
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel naphthalene-heterocycle hybrids with potent antitumor, anti-inflammatory and antituberculosis activities†
Mohamed Ahmed Abozeid a,
Aya Atef El-Sawia,
Mohamed Abdelmotelebbc,
Hanem Awadd,
Marwa Mostafa Abdel-Azize,
Abdel-Rahman Hassan Abdel-Rahmana and
El-Sayed Ibrahim El-Desoky*a
a,
Aya Atef El-Sawia,
Mohamed Abdelmotelebbc,
Hanem Awadd,
Marwa Mostafa Abdel-Azize,
Abdel-Rahman Hassan Abdel-Rahmana and
El-Sayed Ibrahim El-Desoky*a
aDepartment of Chemistry, Faculty of Science, Mansoura University, Mansoura-35516, Egypt. E-mail: prof.deosky.orgchem@gmail.com; desoky199@mans.edu.eg; Tel: +201060614054
bFood Allergy Research & Resource Program (FARRP), Department of Food Science & Technology, University of Nebraska, Lincoln, NE, USA
cDepartment of Botany, Faculty of Science, Mansoura University, Mansoura-35516, Egypt
dDepartment of Tanning Materials and Leather Technology, Chemical Industries Research Division, National Research Centre, 33 EL-Bohouth St., Dokki 12622, Giza, Egypt
eThe Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo 11759, Egypt
First published on 26th November 2020
Abstract
Multitarget-directed drugs (hybrid drugs) constitute an efficient avenue for the treatment of multifactorial diseases. In this work, novel naphthalene hybrids with different heterocyclic scaffolds such as nicotinonitrile, pyran, pyranopyrazole, pyrazole, pyrazolopyridine, and azepine were efficiently synthesized via tandem reactions of 3-formyl-4H-benzo[h]chromen-4-one 1 with different nucleophilic reagents. Analysis of these hybrids using PASS online software indicated different predicted biological activities such as anticancer, antimicrobial, antiviral, antiprotozoal, anti-inflammatory, etc. By focusing on antitumor, anti-inflammatory, and antituberculosis activities, many compounds revealed remarkable activities. While 3c, 3e, and 3h were more potent than doxorubicin in the case of HepG-2 cell lines, 3a–e, 3i, 6, 8, 10, 11, and 12b were more potent in the case of MCF-7. Moreover, compounds 3c, 3h, 8, 10, 3d, and 12b manifested superior activity and COX-2 selectivity to the reference anti-inflammatory Celecoxib. Regarding antituberculosis activity, 3c, 3d, and 3i were found to be the most promising with MIC less than 1 μg mL−1. The molecular docking studies showed strong polar and hydrophobic interactions with the novel naphthalene-heterocycle hybrids that were compatible with experimental evaluations to a great extent.
Introduction
Naphthalene constitutes a flexible and multifaceted platform in medicinal chemistry.1 This scaffold emerges as a promising moiety in drug design due to its diverse biological activities generated from structural modifications. Various antagonistic activities were reported for naphthalene-based compounds such as antimicrobial,2 anticancer,3 antiviral,4 anticonvulsant,5 antitubercular,6 and anti-inflammatory.7 Besides, different naturally occurring compounds including the naphthalene nucleus revealed notable biological activities such as justicidin A (anticancer),8 rifampicin (antitubercular),9 patentiflorin A (anti-HIV),10 and bis-ANS 82 (tubulin polymerization inhibitor).11 In 2018, Kittakoop and his team isolated a set of naphthalene derivatives from Ventilago denticulata which exhibited promising antibacterial and cytotoxic activities in addition to aromatase and phosphodiesterase inhibitory activities.12 Naphthalene nucleus also appeared in different marketed therapeutics such as Nafcillin,13 Bedaquiline,14 Naproxen,15 and Nafimidone16 (Fig. 1).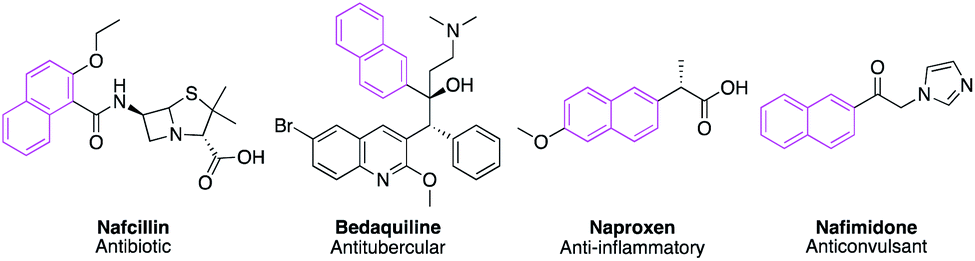 | ||
| Fig. 1 Representative examples for naphthalene containing marketed drugs. | ||
Moreover, naphthalene was known to be metabolized intracellularly into naphthoquinone and naphthalene epoxide that could react with thiol group (SH) of cysteine amino acid residue via nucleophilic substitution and 1,4-Michael addition, respectively.17 1-Naphthol and naphthoquinone were also reported to produce reactive oxygen species (ROS) under the effect of hepatic microsomal enzymes18 that recently achieved promising results in cancer treatment.19 Additionally, naphthoquinone fulfilled Lipinski's rule of five to be an orally active drug.
On the other hand, multitarget-directed compounds are the combination of two or more distinctive biologically active molecules into one entity to achieve dual-drug action.20 This hybrid design would be promising to treat multifactorial diseases such as cancer in addition to neurodegenerative disorders including Parkinson's and Alzheimer's disease. The drug hybridization could also generate new molecules with lower toxicity, improved affinity, and better pharmacokinetics in addition to avoiding common drug–drug interactions observed in drug cocktails.20 As seen in Fig. 1 examples, naphthalene could form very efficient hybrid drugs by merging with different biologically active moieties. Moreover, our previous studies unveiled different promising hybrid molecules that resulted from the combination of naphthalene with pyran, pyridine, pyrazole, and others.2
Herein, new naphthalene hybrids with nicotinonitrile, pyrazole, and pyran moieties were designed (Fig. 2). Moreover, molecular hybridization of naphthalene with pyrazolopyridine21 and azepine22 pharmacophores was reported for the first time (Fig. 2). The anticancer, anti-inflammatory, and antituberculosis activities of the synthesized compounds were evaluated, and the results were analyzed in light of the molecular docking.
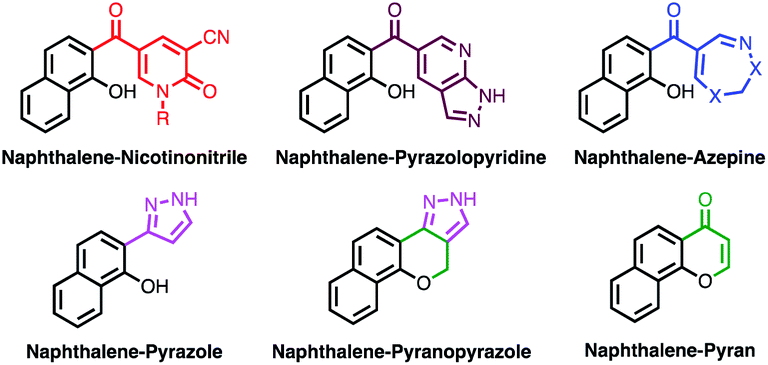 | ||
| Fig. 2 Design of new naphthalene hybrid skeletons. | ||
Results and discussion
Chemistry
The Knoevenagel condensation of formylchromone 123 with different cyanoacetanilide derivatives 2a–h24 as 1,3-C,N-dinucleophiles in the presence of a catalytic amount of triethylamine afforded the corresponding nicotinonitrile derivatives 3a–h in excellent yields (Scheme 1). First, carbon nucleophilic center, which was generated from cyanoacetanilides 2a–h by deprotonation under basic conditions, attacked the formyl group followed by dehydration to give intermediate 1A. After that, NH functionality underwent conjugate addition to α,β-unsaturated carbonyl moiety of chromone nucleus to give intermediate 1B. The ring-opening of chromene intermediate 1B generated nicotinonitrile derivatives 3a–h. Similarly, the condensation of formylchromone 1 with 2-cyano-N′-(1-(2-oxo-2H-chromen-3-yl)-ethylidene)acetohydrazide (4)25 afforded multi-functionalized derivative 3i (Scheme 1).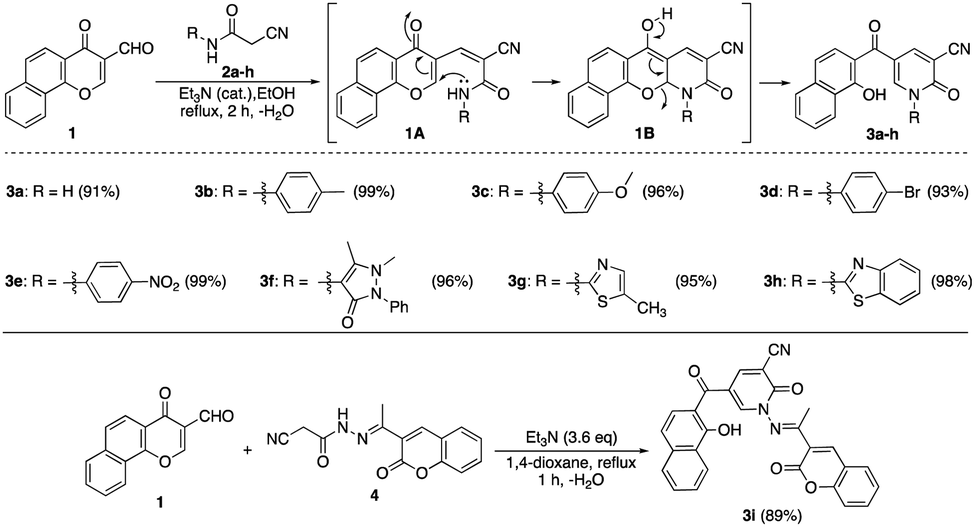 | ||
| Scheme 1 Reaction of formylchromone 1 with cyanoacetanilides 2a–h. | ||
It was found that the reaction of formylchromone 1 with cyanoacetic acid hydrazide instead of cyanoacetanilide 2a by heating in ethanol afforded product 5 as a mixture of hydrazone and azo tautomers. In acetic acid, the reaction proceeded via γ-pyrone ring opening to give the novel naphthyl pyrazolopyridine 6 in 99% yield. Compound 6 gave a dark red color with FeCl3 solution, indicating the presence of the free phenolic OH group. The proposed pathway included 1,4-conjugate addition to the α,β-unsaturated carbonyl moiety of formylchromone 1 followed by γ-pyrone ring opening to give intermediate 1C. After that, hydrazido NH2 attacked the nearby cyano function to afford aminopyrazolone intermediate 1D which transformed into intermediate 1E through the intramolecular condensation of its amino group with the pendant formyl group. Finally, intermediate 1E gave naphthyl pyrazolopyridine ketone 6 by keto to enol tautomerization (Scheme 2).
