
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly active and durable WO3/Al2O3 catalysts for gas-phase dehydration of polyols†
Takeshi Aiharaa,
Katsuya Asazumaa,
Hiroki Miura abd and
Tetsuya Shishido*abcd
abd and
Tetsuya Shishido*abcd
aDepartment of Applied Chemistry for Environment, Graduate School of Urban Environmental Sciences, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
bResearch Center for Hydrogen Energy-based Society, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
cResearch Center for Gold Chemistry, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
dElements Strategy Initiative for Catalysts & Batteries, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8520, Japan
First published on 12th October 2020
Abstract
Gas-phase glycerol dehydration over WO3/Al2O3 catalysts was investigated. WO3 loading on γ-Al2O3 significantly affected the yield of acrolein and the catalyst with 20 wt% WO3 loading showed the highest activity. The WO3/Al2O3 catalyst with 20 wt% WO3 loading showed higher activity and durability than the other supported WO3 catalysts and zeolites. The number of Brønsted acid sites and mesopores of the WO3/Al2O3 catalyst did not decrease after the reaction, suggesting that glycerol has continuous access to Brønsted acid sites inside the mesopores of WO3/Al2O3, thereby sustaining a high rate of formation of acrolein. Dehydration under O2 flow further increased the durability of the WO3/Al2O3 catalyst, enabling the sustainable formation of acrolein. In addition, the WO3/Al2O3 catalyst with 20 wt% WO3 loading showed high activity for the dehydration of various polyols to afford the corresponding products in high yield.
Introduction
The conversion of biomass-derived compounds instead of oil-derived resources to valuable chemicals is essential for the sustainable development of society.1–5 Glycerol is one of the most important biomass-derived compounds. It is produced by the hydrolysis of glycerides such as those in vegetable oils and animal fats.6–8 A wide range of commodity chemicals can be produced from glycerol through various reactions such as dehydration,9–25 hydrogenolysis26,27 and oxidation28–30 over metal oxides, zeolites, metal phosphides and metal–organic frameworks. The dehydration of glycerol is a valuable reaction for the formation of acrolein, which is an important intermediate for the production of valuable compounds in the chemical and agricultural industries, such as acrylic acid and DL-methionine.31–34 Since acrolein is produced by the oxidation of propylene over a Bi/Mo-mixed oxide catalyst,35–40 a method for the production of acrolein from glycerol instead of oil-derived resources should be quite valuable.Furthermore, solid acid-catalyzed dehydration of polyols has been reported to be a useful method for obtaining value-added chemicals (e.g. diols, aldehydes, ketones and alkenes).41–48 Many of these reactions, however, usually require a low substrate feed rate and high reaction temperatures to achieve a high conversion rate. Hence, the development of a catalyst that shows high activity and selectivity to give the target products under mild conditions is still a challenge.
On the other hand, the deposition of coke on the catalyst surface contributes to deactivation of the catalyst in dehydration. The mechanism of coke formation and the relationship between coke formation and acid strength and/or porosity of the catalyst have been widely investigated.49–54 Two deactivation mechanisms due to coke formation have been proposed so far; the direct deposition of coke at catalytically active acid sites and coke formation at the entrance of micropores, which hinders the substrate from accessing the active site inside the pore. A better understanding of the deactivation mechanism could be useful for the design of novel dehydration catalysts with high durability.
In the course of our investigation of the relationship between the structure of a two-dimensional tungsten oxide monolayer supported on metal oxides and their performance as acid catalysts,55,56 we reported that supported WO3 catalysts showed high activity for selective biomass conversion, such as the hydrogenolysis of glycerol and tetrahydrofurfuryl alcohol.57–60 These results encouraged us to further explore the application of selective conversion of biomass-derived chemicals over supported WO3 catalysts, in particular polyols to give value-added chemicals.
In this study, gas-phase glycerol dehydration over WO3/Al2O3 catalysts was investigated. The optimization of WO3 loading on Al2O3 enabled us to devise highly active and durable catalysts for glycerol dehydration. To reveal the effect of coke deposition at the catalyst surface on catalytic activity, the reaction under O2 flow and a detailed characterization were carried out. Furthermore, the application of WO3/Al2O3 catalyst to the dehydration of various polyols was investigated.
