
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot mechanochemical exfoliation of graphite and in situ polymerization of aniline for the production of graphene/polyaniline composites for high-performance supercapacitors
Yulin Jiang†
a,
Jiawen Ji†c,
Leping Huangb,
Chengen He *bd,
Jinlong Zhangd,
Xianggang Wangd and
Yingkui Yang*ad
*bd,
Jinlong Zhangd,
Xianggang Wangd and
Yingkui Yang*ad
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, South-Central University for Nationalities, Wuhan 430074, China. E-mail: ykyang@mail.scuec.edu.cn
bState Key Laboratory of New Textile Materials and Advanced Processing Technology, School of Materials Science and Engineering, Wuhan Textile University, Wuhan 430200, China. E-mail: cehe@wtu.edu.cn
cKey Laboratory of Space Manufacturing Technology, Technology and Engineering Center for Space Utilization, Chinese Academy of Sciences, Beijing 100094, China
dGraphene R&D Center, Guangdong Xigu Tanyuan New Materials Corporation Limited & South-Central University for Nationalities, Foshan 528000, China
First published on 17th December 2020
Abstract
Graphene/polyaniline composites have attracted considerable attention as high-performance supercapacitor electrode materials; however, there are still numerous challenges for their practical applications, such as the complex preparation process, high cost, and disequilibrium between energy density and power density. Herein, we report an efficient method to produce graphene/polyaniline composites via a one-pot ball-milling process, in which aniline molecules act as both the intercalator for the exfoliation of graphite and the monomer for mechanochemical polymerization into polyaniline clusters on the in situ exfoliated graphene sheets. The graphene/polyaniline composite electrode delivered a large specific capacitance of 886 F g−1 at 5 mV s−1 with a high retention of 73.4% at 100 mV s−1. The high capacitance and rate capability of the graphene/polyaniline composite can contribute to the fast electron/ion transfer and dominantly capacitive contribution because of the synergistic effects between the conductive graphene and pseudocapacitive polyaniline. In addition, a high energy density of 40.9 W h kg−1 was achieved by the graphene/polyaniline-based symmetric supercapacitor at a power density of 0.25 kW kg−1, and the supercapacitor also maintained 89.1% of the initial capacitance over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
1. Introduction
During the past decade, graphene has been widely used as an electrode material for supercapacitors due to its unique two-dimensional structure, large theoretical surface area, excellent electrical conductivity, and outstanding mechanical strength.1 As a carbon-based electrode material, graphene stores electric double-layer capacitance (EDLC) via fast ion adsorption on the electrode surface (fast kinetics); hence, it can achieve high rate capability, long cycling stability, and moderate theoretical capacitance of 550 F g−1.2 Nevertheless, graphene sheets tend to aggregate via π–π interactions and van der Waals force, which will seriously hinder the rapid diffusion of electrolyte ions and reduce the available surface area; therefore, the specific capacitance of graphene-based electrodes in the aqueous electrolyte is limited to the range of 100–250 F g−1.3 In comparison, transition metal oxides4,5 and conducting polymers6 have apparent advantages because they can provide high pseudocapacitance via redox reactions with the electrolyte ions on/near the electrode surface. As one of the most promising pseudocapacitive materials, polyaniline (PANI) possesses a large theoretical capacitance (1200 F g−1), relatively good conductivity, easy to synthesize, and low cost.7 Since the electrochemical performance of PANI depends highly on its morphology and thickness, Yue et al.8 prepared well-defined PANI nanotubes, which presented a high specific capacitance of 714.7 F g−1 at a discharge current of 0.5 mA due to the unique morphology; however, the capacitance decreased rapidly to 619.8 F g−1 after doubling the current to 1.0 mA due to the relatively low conductivity and dominantly diffusion-controlled energy-storage process (slow kinetics).9 Moreover, the PANI nanotube-based electrode lost 15% of the initial capacitance after only 500 cycles, demonstrating a poor cycling stability, which can be attributed to the swelling, shrinking, or breaking in the PANI skeleton because of the ion doping/de-doping during the charge/discharge process.10To improve the overall performance of supercapacitors, an effective strategy is to develop composite electrode materials by combining graphene with PANI. Such composites can not only provide high specific capacitance because of the efficient pseudocapacitive reaction on/near the large available surface area but also excellent rate performance owing to the highly conductive graphene network as well as outstanding cycle stability, originating from the mechanically stable graphene substrate.10 Till now, numerous efforts have been made for the preparation of graphene/PANI nanocomposites for high-performance supercapacitors. However, the reported graphene/PANI nanocomposites were mainly fabricated using pre-produced graphene or graphene derivatives (such as graphene oxide, GO) as a substrate, followed by the in situ growth of PANI via the oxidative/electrochemical polymerization of aniline.11 For instance, Yang et al.12 prepared a graphene/PANI composite hydrogel via the in situ polymerization of aniline on the surface of GO in the presence of phytic acid, followed by the chemical reduction of GO to improve its electrical conductivity. The hydrogel electrode exhibited a high specific capacitance up to 866 F g−1 at 1 A g−1 and 678 F g−1 (78.3%) at 10 A g−1, and it also maintained 82% of the initial capacitance after 1000 cycles. Zhang et al.13 employed reduced graphene oxide (rGO) as a support electrode to electrodeposit PANI, and the rGO/PANI composite possessed a porous structure and large specific surface area and hence delivered a high specific capacitance of 710 F g−1 at 2 A g−1 with 73% retention at 100 A g−1 and retained 86% of the initial capacitance after 1000 cycles. Jun et al.14 pre-prepared a 3D graphene framework (GF) via chemical vapor deposition (CVD) on a Ni foam with a subsequent etching of Ni, and then the GF/PANI composite was synthesized via the oxidative polymerization of aniline onto the surface of GF. The GF/PANI composite exhibited ultra-high specific capacitances of 1002 and 695 F g−1 at current densities of 1 and 4 mA cm−2, respectively, and it also maintained 86.5% of the initial capacitance after 5000 cycles. Nevertheless, these production processes are complex, have low efficiency, and high cost, thus unconducive for industrial applications.
Notably, rGO sheets are usually produced via chemical oxidation/reduction processes using hazardous oxidizing and reducing reagents, which would not only cause environmental pollution but also introduce oxygen functional groups and structural defects in the rGO sheets, resulting in low electric conductivity and poor rate capability of the rGO-based electrodes.15 In comparison, the ball-milling of graphite is an efficient and low-cost way to produce graphene sheets with few defects at massive production levels;16 moreover, the ball-milling technique is also a general strategy for the synthesis of inorganic metal oxides and organic compounds.17,18 Nevertheless, very few studies have been reported to produce graphene-based composites directly using graphite as the precursor, and further studies are still required to explore more effective routes for fabricating graphene/polymer composites.