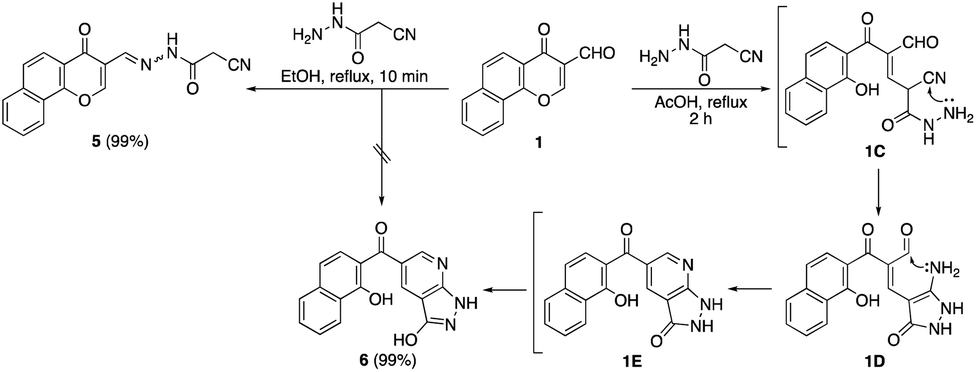 | ||
| Scheme 2 Reaction of formylchromone 1 with cyanoacetic acid hydrazide. | ||
The scope of this tandem process was extended by the reaction of formylchromone 1 with 1,4-N,N- and 1,4-N,S-dinucleophiles as shown in Scheme 3. Initially, the reaction of precursor 1 with thiosemicarbazide afforded the corresponding naphthyl triazepine ketones 7 in excellent yields. Similarly, the reaction with o-aminothiophenol afforded naphthyl thiazepine ketone 8 in 98% yield. Formation of thiosemicarbazone or imine intermediates could be the first step in the formation of products 7 and 8 followed by spontaneous γ-pyrone ring opening via intramolecular cyclization by the aid of the second nucleophilic center whether NH2 or SH.
 | ||
| Scheme 3 Reaction of formylchromone 1 with thiosemicarbazide and o-aminothiophenol. | ||
The reaction of formylchromone 1 with hydrazine hydrate (1,2-N,N-dinucleophile) at room temperature proceeded via an unexpected tandem pathway to give novel chromenopyrazole 10 instead of naphthyl pyrazolyl ketone 9. Moreover, the presence of acetal functionality in compound 10 offers an excellent platform for further functionalization through the reaction with different nucleophiles (Scheme 4).26
 | ||
| Scheme 4 Reaction of formylchromone 1 with hydrazine hydrate. | ||
By performing the reaction of formylchromone 1 with hydrazine in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in boiling ethanol, hydrazineylidenomethylpyrazole 11 was obtained in excellent yield. The intermediacy of chromenopyrazole 10 in this transformation was confirmed by the conversion of 10 into 11 by the reaction with one equivalent hydrazine. Our proposed mechanistic pathway involved the formation of hydrazone intermediate 1F which couldn't be converted into naphthyl pyrazolyl ketone 9 due to the addition of ethanol molecule to give intermediate 1G. The absence of ethanol reversible elimination pathway suggested possible role of hydrogen bonding between NH and carbonyl oxygen to stabilize intermediate 1G. While the formation of chromenopyrazole 10 could be accomplished by dehydration, its over-reaction with one equivalent hydrazine afforded pyrazole 11 via nucleophilic attack at acetal carbon followed by ring-opening.
:2 in boiling ethanol, hydrazineylidenomethylpyrazole 11 was obtained in excellent yield. The intermediacy of chromenopyrazole 10 in this transformation was confirmed by the conversion of 10 into 11 by the reaction with one equivalent hydrazine. Our proposed mechanistic pathway involved the formation of hydrazone intermediate 1F which couldn't be converted into naphthyl pyrazolyl ketone 9 due to the addition of ethanol molecule to give intermediate 1G. The absence of ethanol reversible elimination pathway suggested possible role of hydrogen bonding between NH and carbonyl oxygen to stabilize intermediate 1G. While the formation of chromenopyrazole 10 could be accomplished by dehydration, its over-reaction with one equivalent hydrazine afforded pyrazole 11 via nucleophilic attack at acetal carbon followed by ring-opening.
Finally, the reaction with primary heterocyclic amines namely; 4-aminoantipyrine and 2-benzothiazolamine afforded the corresponding Schiff bases 12a,b respectively in excellent yields. The reaction proceeded as a simple condensation without ring-opening of γ-pyrone, which was confirmed by a negative ferric chloride test to support the absence of a phenolic hydroxyl group (Scheme 5).
 | ||
| Scheme 5 Reaction of formylchromone 1 with 4-aminoantipyrine and 2-aminobenzothiazole. | ||
Biological evaluation
The physicochemical parameters are molecular weight (MW), count of specific atom types (hydrogen donors, hydrogen acceptors, and rotatable bonds), and topological polar surface area (TPSA) that represents the surface associated with polar atoms. Compounds 3c, 3e, 3f, 3i, and 5 with four rotatable bonds suggested their better interaction ability. Moreover, compounds 3e and 3i showed six and seven hydrogen bond acceptors, respectively. Regarding lipophilicity, most of the synthesized compounds gave logP values below five indicating their good cell membrane permeability. Most of the synthesized compounds showed good water solubility suggesting their better delivery even upon using small doses.27 Since the bioactivity score describes the positive medicinal impacts of putative drugs, all compounds were found to have a score of 0.55 suggesting higher biological activities in the clinical trial stage.28
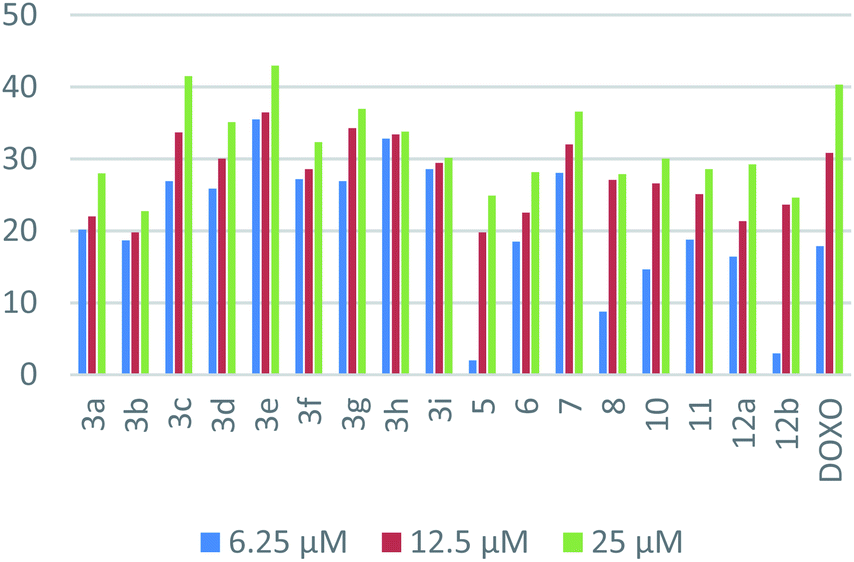 | ||
| Fig. 3 Dose-dependent cytotoxic activities for the synthesized compounds on HepG-2. | ||
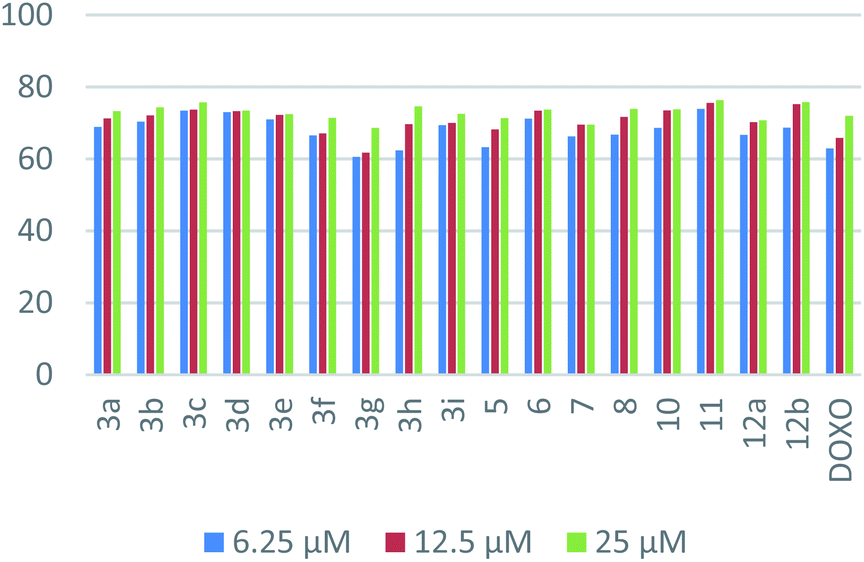 | ||
| Fig. 4 Dose dependent cytotoxic activities for the synthesized compounds on MCF-7. | ||
| Comp. | IC50 (μM) ± SD | PDB 2I4I | PDB 5GWK | |||
|---|---|---|---|---|---|---|
| HepG-2 | MCF-7 | Binding affinity (kcal mol−1) | Amino acid involved in hydrogen bonds | Binding affinity (kcal mol−1) | Amino acid involved in hydrogen bonds | |
| 3a | 34.6 ± 3.3 | 9.6 ± 0.7 | −7.4 | ARG-531, ARG-534 | −7.3 | LYS-197 |