Experimental
Materials
Glycerol was purchased from Nacalai Tesque, Japan. Polyols, including ethylene glycol, 1,2-propanediol, 1,2-butanediol, 1,3-butanediol, 1,4-butanediol and 1,2-pentanediol, were purchased from Wako Pure Chemical Industries and Tokyo Chemical Industry, Japan. γ-Al2O3 (JRC-ALO-8), ZrO2(JRC-ZRO-3, monoclinic), TiO2(JRC-TIO-4, rutile) and Nb2O5 (JRC-NBO-1, orthorhombic) were supplied by Japan Reference Catalyst. (NH4)10W12O41·5H2O and WO3 were purchased from Wako Pure Chemical Industries, Japan. All zeolites such as H-ZSM-5(90), H-β(25), H-Y(5.5) and H-MOR(20), namely, JRC-Z5-90H, JRC-Z-HB25, JRC-Z-HY5.5 and JRC-Z-HM20, where the number in parentheses is the SiO2/Al2O3 ratio, were supplied by Japan Reference Catalyst.Catalyst preparation
Metal oxides and zeolites were calcined at 773 K for 3 h in flowing air. A series of WO3-loaded catalysts were prepared by impregnation of supports with an aqueous solution of (NH4)10W12O42, dried at 353 K for 6 h, and then calcined at 1123 K for 3 h in flowing air.55,56 The surface coverages of Al2O3, ZrO2 and TiO2 with a WO3 monolayer were estimated by using the cross-sectional area of a WO6 octahedral unit (0.22 nm−2)61 and were almost 100% at 20, 10 and 2.5 wt% WO3 loading, respectively.60Catalytic dehydration of polyols
Catalytic polyol dehydration was performed in a fixed-bed down-flow glass reactor with an inner diameter of 6 mm at an ambient pressure of N2 and O2. Prior to the reaction, 100 mg of a catalyst was placed in the catalyst bed, and the catalyst was treated at 588 K with N2 for 1.5 h. A 10 mol% aqueous solution of polyols was fed through the top of the reactor at a prescribed liquid feed rate together with a carrier gas flow of 20 mL min−1. The total substrate feed rate was 5.1 mmol h−1 and the composition of the mixture gases was substrate/H2O/carrier gas = 1/9/10 (molar ratio). The liquid products were collected in an ice trap (273 K) and a dry ice–methanol trap (195 K) every hour, and analysed by a FID-GC (GC2014, Shimadzu) with a 30 m capillary column of Stabilwax (GL Science, Japan). Gaseous products were analysed by on-line TCD-GC (GC-8A, Shimadzu, Japan) with a 3 m packed column (Porapak-Q, GL Science, Japan). For all the catalysts tested, the carbon balance was >90%, with a few exceptions.Characterization
X-ray diffraction (XRD) patterns of the catalyst were recorded by Rigaku SmartLab with Cu Kα radiation. The samples were scanned from 2θ = 10–70° at 10° min−1 and a resolution of 0.01°. X-ray photoelectron spectroscopy (XPS) analysis of the catalysts was performed using a JEOL JPS-9010 MX instrument. The spectra were measured using Mg Kα radiation. All spectra were calibrated using C 1s (284.5 eV) as a reference. The surface properties of samples were obtained from N2 isotherms obtained using a BELSORP-mini II (MicrotracBEL, Japan) at 77 K. The analysed samples were evacuated at 573 K for 3 h prior to the measurement. The surface area was estimated by the Brunauer–Emmett–Teller (BET) method. Micropore size distributions were determined by t-plot and micropore (MP) analysis. The Barrett–Joyner–Halenda (BJH) method was used to determine mesopore size distributions. FT-IR spectra were recorded by FT/IR-4200 typeA (JASCO, Japan) with a resolution of 4 cm−1. A total of 64 scans were averaged for each spectrum. Each sample (30 mg) was pressed into a self-supporting wafer with a diameter of 20 mm. Catalysts were pretreated under 20 kPa of flowing O2 at 773 K for 1.5 h and then evacuated. Catalysts were exposed to pyridine (0.5 kPa) at 303 K for 30 min, and then evacuated at 423 K for 30 min. NH3 temperature programmed desorption (NH3-TPD) was measured using a BELCAT II (MicrotracBEL, Japan). Fifty mg of the sample was loaded in a quartz reactor and pretreated under a He flow at 773 K for 1 h. NH3 adsorption was carried out over 30 min with 5% NH3/He at 373 K followed by purging with He for 15 min. The temperature was linearly increased from 373–873 K (10 K min−1). The outlet flow was analysed by means of a TCD and Q-Mass (BELMASS, MicrotracBEL, Japan). Thermogravimetric analyses (TG) were performed using a DTG-60H (Shimadzu, Japan) The temperature range was 303–1273 K (10 K min−1).Results and discussion
Fig. 1 shows the effect of WO3 loading over γ-Al2O3 on the gas-phase dehydration of glycerol at 588 K with WHSV by glycerol of 4.7 h−1. The catalysts with 0 and 100 wt% WO3 loading were γ-Al2O3 and bulk WO3, respectively. Dehydration over WO3/Al2O3 provided acrolein as a main product together with the formation of hydroxyacetone as a by-product. The selectivity for acrolein was around 80% regardless of the WO3 loading on γ-Al2O3. In contrast, the WO3 loading significantly affected the yield of acrolein. The yield of acrolein increased with an increase in WO3 loading; WO3/Al2O3 catalyst with 20 wt% WO3 loading showed the highest acrolein yield. On the other hand, excess loading of WO3 led to a decrease in the acrolein yield. Notably, γ-Al2O3 and WO3 showed no activity for this reaction, suggesting that WO3 loaded on γ-Al2O3 was the main active site for the dehydration of glycerol to form acrolein.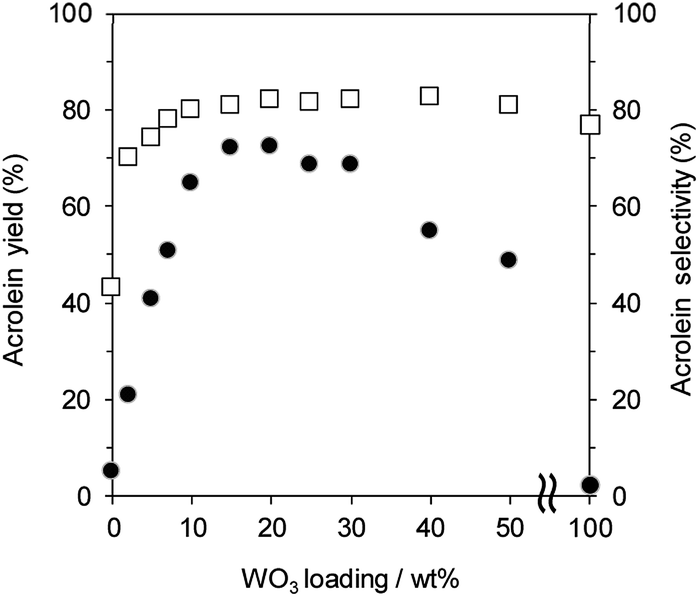 | ||
| Fig. 1 Dehydration of glycerol to acrolein over WO3/Al2O3 catalysts with various WO3 loadings. ●: yield, □: selectivity. Conditions: catalyst (100 mg), WHSV by glycerol (4.7 h−1), T = 588 K. | ||
Since previous studies on the relationship between Brønsted acidity and the activity for glycerol dehydration to form acrolein have revealed that Brønsted acid sites were a main active site for acrolein formation,10,62 the number of Brønsted acid sites (Brønsted acidity) on WO3/Al2O3 catalysts with various WO3 loadings was estimated by pyridine adsorbed FT-IR (Fig. 2 and S2†). Brønsted acidity increased with an increase in WO3 loading, and WO3/Al2O3 catalyst with 20 wt% WO3 loading exhibited the highest Brønsted acidity. In contrast, the Brønsted acidity decreased gradually for catalysts with loadings above 20 wt%. Acrolein was formed in the highest yield with the catalyst with 20 wt% WO3 loading which showed the highest Brønsted acidity. We reported that a two-dimensional WO3 monolayer was formed on γ-Al2O3 when the WO3 loading was below 20 wt% and Brønsted acid sites were generated at the boundaries between the WO3 domains.48,49 The close correlation between the catalytic activity and Brønsted acidity was observed. Some researchers reported that dehydration mechanism of glycerol and the formation of acrolein proceeds over Brønsted acid sites preferentially to over Lewis acid sites.9,10,16,18,62 Similarly, the active sites of this reaction should be the Brønsted acid sites at the boundaries between WO3 domains on γ-Al2O3. After protonation of hydroxyl group at the secondary carbon of glycerol by the Brønsted acid sites at the boundaries between WO3 domains, the dehydration and keto–enol tautomerism takes place to give 3-hydroxypropionaldehyde. The further dehydration of 3-hydroxypropionaldehyde can easily proceed because of the low stability to produce acrolein.
