrossRef CAS; (b) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS; (c) S. Mellouli, L. Bousekkine, A. B. Theberge and W. T. S. Huck, Angew. Chem., Int. Ed., 2012, 51, 7981 CrossRef CAS; (d) J. Han, J.-L. Zhang, W.-Q. Zhang, Z. Gao, L.-W. Xu and Y. Jian, J. Org. Chem., 2019, 84, 7642 CrossRef CAS.
- M. Debiais, A. Hamoud, P. Barthélémy and V. Desvergnes, French patent FR 19 09789. International extension 4/09/2020: PCT/EP2020/074787.
- (a) M. Beigi, S. Waltzer and M. Zarei, J. Biotechnol., 2014, 191, 64 CrossRef CAS; (b) M. Müller, G. A. Sprenger and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261 CrossRef.
- (a) Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103 CrossRef CAS; (b) S. Kobayashi, Water in Organic Synthesis, in Science of Synthesis, Thieme, Stuttgart, Workbench edn, 2012 Search PubMed; (c) R. N. Butler and A. G. Coyne, Org. Biomol. Chem., 2016, 14, 9945 RSC and references cited herein.
- V. K. R. Garapati and M. Gravel, Org. Lett., 2018, 20, 6372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08326g |
| This journal is © The Royal Society of Chemistry 2020 |