This study develops an efficient strategy to prepare graphene/PANI composites via the ball-milling of graphite, aniline, and ammonium persulfate in one pot, where aniline was used as both the intercalator for the exfoliation of graphite. The noncovalent functional reagent stabilizes graphene but also the monomer for mechanochemical polymerization into PANI clusters on the in situ exfoliated graphene sheets. Owing to the synthetic effects between the pseudocapacitive PANI and conductive graphene, the graphene/PANI composites exhibited high specific capacitances and excellent rate performances. The energy storage mechanisms of the graphene/PANI composites were also studied by distinguishing their capacitive contributions and diffusion-controlled contributions. In addition, symmetric supercapacitors were further assembled to evaluate their application prospects. As expected, the devices achieved high energy density, high power density, and outstanding cycling stability.
2. Experimental
2.1 Materials
Graphite powder (≤40 μm), ammonium persulfate (APS, (NH4)2S2O8), aniline (C6H5NH2), sulfuric acid (H2SO4), ethanol, and isopropanol were obtained from Shanghai Sinopharm Chemical Reagent Co., Ltd. Active carbon and polyvinylidene fluoride (PVDF) were purchased from Shanghai Aladdin Bio-Chem Technology Co., LTD. All these chemicals were used directly without any additional treatments. A buckypaper was provided by Nanjing Jicang Nanotechnology Co., LTD and was used as the current collector after washing it with dilute hydrochloric acid and ethanol.2.2 Synthesis of graphene/PANI composites
The graphene/PANI composites were produced via a one-pot ball-milling process. In a typical procedure, 6 g of aniline was dissolved in 60 mL pre-cooled H2SO4 solutions (0.5 mol L−1) and transformed into a stainless-steel jar containing a certain weight of graphite flakes and 500 g stainless steel beads (5 mm in diameter). Then, the jar was sealed and placed in a planetary ball-mill machine to ball mill for 2 h at 300 rpm. After that, 4 g of APS (dissolved in 40 mL H2SO4 solutions) was introduced into the above jar and ball-milled for another 2 h. Finally, the mixture was filtered, washed with ethanol repeatedly, vacuum-dried at 75 °C for 10 h, and dark green powders were obtained. For comparison, a series of graphene/PANI composites were fabricated with different graphite weights of 0.2, 0.4, and 0.6 g, resulting in composites, namely graphene/PANI-1, graphene/PANI-2, and graphene/PANI-3, with the calculated yields up to 72%, 75% and 77%, respectively.Pure PANI was also synthesized via a comparable mechanochemical polymerization of aniline in the absence of graphite. In addition, pure graphene was produced by the ball-milling of graphite for 4 h using aniline as the stripping reagent.
2.3 General characterization
Transmission Electron Microscope (TEM) images were recorded on a TECN G220 S-TWIN system operating at 200 kV. Scanning Electron Microscope (SEM) observations were conducted on a Hitachi SU 8010 field emission microscope. A D/Max 2400 diffractometer was used to record X-ray diffraction (XRD) patterns, a Renishaw spectrometer was employed to record Raman spectra, a PE-100 system was used to record the Fourier transform infrared (FT-IR) spectra, a Diamond TG/f DTG analyzer was used to record thermogravimetric analysis (TGA) curves, and a Multilab 2000 spectrometer was used to perform X-ray photoelectron spectroscopy (XPS).2.4 Electrochemical measurements
The working electrodes were fabricated by grinding the mixture of graphene/PANI composites, active carbon and PVDF (8:1:1 in weight) with an appropriate amount of isopropanol. Subsequently, the as-generated paste was scraped onto bucky papers (1 × 1 cm2), pre-dried at 60 °C for 6 h, compressed at 5 MPa, and vacuum-dried at 100 °C for another 6 h. Based on the weight difference of the bucky paper before and after coating, the active material on each electrode was about 2 mg.
The electrochemical properties of graphene/PANI composites were tested on a CHI760 electrochemical workstation (CH Instruments, Inc., Shanghai). In a three-electrode system, the as-fabricated graphene/PANI electrodes were pre-immersed into an 1.0 mol L−1 H2SO4 electrolyte overnight and used as the working electrodes, where an Ag/AgCl electrode was acted as the reference electrode, and a platinum plate was employed as the counter electrode. Cyclic Voltammetry (CV) measurements were performed in a voltage window of −0.2 to 0.8 V to investigate the energy storage mechanism of the working electrodes.19
Symmetric supercapacitors were further assembled by separating the two working electrodes with a cellulose acetate membrane (soaking with 1.0 mol L−1 H2SO4) and then sealing them into button cells. The electrochemical performances of the symmetric supercapacitors were detected via CV and galvanostatic charge/discharge (GCD) curves. Electrochemical Impedance Spectroscopies (EIS) were also measured in the frequency range of 100 kHz to 0.01 Hz at the open circuit potential. The specific capacitance, energy density and power density of the supercapacitors were calculated from the GCD curves using the equations described in other studies.20,21
3. Results and discussion
3.1 Synthesis and structural characteristics
Mechanical exfoliation of graphite has proved to be the most economical method for the macroscopic preparation of graphene with fewer defects.22 The graphite exfoliation requires to break the enormous van der Waals forces in the graphitic layers; moreover, it also needs to inhibit the aggregation of the as-generated graphene sheets since they tend to re-establish the graphitic structure for minimizing the surface free energy.23 In this study, the graphite flakes were exfoliated by the press and shear forces from the high-energy ball-milling during the first step, and this process was accelerated by the assistance of aniline intercalation since it can effectively reduce the van der Waals force of the graphitic layers by increasing the interlayer spacing.24 In addition, the aromatic aniline molecules can also simultaneously stabilize the as-generated sp2-bonded graphene sheets via π–π non-covalent modification, hence improving the preparation efficiency of graphene.23 In the second step, the addition of APS led to the in situ mechanochemically oxidative polymerization of aniline; therefore, PANI chains were formed on the surface of graphene sheets,25 as illustrated in Fig. 1. Due to the effective method, it achieved a high yield of graphene/PANI composites over 70 wt%. | ||
| Fig. 1 Schematic for the preparation of the graphene/PANI composite in a one-pot ball-milling process. | ||
Pure graphene was successfully produced by the ball-milling of graphite with aniline-assisted intercalation and non-covalent functionalization. The as-exfoliated graphene can be re-dispersed in ethanol and other organic solvents. As shown in Fig. 2a, the TEM image of graphene displays a translucent sheet-like structure, indicating its mono- or few-layer characteristics.26 The pure PANI was synthesized via mechanochemical oxidation polymerization of aniline in the absence of graphite. The TEM image of pure PANI shows a randomly aggregated morphology (Fig. 2b), which is unconducive for the diffusion of electrolyte ions into the polymer blocks, resulting in low electrochemical utilization of PANI.27 Therefore, it is worthwhile to combine the mechanochemical fabrication of graphene and PANI into a one-pot process, which can not only enhance the production efficiency but also inhibit the self-aggregation of graphene and PANI. As shown in Fig. 2c–f, the as-prepared graphene/PANI composites exhibit plate-like structures, in which the locally ordered fringe structures indicate the presence of graphene sheets, and polymer clusters are anchored disorderly on the surface. Moreover, the density of the PANI cluster decreases with the increase in the ratio of graphite, corresponding to the increasing graphene content. Particularly, the polymer clusters and graphene sheets construct an interconnected porous network, as shown in the high magnification TEM image of graphene/PANI-2 (Fig. 2e). This skeleton structure is conducive to enhance electron conduction, ion diffusion, and hence the electrochemical performance of the electrode.28
 | ||
| Fig. 2 TEM images of (a) pure graphene, (b) PANI, and (c) graphene/PANI-1, (d and e) graphene/PANI-2, and (f) graphene/PANI-3. | ||
The microstructures of the pure PANI and graphene/PANI-2 can also be identified from the SEM images. The pure PANI clusters aggregate irregularly into big blocks (Fig. 3a and b), while the graphene/PANI-2 exhibits an obviously sheet-like structure, where PANI nanorods are covered uniformly on the graphene sheets (Fig. 3c and d), consistent with the above TEM images.