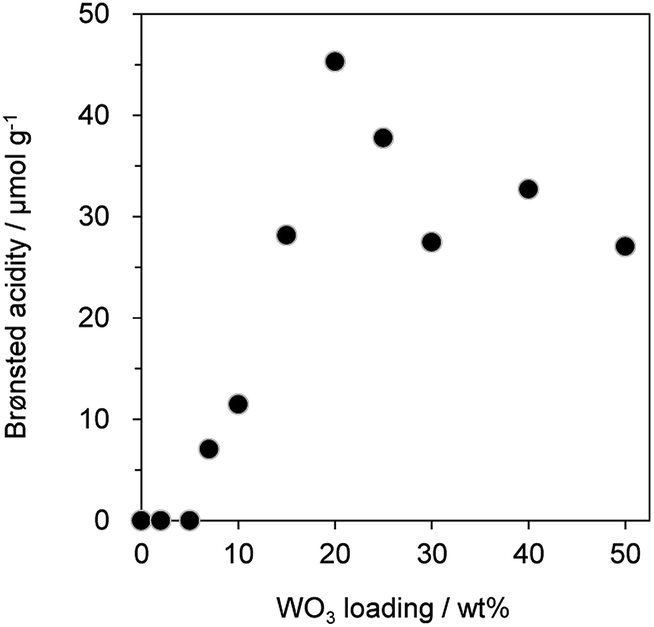 | ||
| Fig. 2 Brønsted acidity of WO3/Al2O3 catalysts with various WO3 loadings. | ||
Fig. 3 shows the durability of various solid acid catalysts in the dehydration of glycerol to acrolein. All the tested catalysts provided acrolein as a main product (Fig. S3†). Although WO3/ZrO2 with 10 wt% WO3,60 WO3/TiO2 with 2.5 wt% WO3, H-ZSM-5(90) and H-β(25) showed high acrolein yield at the initial stage of the reaction, the rapid deactivation of these catalysts was observed. H-Y(5.5), H-MOR(20) and Nb2O5 showed low initial activity. On the other hand, WO3/Al2O3 catalyst with 20 wt% WO3 loading showed high catalytic performance regarding both activity and durability.
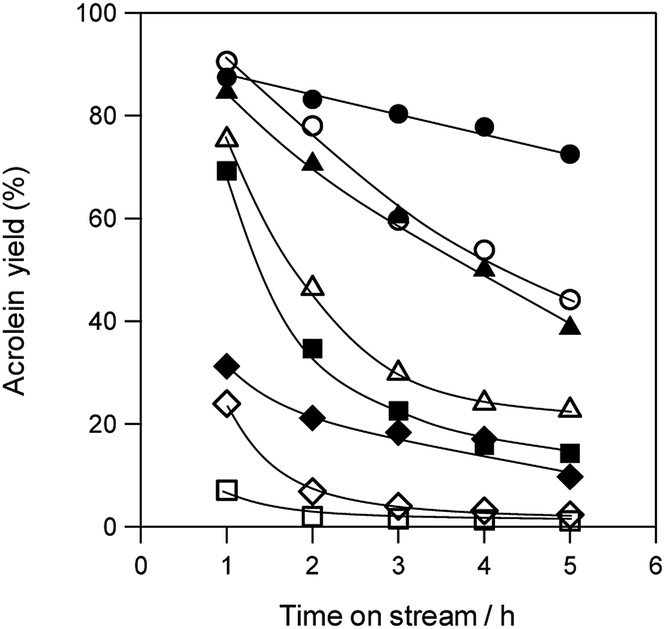 | ||
| Fig. 3 Dehydration of glycerol to acrolein over various acid catalysts. ●: WO3/Al2O3 with 20 wt% WO3 loading, ▲: WO3/ZrO2 with 10 wt% WO3 loading, ■: WO3/TiO2 with 2.5 wt% WO3 loading, ◆: Nb2O5, ○: H-ZSM-5(90), △: H-β(25), □: H-Y(5.5), ◇: H-MOR(20). Conditions: catalyst (100 mg), WHSV by glycerol (4.7 h−1), T = 588 K. | ||
Coke deposition during dehydration is a well-known cause of catalyst deactivation.49–54 Thus, to identify factors that contribute to deactivation, we evaluated the BET surface area of various solid acid catalysts before and after the reaction for 5 h as well as the amount of deposited coke (Table 1). Although coke was deposited on all of the catalyst surfaces, no remarkable relationship was observed between the amount of deposited coke and deactivation rates of catalysts. The BET surface areas of zeolite catalysts were drastically decreased after reaction. On the other hand, the decreases in the surface area of supported WO3 catalysts were much smaller than those of zeolite catalysts. Particularly, WO3/Al2O3 catalyst maintained a larger surface area than the other supported WO3 catalysts. These results suggest that WO3/Al2O3 catalyst maintained a large surface area during the reaction showed the highest acrolein yield and stability in the dehydration of glycerol.