| 3b | 36.8 ± 3.9 | 9.2 ± 0.6 | −8.1 | ARG-531, ARG-534 | −7.8 | LYS-197, GLU-194 |
| 3c | 27.8 ± 2.4 | 8.5 ± 0.7 | −8.3 | ARG-531, ARG-534, ARG-202, LYS-208 | −7.7 | LYS-197 |
| 3d | 30.1 ± 2.6 | 8.9 ± 0.7 | −7.6 | ARG-531, ARG-534 | −7.6 | LYS-197 |
| 3e | 25.3 ± 2.1 | 9.3 ± 1.1 | −8.8 | ARG-531, ARG-534, ARG-202 | −8.7 | SER-361, MET363 |
| 3f | 30.5 ± 3.1 | 10.5 ± 1.1 | −8.8 | ARG-531, ARG-534, ILE-507, HIS-527 | −8.7 | SER-198, ARG-402, ASP-258 |
| 3g | 28.5 ± 2.9 | 12.1 ± 1.2 | −8.2 | SER-228, THR-204 | −7.7 | LYS-197, GLU-194 |
| 3h | 28.1 ± 3.1 | 10.3 ± 1.2 | −8.2 | HIS-527 | −8.6 | ARG-402, GLU-194 |
| 3i | 30.5 ± 2.9 | 9.7 ± 0.8 | −9.2 | HIS-527, ARG-534, GLY-504 | −8.8 | GLU-194, SER-361, SER-198, LEU-362 |
| 5 | 41.6 ± 3.5 | 10.7 ± 1.2 | −7.7 | HIS-527, ARG-202, ARG-531 | −7.3 | LEU-362 |
| 6 | 34.9 ± 2.8 | 9.1 ± 0.5 | −7.2 | — | −7.7 | ARG-402, ASP-258, ASP-257 |
| 7 | 28.8 ± 2.5 | 10.5 ± 0.9 | −6.8 | ARG-531, ARG-534 | −7.7 | ARG-402, ASP-258, SER-400 |
| 8 | 36.5 ± 3.4 | 9.7 ± 1.1 | −7.4 | ARG-531, ARG-534 | −7.8 | — |
| 10 | 34.5 ± 2.6 | 9.3 ± 0.5 | −7.3 | ARG-534, HIS-527 | −6.8 | SER-361 |
| 11 | 34.1 ± 3.1 | 8.2 ± 0.6 | −6.2 | ARG-531, ARG-534 | −6.5 | LYS-197, GLU-194 |
| 12a | 35.5 ± 3.6 | 10.2 ± 1.2 | −7.9 | — | −8.1 | PRO-191 |
| 12b | 40.1 ± 3.9 | 8.9 ± 0.7 | −7.7 | — | −7.7 | GLU-194 |
| DOXO | 28.5 ± 1.9 | 10.3 ± 0.8 | −7.7 | ARG-202, ARG-531, ARG-534, GLY-504, LYS-208 | −7.9 | GLU-194, SER-198 |
In the case of HepG-2, the superiority of naphthalene–nicotinonitrile hybrid as seen in the p-nitrophenyl derivative 3e compared to other nicotinonitrile derivatives emphasized the significance of the nitro group. The antitumor activity decayed gradually by changing the nitrogen substituent as in the order of 3c > 3h > 3g > 3d > 3f > 3i > 3a > 3b. Moreover, macrocyclic segment of the naphthalene–azepine hybrid with three nitrogen atoms (1,2,4-triazepine; 7) revealed enhanced activity compared to its closely related analog with nitrogen and sulfur atoms (1,4-thiazepine; 8). Merging of naphthalene and pyrazole as in compound 11 appeared as a better hybrid than that of naphthalene and pyran (compound 12a). The combination of the three motifs together in one ternary hybrid (compound 10) produced an antitumor activity resembling naphthalene–pyrazole hybrid 11. Finally, the activity of naphthalene–pyran hybrid degraded by changing the attached substituent from imine into hydrazone as seen in the order of 12a > 12b > 5.
In the case of MCF-7, naphthalene–pyrazole hybrid (compound 11) appeared as the most superior antitumor compound followed by the naphthalene–nicotinonitrile bearing p-methoxyphenyl substituent 3c. The activity of naphthalene–nicotinonitrile hybrids decreased by changing the nitrogen substituents from p-substituted phenyl groups (3c > 3d > 3b > 3e) into non-substituted parent (3a), and then coumarylimino (3i) followed by 5-membered heterocyclic substituents (3h > 3f > 3g). Regarding naphthalene–pyran hybrids, the activity was lessened in the view of the attached moiety as seen in the order of 12b > 12a > 5. Finally, the hybrid with 1,4-thiazepine (8) appeared to be more active than the one with 1,2,4-triazepine (7).
In order to assess the selectivity of the synthesized compounds for human liver and breast cancer cell lines, Autodock was used to predict the binding pose with active sites of two protein targets (2I4I and 5GWK) (Table 1). The anticancer Doxorubicin was also docked on the targets for comparison purposes. The lowest energy docking mode and the best conformation pose, having the best affinity for a molecule out of ten docking modes obtained by Lamarckian Genetic Algorithm (LGA cluster analysis with respective predicted IC50), were selected from each docking simulation. The polar interactions with the two protein targets were illustrated (Table 1). However, the binding energies of compounds 3e, 3c, and 3h were in good agreement with the experimental data, docking scores for compounds 3f and 3i were not. For examples, compound 3c showed identical binding modes that involved polar interactions with ARG-531, ARG-534, ARG-202, and LYS-208 residues in 2I4I (Fig. 5A); compound 3e were hydrogen-bonded with ARG-531, ARG-534, and ARG-20 residues in 2I4I (Fig. 5B); while compound 3h revealed hydrogen bonding with HIS-523 residue in 2I4I (Fig. 5C).
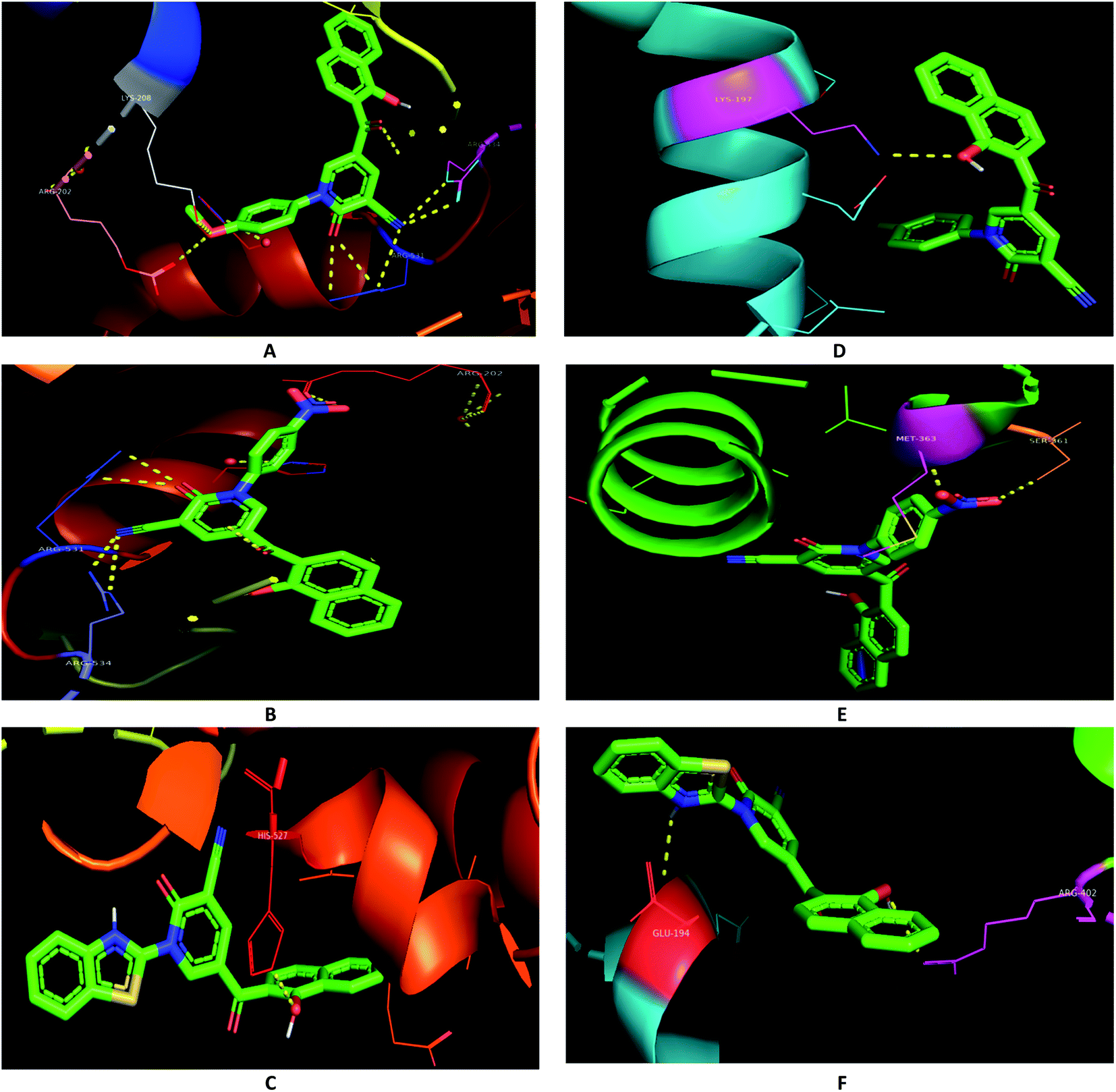 | ||
| Fig. 5 Binding modes (A) 3c and 2I4I; (B) 3e and 2I4I; (C) 3h and 2I4I; (D) 3c and 5GWK; (E) 3e and 5GWK; (F) 3h and 5GWK. | ||
Similarly, the hydroxyl group of naphthalene in compound 3c were included in polar interactions with LYS-197 in 5GWK (Fig. 5D); compound 3e showed polar interactions with SER-361 and MET-363 in 5GWK (Fig. 5E); while compound 3h had polar interactions with ARG-402, and GLU-194 in 5GWK (Fig. 5F). Besides, naphthalene played a major role in the binding affinities of different compounds against the two cancer cell lines. For example, compound 3c was stabilized through π-interactions between the naphthalene with SER-228, GLN-225, GLY-406, and THR-411 in 2I4I; and ALA-350, GLY-347, LEU-362, SER-198, and GLU-194 in 5GWK (ESI 1†).