 | ||
| Fig. 3 SEM images of (a and b) the pure PANI and (c and d) graphene/PANI-2 composite. | ||
The crystalline structures of the samples were confirmed from the XRD patterns by analyzing the peak position and relative peak intensity. As shown in Fig. 4a, the raw graphite demonstrates a sharp and intense peak at 2θ = 26.7°, while the graphene powder exhibits a relatively broad peak at around 26.1°, suggesting a disordered crystal structure with a slightly increased d-spacing.23 In addition to the graphene correlated peak at 2θ = 26.1°, the characteristic peaks of the PANI emeraldine salt at 11.6° (011), 19.8° (020), and 25.3° (200) are also present in the XRD pattern of the graphene/PANI-2 composite, indicating the co-existence of graphene sheets and PANI in the composite.29 The chemical compositions of the samples were also studied via Raman spectroscopy, as shown in Fig. 4b. Compared to graphite that presents a strong G band (sp2 hybridized carbon) at 1578 cm−1 and a wide 2D band at 2713 cm−1, graphene shows an additional D band (disordered graphite structure) at 1334 cm−1, reflecting the size reduction and defect introduction during the ball-milling treatment, which is consistent with the XRD analysis.20 The bands at 1163, 1257, and 1484 cm−1 in the spectrum of pure PANI correspond to the C–H bending of the quinoid ring, C–N stretching of the polaronic group, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching of the quinonoid unit. All the above bands associated with PANI also appear in the spectrum of graphene/PANI-2, demonstrating the successful synthesis of the graphene/PANI composites.30
N stretching of the quinonoid unit. All the above bands associated with PANI also appear in the spectrum of graphene/PANI-2, demonstrating the successful synthesis of the graphene/PANI composites.30
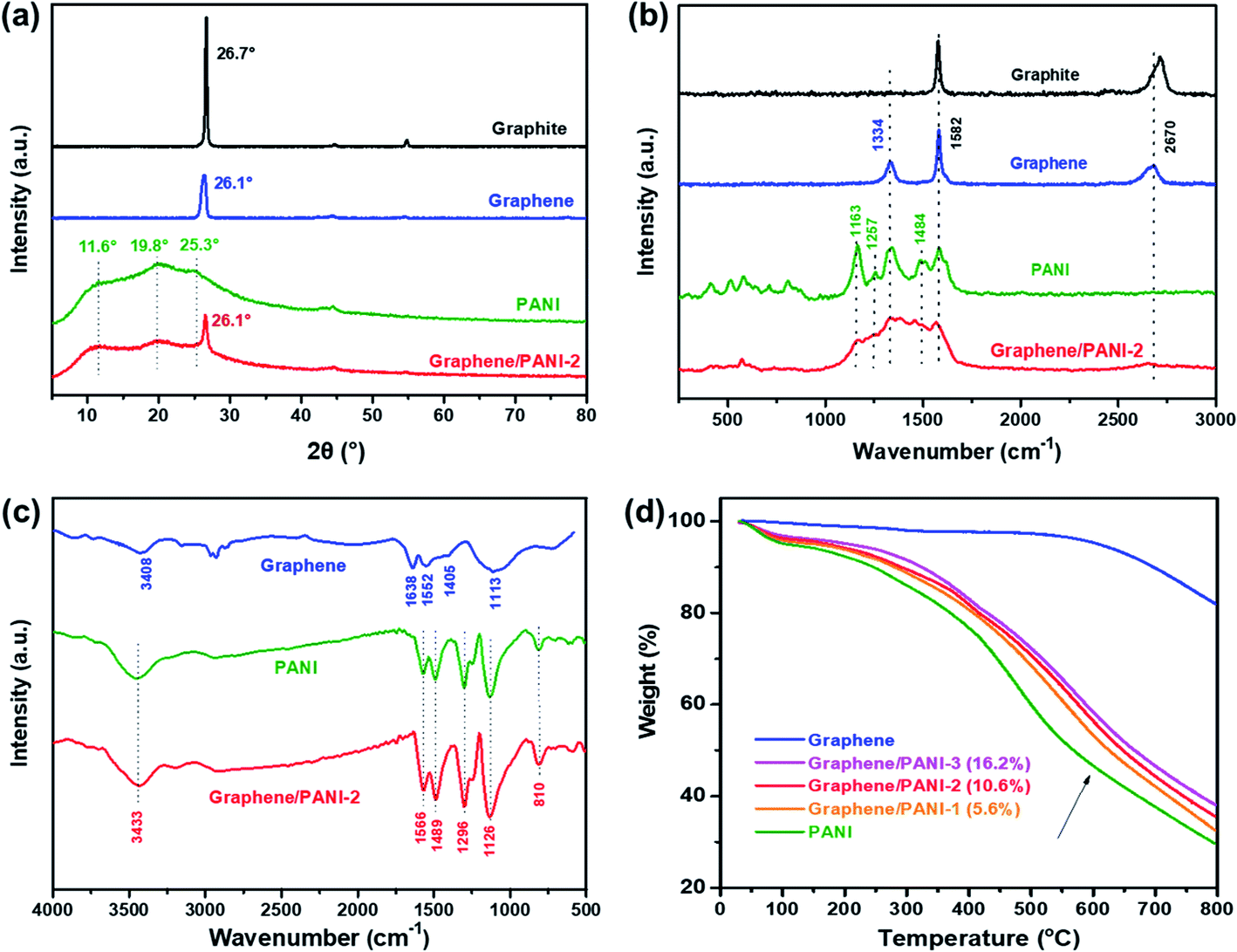 | ||
| Fig. 4 (a) XRD patterns, (b) Raman spectra, and (c) FTIR spectra of the raw graphite, graphene, PANI, and graphene/PANI-2 composite. (d) TGA curves of the pure graphene, PANI, and graphene/PANI composites. | ||
The chemical bond structures of the samples were further studied via FT-IR spectroscopy. As shown in Fig. 4c, the FT-IR spectrum of graphene displays two broad peaks at 3408 and 1113 cm−1, corresponding to O–H and C–O stretching bands caused by the adsorption water and oxidation of graphite skeleton, respectively, due to the oxidation of highly reactive dangling bonds formed by high-energy ball-milling.31 In addition, the characteristic peaks at 1638, 1552, and 1405 cm−1 in the graphene spectrum are attributed to the CC stretching of aromatic rings. For the pure PANI and graphene/PANI-2 composite, a series of characteristic peaks at 3433, 1566, 1489, 1296, 1126, and 810 cm−1 are stretching vibrations originating from N–H, CC in the quinoid rings, CC in the benzenoid rings, C–N in the secondary aromatic amines, NQN (Q corresponds to the quinoid rings), and aromatic C–H, respectively, indicating the combination of PANI in the composites.32 Notably, the intensity ratio of the NQN band at 1126 cm−1, regarded as an electronic-like band relating to the conductivity of PANI, improved in the spectrum of the graphene/PANI-2 composite and is attributed to the noncovalent interactions between the sp2-bonded graphene sheet and conjugated PANI.33 The composition components of the graphene/PANI composites were studied by TGA measurements. As shown in Fig. 4d, the graphene stays thermodynamically stable before 600 °C in Ar atmosphere, and then gives total weightlessness of 17.8% over 800 °C due to the dissociation of the marginal defects of single or few-layer graphene sheets.34 The pure PANI displays a typical thermal behavior as described previous literature reports;29 the curve shows a weight loss of 4% before 150 °C is attributed to the desorption water, and a drastic weight loss of 64% during 200–800 °C is due to the decomposition of PANI. The graphene/PANI composites exhibit similar TGA curves to those of pure PANI, and the thermal stability was enhanced with the increase in the graphite ratio,35 and the weight percents of graphene in the composites were 5.6%, 10.6%, and 16.2% for the graphene/PANI-1, graphene/PANI-2, and graphene/PANI-3 composites, respectively.