| Catalyst | Amount of deposited cokea/mg gcat−1 | BET surface area/m2 gcat−1 | |
|---|---|---|---|
| Before reaction | After reactionb | ||
| a Estimated by TG analysis.b Reaction under flowing N2 after 5 h. | |||
| WO3/Al2O3 (WO3: 20 wt%) | 131 | 109 | 104 |
| WO3/ZrO2 (WO3: 10 wt%) | 82 | 48 | 69 |
| WO3/TiO2 (WO3: 2.5 wt%) | 35 | 14 | 18 |
| H-ZSM-5 (90) | 138 | 460 | 116 |
| H-β (25) | 198 | 660 | 103 |
| H-Y (5.5) | 225 | 749 | 25 |
| H-MOR (20) | 151 | 502 | 14 |
Oxidative treatment is useful for removing the coke deposited at active Brønsted acid sites.63–65 Hence, we examined the effect of the oxidative treatment of WO3/Al2O3 and H-ZSM-5(90) after the catalytic run on the regeneration of catalytic performance. Treatment with O2 flow (at 823 and 923 K for WO3/Al2O3 and H-ZSM-5(90), respectively) successfully recovered the initial catalytic activity for both WO3/Al2O3 with 20 wt% WO3 loading and H-ZSM-5(90) (Fig. 4). This suggests that the coke deposited at active Brønsted acid sites can be removed by oxidative treatment, and WO3/Al2O3 catalyst can be repeatedly used for the dehydration reaction.
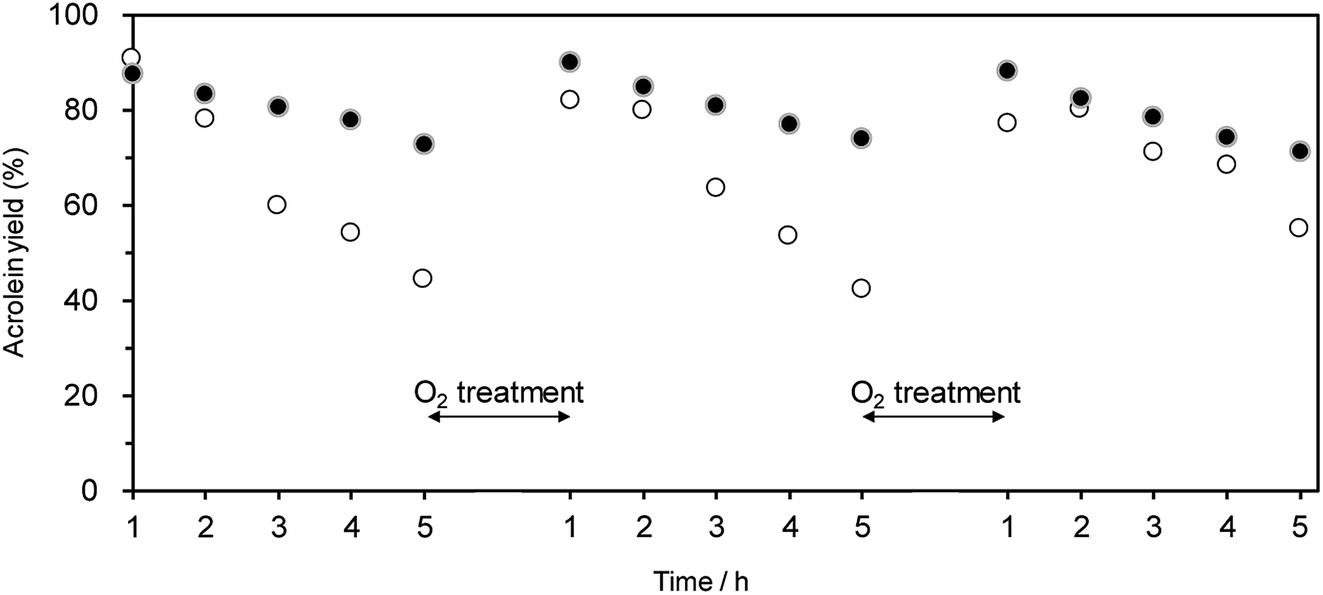 | ||
| Fig. 4 Effect of O2 treatment on acrolein yield over (●) WO3/Al2O3 with 20 wt% WO3 loading and (○) H-ZSM-5(90) catalysts. Conditions: catalyst (100 mg), WHSV by glycerol (4.7 h−1), T = 588 K. O2 treatment was carried out after 5 h at the temperature at which deposited coke completely decomposed; 823 K (WO3/Al2O3) and 923 K (H-ZSM-5), respectively. | ||
Additionally, the dehydration of glycerol over WO3/Al2O3 catalyst with 20 wt% WO3 loading (Fig. 5) and H-ZSM-5 (Fig. S4†) under flowing N2 and O2 gas was investigated. The reaction under O2 flow with both WO3/Al2O3 and H-ZSM-5 catalysts showed constant acrolein yields of around 95% for 5 h, indicating that the reaction under an O2 atmosphere was effective for protecting the catalysts from deactivation. Note that no excess oxidised products (such as acrylic acid) were obtained in this reaction condition.