| Comp. | IC50a (μg mL−1) | Selectivity index (SI)b | COX-2 (PDB 4PH9) | ||
|---|---|---|---|---|---|
| COX-1 | COX-2 | Binding affinity (kcal mol−1) | Amino acid involved in hydrogen bonds | ||
| a It is the compound concentration required to produce 50% inhibition of COX-1 or COX-2.b Selectivity index = (COX-1 IC50/COX-2 IC50). | |||||
| 3a | 124.6 | 8.1 | 15.4 | −8.8 | ARG-345 |
| 3b | 176.4 | 2.3 | 76.7 | −10.6 | GLN-343, ARG-345 |
| 3c | 312.9 | 0.21 | 1490 | −10.2 | ARG-345 |
| 3d | 632.4 | 0.65 | 972.9 | −9.7 | ARG-345 |
| 3e | 116.4 | 0.69 | 168.7 | −11 | GLN-343, ASN-506, ARG-345 |
| 3f | 346.9 | 7.8 | 44.5 | −10.7 | ARG-345 |
| 3g | 1000 | 18.6 | 53.8 | −10.5 | GLN-343, ARG-345 |
| 3h | 289.8 | 0.21 | 1380 | −10.8 | ARG-345 |
| 3i | 712.5 | 6.4 | 111.3 | −12.2 | GLN-343, ARG-345 |
| 5 | 632.4 | 7.5 | 84.3 | −9.5 | ASN506, ASN-344 |
| 6 | 325.6 | 9.3 | 35.0 | −9.5 | ARG-345 |
| 7 | 217.8 | 0.37 | 588.6 | −9.3 | — |
| 8 | 284.5 | 0.25 | 1138 | −9.3 | ARG-345 |
| 10 | 267.1 | 0.27 | 989.3 | −7.7 | GLN-343 |
| 11 | 1000 | 12.1 | 82.6 | −7.7 | GLN-343, ARG-345 |
| 12a | 500 | 10.1 | 49.5 | −10.5 | — |
| 12b | 476.2 | 0.49 | 971.8 | −9 | — |
| CEL | 248.9 | 0.26 | 957.3 | −9 | — |
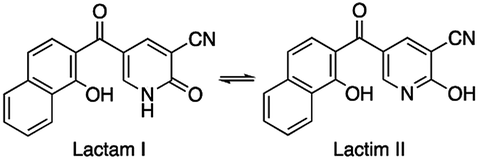
Based upon judicious analysis of the structure–activity relationships (SARs), the utility to cap the NH group in the case of hybrid 3 to suppress the tautomerization of lactam into lactim was highlighted by the potent activity of naphthalene–nicotinonitrile 3c compared to 3a. The anti-inflammatory activities of other naphthalene–nicotinonitrile derivatives were decreased in the order of 3h > 3d > 3e > 3i > 3b > 3g > 3f > 3a. However, the naphthalene–thiazepine hybrid appeared to be more selective than Celecoxib, naphthalene–triazepine revealed inferior selectivity. The formation of a ternary hybrid between naphthalene, pyran, and pyrazole (compound 10) or at least a binary hybrid between naphthalene and pyran (12b) was necessary to achieve high selectivity compared to the binary hybrid between naphthalene and pyrazole (11). Compound 12b as an example of naphthalene–pyran hybrid showed the importance of benzothiazole moiety compared to other analogous compounds 12a and 5. Finally, the combination of naphthalene and pyrazolopyridine moieties was found very deleterious as seen in the least selective anti-inflammatory derivative 6.
The molecular docking of the synthesized compounds in addition to the anti-inflammatory Celecoxib with the drug target COX-2 domains supported the experimental screening in the view of binding energies (Table 2). The binding energies on COX-2 were found in the range of 7.7–12.2 kcal mol−1. As seen in Table 2, compounds 3c and 3h were noted to have the best estimated binding energies for COX-2. However, several amino acid residues were involved in the binding mode, only one polar interaction was shown with ARG-345 residue in 4PH9 (Fig. 6). Besides, the naphthalene skeleton increased the binding affinities against COX-2. For example, compound 3c was stabilized through π-interactions between the naphthalene with PRO-96, GLN-343, ASN-344, ASP-198, SER-112, and GLY-194 in 4PH9 (ESI†).
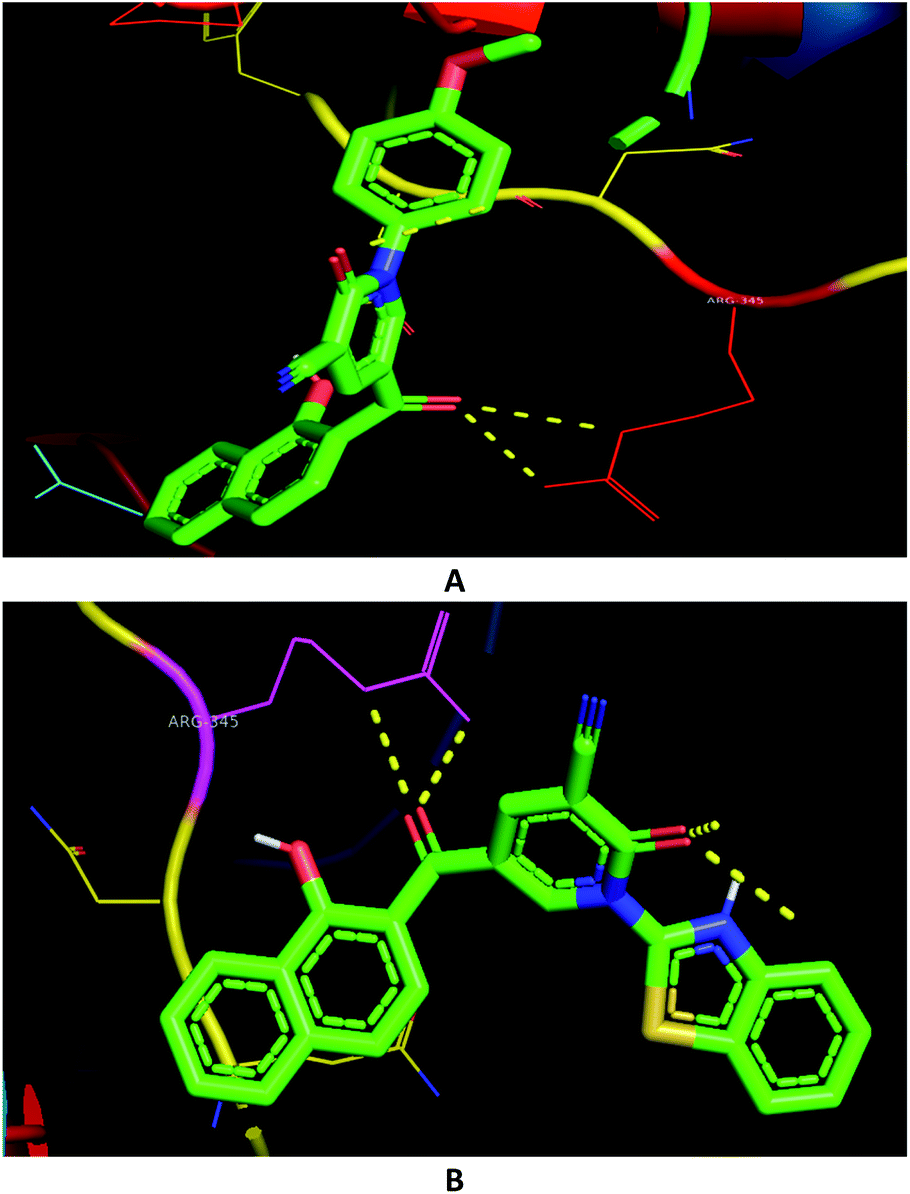
| Comp. | MIC (μg mL−1) | PDB 4DRE | |
|---|---|---|---|
| Binding affinity (kcal mol−1) | Amino acid involved in hydrogen bonds | ||
| 3a | 15.63 | −8.1 | ILE-257 |
| 3b | 3.9 | −9.1 | ILE-257 |
| 3c | 0.48 | −9.0 | ARG-153, ASP-256 |
| 3d | 0.98 | −9.1 | ASP-256 |
| 3e | 1.95 | −9.6 | ARG-153, ASP-256, |
| 3f | 1.95 | −10.5 | ARG-173, GLU-169, TYR-259 |
| 3g | 3.9 | −8.9 | ASP-256 |
| 3h | 1.95 | −9.3 | ARG-153, ARG-173 |
| 3i | 0.98 | −10.7 | ARG-153, ARG-173 |
| 5 | 3.9 | −8.3 | GLU-169, ARG-173, SER-166 |
| 6 | 1.95 | −8.5 | HIS-265, ARG-153 |
| 7 | 1.95 | −7.6 | ASP-256, GLU-169 |
| 8 | 7.81 | −8.1 | TYR-259 |
| 10 | 7.81 | −7.3 | ASP-256 |
| 11 | 31.25 | −7.6 | ARG-173, SER-152, SER-166 |
| 12a | 62.5 | −9.4 | ARG-153 |
| 12b | 15.63 | −9.1 | ARG-153 |
| IN | 0.24 | −4.8 | GLU-169, SER-166, SER-152 |
The excellence of the naphthalene–nicotinonitrile hybrid scaffold was assured by the higher activity of compounds 3c, 3d, and 3i. Moreover, the poor antituberculosis activity of compound 3a, highlighted the need to cap the NH group to retain high antituberculosis activity levels. Protection with p-methoxyphenyl group appeared to be the optimum compared to other protecting groups in derivatives 3d, 3i, 3e, 3f, 3h, 3b, and 3g. Guided by the equal MICs of compounds 3e–h, 6, and 7, pyrazolopyridine or triazepine moieties produced the same activity observed in substituted nicotinonitrile derivatives 3e–h. Moreover, the activity of compound 7 was significantly decayed by replacement of triazepine with thiazepine moiety as seen in compound 8. The combination of naphthalene and pyran moieties as in compound 5 appeared to be superior to compound 10 with the naphthalene–pyranopyrazole and compound 11 with the naphthalene–pyrazole skeleton.