XPS spectra were recorded to analyze the surface elements, particularly the electronic structure and chemical bond information of the samples. As shown in Fig. 5a, the survey XPS spectrum of graphene shows two peaks for C 1s and O 1s at 284.8 and 532.2 eV, respectively, with a C/O atom ratio of 15.8.36 The core-level spectrum of C 1s (Fig. 5b) can be further fitted into the main peak and two weak peaks at 284.8 eV (CC/C–C), 286.4 (C–O), and 287.4 eV (CO), respectively, revealing a low content of oxygen-containing groups or defects in the graphene skeleton, and hence a high conductivity is expected. For the survey spectra of PANI and the graphene/PANI-2 composite, additional peaks of N 1s, S 1s, and S 2p are found at 399.8, 232, and 169.0 eV, respectively, demonstrating the combination of doped PANI in the composite.37 In addition, the C, O, N, and S content in the graphene/PANI-2 composite is 75.1%, 13.8, 9.2, and 1.9% by atom, respectively; thus, the weight percentage of PANI in the composite can be calculated to be about 11.2%, which is close to that obtained in the above TGA results. Fig. 5c presents the N 1s core-level spectrum of the graphene/PANI-2 composite, which can be deconvoluted into three peaks at 401.3, 399.8, and 398.8 eV, corresponding to positively charged nitrogen (–NH+), benzenoid amine (–NH–), and quinoid imine (N–), revealing that some N had been transmuted into protonated N+ species.13 Moreover, the CN and C–N bands (285.8 eV) can also be traced in the C 1s core-level spectrum of the graphene/PANI-2 composite, as shown in Fig. 5d.
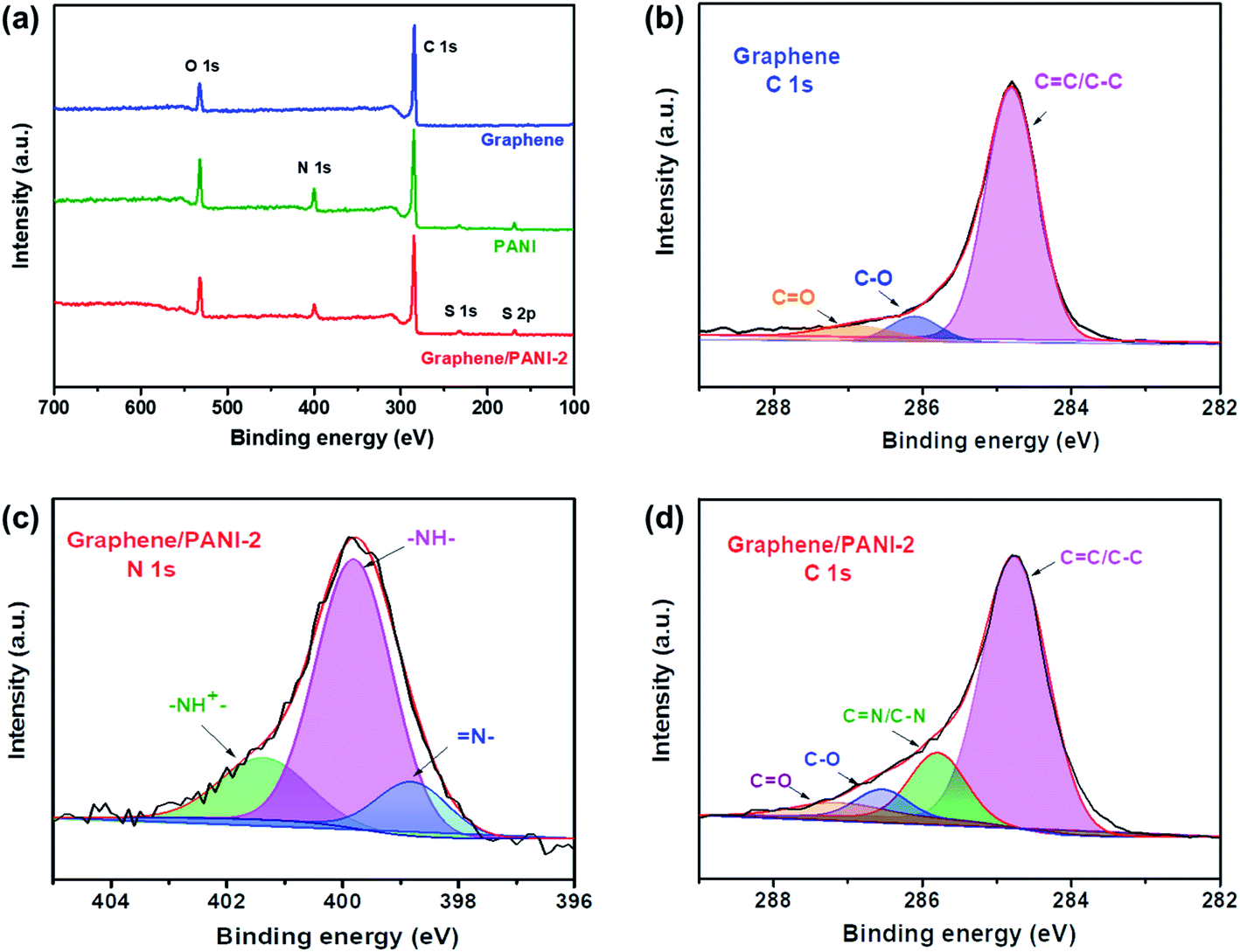 | ||
| Fig. 5 (a) XPS survey spectra of the pure graphene, PANI, and graphene/PANI-2 composite, (b) the C 1s core level spectrum of the graphene, (c) N 1s and (d) C 1s core-level spectra of the graphene/PANI-2 composite. | ||
3.2 Electrochemical properties in a three-electrode system
To evaluate the electrochemical properties of the graphene/PANI composites, the composite-based electrodes were first tested with a three-electrode system. Fig. 6a presents the CV curves of the pure graphene, PANI, and graphene/PANI composites at 20 mV s−1. The CV curve of the graphene electrode shows a nearly rectangular shape in the potential window of −0.2 to 0.8 V, indicating a typical EDLC characteristic.38,39 In comparison, two pairs of redox peaks (A1/C1 and A2/C2) appear in the CV curves of pure PANI and graphene/PANI composites, representing their pseudocapacitive characteristics. The A1/C1 peaks are attributed to the redox transitions of PANI between the leucoemeraldine and polaronic emeraldine, while the A2/C2 peaks are due to the faradaic transformations between the emeraldine and pernigraniline states.30 It is also noted that the current densities of the graphene/PANI composite electrodes are significantly larger than the pure PANI electrode at the same scan rate of 20 mV s−1; notably, the graphene/PANI-2 presented the highest current density among them, suggesting the maximum specific capacitance for it. Moreover, the current density of the graphene/PANI-2 increases accordingly as the scan rate increases from 5 to 100 mV s−1 without recognizable shape deformation (Fig. 6b), demonstrating an outstanding rate capability and high reversibility of the electrode.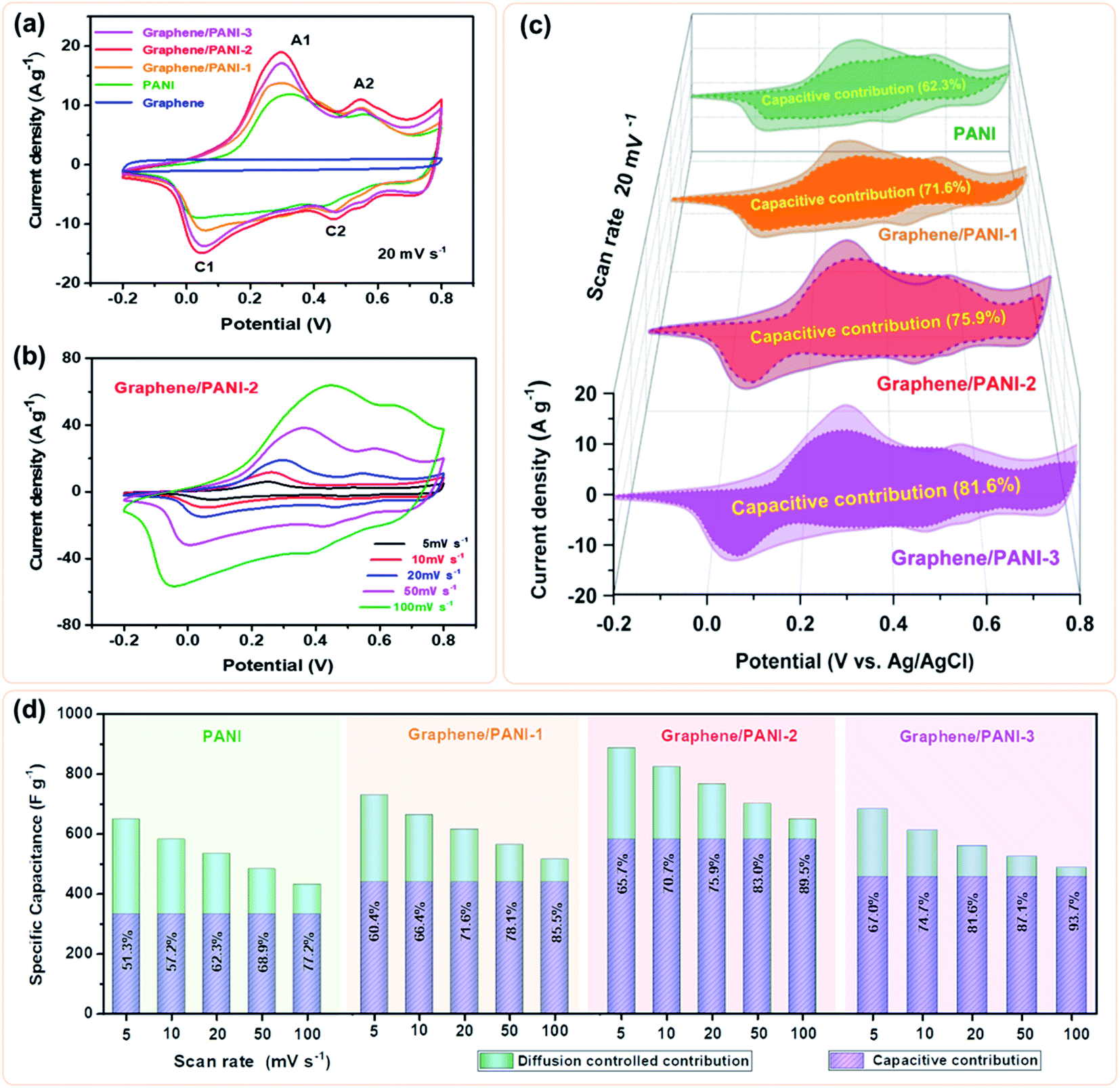 | ||