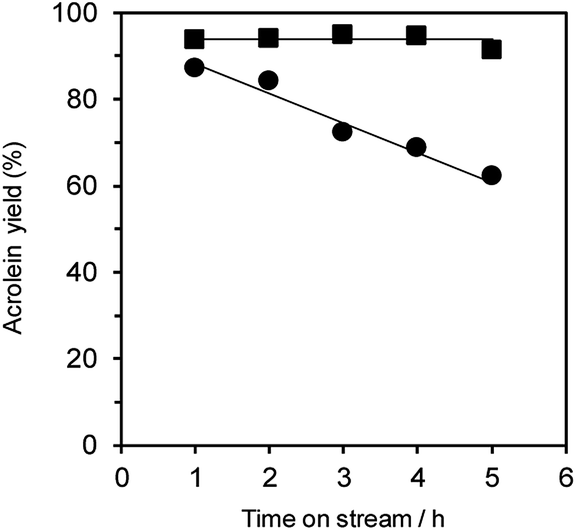 | ||
| Fig. 5 Dehydration of glycerol to acrolein over WO3/Al2O3 catalysts with 20 wt% WO3 loading and under flowing N2 (circle) and O2 (square). Conditions: catalyst (100 mg), WHSV by glycerol (4.7 h−1), T = 588 K. | ||
Table 2 shows the effect of the feed gas on the physical properties of WO3/Al2O3 and H-ZSM-5 catalysts after the catalytic run. The amount of deposited coke in the reaction under O2 was greater than that under N2 over both catalysts. The acidity of catalysts after the reaction with flowing N2 and O2 was also estimated by NH3-TPD (Fig. S6†). Note that pyridine adsorbed FT-IR measurement could not be used to estimate the acidity because coke deposition on the catalyst caused quite low IR transmittance. No difference in the acidity of the WO3/Al2O3 catalyst between before and after the reaction was observed regardless of the reaction atmosphere. In contrast, the area of NH3 desorption peak at 673 K was drastically decreased, indicating that the number of strong acid sites of H-ZSM-5 was decreased by the reaction. (Fig. S6(B)).† The micropore and mesopore distributions of WO3/Al2O3 and H-ZSM-5 catalysts are shown in Fig. 6. No micropores were observed in the WO3/Al2O3 catalyst. Furthermore, no significant change in pore size distribution after the reaction under both N2 and O2 was observed. On the other hand, H-ZSM-5 showed large number of micropores and mesopores. The catalytic runs drastically decreased the number of micropores in H-ZSM-5. These results suggest that coke deposition at the entrance of the H-ZSM-5 pore reduced the ability of glycerol to access the acid sites inside the micropores. Based on these results, we can conclude that the high activity and durability of WO3/Al2O3 are responsible for the fact that the mesoporous nature of WO3/Al2O3 was not prevented by coke formation at the entrance of pores and Brønsted acid sites.
| Catalyst | Carrier gas | Amount of deposited cokea/mg gcat−1 | BET surface area/m2 gcat−1 | Acidityb/μmol gcat−1 | ||
|---|---|---|---|---|---|---|
| Before reaction | After reactionc | Before reaction | After reactionc | |||
| a Estimated by TG analysis.b Estimated by NH3-TPD.c Reaction after 5 h. | ||||||
| WO3/Al2O3 (WO3: 20 wt%) | N2 | 131 | 109 | 104 | 296 | 104 |
| O2 | 221 | 69 | 297 | |||
| H-ZSM-5 (90) | N2 | 138 | 460 | 116 | 158 | 48 |
| O2 | 170 | 131 | 30 | |||
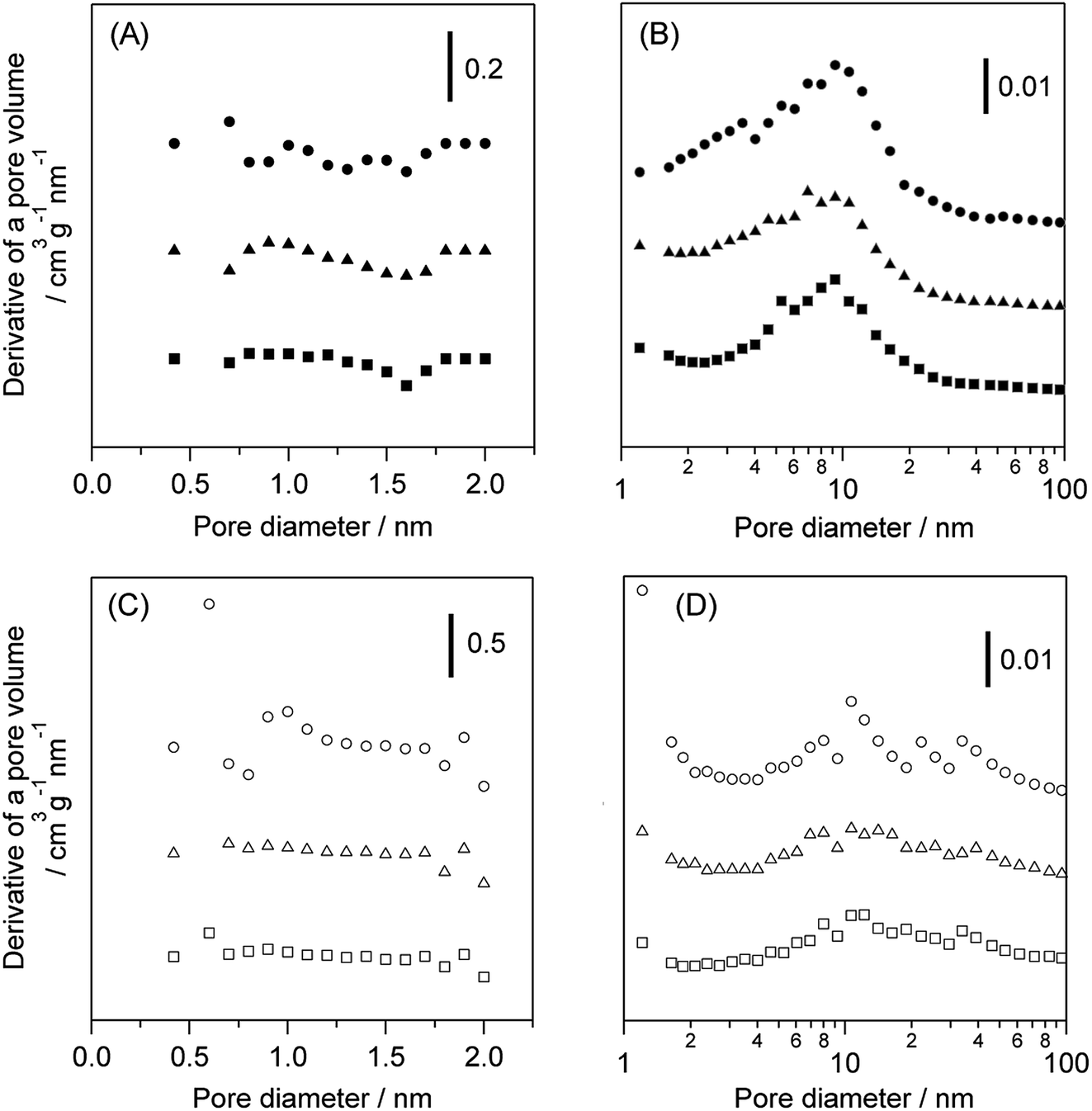 | ||
| Fig. 6 Pore size distribution of the (A) micropore and (B) mesopore regions in WO3/Al2O3 with 20 wt% WO3 loading and (C) micropore and (D) mesopore regions in H-ZSM-5(90). Fresh catalyst (circle), after the reaction with N2 (triangle) and O2 (square) for 5 h. | ||