Based on molecular docking analysis, compound 3c was seen to have two polar interactions with ARG-153, ASP-256 residues (Fig. 7), and π-interactions between the naphthalene skeleton with ILE-257, HIS-265, and SER-152 in 4DRE (ESI†). In addition, compounds 3d, 3e, 3f, 3h, and 3i appeared to be good candidates.
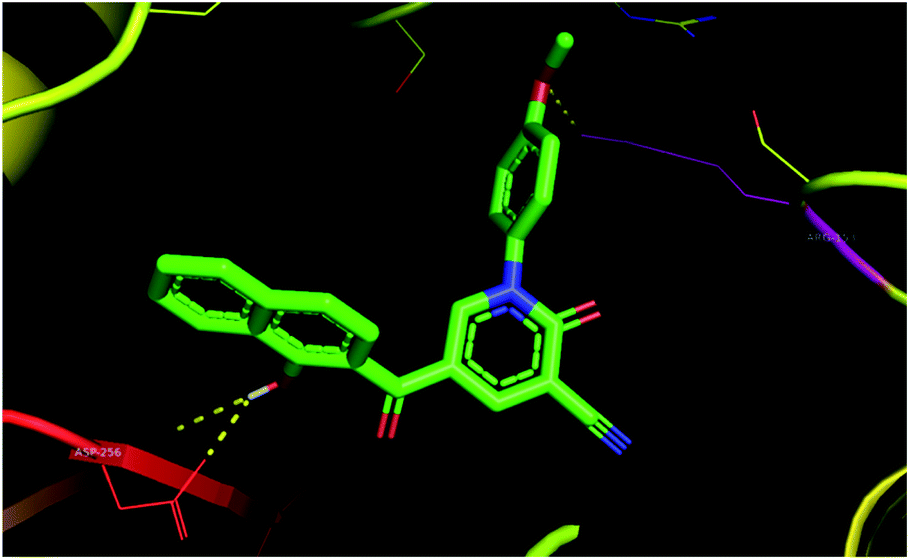 | ||
| Fig. 7 Binding of 3c with M. tuberculosis InhA (PDB 4DRE). | ||
Conclusion
Novel hybrid naphthalene compounds with different heterocyclic scaffolds have been synthesized via tandem reactions of 3-formylchromone with different nucleophilic reagents such as cyanoacetanilides, cyanoacetic acid hydrazide and its hydrazone, thiosemicarbazide, o-aminothiophenol, hydrazine, and heterocyclic °1-amines. Different predicted biological activities have been disclosed based upon analysis using PASS online software. Experimentally, different compounds revealed potent antitumor, anti-inflammatory and antituberculosis activities. Compared to the antitumor Doxorubicin, compounds 3c, 3e, and 3h were more cytotoxic in the case of HepG-2, and compounds 3a–e, 3i, 6, 8, 10, 11, and 12b were more cytotoxic against MCF-7. While compounds 3c, 3h, 8, 10, 3d, and 12b appeared as potent COX-2 selective anti-inflammatory agents, compounds 3c, 3d, and 3i revealed promising antituberculosis activity with MIC less than 1 μg mL−1. A strong agreement between PASS predictions, pharmacokinetic properties, experimental biological evaluations, and docking results were noticed. Molecular docking showed strong polar and hydrophobic interactions between the novel naphthalene-heterocycle hybrids and multifunctional drug targets. Further extensive studies including in vivo studies and approaching more optimized hybrid naphthalene skeletons in addition to the unveiling of the mechanism of action (MOA) of potent compounds are currently underway.Experimental section
General
All melting points in this study were measured by using the Gallenkamp electrical melting point device and reported using the celsius scale. The infrared absorptions of new compounds as potassium bromide pellets were measured on a Mattson 5000 FT-IR spectrophotometer (Faculty of Pharmacy, Mansoura University, Egypt) or Thermo Fisher Nicolete iS 10, USA (Faculty of Science, Mansoura University, Egypt). The 1H- and 13C-NMR spectra were measured on Bruker Avance III 400 MHz (Faculty of Pharmacy, Mansoura University or Faculty of Pharmacy, Beni Suef University, Egypt), and/or JEOL ECA II 500 MHz (Faculty of Science, Mansoura University, Egypt) using tetramethylsilane as an internal reference. The multiplicities of NMR signals were reported as: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet and m = multiplet. 13C NMR spectra were measured on JEOL ECA II 125 MHz (Faculty of Science, Mansoura University, Egypt). The EI-Mass analyses were performed on Thermo Fisher scientific DSQ II GC/MS with Kratos MS-70 ev (Faculty of Pharmacy, Al-Azhar University, Egypt). C, H, and N analyses were carried at the microanalytical unit, Cairo University, Egypt. For high-resolution mass spectrometry (HRMS) was performed using Thermo Fisher Scientific Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (College of Pharmacy, Chung-Ang University, South Korea) and m/z ratios were reported as values in atomic mass units.Synthesis
It is a tautomeric mixture in 4
 :1 ratio. Yield (263 mg, 91%); canary yellow crystals; m.p. = 346–347 °C; IR (KBr, ν/cm−1) = 3375, 3367, 3175 (2× OH, NH), 2231 (CN), 1706 (COKetone), 1630 (COPyridone); 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 7.48–7.46 (m, 1H, Ar–H, I and II), 7.62–7.57 (m, 2H, Ar–H, I and II), 7.71–7.68 (m, 1H, Ar–H, I and II), 7.77 (d, 1H, Ar–H, J = 4.50 Hz, I or II), 7.94 (d, 1H, Ar–H, J = 8.30 Hz, I or II), 8.15 (d, 1H, Ar–H (pyridyl), J = 2.70 Hz, I or II), 8.21 (d, 1H, Ar–H (pyridyl), J = 2.00 Hz, I or II), 8.35 (d, 1H, Ar–H, J = 8.30 Hz, I or II), 8.49 (d, 1H, Ar–H (pyridyl), J = 2.00 Hz, I or II), 8.67 (d, 1H, Ar–H (pyridyl), J = 2.70 Hz), 8.80 (d, 1H, Ar–H, J = 4.50 Hz, I or II), 12.26 (brs, 1H, OH or NH, exchangeable with D2O), 13.01 (brs, 1H, OH or NH, exchangeable with D2O); HRMS (ESI): m/z calcd for C17H10N2NaO3 [(M + Na)+] 313.0584, found 313.0576; anal. calcd for C17H10N2O3 (290.07): C, 70.34; H, 3.47; N, 9.65%. Found: C, 70.54; H, 3.49; N, 9.61%.:1 mixture. Yield (301 mg, 98.69%); buff crystals; m.p. = 356–357 °C; IR (KBr, ν/cm−1) = 3206 (NH), 2262 (CN), 1690 (COChromone), 1639 (COHydrazide), 1624 (C
:1 ratio. Yield (263 mg, 91%); canary yellow crystals; m.p. = 346–347 °C; IR (KBr, ν/cm−1) = 3375, 3367, 3175 (2× OH, NH), 2231 (CN), 1706 (COKetone), 1630 (COPyridone); 1H-NMR (500 MHz, DMSO-d6) δ (ppm): 7.48–7.46 (m, 1H, Ar–H, I and II), 7.62–7.57 (m, 2H, Ar–H, I and II), 7.71–7.68 (m, 1H, Ar–H, I and II), 7.77 (d, 1H, Ar–H, J = 4.50 Hz, I or II), 7.94 (d, 1H, Ar–H, J = 8.30 Hz, I or II), 8.15 (d, 1H, Ar–H (pyridyl), J = 2.70 Hz, I or II), 8.21 (d, 1H, Ar–H (pyridyl), J = 2.00 Hz, I or II), 8.35 (d, 1H, Ar–H, J = 8.30 Hz, I or II), 8.49 (d, 1H, Ar–H (pyridyl), J = 2.00 Hz, I or II), 8.67 (d, 1H, Ar–H (pyridyl), J = 2.70 Hz), 8.80 (d, 1H, Ar–H, J = 4.50 Hz, I or II), 12.26 (brs, 1H, OH or NH, exchangeable with D2O), 13.01 (brs, 1H, OH or NH, exchangeable with D2O); HRMS (ESI): m/z calcd for C17H10N2NaO3 [(M + Na)+] 313.0584, found 313.0576; anal. calcd for C17H10N2O3 (290.07): C, 70.34; H, 3.47; N, 9.65%. Found: C, 70.54; H, 3.49; N, 9.61%.:1 mixture. Yield (301 mg, 98.69%); buff crystals; m.p. = 356–357 °C; IR (KBr, ν/cm−1) = 3206 (NH), 2262 (CN), 1690 (COChromone), 1639 (COHydrazide), 1624 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 4.27 (s, 2H, CH2, E or Z), 3.88 (s, 2H, CH2, E or Z), 7.83–7.82 (m, 2H, Ar–H, E and Z), 8.04–8.00 (m, 2H, Ar–H, E and Z), 8.13 (d, 1H, Ar–H, J = 7.68 Hz, E and Z), 8.19 (d, 1H, H-10, J = 7.68 Hz, E and Z), 8.48 (s, 1H, pyrone–H, E or Z), 8.32 (s, 1H, pyrone–H, E or Z), 8.95 (s, 1H,
N); 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 4.27 (s, 2H, CH2, E or Z), 3.88 (s, 2H, CH2, E or Z), 7.83–7.82 (m, 2H, Ar–H, E and Z), 8.04–8.00 (m, 2H, Ar–H, E and Z), 8.13 (d, 1H, Ar–H, J = 7.68 Hz, E and Z), 8.19 (d, 1H, H-10, J = 7.68 Hz, E and Z), 8.48 (s, 1H, pyrone–H, E or Z), 8.32 (s, 1H, pyrone–H, E or Z), 8.95 (s, 1H, 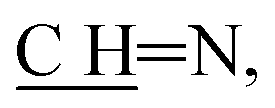 E or Z), 9.06 (s, 1H,
E or Z), 9.06 (s, 1H, 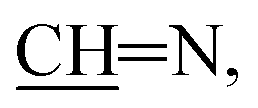 E or Z), 11.89 (s, 1H, NH, exchangeable with D2O, E or Z), 11.92 (s, 1H, NH, exchangeable with D2O, E or Z); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 174.5, 164.9, 153.9, 153.1, 136.6, 135.4, 129.9, 128.3, 128.0, 126.0, 123.2, 121.8, 120.0, 119.7, 119.4, 116.1, 24.2; EI-MS m/z (%): 40.19 (100.00), 44.09 (21.59%), 69.19 (1.58%), 113.89 (1.57%), 299.48 (1.42%), 305.20 (3.90%), 306.12 (2.18%); HRMS (ESI): m/z calcd for C17H11N3NaO3 [(M + Na)+] 328.0693, found 328.0682; anal. calcd for C17H11N3O3 (305.08): C, 66.88; H, 3.63; N, 13.76%. Found: C, 66.94; H, 3.62; N, 13.85%.N); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 7.48 (d, 1H, naphthyl–H, J = 8.65 Hz), 7.60 (d, 1H, naphthyl–H, J = 8.65 Hz), 7.65–7.63 (m, 1H, naphthyl–H), 7.75–7.73 (m, 1H, naphthyl–H), 7.96 (d, 1H, naphthyl–H, J = 8.15 Hz), 8.39 (d, 1H, naphthyl–H, J = 8.30 Hz), 8.49 (d, 1H, pyridine, J = 1.95 Hz), 8.82 (d, 1H, pyridine, J = 1.95 Hz), 11.30 (brs, 1H, OH or NH, exchangeable with D2O), 12.75 (brs, 1H, OH or NH, exchangeable with D2O), 12.88 (s, 1H, NH or OH, exchangeable with D2O); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 197.7, 159.6, 150.6, 136.5, 132.5, 130.0, 127.7, 126.9, 126.3, 125.0, 124.6, 123.5, 118.7, 114.6; HRMS (ESI): m/z calcd for C17H11N3NaO3 [(M + Na)+] 328.0693, found 328.0691; anal. calcd for C17H11N3O3 (305.08): C, 66.88; H, 3.63; N, 13.76%. Found: C, 66.83; H, 3.58; N, 13.72%.N); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37 (d, 1H, Ar–H, J = 10.80 Hz), 7.64–7.55 (m, 3H, Ar–H), 7.74 (s, 1H, thiazepine–H), 7.81 (d, 1H, Ar–H, J = 10.00 Hz), 7.91 (dd, 2H, Ar–H, J = 11.00, 11.60 Hz), 8.05 (d, 1H, Ar–H, J = 11.00 Hz), 8.16 (d, 1H, Ar–H, J = 10.50 Hz), 8.19 (s, 1H, thiazepine–H), 8.45 (d, 1H, Ar–H, J = 10.00 Hz), 12.50 (s, 1H, OH, exchangeable with D2O); HRMS (ESI): m/z calcd for C20H13NNaO2S [(M + Na)+] 354.0559, found 354.0575; anal. calcd for C20H13NO2S (331.07): C, 72.49; H, 3.95; N, 4.23, S, 9.67%. Found: C, 72.42; H, 3.99; N, 4.31, S, 9.65%.N); 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.06 (t, 3H, CH2CH3, J = 6.80 Hz), 3.45 (q, 2H, CH2CH3, J = 6.80 Hz), 4.39 (br. s, 1H, NH, exchangeable with D2O), 6.94 (s, 1H, H-2Chromene), 7.57–7.55 (m, 2H, H-Ar–H), 7.61–7.59 (m, 2H, Ar–H), 7.92 (d, 1H, Ar–H, J = 8.01 Hz), 8.32 (d, 1H, Ar–H, J = 8.01 Hz), 8.66 (brs, 1H, CHPyrazole); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 154.4, 151.0, 134.3, 127.6, 127.0, 125.5, 125.4, 122.6, 119.3, 114.7, 111.5, 56.1, 18.6; EI-MS m/z (%): 50.92 (46.23%), 58.47 (57.12%), 98.4 (74.18%), 106.85 (70.21%), 195.09 (68.37%), 214.05 (100.00%), 238.49 (37.40%); HRMS (ESI): m/z calcd for C16H15N2O2 [(M + H)+] 267.1128, found 267.11.21; anal. calcd for C16H14N2O2 (266.11): C, 70.12; H, 5.30; N, 10.52%. Found: C, 70.21; H, 5.36; N, 10.42%.N), 1574 (CC); 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 7.19 (br. s, 2H, NH2), 7.75–7.54 (m, 5H, Ar–H, CHN), 7.92 (d, 1H, Ar–H, J = 8.00 Hz), 8.32 (d, 1H, Ar–H, J = 8.00 Hz), 8.67 (brs, 1H, CHPyrazole), 10.61 (s, 1H, OH or NH), 13.6 (s, 1H, OH or NH); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 115.10, 116.63, 119.81, 123.08, 125.79, 126.02, 127.46, 127.84, 128.06, 128.69, 129.38, 134.73, 151.36, 154.84; EI-MS m/z (%): 77.24 (72.11%), 122.06 (82.14%), 151.26 (100.00%), 152.19 (62.05%), 166.96 (48.52%), 202.12 (56.76%), 216.92 (57.41%), 252.00 (11.34%); HRMS (ESI): m/z calcd for C14H12N4NaO [(M + Na)+] 275.0903, found 275.0901; anal. calcd for C14H12N4O (252.10): C, 66.65; H, 4.79; N, 22.21%. Found: C, 70.53; H, 4.37; N, 11.72%.N); 1H NMR (500 MHz, CDCl3) δ (ppm): 2.49 (s, 3H, CH3), 3.15 (s, 3H, NCH3), 7.30 (t, 1H, Ar–H, J = 7.25 Hz), 7.38 (d, 2H, Ar–H, J = 7.55 Hz), 7.45 (dd, 2H, Ar–H, J = 7.25, 7.55 Hz), 7.69–7.63 (m, 2H, Ar–H) 7.76 (d, 1H, Ar–H, J = 8.65 Hz), 7.90 (d, 1H, Ar–H, J = 7.60 Hz), 8.19 (d, 1H, Ar–H, J = 8.65 Hz), 8.46 (d, 1H, Ar–H, J = 7.60 Hz), 8.93 (s, 1H, chromone–H), 9.87 (s, 1H, CHN); 13C NMR (150 MHz, CDCl3) δ (ppm): 188.8, 175.6, 167.8, 160.3, 159.5, 153.6, 150.7, 135.9, 134.6, 129.3, 129.2, 128.1, 127.2, 127.0, 125.6, 124.5, 124.0, 122.2, 122.1, 121.1, 121.0, 35.8, 10.3; EI-MS m/z (%): 73.13 (50.42%), 77.15 (65.69%), 267.18 (32.21%), 295.22 (58.23%), 336.25 (100.00%), 354.20 (48.79%), 409.02 (3.45%); HRMS (ESI): m/z calcd for C25H19N3NaO3 [(M + Na)+] 432.1319, found 432.1319; anal. calcd for C25H19N3O3 (409.14): C, 73.34; H, 4.68; N, 10.26%. Found: C, 73.32; H, 4.71; N, 10.31%.N); HRMS (ESI): m/z calcd for C21H12N2NaO2S [(M + Na)+] 379.0512, found 379.0506; anal. calcd for C21H12N2O2S (356.06): C, 70.77; H, 3.39; N, 7.86; S, 9.00%. Found: C, 70.72; H, 3.41; N, 7.81; S, 9.07%.
E or Z), 11.89 (s, 1H, NH, exchangeable with D2O, E or Z), 11.92 (s, 1H, NH, exchangeable with D2O, E or Z); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 174.5, 164.9, 153.9, 153.1, 136.6, 135.4, 129.9, 128.3, 128.0, 126.0, 123.2, 121.8, 120.0, 119.7, 119.4, 116.1, 24.2; EI-MS m/z (%): 40.19 (100.00), 44.09 (21.59%), 69.19 (1.58%), 113.89 (1.57%), 299.48 (1.42%), 305.20 (3.90%), 306.12 (2.18%); HRMS (ESI): m/z calcd for C17H11N3NaO3 [(M + Na)+] 328.0693, found 328.0682; anal. calcd for C17H11N3O3 (305.08): C, 66.88; H, 3.63; N, 13.76%. Found: C, 66.94; H, 3.62; N, 13.85%.N); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 7.48 (d, 1H, naphthyl–H, J = 8.65 Hz), 7.60 (d, 1H, naphthyl–H, J = 8.65 Hz), 7.65–7.63 (m, 1H, naphthyl–H), 7.75–7.73 (m, 1H, naphthyl–H), 7.96 (d, 1H, naphthyl–H, J = 8.15 Hz), 8.39 (d, 1H, naphthyl–H, J = 8.30 Hz), 8.49 (d, 1H, pyridine, J = 1.95 Hz), 8.82 (d, 1H, pyridine, J = 1.95 Hz), 11.30 (brs, 1H, OH or NH, exchangeable with D2O), 12.75 (brs, 1H, OH or NH, exchangeable with D2O), 12.88 (s, 1H, NH or OH, exchangeable with D2O); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 197.7, 159.6, 150.6, 136.5, 132.5, 130.0, 127.7, 126.9, 126.3, 125.0, 124.6, 123.5, 118.7, 114.6; HRMS (ESI): m/z calcd for C17H11N3NaO3 [(M + Na)+] 328.0693, found 328.0691; anal. calcd for C17H11N3O3 (305.08): C, 66.88; H, 3.63; N, 13.76%. Found: C, 66.83; H, 3.58; N, 13.72%.N); 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37 (d, 1H, Ar–H, J = 10.80 Hz), 7.64–7.55 (m, 3H, Ar–H), 7.74 (s, 1H, thiazepine–H), 7.81 (d, 1H, Ar–H, J = 10.00 Hz), 7.91 (dd, 2H, Ar–H, J = 11.00, 11.60 Hz), 8.05 (d, 1H, Ar–H, J = 11.00 Hz), 8.16 (d, 1H, Ar–H, J = 10.50 Hz), 8.19 (s, 1H, thiazepine–H), 8.45 (d, 1H, Ar–H, J = 10.00 Hz), 12.50 (s, 1H, OH, exchangeable with D2O); HRMS (ESI): m/z calcd for C20H13NNaO2S [(M + Na)+] 354.0559, found 354.0575; anal. calcd for C20H13NO2S (331.07): C, 72.49; H, 3.95; N, 4.23, S, 9.67%. Found: C, 72.42; H, 3.99; N, 4.31, S, 9.65%.N); 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.06 (t, 3H, CH2CH3, J = 6.80 Hz), 3.45 (q, 2H, CH2CH3, J = 6.80 Hz), 4.39 (br. s, 1H, NH, exchangeable with D2O), 6.94 (s, 1H, H-2Chromene), 7.57–7.55 (m, 2H, H-Ar–H), 7.61–7.59 (m, 2H, Ar–H), 7.92 (d, 1H, Ar–H, J = 8.01 Hz), 8.32 (d, 1H, Ar–H, J = 8.01 Hz), 8.66 (brs, 1H, CHPyrazole); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 154.4, 151.0, 134.3, 127.6, 127.0, 125.5, 125.4, 122.6, 119.3, 114.7, 111.5, 56.1, 18.6; EI-MS m/z (%): 50.92 (46.23%), 58.47 (57.12%), 98.4 (74.18%), 106.85 (70.21%), 195.09 (68.37%), 214.05 (100.00%), 238.49 (37.40%); HRMS (ESI): m/z calcd for C16H15N2O2 [(M + H)+] 267.1128, found 267.11.21; anal. calcd for C16H14N2O2 (266.11): C, 70.12; H, 5.30; N, 10.52%. Found: C, 70.21; H, 5.36; N, 10.42%.N), 1574 (CC); 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 7.19 (br. s, 2H, NH2), 7.75–7.54 (m, 5H, Ar–H, CHN), 7.92 (d, 1H, Ar–H, J = 8.00 Hz), 8.32 (d, 1H, Ar–H, J = 8.00 Hz), 8.67 (brs, 1H, CHPyrazole), 10.61 (s, 1H, OH or NH), 13.6 (s, 1H, OH or NH); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 115.10, 116.63, 119.81, 123.08, 125.79, 126.02, 127.46, 127.84, 128.06, 128.69, 129.38, 134.73, 151.36, 154.84; EI-MS m/z (%): 77.24 (72.11%), 122.06 (82.14%), 151.26 (100.00%), 152.19 (62.05%), 166.96 (48.52%), 202.12 (56.76%), 216.92 (57.41%), 252.00 (11.34%); HRMS (ESI): m/z calcd for C14H12N4NaO [(M + Na)+] 275.0903, found 275.0901; anal. calcd for C14H12N4O (252.10): C, 66.65; H, 4.79; N, 22.21%. Found: C, 70.53; H, 4.37; N, 11.72%.N); 1H NMR (500 MHz, CDCl3) δ (ppm): 2.49 (s, 3H, CH3), 3.15 (s, 3H, NCH3), 7.30 (t, 1H, Ar–H, J = 7.25 Hz), 7.38 (d, 2H, Ar–H, J = 7.55 Hz), 7.45 (dd, 2H, Ar–H, J = 7.25, 7.55 Hz), 7.69–7.63 (m, 2H, Ar–H) 7.76 (d, 1H, Ar–H, J = 8.65 Hz), 7.90 (d, 1H, Ar–H, J = 7.60 Hz), 8.19 (d, 1H, Ar–H, J = 8.65 Hz), 8.46 (d, 1H, Ar–H, J = 7.60 Hz), 8.93 (s, 1H, chromone–H), 9.87 (s, 1H, CHN); 13C NMR (150 MHz, CDCl3) δ (ppm): 188.8, 175.6, 167.8, 160.3, 159.5, 153.6, 150.7, 135.9, 134.6, 129.3, 129.2, 128.1, 127.2, 127.0, 125.6, 124.5, 124.0, 122.2, 122.1, 121.1, 121.0, 35.8, 10.3; EI-MS m/z (%): 73.13 (50.42%), 77.15 (65.69%), 267.18 (32.21%), 295.22 (58.23%), 336.25 (100.00%), 354.20 (48.79%), 409.02 (3.45%); HRMS (ESI): m/z calcd for C25H19N3NaO3 [(M + Na)+] 432.1319, found 432.1319; anal. calcd for C25H19N3O3 (409.14): C, 73.34; H, 4.68; N, 10.26%. Found: C, 73.32; H, 4.71; N, 10.31%.N); HRMS (ESI): m/z calcd for C21H12N2NaO2S [(M + Na)+] 379.0512, found 379.0506; anal. calcd for C21H12N2O2S (356.06): C, 70.77; H, 3.39; N, 7.86; S, 9.00%. Found: C, 70.72; H, 3.41; N, 7.81; S, 9.07%.Biological evaluation
Antitumor activity
The cells of human liver carcinoma (HepG-2), and human breast adenocarcinoma (MCF-7) were purchased from the American Type Culture Collection (Rockville, MD). All cells were maintained in a DMEM medium, which was supplemented with 10% of heat-inactivated fetal bovine serum (FBS), 100 U ml−1 of each of penicillin and streptomycin. The cells were grown at 37 °C in a humidified atmosphere of 5% CO2. The cytotoxicity activities on the HepG-2, and MCF-7 human cancer cell lines were estimated, employing the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, which was grounded on the reduction of the tetrazolium salt by the mitochondrial dehydrogenases in viable cells.30 The cells were dispensed in a 96 well sterile microplate (3 × 104 cells per well), followed by their incubation at 37 °C with a series of different concentrations of 10 μL of each compound or Doxorubicin® (positive control, in DMSO) for 48 h in a serum-free medium prior to the MTT assay. Subsequently, the media were carefully removed, 40 μL of MTT (2.5 mg mL−1) was added to each well, and then incubated for an additional 4 h. The purple formazan dye crystals were solubilized by the addition of 200 μL of DMSO. The absorbance was measured at 570 nm applying a SpectraMax® Paradigm® Multi-Mode microplate reader. The relative cell viability was expressed as the mean percentage of viable cells relative to the untreated control cells. All experiments were conducted in triplicate and were repeated on three different days. All the values were represented as mean ± SD. The IC50s were determined by the SPSS probit analysis software program (SPSS Inc., Chicago, IL).Anti-inflammatory activity
The ability of the synthesized compounds to inhibit ovine COX-1 and COX-2 (IC50 values) was determined using an enzyme immunoassay (EIA) kit (Catalog number 560101, Cayman Chemical, Ann Arbor, MI, USA). Briefly, to a series of supplied reaction buffer, solutions (960 mL, 0.1 M Tris HCl pH 8.0 containing 5 mM EDTA and 2 mM phenol) with either COX-1 or COX-2 (10 mL) enzyme in the presence of heme (10 mL) were added 10 mL of various concentrations of test drug solutions (1000–0.49 μg mL−1 in a final volume of 1 mL). These solutions were incubated for 5 min at 37 °C after which 10 mL of AA (100 mM) solution were added and the COX reaction was stopped by the addition of 50 mL of 1 M HCl after 2 min. This assay is based on the competition between PGs and a PG-acetylcholinesterase conjugate (PG tracer) for a limited amount of PG antiserum. The amount of PG tracer that can bind to the PG antiserum is inversely proportional to the concentration of PGs in the wells since the concentration of PG tracer is held constant while the concentration of PGs varies. This antibody ePG complex binds to a mouse anti-rabbit monoclonal antibody that had been previously attached to the well. The plate is washed to remove any unbound reagents and then Ellman's reagent, which contains the substrate to acetylcholine esterase, is added to the well. The product of this enzymatic reaction produces a distinct yellow color that absorbs at 406 nm. IC50 value is the compound concentration required to produce 50% inhibition of COX-1 or COX-2 for means of two determinations using an ovine COX-1/COX-2 assay kit (USA) (triplicate determinations). Selectivity index (COX-1 IC50/COX-2 IC50).Antituberculosis activity
The Mycobacterium tuberculosis (RCMB 010126) strain was provided from the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University (Cairo, Egypt). Antimycobacterial activity of the synthesized compounds was performed using the microplate Alamar blue assay (MABA) which was performed in black, clear-bottomed, 96 well microplates to minimize background effects.31 Outer perimeter wells were filled with sterile water to prevent dehydration in experimental wells. Initial compounds dilutions were prepared in dimethyl sulfoxide and subsequent twofold dilutions were performed in the microplates. 0.1 mL of 105 CFU mL−1 Mycobacterium tuberculosis inoculum was added to wells, additional control wells consisted of bacteria only (B). Isoniazid was used as a reference drug. Plates were incubated at 37 °C. Starting at day 4 of incubation, 20 μL of alamar Blue solution (Alamar Biosciences/Accumed, Westlake, OH, USA) and 12.5 μL of 20% Tween 80 were added to the entire plate. Plates were then incubated at 37 °C, and results were recorded at 24 h post-reagent addition at 590 nm. Percent inhibition was defined as [1 − (mean of test well/mean of B wells) × 100]. Visual MICs were defined as the lowest concentration of drug that prevented a color change.Computational analysis
Molecular docking
To predict the inhibition mechanisms and the most suitable targets for the synthesized compounds, several biological targets related to anticancer, anti-inflammatory, and antituberculosis activities were screened. The crystal structures Human DEAD-box RNA helicase DDX3X (PDB 2I4I) and Human topoisomerase II (PDG 5GWK) as cancer targets; COX-1 (PDB 1EGQ) and COX-2 (PDB 4PH9) as anti-inflammatory targets; M. tuberculosis InhA enzyme (PDB 4DRE) as tuberculosis target were selected and fetched from Protein Data Bank (www.rcsb.org/pdb). Chemical structures of ligands were drawn into Marvin Sketch (ChemAxon) and the most energetically favored conformer was saved as (*.sdf) file format for docking. Molecular AutoDock 4.2 (MGL tools-1.5.6) docking protocol was performed using PYRX software.32 Prior to the semi-flexible docking, the target proteins were cleaned up by removing water molecules and adding hydrogen atoms to obtain correct ionization and tautomeric states of amino acid residues. All rotatable bonds could rotate during the docking process, and protein-ligand interactions were saved in the PDBQT format suitable for calculating binding energy. The structure of ligand and its complexes were transformed into PDB file format using PYMOL software (Version 2.2.3).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We would like to deeply thank professor Kyungsoo Oh and professor Hun Young Kim (College of Pharmacy, Chung-Ang University, South Korea) for their assistance in HR-MS analyses.References
- S. Makar, T. Saha and S. K. Singh, Eur. J. Med. Chem., 2019, 161, 252–276 CrossRef CAS.
- (a) S. I. El-Desoky, E. M. Keshk, A. A. El-Sawi, M. A. Abozeid, L. A. Abouzeid and A. H. Abdel-Rahman, Saudi Pharm. J., 2018, 26, 852–859 CrossRef; (b) M. A. Abozeid, M. R. El-Kholany, A. H. Abdel-Rahman and S. I. El-Desoky, Int. J. Mod. Org. Chem., 2018, 5, 1–11 CAS; (c) M. A. Abozeid, M. R. El-Kholany, L. A. Abouzeid, A. H. Abdel-Rahman and S. I. El-Desoky, J. Heterocycl. Chem., 2019, 56, 2922–2933 CrossRef CAS; (d) S. I. El-Desoky, A. A. El-Sawi, M. A. Abozeid, M. Abdelmoteleb, M. Shaaban, E. M. Keshk and A. H. Abdel-Rahman, Med. Chem. Res., 2019, 28, 1601–1617 CrossRef; (e) M. A. Abozeid, A. A. El-Sawi, M. R. Elmorsy, M. Abdelmoteleb, A. H. Abdel-Rahman and S. I. El-Desoky, RSC Adv., 2019, 9, 27996–28005 RSC.
- (a) S. Valente, D. Trisciuoglio, T. De Luca, A. Nebbioso, D. Labella, A. Lenoci, C. Bigogno, G. Dondio, M. Miceli and G. Brosch, J. Med. Chem., 2014, 57, 6259–6265 CrossRef CAS; (b) C. Abate, M. Niso, E. Lacivita, P. D. Mosier, A. Toscano and R. Perrone, J. Med. Chem., 2011, 54, 1022–1032 CrossRef CAS.
- R. Perrone, F. Doria, E. Butovskaya, I. Frasson, S. Botti, M. Scalabrin, S. Lago, V. Grande, M. Nadai, M. Freccero and S. N. Richter, J. Med. Chem., 2015, 58, 9639–9652 CrossRef CAS.
- Z. Özdemir, S. Sari, A. Karakurt and S. Dalkara, Drug Dev. Res., 2019, 80, 269–280 CrossRef.
- S. K. Das, G. Panda, V. Chaturvedi, Y. S. Manju, A. K. Gaikwad and S. Sinha, Bioorg. Med. Chem. Lett., 2007, 17, 5586–5589 CrossRef CAS.
- E. A. Boyle, P. C. Freemanm, F. R. Mangan and M. J. Thomson, J. Pharm. Pharmacol., 1982, 34, 562–569 CrossRef CAS.
- T. A. Unzner, A. S. Grossmann and T. Magauer, Angew. Chem., Int. Ed., 2016, 55, 9763–9767 CrossRef CAS.
- D. Quan, G. Nagalingam, R. Payne and J. A. Triccas, Int. J. Infect. Dis., 2017, 56, 212–220 CrossRef CAS.
- H.-J. Zhang, E. Rumschlag-Booms, Y.-F. Guan, D.-Y. Wang, K.-L. Liu, W.-F. Li, V. H. Nguyen, N. M. Cuong, D. D. Soejarto, H. H. S. Fong and L. Rong, J. Nat. Prod., 2017, 80, 1798–1807 CrossRef CAS.
- R. F. Luduena, M. C. Roach and P. Horowitz, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 873, 143–146 CrossRef CAS.
- W. Molee, A. Phanumartwiwath, C. Kesornpun, S. Sureram, N. Ngamrojanavanich, K. Ingkaninan, C. Mahidol, S. Ruchirawat and P. Kittakoop, Chem. Biodiversity, 2018, 15, e1700537 CrossRef.
- F.-Y. Chang, J. E. Peacock Jr, D. M. Musher, P. Triplett, B. B. MacDonald, J. M. Mylotte, A. O'Donnell, M. M. Wagener and L. Y. Victor, Medicine, 2003, 82, 333–339 CrossRef CAS.
- M. V. Worley and S. J. Estrada, Bedaquiline, Pharmacotherapy, 2014, 34, 1187–1197 CrossRef CAS.
- F. K. Chan, N. S. Abraham, J. M. Scheiman and L. Laine, Am. J. Gastroenterol., 2008, 103, 2908–2918 CrossRef CAS.
- D. W. Robertson, J. H. Krushinski, E. Beedle, J. D. Leander, D. T. Wong and R. Rathbun, J. Med. Chem., 1986, 29, 1577–1586 CrossRef CAS.
- J. Zheng, M. Cho, A. D. Jones and B. D. Hammock, Chem. Res. Toxicol., 1997, 10, 1008–1014 Search PubMed.
- P. J. Thornalley, M. d. A. Doherty, M. T. Smith, J. V. Bannister and G. M. Cohen, Chem.-Biol. Interact., 1984, 48, 195–206 CrossRef CAS.
- P. Jia, C. Dai, P. Cao, D. Sun, R. Ouyang and Y. Miao, RSC Adv., 2020, 10, 7740–7750 RSC.
- M. Decker, Design of hybrid molecules for drug development, Elsevier, Amsterdam, 2017 Search PubMed.
- S. V. Kumar, S. Muthusubramanian and S. Perumal, Org. Prep. Proced. Int., 2019, 51, 1–89 CrossRef CAS.
- (a) K. M. Elattar, M. A. Abozeid, I. A. Mousa and A. El-Mekabaty, RSC Adv., 2015, 5, 106710–106753 RSC; (b) K. A. M. El-Bayouki, J. Sulphur Chem., 2011, 32, 623–690 CrossRef CAS.
- X.-J. Huang, Y. Tao, Y.-K. Li, X.-Y. Wu and F. Sha, Tetrahedron, 2016, 72, 8565–8577 CrossRef CAS.
- (a) W. Ried and B. Schleimer, Angew. Chem., 1958, 70, 164 Search PubMed; (b) N. Y. Gorobets, B. H. Yousefi, F. Belaj and C. O. Kappe, Tetrahedron, 2004, 60, 8633–8644 CrossRef CAS; (c) S. M. Gomha, K. D. Khalil, A. M. El-Zanaty and S. M. Riyadh, Heterocycles, 2013, 87, 1109–1120 CrossRef CAS.
- S. M. Gomha and K. D. Khalil, Molecules, 2012, 17, 9335–9347 CrossRef CAS.
- O. Ahmed, V. Cherkadu, P. K. Kalavagunta and J. Shang, RSC Adv., 2019, 9, 20573–20581 RSC.
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, ISRN Pharm., 2012, 2012, 195727 Search PubMed.
- Y. C. Martin, J. Med. Chem., 2005, 48, 3164–3170 CrossRef CAS.
- N. Mohsin and M. Irfan, Med. Chem. Res., 2020, 29, 809–830 CrossRef CAS.
- (a) A. F. Kassem, I. F. Nassar, M. T. Abdel-Aal, H. M. Awad and W. A. El-Sayed, Chem. Pharm. Bull., 2019, 67, 888–895 CrossRef CAS; (b) F. M. Alminderej, H. H. Elganzory, M. N. El-Bayaa, H. M. Awad and W. A. El-Sayed, Molecules, 2019, 24, 3738 CrossRef CAS; (c) M. E. Haiba, E. S. Al- Abdullah, N. S. Ahmed, H. A. Ghabbour and H. M. Awad, J. Mol. Struct., 2019, 1195, 702–711 CrossRef CAS.
- (a) M. A. Abdelrahman, I. Salama, M. S. Gomaa, M. M. Elaasser, M. M. Abdel-Aziz and D. H. Soliman, Eur. J. Med. Chem., 2017, 138, 698–714 CrossRef CAS; (b) M. M. Ghorab, M. S. A. El-Gaby, A. M. Soliman, M. S. Alsaid, M. M. Abdel-Aziz and M. M. Elaasser, Chem. Cent. J., 2017, 11, 42 CrossRef.
- O. Trott, A. J. Olson and J. Comput, Chem, 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08526j |
| This journal is © The Royal Society of Chemistry 2020 |