| Fig. 6 (a) CV curves of the pure graphene, PANI, graphene/PANI-1, graphene/PANI-2, and graphene/PANI-3 electrodes at a scan rate of 20 mV s−1, (b) CV curves of the graphene/PANI-2 at different scan rates, (c) the deconvolutions of the total capacitances into capacitive contributions (dark parts) and diffusion-controlled contributions (light parts) for different electrodes at 20 mV s−1, (d) bar graphs represent the ratios of capacitive capacitance for the above electrodes at various scan rates. | ||
In fact, the relationship between the response current density (i) and scan rate (ν) can be expressed as:40
| i = kνa | (1) |
| i = kcν + kdν1/2 | (2) |
| i/ν1/2 = kcν1/2 + kd | (3) |
Hence, the kc at a certain potential can be determined by the slope of the line by plotting i/ν1/2 as a function of ν1/2 at the given scan rates. By repeated fitting calculations, the kc values at different potentials can be obtained. Fig. 6c exhibits the fitted capacitive currents (ic = kcν) of the pure PANI, graphene/PANI-1, graphene/PANI-2, and graphene/PANI-3 electrodes at 20 mV s−1 (the dark regions); accordingly, their capacitive contribution rates in the total capacitances have also been calculated, which are 62.3%, 71.6%, 75.9%, and 81.6%, respectively. It is clear that the capacitive contribution rate of electrodes, at a certain scan rate, was enhanced with an increase in the graphene content due to the large specific surface area and excellent conductivity of graphene, which accelerated the electron conduction, ion transfer, and facilitated the fast energy-storage kinetics.42 The complete results of capacitive contribution rates for each of the electrodes at various scan rates can be seen in Fig. 6d. The capacitive capacitances of each electrode remain unchanged, while the diffusion-controlled capacitances decrease with an increase in the scan rate, such that the capacitive contribution rate of the pure PANI enhances from 51.3% at 5 mV s−1 to 77.2% at 100 mV s−1. In comparison, the capacitive contribution rates of the composite electrodes were greatly enhanced due to the coupling of graphene sheets, which helps to improve the electric conductivity and electrolyte-ion accessibility for the fast surface-dependent energy storage.43 Hence, the capacitive contribution of graphene/PANI-3 is enhanced to 67.0% and 93.7% at 5 and 100 mV s−1, respectively, demonstrating that the energy-storage kinetics of the composite is dominated by capacitive contribution, which helps to promote a better rate performance. The graphene/PANI-2 electrode displays a high specific capacitance of 886 F g−1 at 5 mV s−1 and a good rate capability of 73.4% up to 100 mV s−1. The overall electrochemical performance of graphene/PANI-2 is obviously better than that of the pure PANI, which exhibited 649 F g−1 at 5 mV s−1 with a capacitance retention of 66.6% at 100 mV s−1, and it is also superior to that of numerous previously reported PANI-based materials.44,45 The excellent performance of graphene/PANI-2 is attributed to the synergistic effects between the high-quality graphene sheets and pseudocapacitive PANI nanostructures. Aniline molecules promoted the mechanochemical exfoliation of graphite, generating few-layer graphene sheets with a regular lattice structure and excellent electrical conductivity. The PANI clusters were loaded uniformly on graphene sheets to prevent their face-to-face aggregation, thus enhancing the specific surface area and utilization efficiency of the graphene/PANI composites, leading to a large specific capacitance. The excellent conductivity and porous structure also promoted the charge transfer, hence providing the composites with high capacitive contributions and outstanding rate performances.