The dehydration of polyols other than glycerol can also provide useful chemicals. Therefore, the dehydration of various polyols over WO3/Al2O3 catalyst with 20 wt% WO3 loading was investigated (Fig. 7). The reactions of polyols gave the corresponding aldehydes, dienes, ketones and ethers. In particular, the dehydration of polyols with hydroxyl groups at the 1,2-position such as glycerol, ethylene glycol, 1,2-propanediol, 1,2-butanediol and 1,2-pentanediol afforded the aldehydes (e.g. acrolein, acetaldehyde, propionaldehyde, butyraldehyde and valeraldehyde) as main products. On the other hand, the reaction of 1,3-butanediol mainly produced 1,3-butadiene through double dehydration. In contrast, the dehydration of 1,4-butanediol gave tetrahydrofuran as a sole product. These results indicate that WO3/Al2O3 catalyst with 20 wt% WO3 loading shows high activity and durability for the dehydration of various polyols.
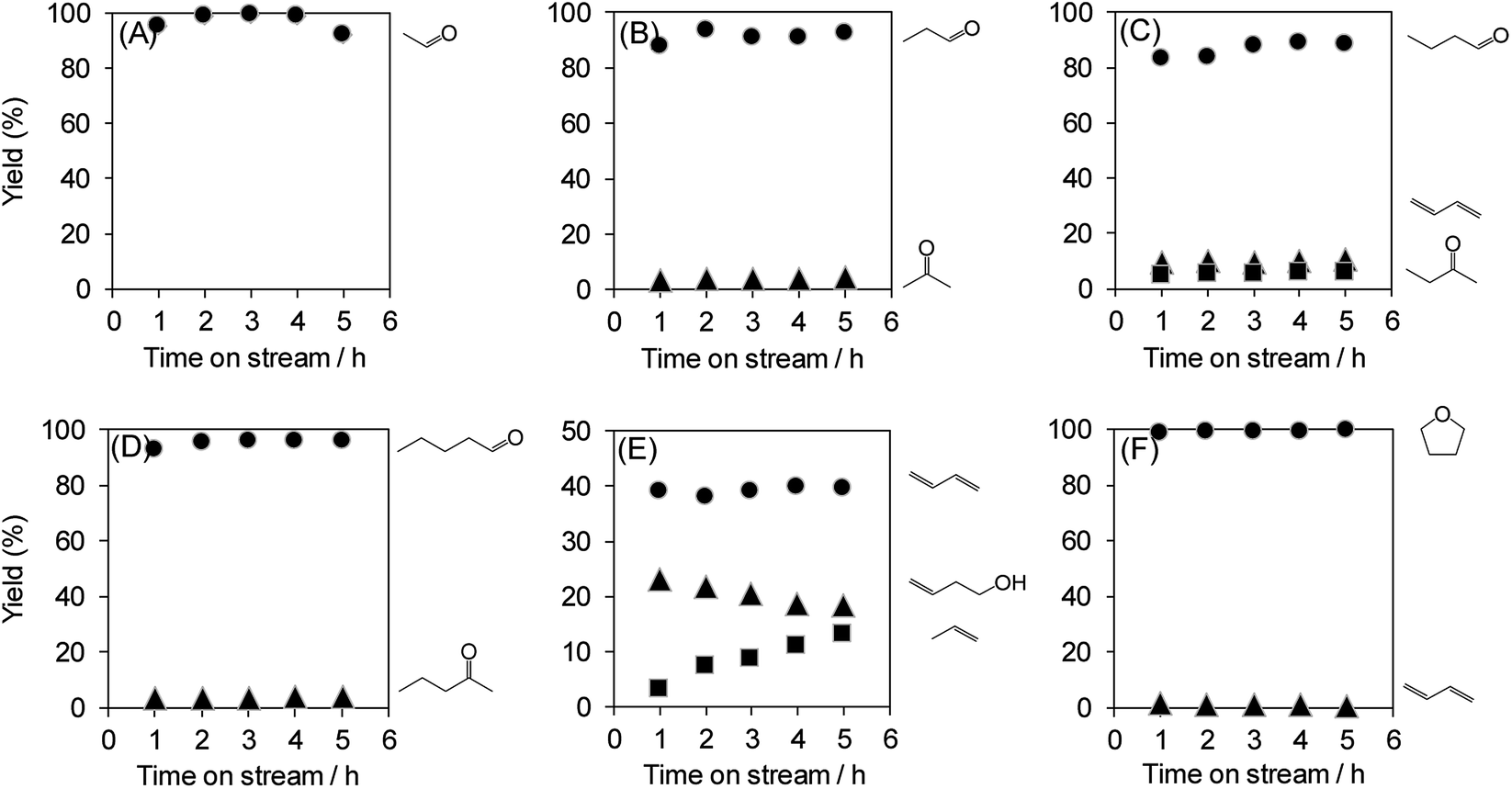 | ||
| Fig. 7 Dehydration of various polyols over WO3/Al2O3 catalysts with 20 wt% WO3 loading. Conditions: catalyst (100 mg), WHSV by substrate (4.7 h−1), T = 588 K. (A) Ethylene glycol, (B) 1,2-propanediol, (C) 1,2-butanediol, (D) 1,2-pentanediol, (E) 1,3-butanediol and (F) 1,4-butanediol. | ||
Conclusions
Gas-phase dehydration of polyols over WO3/Al2O3 catalysts was investigated. WO3 loading on γ-Al2O3 strongly affected the activity for the dehydration of glycerol to acrolein, and WO3/Al2O3 catalyst with 20 wt% WO3 loading showed the highest acrolein yield. Brønsted acid site on the WO3/Al2O3 surface catalysed the formation of acrolein with high selectivity. WO3/Al2O3 catalyst with 20 wt% WO3 loading showed a higher product yield than the other supported WO3 catalysts and zeolites. H-ZSM-5 has both micro- and mesopores and coke deposits at the entrances of those pores. On the other hand, WO3/Al2O3 catalysts have no micropores, suggesting that glycerol can access surface Brønsted acid sites at the inside of the mesopores of WO3/Al2O3 and acrolein is continuously formed with high durability during the reaction. No decrease in acrolein yield was observed under O2 flow during dehydration over WO3/Al2O3 catalyst. On the other hand, the amount of deposited coke under O2 flow was greater than that under N2, implying that coke deposited at the catalyst surface was removed by O2. Furthermore, WO3/Al2O3 catalyst with 20 wt% WO3 loading showed high activity and durability for the dehydration of various polyols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially supported by the Program for Elements Strategy Initiative for Catalysts & Batteries (ESICB) (Grant JPMXP0112101003). This work was also supported in part by Grants-in-Aid for Scientific Research (B) (Grant 17H03459) and Scientific Research on Innovative Areas (Grant 17H06443) commissioned by MEXT of Japan. The XAFS experiments at SPring-8 were conducted with the approval (No. 2019A1717) of the Japan Synchrotron Radiation Research Institute (JASRI).Notes and references
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas; 2004 Search PubMed.
- M. Pagliaro and M. Rossi, The Future of Glycerol, 2nd edn, RSC Publishing, Cambridge, 2010 Search PubMed.
- C.-H. Clayton Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Max Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- J. ten dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef CAS.
- A. Corma, G. Huber, L. Sauvanaud and P. Oconnor, J. Catal., 2008, 257, 163–171 CrossRef CAS.
- S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Green Chem., 2007, 9, 1130–1136 RSC.
- S. Chai, H. Wang, Y. Liang and B. Xu, J. Catal., 2007, 250, 342–349 CrossRef CAS.
- S.-H. Chai, B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, Catal. Today, 2014, 234, 215–222 CrossRef CAS.
- L.-Z. Tao, S.-H. Chai, Y. Zuo, W.-T. Zheng, Y. Liang and B.-Q. Xu, Catal. Today, 2010, 158, 310–316 CrossRef CAS.
- L. H. Vieira, K. T. G. Carvalho, E. A. Urquieta-González, S. H. Pulcinelli, C. V. Santilli and L. Martins, J. Mol. Catal. A: Chem., 2016, 422, 148–157 CrossRef CAS.
- M. H. Haider, N. F. Dummer, D. Zhang, P. Miedziak, T. E. Davies, S. H. Taylor, D. J. Willock, D. W. Knight, D. Chadwick and G. J. Hutchings, J. Catal., 2012, 286, 206–213 CrossRef CAS.
- B. Katryniok, S. Paul, V. Bellière-Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079–2098 RSC.
- E. Tsukuda, S. Sato, R. Takahashi and T. Sodesawa, Catal. Commun., 2007, 8, 1349–1353 CrossRef CAS.
- M. R. Nimlos, S. J. Blanksby, X. Qian, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 6145–6156 CrossRef CAS.
- B. Ali, X. Lan, M. T. Arslan, S. Z. A. Gilani, H. Wang and T. Wang, J. Ind. Eng. Chem., 2020, 88, 127–136 CrossRef CAS.
- G. Ruoppolo, G. Landi and A. D. Benedetto, Catalysts, 2020, 10, 673 CrossRef CAS.
- H. Gong, C. Zhou, Y. Cui, S. Dai, X. Zhao, R. Luo, P. An, H. Li, H. Wang and Z. Hou, ChemSusChem, 2020, 13, 4954–4966 CrossRef CAS.
- X. Li, L. Huang, A. Kochubei, J. Huang, W. Shen, H. Xu and Q. Li, ChemSusChem, 2020, 13, 1–8 CrossRef.
- T. Ma, J. Ding, X. Liu, G. Chen and J. Zheng, Korean J. Chem. Eng., 2020, 37, 955–960 CrossRef CAS.
- T. Ma, J. Ding, R. Shao, W. Xu and Z. Yun, Chem. Eng. J., 2017, 316, 797–806 CrossRef CAS.