3.3 Electrochemical performances of symmetric supercapacitors
Symmetric supercapacitors of the graphene/PANI composites were also assembled by sandwiching an H2SO4 soaked cellulose acetate membrane with two composite electrodes. Fig. 7a shows the CV curves of pure PANI and graphene/PANI composite-based symmetric supercapacitors at 20 mV s−1. All these plots display a roughly rectangular shape with two pairs of redox waves at around 0.28/0.15 and 0.65/0.53 V, respectively, demonstrating that the energy storage mechanisms of these symmetric supercapacitors are a combination of EDLC and pseudocapacitance.29 Among these symmetric supercapacitors, the graphene/PANI-2-based supercapacitor possesses the largest area of surrounded CV loops, indicating the largest specific capacitance. Moreover, the CV curves of the graphene/PANI-2 based supercapacitor at different scan rates are shown in Fig. 7b; the response current densities increased proportionally with the increase in the scan rate even up to 100 mV s−1, representing an outstanding rate capability. In addition, the graphene/PANI-2 supercapacitor also presents the longest discharge time among these GCD profiles at the same current density of 0.5 A g−1 (Fig. 7c), corresponding to the highest specific capacitance of 294 F g−1 for it compared to 200, 263, and 274 F g−1 obtained for the pure PANI, graphene/PANI-1, and graphene/PANI-3 based supercapacitors, respectively. It is rational that an appropriate content of graphene would enhance the electric conductivity and specific surface area of the composite, thus improving the rate capability and specific capacitance of the composite-based supercapacitor. Furthermore, the GCD curves of the graphene/PANI-2 supercapacitor at different current densities show nearly symmetrical triangle shapes, without obvious potential drops (Fig. 7d), indicating excellent reversibility due to the excellent electric conductivity of graphene.46 Therefore, the graphene/PANI-2-based supercapacitor maintained high specific capacitances of 284, 277, 266, and 256 F g−1 at 1, 2, 5, and 10 A g−1, respectively, with the capacitance retention reaching to 87% after the current density increased by 20 times. The high specific capacitance and rate capability of the graphene/PANI-2 based supercapacitor are obviously higher than those of pure PANI (139 F g−1 with 71.5% retention at 10 A g−1), and also superior to numerous PANI-based symmetric supercapacitors, such as graphene/polyaniline nanofibers (G/PNF),47 polyaniline–polyvinyl alcohol hydrogel (PPH),48 macroporous carbon/PANI (MC/PANI),49 and manganese dioxide/polyaniline (MnO2/PANI),50 as shown in Fig. 8a. | ||
| Fig. 7 (a) CV comparison of the pure PANI and graphene/PANI composite-based symmetric supercapacitors at a scan rate of 20 mV s−1; (b) CV curves of the graphene/PANI-2-based supercapacitor at various scan rates; (c) GCD comparison of different supercapacitors at a current density of 1 A g−1; (d) GCD curves of the graphene/PANI-2-based supercapacitor at various current densities. | ||
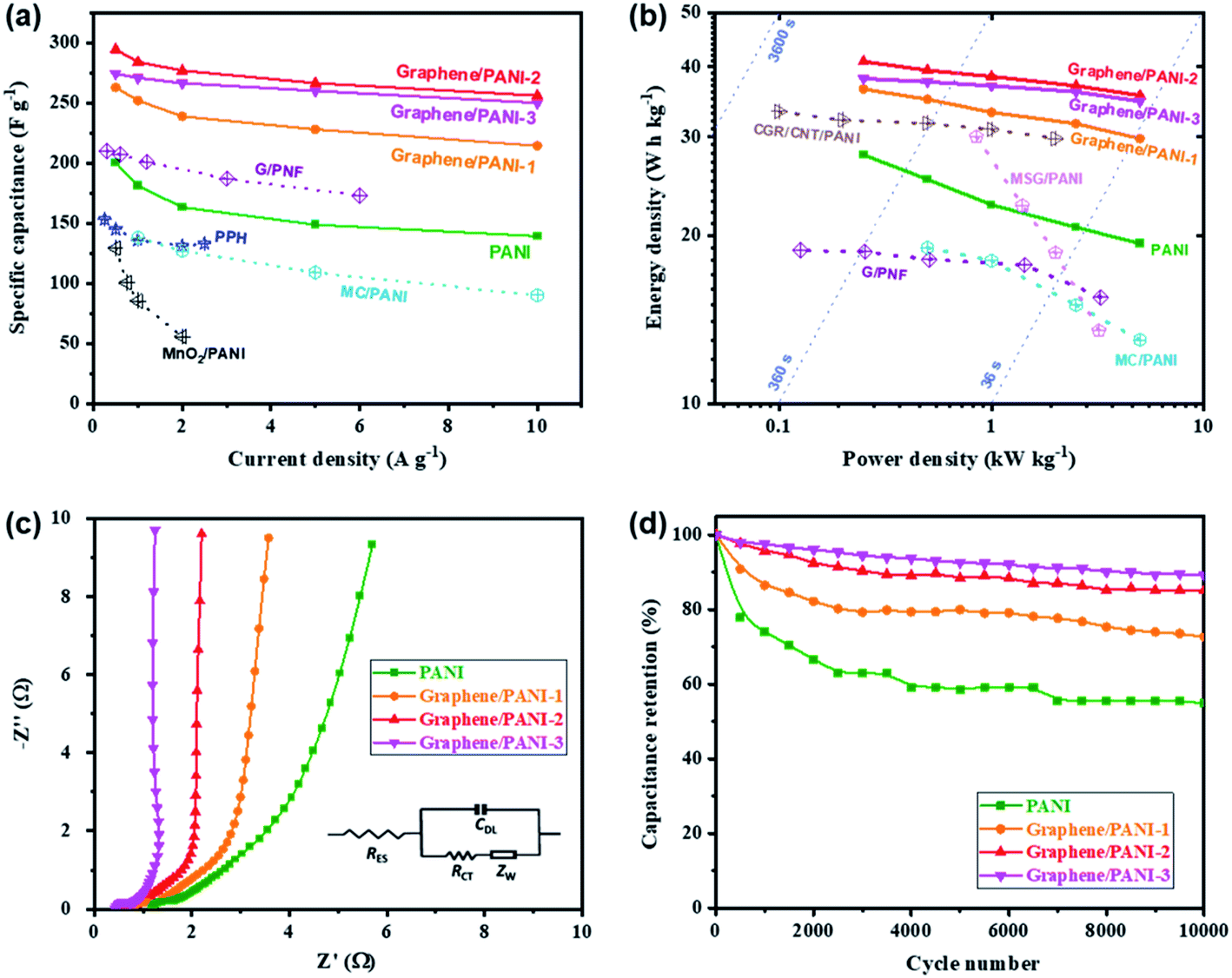 | ||
| Fig. 8 (a) Specific capacitances, (b) Ragone plots, (c) Nyquist plots, and (d) long-term cycling stabilities of the pure PANI and graphene/PANI composite-based symmetric supercapacitors. | ||
The energy density and power density are also presented to reveal the application prospect of the supercapacitors. Fig. 8b shows the Ragone plots of the pure PANI and graphene/PANI composite-based symmetric supercapacitors; among them, the graphene/PANI-2 based supercapacitor delivered the largest energy density of 40.9 W h kg−1 at a power density of 0.25 kW kg−1 and maintained 35.6 W h kg−1 at 5.0 kW kg−1 (within a discharge time of ∼25 s). The energy storage property of the graphene/PANI-2-based supercapacitor is obviously better than that of pure PANI (27.8 W h kg−1 at 0.25 kW kg−1 and 19.3 W h kg−1 at 5.0 kW kg−1), and those of PANI-based symmetric supercapacitors reported previously, such as MC/PANI (19 W h kg−1 at 0.5 W kg−1 and 13 W h kg−1 at 5 kW kg−1),49 crumpled graphene/carbon nanotube/polyaniline (CGR/CNT/PANI, 33.3 W h kg−1 at 0.1 W kg−1),51 multi-growth site graphene/polyaniline (MSG/PANI, 30 W h kg−1 at 0.85 W kg−1 and 13.5 W h kg−1 at 3.2 kW kg−1).13 