- J. Mazarío, P. Concepción, M. Ventura and M. E. Domine, J. Catal., 2020, 385, 160–175 CrossRef.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- R. Arundhathi, T. Mizugaki, T. Mitsudome, K. Jitsukawa and K. Kaneda, ChemSusChem, 2013, 6, 1345–1347 CrossRef CAS.
- S. Feng, K. Takahashi, H. Miura and T. Shishido, Fuel Process. Technol., 2020, 197, 106202 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329–1336 RSC.
- S. T. Wu, Q. M. She, R. Tesser, M. D. Serio and C. H. Zhou, Catal. Rev.: Sci. Eng., 2020, 1–43 Search PubMed.
- B. Katryniok, S. Paul, M. Capron and F. Dumeignil, ChemSusChem, 2009, 2, 719–730 CrossRef CAS.
- T. V. Andrushkevich, Catal. Rev., 1993, 35, 213–259 CrossRef CAS.
- A. Yamamoto, Encyclopedia of Chemical Technology, 3rd edn, Wiley, New York, 1978; vol. 2 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, 3rd Completely Revised edn, Wiley, Hoboken, 2008 Search PubMed.
- B. Grzybowska-Świerkosz, Top. Catal., 2002, 21, 35–46 CrossRef.
- B. Grzybowska-Świerkosz, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2000, 96, 297–334 RSC.
- B. Grzybowska-Świerkosz, Top. Catal., 2000, 11/12, 23–42 CrossRef.
- Y. Moro-Oka and W. Ueda, Adv. Catal., 1994, 40, 233–273 CAS.
- G. W. Keulks, L. D. Krenzke and T. M. Notermann, Adv. Catal., 1979, 27, 183–225 Search PubMed.
- R. Otomo, C. Yamaguchi, D. Iwaisako, S. Oyamada and Y. Kamiya, ACS Sustainable Chem. Eng., 2019, 7, 3027–3033 CrossRef CAS.
- L. Li, K. J. Barnett, D. J. McCelland, D. Zhao and G. Liu, Appl. Catal., B, 2019, 245, 62–70 CrossRef CAS.
- Y. Morita, S. Furusato, A. Takagaki, S. Hayashi, R. Kikuchi and S. T. Oyama, ChemSusChem, 2014, 7, 748–752 CrossRef CAS.
- S. Sato, R. Takahashi, N. Yamamoto, E. Kaneko and H. Inoue, Appl. Catal., A, 2008, 334, 84–91 CrossRef CAS.
- N. Yamamoto, S. Sato, R. Takahashi and K. Inui, J. Mol. Catal. A: Chem., 2006, 243, 52–59 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodesawa and N. Honda, J. Mol. Catal. A: Chem., 2004, 221, 177–183 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodekawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodesawa, N. Honda and H. Shimizu, Catal. Commun., 2003, 4, 77–81 CrossRef CAS.
- H. An, F. Zhang, Z. Guan, X. Liu, F. Fan and C. Li, ACS Catal., 2018, 8, 9207–9215 CrossRef CAS.
- C. D. Lago, H. P. Decolatti, L. G. Tonutti, B. O. Dalla Costa and C. A. Querini, J. Catal., 2018, 366, 16–27 CrossRef CAS.
- Y. Choi, H. Park, Y. S. Yun and J. Yi, ChemSusChem, 2015, 8, 974–979 CrossRef CAS.
- M. V. Rodrigues, C. Vignatti, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, Appl. Catal., A, 2015, 495, 84–91 CrossRef CAS.
- J. A. Cecilia, C. García-Sancho, J. M. Mérida-Robles, J. Santamaría-González, R. Moreno-Tost and P. Maireles-Torres, Catal. Today, 2015, 254, 43–52 CrossRef CAS.
- H. Zhang, Z. Hu, L. Huang, H. Zhang, K. Song, L. Wang, Z. Shi, J. Ma, Y. Zhuang and W. Shen, et al., ACS Catal., 2015, 5, 2548–2558 CrossRef CAS.
- T. Kitano, T. Hayashi, T. Uesaka, T. Shishido, K. Teramura and T. Tanaka, ChemCatChem, 2014, 6, 2011–2020 CrossRef CAS.
- M. Saito, T. Aihara, H. Miura and T. Shishido, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.02.009.
- T. Aihara, H. Kobayashi, S. Feng, H. Miura and T. Shishido, Chem. Lett., 2017, 46, 1497–1500 CrossRef CAS.
- T. Aihara, H. Miura and T. Shishido, Catal. Sci. Technol., 2019, 9, 5359–5367 RSC.
- T. Aihara, H. Miura and T. Shishido, Catal. Today, 2020, 352, 73–79 CrossRef.
- S. Feng, A. Nagao, T. Aihara, H. Miura and T. Shishido, Catal. Today, 2018, 303, 207–212 CrossRef CAS.
- C. Pfaff, M. J. P. Zurita, C. Scott, P. Patiño, M. R. Goldwasser, J. Goldwasser, F. M. Mulcahy, M. Houalla and D. M. Hercules, Catal. Lett., 1997, 49, 13–16 CrossRef CAS.
- S. R. Ginjupalli, S. Mugawar, P. Rajan N., P. K. Balla and V. R. C. Komandur, Appl. Surf. Sci., 2014, 309, 153–159 CrossRef.
- M. Massa, A. Andersson, E. Finocchio and G. Busca, J. Catal., 2013, 307, 170–184 CrossRef CAS.
- J. Deleplanque, J.-L. Dubois, J.-F. Devaux and W. Ueda, Catal. Today, 2010, 157, 351–358 CrossRef CAS.
- F. Wang, J.-L. Dubois and W. Ueda, J. Catal., 2009, 268, 260–267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08340b |
| This journal is © The Royal Society of Chemistry 2020 |