The fast charging/discharging capability of the graphene/PANI-2-based supercapacitor can be attributed to its fast charge-transport kinetics, which is further confirmed by EIS detections. As shown in Fig. 8c, each symmetric supercapacitors present typical Nyquist plots with a semicircle in the high-frequency region, a 45° Warburg section in the intermediate frequency region, and a straight line in the low frequency region, related to the interfacial charge-transfer resistance (RCT), Warburg impedance (ZW), and capacitive behavior (CDL) of the electrodes, respectively. Moreover, the intercept on the real axis at the high frequency corresponds to the equivalent series resistance (RES) of the supercapacitor.52 The impedance spectra were then fitted according to the equivalent circuit, as shown inset Fig. 8c. The graphene/PANI-3-based supercapacitor displays the lowest RES (0.42 Ω) and RCT (0.44 Ω) compared to pure PANI (1.18 and 0.61 Ω), graphene/PANI-1 (0.78 and 0.55 Ω), and graphene/PANI-2 (0.60 and 0.46 Ω), respectively due to the increased graphene content in the composite electrode, resulting in fast charge-transfer reactions during the electrochemical processes. In addition, with an increase in the graphene content, the Warburg region becomes shorter, while the straight line becomes more vertical, indicating faster ion diffusion into the porous electrodes and hence better rate performance and power capacity for the composite-based supercapacitors. Furthermore, the cycling stability of the graphene/PANI-3 based supercapacitor has also been enhanced to 89.1%, from 54.9% for pure PANI, after 10000 charges/discharge cycles at 10 A g−1, as shown in Fig. 8d. The greatly improved stabilities of the supercapacitors are mainly due to the strong π–π interactions between PANI and graphene because the graphene serves as a strong scaffold to suppress the swelling and shrinking of PANI during the doping–dedoping process.10
4. Conclusions
Graphene/PANI composites were fabricated via the mechanochemical polymerization of aniline onto in situ exfoliated graphene sheets by a one-pot ball-milling process. The as-generated PANI clusters were absorbed tightly on the few-layered graphene sheets via π–π interactions, which then stimulated synergistic effects to accelerate the electron conduction, ion diffusion, and utilization efficiency of the active materials. As a result, the graphene/PANI-based electrodes exhibited a large specific capacitance of 886 F g−1 at 5 mV s−1, with high rate retention of 73.4% at 100 mV s−1 due to the dominantly capacitive contribution. In addition, the graphene/PANI-based symmetric supercapacitor delivered a high energy density of 40.9 W h kg−1 at a power density of 0.25 kW kg−1; moreover, it also achieved excellent cycling stability with capacitance retention of 89.1% after 10000 charge/discharge cycles. This study provides a convenient and efficient approach to produce graphene/polymer composites for high-performance supercapacitors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51973235 and 51673061), Fundamental Research Funds for the Central Universities (CZP19001), and Project for Wuhan Science and Technology Bureau (2020010601012198).References
- H. X. Chang and H. K. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 Search PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 Search PubMed.
- Y. Yang, C. Han, B. Jiang, J. Iocozzia, C. He, D. Shi, T. Jiang and Z. Lin, Mater. Sci. Eng., 2016, 102, 1–72 Search PubMed.
- X. Wang, A. Dong, Y. Hu, J. Qian and S. Huang, Chem. Commun., 2020, 56, 10809–10823 Search PubMed.
- Z. Zhu, C. Han, T.-T. Li, Y. Hu, J. Qian and S. Huang, CrystEngComm, 2018, 20, 3812–3816 Search PubMed.
- Q. Meng, K. Cai, Y. Chen and L. Chen, Nano Energy, 2017, 36, 268–285 Search PubMed.
- X. Wang, D. Wu, X. Song, W. Du, X. Zhao and D. Zhang, Molecules, 2019, 24, 2263 Search PubMed.
- M. M. Sk and C. Y. Yue, J. Mater. Chem. A, 2014, 2, 2830–2838 Search PubMed.
- H. Kwon, D. Hong, I. Ryu and S. Yim, ACS Appl. Mater. Interfaces, 2017, 9, 7412–7423 Search PubMed.
- P. B. Liu, J. Yan, Z. X. Guang, Y. Huang, X. F. Li and W. H. Huang, J. Power Sources, 2019, 424, 108–130 Search PubMed.
- N. A. Kumar and J.-B. Baek, Chem. Commun., 2014, 50, 6298–6308 Search PubMed.
- J. W. Ji, R. Li, H. Y. Li, Y. Shu, Y. Li, S. Q. Qiu, C. E. He and Y. K. Yang, Composites, Part B, 2018, 155, 132–137 Search PubMed.
- X. Zheng, H. Yu, R. Xing, X. Ge, H. Sun, R. Li and Q. Zhang, Electrochim. Acta, 2018, 260, 504–513 Search PubMed.
- S. B. Kulkarni, U. M. Patil, I. Shackery, J. S. Sohn, S. Lee, B. Park and S. Jun, J. Mater. Chem. A, 2014, 2, 4989–4998 Search PubMed.
- J. Azadmanjiri, V. K. Srivastava, P. Kumar, M. Nikzad, J. Wang and A. Yu, J. Mater. Chem. A, 2018, 6, 702–734 Search PubMed.
- C. Teng, D. Xie, J. Wang, Z. Yang, G. Ren and Y. Zhu, Adv. Funct. Mater., 2017, 27, 1700240 Search PubMed.
- C. Chen, Y. Xue, Z. Li, Y. Wen, X. Li, F. Wu, X. Li, D. Shi, Z. Xue and X. Xie, Chem. Eng. J., 2019, 369, 1150–1160 Search PubMed.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 Search PubMed.
- C. He, S. Qiu, H. Peng, Q. Zhang, X. Han, Y. Yang, D. Shi and X. Xie, Compos. Sci. Technol., 2018, 167, 155–163 Search PubMed.
- Y. Jiang, C. He, S. Qiu, J. Zhang, X. Wang and Y. Yang, Chem. Eng. J., 2020, 397, 125503 Search PubMed.
- K. Ghosh, C. Y. Yue, M. M. Sk, R. K. Jena and S. Bi, Sustainable Energy Fuels, 2018, 2, 280–293 Search PubMed.
- V. Leon, M. Quintana, M. A. Herrero, J. L. Fierro, A. de la Hoz, M. Prato and E. Vazquez, Chem. Commun., 2011, 47, 10936–10938 Search PubMed.
- V. Leon, A. M. Rodriguez, P. Prieto, M. Prato and E. Vazquez, ACS Nano, 2014, 8, 563–571 Search PubMed.
- N. I. Kovtyukhova, Y. Wang, A. Berkdemir, R. Cruz-Silva, M. Terrones, V. H. Crespi and T. E. Mallouk, Nat. Chem., 2014, 6, 957–963 Search PubMed.
- X. Du, H. Y. Liu, G. Cai, Y. W. Mai and A. Baji, Nanoscale Res. Lett., 2012, 7, 111 Search PubMed.
- L. Liang, P. Xu, Y. Wang, Y. Shang, J. Ma, F. Su, Y. Feng, C. He, Y. Wang and C. Liu, Chem. Eng. J., 2020, 395, 125209 Search PubMed.
- J. Xing, M. Liao, C. Zhang, M. Yin, D. Li and Y. Song, Phys. Chem. Chem. Phys., 2017, 19, 14030–14041 Search PubMed.
- Q. Zhao, D. Yang, C. Zhang, X. Liu, X. Fan, A. K. Whittaker and X. S. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 43730–43739 Search PubMed.
- A. G. Tabrizi, N. Arsalani, A. Mohammadi, L. S. Ghadimi, I. Ahadzadeh and H. Namazi, Electrochim. Acta, 2018, 265, 379–390 Search PubMed.
- F. Ke, Y. Liu, H. Xu, Y. Ma, S. Guang, F. Zhang, N. Lin, M. Ye, Y. Lin and X. Liu, Compos. Sci. Technol., 2017, 142, 286–293 Search PubMed.
- F. Beckert, S. Bodendorfer, W. Zhang, R. Thomann and R. Mülhaupt, Macromolecules, 2014, 47, 7036–7042 Search PubMed.
- Y. Zou, R. Liu, W. Zhong and W. Yang, J. Mater. Chem. A, 2018, 6, 8568–8578 Search PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 Search PubMed.
- C. Liu, X. Liu, J. Tan, Q. Wang, H. Wen and C. Zhang, J. Power Sources, 2017, 342, 157–164 Search PubMed.
- Z. Tong, Y. Yang, J. Wang, J. Zhao, B.-L. Su and Y. Li, J. Mater. Chem. A, 2014, 2, 4642–4651 Search PubMed.
- L. Li, B. Zhou, G. Han, Y. Feng, C. He, F. Su, J. Ma and C. Liu, Compos. Sci. Technol., 2020, 197, 108229 Search PubMed.
- H. Yu, G. Xin, X. Ge, C. Bulin, R. Li, R. Xing and B. Zhang, Compos. Sci. Technol., 2018, 154, 76–84 Search PubMed.
- S. Deng, C. He, S. Qiu, J. Zhang, X. Wang and Y. Yang, J. Mater. Sci.: Mater. Electron., 2020, 31, 10113–10122 Search PubMed.
- C. He, S. Qiu, S. Sun, Q. Zhang, G. Lin, S. Lei, X. Han and Y. Yang, Energy Environ. Mater., 2018, 1, 88–95 Search PubMed.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1703043 Search PubMed.
- Y. Lu, J. Liang, S. Deng, Q. He, S. Deng, Y. Hu and D. Wang, Nano Energy, 2019, 65, 103993 Search PubMed.
- X. Zhang, X. Cui, C.-H. Lu, H. Li, Q. Zhang, C. He and Y. Yang, Chem. Eng. J., 2020, 401, 126031 Search PubMed.
- K. A. Owusu, L. Qu, J. Li, Z. Wang, K. Zhao, C. Yang, K. M. Hercule, C. Lin, C. Shi, Q. Wei, L. Zhou and L. Mai, Nat. Commun., 2017, 8, 14264 Search PubMed.
- L. Liu, Y. Wang, Q. Meng and B. Cao, J. Mater. Sci., 2017, 52, 7969–7983 Search PubMed.
- D. D. Potphode, L. Sinha and P. M. Shirage, Appl. Surf. Sci., 2019, 469, 162–172 Search PubMed.
- H. Luo, J. Dong, Y. Zhang, G. Li, R. Guo, G. Zuo, M. Ye, Z. Wang, Z. Yang and Y. Wan, Chem. Eng. J., 2018, 334, 1148–1158 Search PubMed.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 Search PubMed.
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., Int. Ed. Engl., 2016, 55, 9196–9201 Search PubMed.
- J. Li, Y. Ren, Z. Ren, S. Wang, Y. Qiu and J. Yu, J. Mater. Chem. A, 2015, 3, 23307–23315 Search PubMed.
- K. Ghosh, C. Y. Yue, M. M. Sk and R. K. Jena, ACS Appl. Mater. Interfaces, 2017, 9, 15350–15363 Search PubMed.
- E. H. Jo, H. D. Jang, H. Chang, S. K. Kim, J. H. Choi and C. M. Lee, ChemSusChem, 2017, 10, 2210–2217 Search PubMed.
- Y. He, H. Li, Q. Zhang, C. He, X. Zhang and Y. Yang, RSC Adv., 2019, 9, 40933–40939 Search PubMed.
Footnote |
| † These two authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |