
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized heterocycle-appended porphyrins: catalysis matters†
Inna A. Abdulaeva a,
Kirill P. Birin*a,
Daria A. Polivanovskaiaa,
Yulia G. Gorbunovaab and
Aslan Yu Tsivadzeab
a,
Kirill P. Birin*a,
Daria A. Polivanovskaiaa,
Yulia G. Gorbunovaab and
Aslan Yu Tsivadzeab
aA.N. Frumkin Institute of Physical Chemistry and Electrochemistry RAS, Leninsky pr., 31, building 4, Moscow, 119071, Russia. E-mail: kirill.birin@gmail.com
bN.S. Kurnakov Institute of General and Inorganic Chemistry RAS, Leninsky pr., 31, Moscow, 119991, Russia
First published on 23rd November 2020
Abstract
The scope and limitations of the condensation of labile 2,3-diaminoporphyrin derivatives with aromatic aldehydes to provide functionalized imidazole- and pyrazine-appended porphyrins were investigated in detail. The presence of an acidic catalyst in the reaction was found to be a tool that allows the reaction path to be switched. The influence of the electronic origin of the substituents in the carbonyl components of the condensation on the yields and selectivity of the reaction was revealed. Metal-promoted cross-coupling transformations were found to be convenient for the further targeted construction of functional derivatives based on the prepared bromo-substituted pyrazinoporphyrins. Overall, these strategies provide a versatile technique for the elaboration of a variety of functionalized heterocycle-appended porphyrins for further application in the development of hybrid materials.
Introduction
Currently, the research area of materials chemistry has attracted growing interest from scientists in various academic and applied fields since novel advanced materials are aimed to solve the scientific and technological problems in the different spheres of human life.1–9 Among these materials, the most promising are multifunctional hybrid materials, which are characterized by a set of different properties, and thus can be applied in various fields. They can be achieved by the combination of components of diverse origin, for instance, organic and inorganic moieties.10–14 It is worth noting that this methodology requires a detailed and comprehensive search for the mentioned components.Porphyrins, which possess broad absorptions, and unique electrochemical and exceptional photophysical properties, have been shown to be efficient active sites for these materials for multiple applications. For example, a series of hybrid materials was constructed with tetrapyrrole active sites and inert inorganic supports for the resolution of complicated problems.15–22
It should be noted that solving applied problems using the material chemistry approach always requires extensive preliminary investigations. This inevitably leads to the need to elaborate novel synthetic pathways and the extension of series of known classes of macroheterocyclic compounds with optimized electronic, chemical and physicochemical properties.
In the framework of our ongoing projects, our investigations are focused on tetrapyrrolic compounds, namely porphyrins and phthalocyanines, and their synthetic modifications and search for new types of applications.17,23–33 Recently, we developed synthetic strategies towards imidazole- and pyrazine-annulated porphyrins starting from tetrapyrrole dioxo- or diamino-derivatives and aromatic aldehydes.28,34–38 The sterically hindered tetrapyrrole was used as a synthetic platform for the development of bifunctionalized linear and angular annulated polyaromatic compounds, water soluble derivatives for biomimetic applications and macrocycles with terminal anchoring groups.34,38 Furthermore, the latter methodology was implemented for the elaboration of a novel class of heteroleptic sandwich-type lanthanide (porphyrinato)(phthalocyaninates)39,40 based on heterocycle-appended porphyrin ligands.36 In this case, the influence of the annulated N-cycle on the electronic structure of the entire molecule was revealed by UV-vis spectroscopy.
The most promising synthetic strategy giving access to porphyrins annulated with 5- and 6-membered N-heterocycles implies the use of porphyrin diamines as starting compounds. However, this approach has scarcely been studied applying only 4-bromobenzaldehyde as the carbonyl component of the condensation reaction. Meanwhile, the utilization of different substituted aldehydes allows the direct introduction of various desired functions onto the periphery of the porphyrin core. Accordingly, the scope and limitations of this condensation are of interest, especially in terms of the carbonyl reagent. Moreover, opportunities for the transformation of derivatives bearing active terminal groups are also a topic of the current investigation.
Thus, in the present work, we revealed the applicability of the mentioned synthetic approach towards heterocycle-appended porphyrins. The crucial role of the acidic catalyst in the condensation of 2,3-diaminoporphyrins with aromatic aldehydes is revealed, allowing switching of the direction of the reaction path. Further modification of the obtained heterocycle-porphyrins is shown to be efficiently achieved with the application of metal-promoted transformations. Accordingly, the functionalized imidazo- and pyrazino-porphyrins can be considered as readily available organic precursors for the elaboration of the functional hybrid materials. Consequently, molecules with functional peripheral fragments that can be converted into anchoring groups are of special interest.
Results and discussion
Previously, we reported the selective formation of imidazole-annulated derivatives upon equimolar interaction of sterically hindered nickel(II) diaminoporphyrin bearing meso-mesityl groups with 4-bromobenzaldehyde (Scheme 1).35 Similar condensation with 5-fold molar excess of aldehyde decreased the selectivity of the reaction and provided a mixture of imidazo- and pyrazinoporphyrins.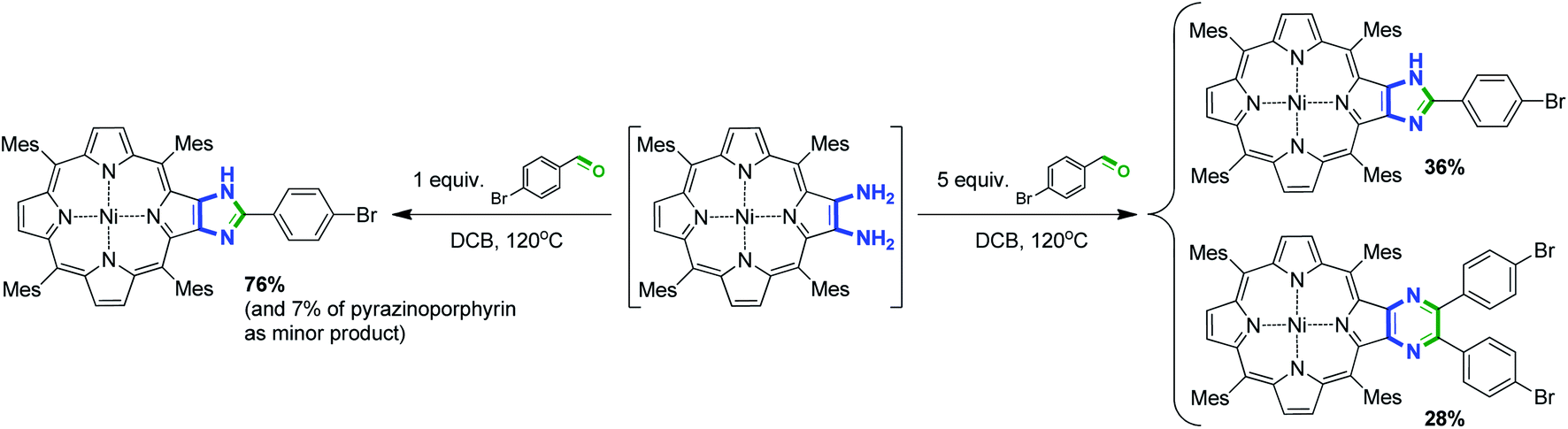 | ||
| Scheme 1 Condensation of 2,3-diaminotetramesitylporphyrinato nickel(II) and 4-bromobenzaldehyde upon variation of the aldehyde ratio.35 | ||
In the present investigation, we chose tetrakis(4-butoxyphenyl)porphyrin as the tetrapyrrolic platform for several reasons. Firstly, the bulkiness of these aryl substituents is negligible in comparison with 2,4,6-trimethylphenyl groups, which allowed any steric effect of the meso-groups on the reaction course to be avoided. Next, the relatively long alkyl chain in the aromatic fragments is expected to provide sufficient solubility to all the semi-products and the final fused compounds. Finally, the electron donor effect of the alkoxy-groups is supposed to promote the condensation reaction due to the increase in the nucleophilicity of the amino-groups in the porphyrin precursor.
These peculiarities of tetrakis(4-butoxyphenyl)porphyrin have already been successfully used for the elaboration of a new class of sandwich-type complexes formed from phthalocyanine and heterocycle-appended porphyrin ligands bearing terminal reactive halogen atoms.36 Accordingly, the extension of the series of functionalized porphyrins fused with different heterocycles is assumed to be of interest for the further design of hybrid materials based not only on porphyrin derivatives, but also on the sophisticated tetrapyrrolic polyaromatic systems.
Firstly, a series of condensations of porphyrin diamine-derivative 2 with different 4-substituted benzaldehydes was performed based on our reported methodology (Scheme 2).35 We found that depending on the stoichiometry of the reaction and the nature of the carbonyl component, three types of compounds may form, namely, imidazoporphyrins 4, pyrazinoporphyrins 5 and pyrazine-fused porphyrin dimer 6. Notably, the mechanism of dimer formation, method for its targeted preparation, and complete investigation of its spectral features were reported by us previously.28 The results of the performed series of condensation reactions are summarized in Table 1.
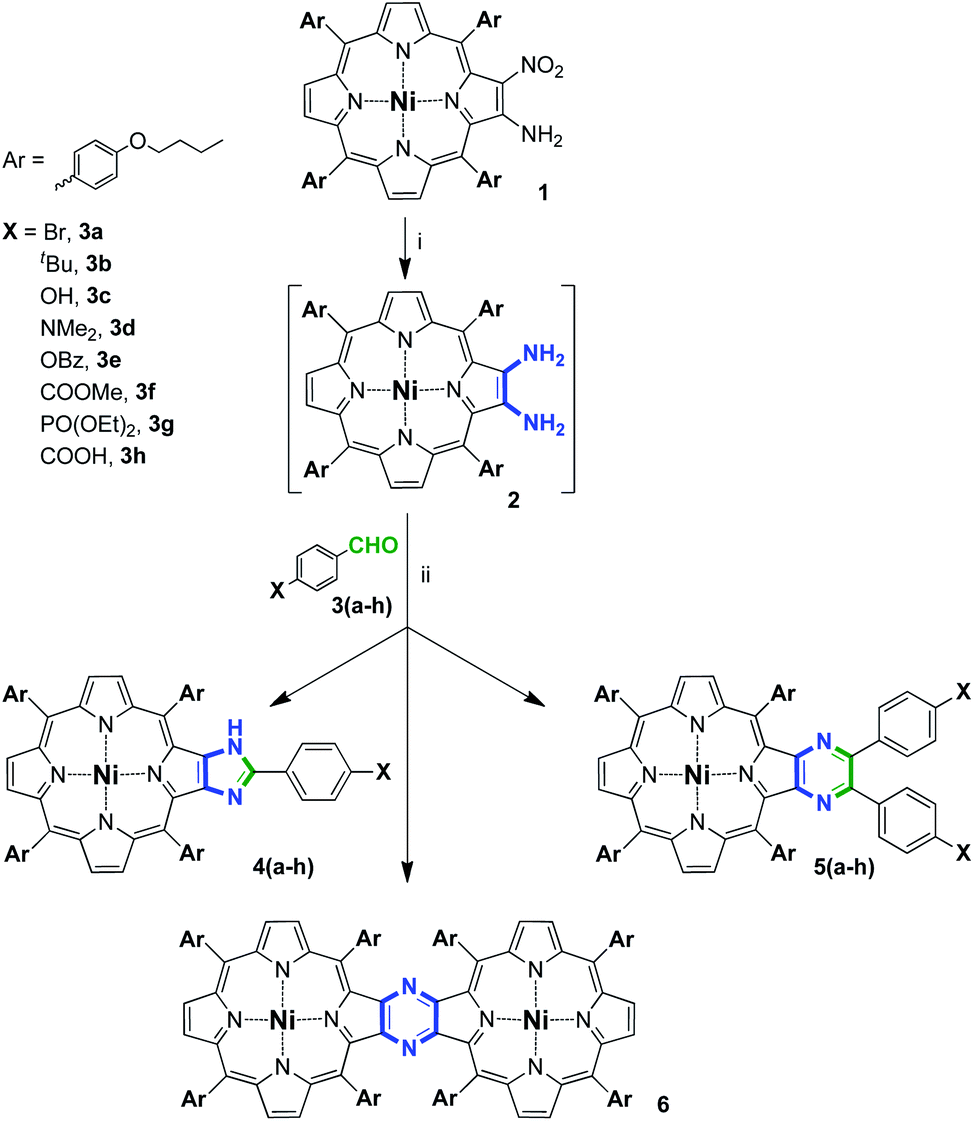 | ||
| Scheme 2 Condensation of nickel(II) 2,3-diamino-tetrakis(4-butoxyphenyl)porphyrin with aromatic aldehydes. Reaction conditions: (i) NaBH4, Pd/C, CH2Cl2/MeOH, inert atm, rt and (ii) 1,2-dichlorobenzene (DCB), 120 °C, inert atm, 18 h. | ||
| Entry | Aldehyde X, (ratio) | Yields (%) | |
|---|---|---|---|
| 4 | 5a | ||
| a The yields of compounds are calculated based on the deficient reagent.b 6 was also isolated in 49% yield. | |||
| 1 | Br, 1 | 73 | 12 |
| 2 | Br, 5 | — | 82 |
| 3 | Br, 25 | — | 86 |
| 4 | tBu, 1 | 53 | 16 |
| 5 | tBu, 5 | — | 78 |
| 6b | OH, 1 | 28 | — |
| 7 | OH, 5 | 11 | 70 |
| 8 | OBz, 1 | 24 | 73 |
| 9 | OBz, 5 | — | 78 |
| 10 | CO2Me, 1 | 42 | 69 |
| 11 | CO2Me, 5 | 80 | — |
| 12 | PO(OEt)2, 5 | 60 | 13 |
| 13 | COOH, 1 | 14 | 80 |
| 14 | COOH, 5 | — | 82 |
4-Bromobenzaldehyde (3a) was used as a reference aldehyde for comparison of the obtained results with the previously reported data.35 Thus, the interaction of 2 with 3a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio provided a mixture of 4a and 5a with 73% and 12% yield, respectively (Table 1, entry 1), that is consistent with the result found for the related tetramesitylporphyrin (Scheme 1).
:1 molar ratio provided a mixture of 4a and 5a with 73% and 12% yield, respectively (Table 1, entry 1), that is consistent with the result found for the related tetramesitylporphyrin (Scheme 1).
The designation of the yields of the reaction products in our opinion demands an additional explanation. The required aldehyde to diamine ratio for the formation of the imidazoporphyrin (1:1) differs from that of pyrazinoporphyrin (2:1). Accordingly, in the latter case, the maximum conversion of diamine that could be reached under equimolar conditions equals 1/2 of its initial quantity. This corresponds to the theoretical 100% yield of the pyrazinoporphyrin upon calculations, starting from the deficient reagent, aldehyde. In the case of imidazoporphyrin and when excess aldehyde is applied, this peculiarity of calculations disappears. Accordingly, similar speculations are valid for the calculation of the yield of 6.
The utilization of excess 4-bromobenzaldehyde (5 equiv.) expectedly resulted in a considerable increase in the yield of pyrazinoporphyrin 5a and complete suppression of the formation of imidazoporphyrin 4a (Table 1, entry 2). However, a further increase in aldehyde ratio had a negligible influence on the yield of 5a (Table 1, entry 3). It is worth noting that in the case of tetramesitylpoprhyrin, the excess aldehyde did not lead to the favorable formation of the pyrazine derivative, and both products formed in comparable yields (Scheme 1).35 This observation can be attributed to the steric effect of the meso-substituents promoting the formation of a less hindered annulated compound. In contrast, the steric hindrance induced by the 4-butoxyphenyl groups is considerably less, which allows the formation of a bulkier pyrazine product.
Changing 4-bromobenzaldehyde to 4-tert-butylbenzaldehyde (3b) did not alter the selectivity of the condensation (Table 1, entry 4). Thus, the equimolar interaction provided the imidazole-fused derivative 4b in 53% yield. The lower yield of the condensation product can be attributed to the decrease in electrophilicity of the formyl group of 3b. The increase in the aldehyde ratio in this case also changed the reaction path to the formation of pyrazine-appended product 5b (Table 1, entry 5).
A further increase in the electron-donor properties of the 4-substituent in the reagent and performing the condensation of porphyrin diamine 2 with 4-hydroxybenzaldehyde (3c) significantly altered the reaction path. Obviously, the presence of the electron-rich hydroxy-group in the reagent molecule significantly decreased the electrophilicity of the carbonyl moiety, and consequently decreased the reactivity of the aldehyde in the condensation. Thus, the interaction with a porphyrin/aldehyde ratio of 1:1 provided only 28% of 4c (Table 1, entry 6). Accordingly, the majority of starting 2 did not interact with 3c and underwent dimerization, providing 6 in 49% yield. The application of excess 3c resulted in a decrease in the yields of 4c and 6, while the yield of 5c changed drastically and reached 70% (Table 1, entry 7).
In the case of 3c, its low reactivity also originated from the possible tautomeric enol-type transitions. Nevertheless, the alkylation of the OH-group prevented tautomerism, and thus did not allow an increase in condensed product yield. Moreover, the decrease in the reactivity of the aromatic aldehyde and the introduction of more electron-rich 4-Me2N substituent using 3d aldehyde completely suppressed the condensation and neither 4d nor 5d was detected in the reaction medium under any conditions.
The crucial influence of the electron-donating substituents in the aromatic aldehyde, and thus the electrophilicity of the formyl group, was proven by involving 4-benzoyloxybenzaldehyde (3e) in the condensation (Table 1, entries 8 and 9). In this case, the electron-donating properties of the oxygen atom are diminished by the conjugation with the electron-withdrawing benzoyl fragment. Accordingly, the reactivity of 3e was found to significantly exceed that of 3c and its interaction with 2 in an equimolar ratio gave rise to a mixture of 4e and 5e (Table 1, entry 8). The increase in the aldehyde/porphyrin ratio resulted in the preferable formation of pyrazinoporphyrin 5e with 78% yield (Table 1, entry 9).
The introduction of the electron-withdrawing groups to the benzaldehyde is obviously expected to increase the electrophilicity of the formyl moiety, and thus facilitate the condensation. Thus, the involvement of 4-methoxycarbonylbenzaldehyde (3f) in the condensation with 2 in an equimolar ratio provided 4f and 5f in high overall yield (Table 1, entry 10). Surprisingly, the increase in the ratio of 3f resulted predominantly in the formation of 4f with 80% yield (Table 1, entry 11). A similar effect was observed with the application of 5 equiv. of 3g in the condensation, resulting in the formation of 60% of imidazoporphyrin 4g together with 13% of 5g (Table 1, entry 12).
In contrast to the 4-methoxycarbonyl- and 4-diethoxyphosphoryl derivatives, 3f and 3g, respectively, 4-carboxybenzaldehyde (3h) exhibited different reactivity. The interaction of 2 and 3f in a 1:5 molar ratio smoothly provided the corresponding pyrazinoporphyrin 5f in high yield. This specific behavior requires particular explanation, which can be provided from a mechanistic point of view.
Previously, we proposed the mechanisms for the formation of imidazo- and pyrazinoporphyrins upon condensation of a diaminoporphyrin with aromatic aldehydes.35 The key step in both cases is the interaction of the aldehyde carbonyl group with one of the amino-groups of the porphyrin precursor (Scheme 3). However, some prerequisites have to be mentioned to analyze the further reaction steps.
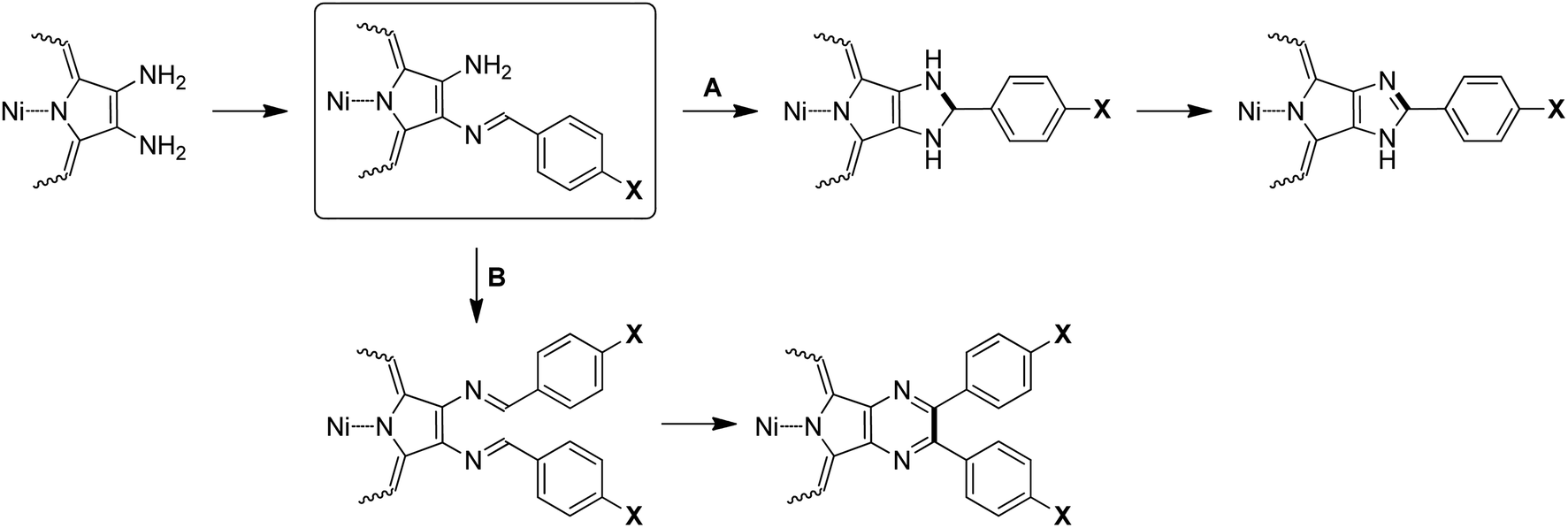 | ||
| Scheme 3 Possible mechanisms for the formation of porphyrins fused with 5- and 6-membered N-heterocycles. | ||
The first condensation step decreases the nucleophilicity of the remaining amino-group and the electrophilicity of the CH-imine fragment in comparison with that of the starting carbonyl compound. Further, the reaction pathway is expected to be dependent on the relative reactivity of the CH-imine group and formyl group of the aldehyde. It is also reasonable to assume that the porphyrin/aldehyde ratio should affect both the first condensation step and the possible condensation with the second molecule of the aldehyde. Moreover, the preorganization of the intermediate favors the formation of a 5-membered heterocycle. The last oxidative steps of both paths are expected to be influenced similarly by the presence of electron-donor or withdrawing groups, and consequently cannot change the course of the reaction.
Thus, based on these considerations, the observed difference in the reaction paths can be explained. The electron-donor hydroxy-group causes the low reactivity of 3c and also defines the decreased electrophilicity of the intermediate CH-imino-fragment, even in comparison with the formyl group of the aldehyde, which disfavors the intramolecular condensation (Scheme 3, path A). Accordingly, the increase in the aldehyde ratio results in the realization of path B through the kinetic facilitation of the second condensation step.
Changing the aldehyde to 3f bearing an electron-withdrawing CO2Me group facilitates the electrophilic steps of the reaction. In this case, the balance between paths A and B depends on the rates of all the stages. Since the intramolecular addition forming the 5-membered ring is facilitated, it can be presumed that the application of excess 3f results in the fast formation of the imine intermediate, which further undergoes intramolecular addition and gives imidazoporphyrin 4f as the main product. This explanation is consistent with the experiment employing 3g in the condensation.
The surprising contrary result obtained with 3h can be explained based another point of view. Previously, we showed that the use of a catalytic amount of acid, namely TsOH, in the condensation reaction allows the reaction pathway to be switched and the preparation of the pyrazinoporphyrin instead of imidazoporphyrin.35 This can be achieved through the increase in its electrophilicity by the protonation equilibrium of the aldehyde carbonyl group. In the case of 3h, the applied aldehyde contains an acidic group itself. The acidity of the aryl carboxylic acid is definitely lower than that of TsOH. Nevertheless, in the case of TsOH, only 20 mol% was sufficient for the reaction, while in the case of 3h, the reaction mixture formally contains an equimolar amount or high excess of the acid. We suppose that this may be the reason for the reverse selectivity of the reactions in this case.
Thus, to prove this assumption, several condensation experiments were performed in the presence of a catalytic amount of TsOH (Table 2). Firstly, we attempted to involve 2 in the condensation under previously reported conditions.35 The interaction of 2 with 5 equiv. of 3a in DMF at 80 °C in the presence of 20 mol% of TsOH unexpectedly provided 5a with only 6% yield and mainly the degradation of 2 was observed (Table 2, entry 1). This behavior can be assumed to be a particular feature of the used porphyrin. Nevertheless, performing the condensation in dichlorobenzene at 120 °C successfully yielded 64% of 5a (Table 2, entry 2). Application of these conditions in the reaction with aldehydes 3f and 3g also allowed the preparation of 5f and 5g with 88% and 42% yield (Table 2, entries 3 and 4), respectively, in contrast to Table 1, entry 12. The decrease in the yield of 5g in the comparison with 5f can be reasonably attributed to the partial lability of the phosphoryl moiety under the reaction conditions.
|
||||
|---|---|---|---|---|
| Entry | Porphyrin | Aldehyde | Solvent, temp. (°C) | Product, yield (%) |
| a 25% of 4a and 15% of 6 were also isolated.b 15% of the corresponding imidazole-fused product was also isolated. | ||||
| 1 | 2 | 3a | DMF, 80 | 5a, 6a |
| 2 | 2 | 3a | DCB, 100 | 5a, 64 |
| 3 | 2 | 3f | DCB, 100 | 5f, 88 |
| 4 | 2 | 3g | DCB, 100 | 5g, 42 |
| 5 | 7 | 3a | DMF, 80 | 8a, 84 (ref. 35) |
| 6 | 7 | 3a | DCB, 100 | 8a, 66 |
| 7 | 7 | 3f | DCB, 100 | 8f, 70 |
| 8 | 7 | 3g | DCB, 100 | 8g, 48b |
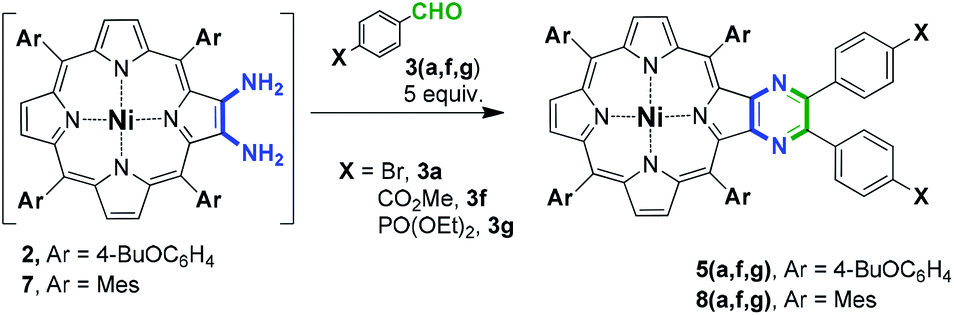
We also applied the currently found condensation conditions to the tetramesitylporphyrin to establish the influence of porphyrin type on the reaction selectivity (Table 2, entries 5 and 6). It was found that the condensation of 7 and 3a in DCB gave rise to the pyrazine-appended porphyrin 8a with a yield of four times less than in DMF (Table 2, compare entry 5 with entry 4). The introduction of 3f in this reaction also allowed the preparation of pyrazinoporphyrin 8f with high yield (Table 2, entry 7). However, the application of aldehyde 3g led to a lower product yield (Table 2, entry 8), which can be attributed to the decreased stability of diethoxyphosphoryl aldehyde under acidic conditions. This is consistent with the results obtained for the 4-butoxyphenyl-substituted porphyrin.
Thus, based on the performed experiments summarized in Tables 1 and 2, it can be concluded that the reaction path of the condensation of 2,3-diaminoporphyrins with aromatic aldehydes strongly depends on the origin of the carbonyl component of the reaction. The selectivity of the reaction is actually a fine balance among multiple effects. Nevertheless, in most cases, a variation in the reaction conditions allowed the determination of the optimal conditions for the selective preparation of the porphyrin derivatives fused with a 5- or 6-membered N-heterocycle. The acidic catalysis was found to be a valuable tool for switching the reaction pathway to the selective formation of imidazole- or pyrazine-appended porphyrins.
The comparison of the UV-vis absorption spectra of the obtained compounds illustrates the difference in the electronic structures of the macrocycles upon fusion with 5- and 6-membered heterocycles (Fig. 1). It can be clearly seen that the imidazole-fused porphyrin 4b demonstrates a Soret band absorption with a maximum in a typical position of 423 nm. In contrast, the fusion with a pyrazine heterocycle results in a significant change in the electronic structure of the macrocycle, as revealed the considerable red shift of the absorption bands.
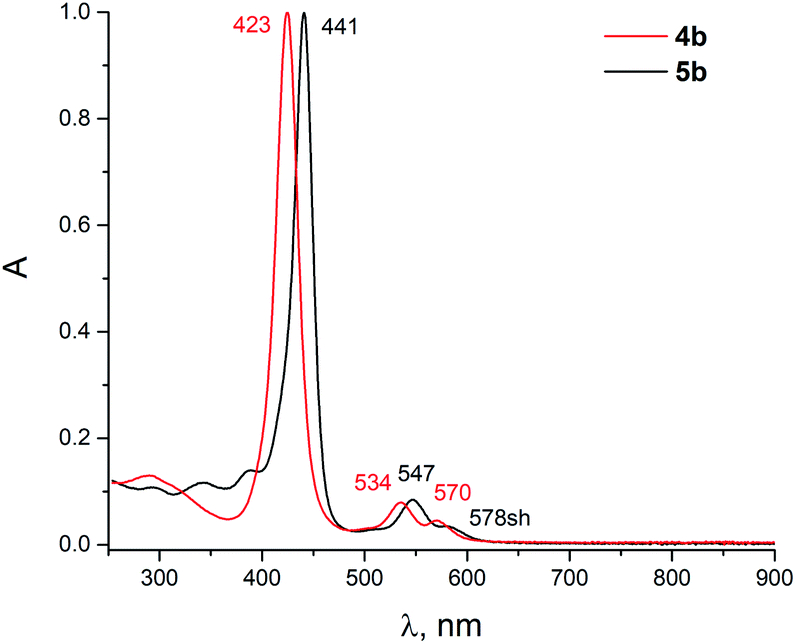 | ||
| Fig. 1 UV-vis spectra of 4b and 5b, normalized at the Soret band (CHCl3). | ||
Porphyrins bearing an appended diaryl-substituted pyrazine unit can be prepared via the interaction of 2,3-diaminoporphyrins and 4,4′-disubstituted benzils. The only reported example of this type of interaction operated with an unsubstituted benzil provided 64% yield of the target compound.41 The main obvious disadvantage of this approach is that 4,4′-disubstituted benzils are less available in comparison with 4-substituted benzaldehydes. Nevertheless, we attempted to perform the condensation of 2 with 4,4′-dibromobenzil (Scheme 4). The interaction at ambient temperature successfully provided the desired pyrazinoporphyrin 5a, but the obtained yield was only 47%. Moreover, this approach did not overcome the difficulty related with the preparation of fused derivatives containing electron donor groups. Accordingly, the approach with the application of functionalized aldehydes can be considered as the more preferable method.
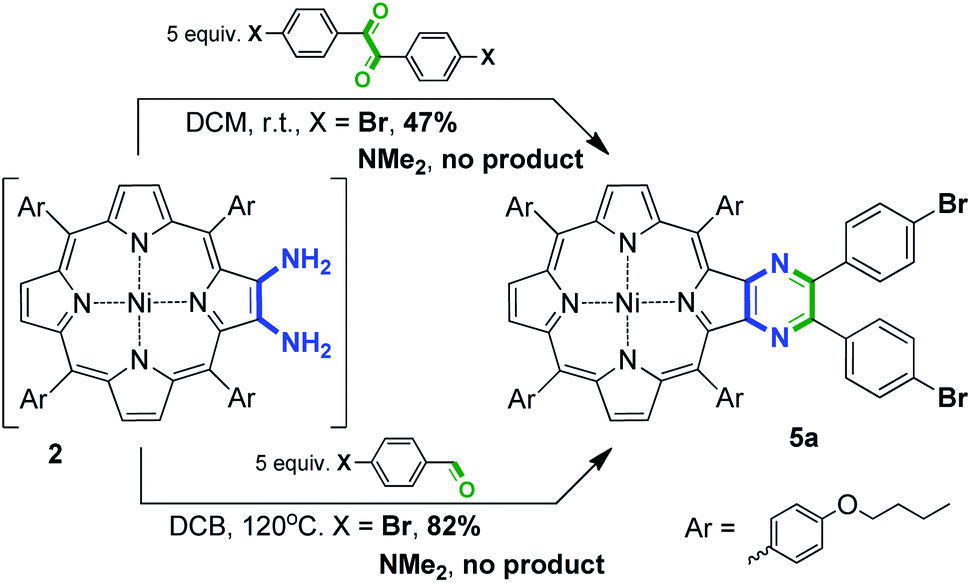 | ||
| Scheme 4 Two pathways to disubstituted pyrazine-appended porphyrins. | ||
Pyrazinoporphyrin 5a, containing two bromo-substituents, appears to be a convenient platform for the design of highly symmetrical difunctionalized porphyrin building blocks. Moreover, recently, we demonstrated the efficient immobilization of dicarboxy-substituted pyrazinoporphyrin on the surface of the UiO-66 and UiO-67 MOFs.42 Accordingly, we performed several metal-promoted transformations to evaluate the reactivity of bromine atoms at the appended pyrazine fragment (Scheme 5).
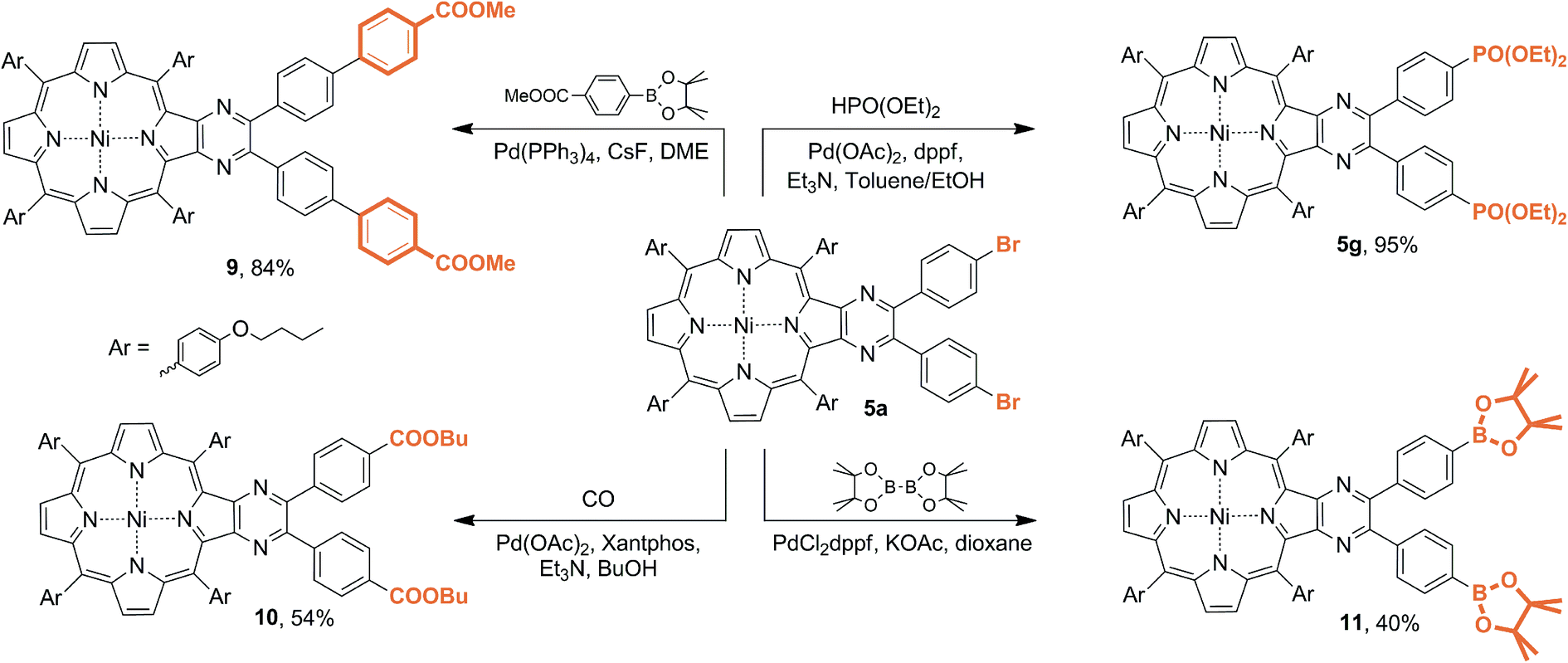 | ||
| Scheme 5 Metal-promoted transformations of 5a. | ||
Firstly, the C–C-cross coupling reaction was performed with 4-methoxycarbonylphenyl pinacolborane. The interaction of the reagents in the presence of the Pd(PPh3)4/CsF catalytic system smoothly provided 9 in high yield. This approach allows the introduction of the peripheral anchoring groups and their analogues, which provides the opportunity for fine tuning the geometry of the prepared molecular building blocks related with 5h.
The bromine atoms in 5a also could be successfully substituted under the carbonylation conditions. Thus, the interaction of 5a with carbon monoxide in the presence of Pd(OAc)2 and xantphos ligand provided double ester 10. Despite the fact that the hydrolysis of this ester lead to 5h, this approach simplifies the purification of the intermediate compounds, avoiding the low solubility of 5h.
Phosphonylation of bromoarenes is another pathway for the introduction of peripheral groups capable of anchoring or providing solubilizing function. The interaction of 5a with diethylphosphite under the reaction conditions previously found for the imidazole porphyrin derivative allowed the preparation of 5g with virtually quantitative yield.38,43,44 This approach appears to be more convenient than the direct synthesis of the phosphoryl-substituted pyrazine porphyrins since the corresponding aldehyde is not commercially available and has to be preliminary prepared. Moreover, its lower stability under the conditions of condensation leads to a decrease in the pyrazinoporphyrin yield, although this drawback is not peculiar to the bromo-derivative.
The substitution of bromine atoms with pinacolborane moieties allowed the preparation of 11, which can be considered as a counterpart in the Suzuki–Miyaura cross-coupling reaction. The availability of these bromo- and boron-derivatives provided by the developed methodology opens access to a valuable class of porphyrinic platforms, which are promising for the further targeted design of functionalized derivatives for multiple applications.
The performed set of catalytic metal-promoted transformations together with that reported before38 reveals the general paths for further modification of the imidazo- and pyrazinoporphyrins. Accordingly, these heterocyclic porphyrin derivatives can be considered as a valuable class of macrocyclic compounds suitable for the design and targeted elaboration of molecules with desired multiple functionality.
Conclusions
The expansion of the scope of the approach towards porphyrins fused with functionalized 5- and 6-membered N-heterocycles was demonstrated through the variation of the carbonyl reagent in the condensation with diaminoporphyrins. It was found that the interaction of diaminoporphyrins with 4-substituted benzaldehydes bearing electron-withdrawing groups successfully gave rise to functionalized imidazo- or pyrazino-fused derivatives, depending on the ratio of the reagents. The limitations of this transformation were also revealed. It was shown that a decrease in the electrophilicity of the formyl group in the substituted benzaldehyde suppressed the interaction with porphyrinic amino-groups. The influence of the acidic catalyst in the condensation was revealed to be a trigger to switch the direction of the reaction path.Other approaches for the introduction of functional groups to the appended disubstituted pyrazine fragment were studied. However, the condensation of a diaminoporphyrin with 4,4′-disubstituted benzils did not reveal any valuable advantages compared to the interaction with functionalized benzaldehydes.
A series of palladium-promoted cross-coupling reactions was performed, allowing the introduction of various functional groups to the pyrazine fragment. The wide diversity of reaction types, and thus the available derivatives, makes this approach a powerful method for the elaboration of tetrapyrrolic platforms, which are promising for further applications.
Experimental
All the used reagent grade chemicals were purchased from commercial suppliers, unless otherwise stated. The solvents were purified according to conventional methods.45 The starting 2-amino-3-nitro-tetrakis(4-butoxyphenyl)porphyrinato nickel(II) 1,28,36,46 2-amino-3-nitro-tetramesitylporphyrinato nickel(II),35 and 4-diethoxyphosphorylbenzaldehyde 3g44,47 were prepared following the conventional procedures. 2,3-Diamino-tetramesitylporphyrinato nickel(II) 7 was generated following the published procedure.35 Chromatographic purification was performed using Macherey-Nagel Silica 60, 0.063–0.2 mm. Merck aluminum plates (TLC Silica 60 F254) were used for TLC analysis with hexane/dichloromethane mixtures as the eluents. Gel-permeation chromatography was performed using Bio-Beads SX-1 sorbent (Bio-Rad) in a CHCl3/EtOH (97.5:2.5) mixture.
MALDI-TOF mass-spectra were recorded on a Bruker Daltonics Ultraflex spectrometer in positive ions mode without a matrix. UV-vis spectra were recorded on a Unicam UV-4 spectrophotometer in rectangular quartz cells with an optical path of 0.1–10 mm in the range of 250–900 nm. 1H NMR spectra were recorded on a Bruker Avance III spectrometer with 600 MHz proton frequency in CDCl3 or a mixture of CDCl3/MeOD at ambient temperature with the use of the residual solvent resonance as the internal reference. The complete NMR spectra are presented in the ESI.†
4-Benzoyloxybenzaldehyde 3e
4-Hydroxybenzaldehyde (4 g, 32.7 mmol) was dissolved in DCM (30 mL) under argon and Et3N (4.6 mL, 32.7 mmol) was added. The solution was cooled in ice bath to ∼0 °C and a solution of BzCl (4.00 g, 32.7 mmol) in DCM (10 mL) was added dropwise with stirring. The resulting mixture was stirred at 0 °C for 3 h. Afterwards the reaction mixture was washed with water (2 × 50 mL), dried over Na2SO4 and evaporated to give a pale solid (6.543 g, 88%).1H NMR (CDCl3; δ, ppm; J, Hz): 10.05 (s, 1H, CHO), 8.23 (d, 2H, 3J = 7.7, CHo-Bz), 8.00 (d, 2H, 3J = 8.2, CHm-Ar), 7.69 (t, 1H, 3J = 7.5, CHp-Bz), 7.56 (d, 2H, 3J = 7.7, CHm-Bz), 7.44 (d, 2H, 3J = 8.3, CHo-Ar).
13C NMR (CDCl3; δ, ppm): 191.07 (CHO), 164.66 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 155.88 (C–O), 134.27 (
O), 155.88 (C–O), 134.27 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –CHO), 134.18 (CHp-Bz), 131.43 (CHo-Ar), 130.44 (CHo-Bz), 129.11 (CHm-Bz), 128.88 (–CO), 122.70 (CHm-Ar).
–CHO), 134.18 (CHp-Bz), 131.43 (CHo-Ar), 130.44 (CHo-Bz), 129.11 (CHm-Bz), 128.88 (–CO), 122.70 (CHm-Ar).
4-Methoxycarbonylphenyl pinacolborane
4-Methoxycarbonylphenyl boronic acid (900 mg, 5 mmol) and pinacol (708 mg, 6 mmol) were refluxed in freshly distilled THF (15 mL) under an argon atmosphere for 6 h. Afterwards, the reaction mixture was cooled to ambient temperature, diluted with EtOAc (20 mL) and extracted with water (3 × 20 mL). The organic phase was evaporated to provide the target compound as a white solid (1.26 g, 96%).1H NMR (CDCl3; δ, ppm; J, Hz): 8.02 (d, 2H, 3J = 7.8, o-CHAr), 7.87 (d, 2H, 3J = 7.8, m-CHAr), 3.92 (s, 3H, COOCH3), 1.35 (s, 12H, CH3Pin).
13C NMR (CDCl3; δ, ppm): 167.30 (CO), 134.81 (CHAr), 132.47 (–CO), 128.74 (CHAr), 84.32 (CPin), 52.21 (OCH3), 25.02 (CH3Pin). The resonance of –Bpin was not observed.
General procedure 1
The preparation of 2,3-diaminoporphyrins. In a typical experiment, 2-amino-3-nitroporphyrin (0.05 mmol) was dissolved in a mixture of freshly distilled dry DCM (16 mL) and MeOH (0.6 mL). The resulting solution was purged with argon, and 10% Pd/C (25 mg) was added upon vigorous stirring. Afterwards, NaBH4 (28 mg, 0.75 mmol) was added and the mixture was stirred at ambient temperature for ca. 15 min until complete consumption of the starting material was observed by TLC (silica gel, DCM/hexane = 2:1). After complete reduction, the mixture was filtered through Celite-545 under an argon atmosphere, the obtained solution was immediately evaporated in vacuo, and the obtained 2,3-diaminoporphyrin was employed in the subsequent reaction without purification and characterization because of its high susceptibility to oxidation.
General procedure 2
The condensation of 2,3-diaminoporphyrins with aromatic aldehydes under catalyst-free conditions. The freshly prepared 2,3-diaminoporphyrin (0.05 mmol) was dissolved under argon in dry dichlorobenzene (DCB) (8 mL) and the required amount of the aromatic aldehyde was added (Table 1). The obtained solution was heated at 120 °C under an argon atmosphere overnight. After the complete consumption of diamine according to TLC, the reaction mixture was evaporated to dryness and the residue was applied to a column packed with silica gel in hexane. The column was eluted with hexane/DCM mixtures (0 → 100% of DCM) and further with DCM/MeOH mixtures (0 → 5% of MeOH) depending on the polarity of the functional groups in the appended heterocyclic fragment. The isolated yields of the annulated compounds are summarized in Table 1.General procedure 3
The condensation of 2,3-diaminoporphyrins with aromatic aldehydes under acid catalyzed conditions. The condensation and treatment of the reaction mixture were generally the same as that described in general procedure 2. The difference is the addition of 20 mol% (based on the amount of the 2-amino-3-nitroporphyrin in the reduction) of TsOH before heating of the reaction mixture and reaction temperature of 100 °C. The isolated yields of the condensed products are summarized in Table 2.4a
1H NMR (CDCl3; δ, ppm; J, Hz): 8.84–8.79 (m, 4H, Hβ), 8.77 (d, 1H, 3J = 4.9, Hβ), 8.76 (d, 1H, 3J = 4.9, Hβ), 8.65 (s, 1H, NH), 7.98 (d, 2H, 3J = 8.2, CHo-Ar), 7.95 (d, 2H, 3J = 8.2, CHo-Ar), 7.91 (d, 4H, 3J = 6.4, CHo-Ar), 7.65 (d, 2H, 3J = 8.2, o-CH), 7.59 (d, 2H, 3J = 8.2, m-CH), 7.35 (d, 2H, 3J = 8.2, CHm-Ar), 7.28 (d, 2H, 3J = 8.2, CHm-Ar), 7.20 (d, 4H, 3J = 8.2, CHm-Ar), 4.27 (t, 4H, 3J = 6.5, CH2O), 4.20 (t, 4H, 3J = 6.5, CH2O), 2.04–1.89 (m, 8H, CH2), 1.74–1.60 (m, 8H, CH2), 1.06–1.15 (m, 12H, CH3).13C NMR (CDCl3; δ, ppm): 159.76, 159.23, 159.14, 151.95, 150.62, 144.38, 142.58, 142.44, 142.31, 141.77, 141.66, 139.17, 134.88, 134.81, 134.78, 133.79, 133.17, 133.13, 132.94, 132.74, 132.31, 132.26, 131.87, 131.81, 131.64, 131.48, 129.95, 129.84, 127.33, 126.83, 123.50, 119.67, 119.59, 116.12, 114.36, 113.58, 113.30, 113.07, 68.42, 68.11, 31.70, 31.67, 31.54, 19.58, 19.55, 14.13, 14.10, 14.09.
MALDI-TOF MS: m/z calcd for C67H63BrN6NiO4 [M]+ 1152.34, found 1152.25.
UV-vis (CHCl3; λmax, nm (logε)): 308 (4.51), 422 (5.37), 534 (4.28), 569 (4.05).
4b
1H NMR (CDCl3; δ, ppm; J, Hz): 8.82 (s, 2H, Hβ), 8.79 (s, 2H, Hβ), 8.76 (d, 1H, 3J = 4.9, Hβ), 8.75 (d, 1H, 3J = 5.0, Hβ), 8.67 (s, 1H, NH), 7.99 (d, 2H, 3J = 8.1, CHo-Ar), 7.97 (d, 2H, 3J = 7.9, CHo-Ar), 7.91 (d, 4H, 3J = 8.2, CHo-Ar), 7.74 (d, 2H, 3J = 8.0, o-CH), 7.50 (d, 2H, 3J = 8.0, m-CH), 7.37 (d, 2H, 3J = 8.0, CHm-Ar), 7.28 (d, 2H, 3J = 8.2, CHm-Ar), 7.20 (d, 4H, 3J = 8.1, CHm-Ar), 4.33–4.25 (m, 4H, CH2O), 4.21 (t, 4H, 3J = 6.5, CH2O), 2.05–1.97 (m, 4H, CH2), 1.97–1.90 (m, 4H, CH2), 1.75–1.60 (m, 8H, CH2), 1.39 (s, 9H, tBu), 1.16–1.10 (m, 6H, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 159.74, 159.19, 159.12, 152.75, 152.05, 151.95, 144.42, 142.59, 142.33, 142.22, 141.68, 141.55, 139.05, 134.96, 134.80, 133.81, 133.27, 132.87, 132.63, 132.38, 131.75, 131.69, 131.62, 129.91, 128.13, 127.10, 126.03, 125.81, 119.52, 119.42, 116.17, 114.37, 113.52, 113.33, 113.06, 68.40, 68.12, 31.73, 31.68, 31.58, 31.38, 29.85, 19.56, 14.11.
MALDI-TOF MS: m/z calcd for C71H72N6NiO4 [M]+1130.50, found 1130.59.
UV-vis (CHCl3; λmax, nm; log(ε)): 289 (4.54), 423 (5.36), 534 (4.32), 570 (4.06).
4c
1H NMR (CDCl3; δ, ppm; J, Hz): 8.86–8.72 (br.m, 6H, Hβ), 8.55 (br.s, 1H, NH), 7.96 (br.s, 4H, CHo-Ar), 7.91 (d, 4H, 3J = 8.1, CHo-Ar), 7.62 (d, 2H, 3J = 8.2, o-CH), 7.32 (br.s, 4H, CHm-Ar), 7.19 (d, 4H, 3J = 8.2, CHm-Ar), 6.83 (d, 2H, 3J = 8.1, m-CH), 4.23 (t, 4H, 3J = 6.5, CH2O), 4.20 (t, 4H, 3J = 6.5, CH2O), 1.98–1.90 (m, 8H, CH2), 1.68–1.59 (m, 8H, CH2), 1.11–1.04 (m, 12H, CH3).Satisfactory 13C NMR spectrum was not obtained for 4c as a result of broadening of the most of its signals. The acquired spectrum is presented in the ESI.†
MALDI-TOF MS: m/z calcd for C67H64N6NiO5 [M]+ 1090.43, found m/z 1090.43.
UV-vis (CHCl3; λmax, nm (logε)): 281 (4.49), 421 (5.38), 534 (4.26), 570 (4.00).
4e
1H NMR (CDCl3; δ, ppm; J, Hz): 8.85–8.79 (m, 4H, Hβ), 8.79–8.74 (m, 2H, Hβ), 8.70 (s, 1H, NH), 8.26 (d, 2H, 3J = 7.6, CHo-Bz), 8.03–7.96 (m, 4H, CHo-Ar) 7.92 (d, 4H, 3J = 8.1, CHo-Ar), 7.86 (d, 2H, 3J = 8.2, o-CH), 7.68 (t, 1H, 3J = 7.6, CHp-Bz), 7.56 (t, 2H, 3J = 7.6, CHm-Bz), 7.38 (d, 2H, 3J = 8.0, CHm-Ar), 7.34 (d, 2H, 3J = 8.3, o-CHAr), 7.29 (d, 2H, 3J = 8.1, CHm-Ar), 7.21 (d, 4H, 3J = 8.1, CHm-Ar), 4.32–4.25 (m, 4H, CH2O), 4.21 (t, 4H, 3J = 6.5, CH2O), 2.03–1.96 (m, 4H, CH2), 1.94 (quint, 4H, 3J = 6.5, CH2), 1.72–1.60 (m, 8H, CH2) 1.15–1.06 (m, 12H, CH3).13C NMR (CDCl3; δ, ppm): 165.04, 159.67, 159.10, 159.01, 151.89, 151.77, 150.92, 144.28, 142.50, 142.28, 142.15, 141.62, 141.51, 139.09, 134.80, 134.68, 133.81, 133.67, 133.10, 133.02, 132.79, 132.57, 132.23, 131.69, 131.64, 131.51, 131.46, 130.30, 129.84, 129.42, 128.69, 128.62, 127.10, 126.84, 122.35, 119.48, 119.40, 116.04, 114.25, 113.48, 113.20, 112.94, 68.29, 68.00, 31.55, 31.44, 19.43, 13.98.
MALDI-TOF MS: m/z calcd for C74H68N6NiO6 [M]+ 1194.46, found m/z 1194.50.
UV-vis (CHCl3; λmax, nm (logε)): 312 (4.70), 422 (5.65), 534 (4.54), 571 (4.28).
4f
1H NMR (CDCl3; δ, ppm; J, Hz): 8.84–8.79 (m, 4H, Hβ), 8.78–8.74 (m, 3H, Hβ + NH), 8.12 (d, 2H, 3J = 8.0, CHAr), 7.98 (d, 2H, 3J = 8.4, CHAr), 7.97 (d, 2H, 3J = 8.4 Hz, CHAr), 7.91 (br.d, 4H, 3J = 8.4, CHAr), 7.84 (d, 2H, 3J = 8.0, CHAr), 7.38 (d, 2H, 3J = 8.1, CHm-Ar), 7.29 (d, 2H, 3J = 8.1, CHm-Ar), 7.20 (d, 4H, 3J = 8.1, CHm-Ar), 4.32–4.26 (m, 4H, CH2O), 4.20 (br.t, 4H, J = 5.8 Hz, CH2O), 3.98 (s, 3H, COOCH3), 2.05–1.97 (m, 4H, CH2), 1.94 (quint., 4H, 3J = 6.8, CH2), 1.74–1.61 (m, 8H, CH2), 1.14 (t, 3H, 3J = 7.4, CH3), 1.13 (t, 3H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 166.76, 159.83, 159.29, 159.16, 152.11, 150.41, 144.39, 142.58, 142.49, 142.33, 141.80, 141.70, 139.38, 134.87, 134.80, 133.79, 133.14, 133.08, 132.98, 132.79, 132.25, 131.91, 131.84, 131.66, 131.48, 130.49, 130.39, 129.97, 126.75, 125.60, 119.73, 119.68, 116.13, 114.42, 113.67, 113.34, 113.08, 68.47, 68.15, 68.12, 52.39, 31.71, 31.67, 31.56, 19.60, 19.57, 19.55, 14.14, 14.10.
MALDI-TOF MS: m/z calcd for C69H66N6NiO6 [M]+ 1132.44, found 1132.36.
UV-vis (CHCl3; λmax, nm (logε)): 328 (4.42), 424 (5.35), 534 (4.25), 570 (3.98).
4g
1H NMR (CDCl3; δ, ppm; J, Hz): 8.88–8.81 (br.m, 4H, Hβ), 8.79 (br.s, 3H, Hβ + NH), 8.00 (d, 4H, 3J = 8.2, CHo-Ar), 7.97–7.88 (m, 8H, CHo-Ar + o- and m-CH) 7.41 (br.d, 2H, 3J = 7.9, CHm-Ar), 7.31 (br.d, 2H, 3J = 8.1, CHm-Ar), 7.23 (d, 4H, 3J = 8.2, CHm-Ar), 4.35–4.28 (br.m, 4H, CH2O), 4.23 (t, 4H, 3J = 6.6, CH2O), 4.25–4.12 (m, 4H, CH3C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2O), 2.03 (br.s, 4H, CH2), 1.97 (quint, 4H, 3J = 6.7, CH2), 1.76–1.70 (br.m, 4H, CH2), 1.66 (sext, 4H, 3J = 7.5, CH2), 1.40 (t, 6H, 3J = 7.1, C3CH2O), 1.19–1.13 (br.m, 6H, CH3), 1.11 (t, 6H, 3J = 7.4, CH3).
2O), 2.03 (br.s, 4H, CH2), 1.97 (quint, 4H, 3J = 6.7, CH2), 1.76–1.70 (br.m, 4H, CH2), 1.66 (sext, 4H, 3J = 7.5, CH2), 1.40 (t, 6H, 3J = 7.1, C3CH2O), 1.19–1.13 (br.m, 6H, CH3), 1.11 (t, 6H, 3J = 7.4, CH3).
13C NMR (CDCl3; δ, ppm): 159.72, 159.15, 159.04, 151.94, 150.19, 144.26, 142.45, 142.37, 142.21, 141.68, 141.58, 139.24, 134.74, 134.67, 134.37 (d, 1JC–P = 3.2 Hz), 133.66, 133.01, 132.96, 132.87, 132.67, 132.53, 132.47, 132.14, 131.80, 131.73, 131.53, 131.34, 129.85, 129.56, 128.30, 126.61, 125.62, 125.52, 119.59 (d, 2JC–P = 7.5 Hz), 115.99, 114.28, 113.54, 113.21, 112.96, 68.35, 68.00, 62.30 (d, 2JC–P = 5.3 Hz), 31.59, 31.55, 31.44, 19.46, 19.42, 16.38 (d, 3JC–P = 6.4 Hz), 14.01, 13.97.
31P NMR (CDCl3; δ, ppm): 18.04 (s).
MALDI-TOF MS: m/z calcd for C71H73N6NiO7P [M]+ 1210.46, found 1210.27.
UV-vis (CHCl3; λmax, nm (logε)): 319 (4.51), 424 (5.43), 534 (4.32), 569 (4.05).
4h
1H NMR (20% MeOD in CDCl3; δ, ppm; J, Hz): 8.71–8.66 (br.s, 4H, Hβ), 8.64 (s, 2H, Hβ), 8.04 (d, 2H, 3J = 8.0, m-CH), 7.86 (d, 4H, 3J = 8.1, CHo-Ar), 7.79 (d, 4H, 3J = 8.2, CHo-Ar), 7.75 (d, 2H, 3J = 8.0, o-CH), 7.27 (br.s, 2H, CHm-Ar), 7.18 (br.s, 2H, CHm-Ar), 7.09 (d, 4H, 3J = 8.2, CHm-Ar), 4.18 (br.t, 4H, 3J = 6.5, CH2O), 4.10 (t, 4H, 3J = 6.5, CH2O), 1.93–1.85 (br.m, 4H, CH2), 1.82 (quint, 4H, 3J = 6.5, CH2), 1.57 (sext, 4H, 3J = 7.6, CH2), 1.52 (sext, 4H, 3J = 7.5, CH2), 1.01 (t, 6H, 3J = 7.4, CH3), 0.97 (t, 6H, 3J = 7.4, CH3).13C NMR (20% MeOD in CDCl3; δ, ppm): 168.28, 158.93, 150.54, 142.23, 141.54, 134.59, 134.52, 133.66, 132.97, 132.67, 131.64, 130.86, 130.44, 125.54, 119.49, 114.21, 113.19, 112.91, 68.00, 31.42, 19.31, 19.29, 13.76.
MALDI-TOF MS: m/z calcd for C68H64N6NiO6 [M]+ 1118.42, found 1117.90.
UV-vis (CHCl3; λmax, nm (logε)): 329 (4.32), 425 (5.13), 534 (4.15), 569 (3.90).
5a
1H NMR (CDCl3; δ, ppm; J, Hz): 8.86 (d, 2H, 3J = 4.9, Hβ), 8.76 (d, 2H, 3J = 4.9, Hβ), 8.72 (s, 2H, Hβ), 7.88 (d, 4H, 3J = 8.5, CHo-Ar), 7.79 (d, 4H, 3J = 8.5, CHo-Ar), 7.41 (d, 4H, 3J = 8.5, o-CH), 7.30 (d, 4H, 3J = 8.4, m-CH), 7.20 (d, 4H, 3J = 8.6, CHm-Ar), 7.18 (d, 4H, 3J = 8.5, CHm-Ar), 4.23–4.16 (m, 8H, CH2O), 2.02 (quint, 4H, 3J = 6.6, CH2), 1.94 (quint, 4H, 3J = 6.6, CH2), 1.72 (sext, 4H, 3J = 6.6, CH2), 1.64 (sext, 4H, 3J = 6.6, CH2), 1.15 (t, 6H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 159.39, 159.29, 147.57, 144.18, 142.79, 142.01, 138.37, 134.75, 134.28, 133.03, 132.93, 132.62, 132.30, 132.12, 131.66, 131.24, 123.41, 120.55, 116.55, 113.24, 113.16, 68.15, 68.13, 31.90, 31.66, 19.78, 19.55, 14.23, 14.10.
MALDI-TOF MS: m/z calcd for C74H66Br2N6NiO4 [M]+ 1318.29, found 1318.08.
UV-vis (CHCl3; λmax, nm (logε)): 347 (4.30), 403sh (4.42), 447 (5.10), 551 (4.10), 581sh (3.69).
5b
1H NMR (CDCl3; δ, ppm; J, Hz): 8.82 (d, 2H, 3J = 4.9, Hβ), 8.75 (d, 2H, 3J = 4.9, Hβ), 8.71 (s, 2H, Hβ), 7.89 (d, 4H, 3J = 8.2, CHo-Ar), 7.83 (d, 4H, 3J = 8.1, CHo-Ar), 7.43 (d, 4H, 3J = 8.1, o-CH), 7.27 (d, 4H, 3J = 8.3, m-CH), 7.23 (d, 4H, 3J = 8.2, CHm-Ar), 7.20 (d, 4H, 3J = 8.2, CHm-Ar), 4.26 (t, 4H, 3J = 6.5, CH2O), 4.20 (t, 4H, 3J = 6.5, CH2O), 2.04 (quint, 3J = 6.6, 4H, CH2), 1.93 (quint, 4H, 3J = 6.7, CH2), 1.72 (sext, 4H, 3J = 7.4, CH2), 1.64 (sext, 4H, 3J = 7.4, CH2), 1.37 (s, 18H, tBu), 1.14 (t, 6H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 159.30, 159.23, 151.74, 148.78, 147.35, 144.29, 142.55, 141.71, 136.88, 134.74, 134.37, 133.44, 132.85, 132.77, 132.75, 131.86, 131.60, 130.40, 124.72, 120.31, 116.33, 113.20, 113.15, 68.13, 68.10, 32.04, 31.67, 31.46, 19.82, 19.55, 14.24, 14.10.
MALDI-TOF MS: m/z calcd for C82H84N6NiO4 [M]+ 1274.59, found 1274.38.
UV-vis (CHCl3; λmax, nm (logε)): 294 (4.71), 346 (4.74), 389 (4.82), 441 (5.67), 547 (4.60), 578sh (4.19).
5c
1H NMR (CDCl3; δ, ppm; J, Hz): 8.82 (d, 2H, 3J = 4.9, Hβ), 8.75 (d, 2H, 3J = 4.9, Hβ), 8.71 (s, 2H, Hβ), 7.89 (d, 4H, 3J = 8.3, CHo-Ar), 7.81 (d, 4H, 3J = 8.2, CHo-Ar), 7.36 (d, 4H, 3J = 8.3, o-CH), 7.20 (d, 8H, 3J = 8.2, CHm-Ar), 6.70 (d, 4H, 3J = 8.3, m-CH), 4.91 (br.s, 2H, OH), 4.25–4.18 (m, 8H, CH2O), 2.00 (quint, 4H, 3J = 6.7, CH2), 1.93 (quint, 4H, 3J = 6.7, CH2), 1.70 (sext, 4H, 3J = 7.4, CH2), 1.64 (sext, 4H, 3J = 7.5, CH2), 1.12 (t, 6H, 3J = 7.4, CH3), 1.08 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 159.13, 159.09, 156.05, 148.13, 147.12, 144.13, 142.46, 141.62, 134.61, 134.22, 133.28, 132.71, 132.69, 132.63, 132.49, 132.15, 131.74, 131.42, 120.19, 116.15, 114.71, 113.07, 113.02, 68.00, 67.95, 31.77, 31.53, 19.57, 19.41, 14.06, 13.96.
MALDI-TOF MS: m/z calcd for C74H68N6NiO6 [M]+ 1194.46, found 1194.39.
UV-vis (CHCl3; λmax, nm (logε)): 293 (4.46), 345 (4.23), 394 (4.57), 441 (5.38), 547 (4.33), 580 (3.92).
5e
1H NMR (CDCl3; δ, ppm; J, Hz): 8.87 (d, 2H, 3J = 4.9, Hβ), 8.77 (d, 2H, 3J = 4.9, Hβ), 8.73 (s, 2H, Hβ), 8.25 (d, 4H, 3J = 7.7, CHo-Bz), 7.90 (d, 4H, 3J = 8.3, CHo-Ar), 7.84 (d, 4H, 3J = 8.2, CHo-Ar), 7.67 (t, 2H, 3J = 7.5, CHp-Bz), 7.59–7.53 (m, 8H, CHm-Bz + m-CH), 7.21 (t, 8H, 3J = 8.3, CHm-Ar), 7.18 (d, 4H, 3J = 8.4, o-CH), 4.21 (t, 8H, 3J = 6.5, CH2O), 1.99–1.90 (m, 8H, CH2), 1.69–1.60 (m, 8H, CH2), 1.09 (t, 6H, 3J = 7.4, CH3), 1.00 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 165.01, 159.41, 159.27, 151.60, 147.99, 147.56, 144.25, 142.71, 141.92, 137.19, 134.76, 134.34, 133.78, 133.18, 132.96, 132.72, 132.67, 132.01, 131.97, 131.65, 130.33, 129.79, 128.77, 121.23, 120.45, 116.56, 113.23, 113.21, 68.14, 68.10, 31.93, 31.67, 19.68, 19.55, 14.12, 14.10.
MALDI-TOF MS: m/z calcd for C88H76N6NiO8 [M]+ 1402.51, found m/z 1402.50.
UV-vis (CHCl3; λmax, nm (logε)): 341 (4.50), 398 (4.60), 445 (5.34), 546 (4.32), 579 (3.93).
5f
1H NMR (CDCl3; δ, ppm; J, Hz): 8.87 (d, 2H, 3J = 4.9, Hβ), 8.77 (d, 2H, 3J = 4.9, Hβ), 8.73 (s, 2H, Hβ), 7.95 (d, 4H, 3J = 8.0, CHo-Ar), 7.89 (d, 4H, 3J = 8.1, CHo-Ar), 7.81 (d, 4H, 3J = 8.0, m-CH), 7.50 (d, 4H, 3J = 8.0, o-CH), 7.20 (d, 8H, 3J = 8.1, CHm-Ar), 4.25–4.17 (m, 8H, CH2O), 3.97 (s, 6H, COOCH3), 2.04 (quint, 4H, 3J = 8.2, CH2), 1.94 (quint, 4H, 3J = 8.2, CH2), 1.73 (sext, 4H, 3J = 7.6, CH2), 1.64 (sext, 4H, 3J = 7.6, CH2), 1.15 (t, 6H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 166.95, 159.47, 159.29, 147.94, 147.66, 144.20, 143.81, 142.85, 142.08, 134.74, 134.28, 133.05, 132.85, 132.58, 132.53, 132.17, 131.73, 130.75, 130.18, 129.27, 120.59, 116.70, 113.24, 113.19, 68.15, 68.13, 52.25, 31.85, 31.65, 19.70, 19.54, 14.22, 14.09.
MALDI-TOF MS: m/z calcd for C78H72N6NiO8 [M]+ 1278.48, found 1278.38.
UV-vis (CHCl3; λmax, nm (logε)): 347 (4.57), 447 (5.26), 550 (4.32), 578sh (3.92).
5g
1H NMR (CDCl3; δ, ppm; J, Hz): 8.85 (d, 2H, 3J = 4.9, Hβ), 8.77 (d, 2H, 3J = 4.9, Hβ), 8.72 (s, 2H, Hβ), 7.89 (d, 4H, 3J = 8.2, CHo-Ar), 7.82 (d, 4H, 3J = 8.2, CHo-Ar), 7.71 (dd, 4H, 3JH–P = 12.9, 3JH–H = 7.9, m-CH), 7.54 (dd, 4H, 3JH–P = 12.9, 3JH–H = 7.9, o-CH), 7.22 (d, 4H, 3J = 8.2, CHm-Ar), 7.20 (d, 4H, 3J = 8.2, CHm-Ar), 4.25 (t, 4H, 3J = 6.5, CH2O), 4.23–4.12 (m, 12H, CH2O + CH3C2O), 2.04 (quint, 4H, 3J = 6.7, CH2), 1.94 (quint, 4H, 3J = 6.7, CH2), 1.73 (sext, 4H, 3J = 7.4, CH2), 1.64 (sext, 4H, 3J = 7.4, CH2), 1.39 (t, 12H, 3J = 7.1, C3CH2O), 1.15 (t, 6H, 3J = 7.4, CH3), 1.08 (t, 6H, 3J = 7.4, CH3).
13C NMR (CDCl3; δ, ppm): 159.46, 159.31, 144.24, 143.18 (d, 3JC–P = 3.3 Hz), 142.85, 142.07, 134.74, 134.32, 133.08, 132.71, 132.54 (d, 3JC–P = 2.3 Hz), 132.20, 131.78, 131.45, 131.38, 130.73, 130.63, 129.45, 128.65, 128.57, 128.20, 120.62, 116.64, 113.26, 113.22, 68.16 (d, 3JC–P = 4.4 Hz), 62.35, 62.31, 31.87, 31.65, 19.71, 19.54, 16.55 (d, 3JC–P = 6.2 Hz), 14.20, 14.09.
31P NMR (CDCl3; δ, ppm): 18.48 (s).
MALDI-TOF MS: m/z calcd for C82H86N6NiO10P2 [M]+ 1434.52, found 1434.64.
UV-vis (CHCl3; λmax, nm (logε)): 346 (4.41), 403 (4.53), 447 (5.13), 550 (4.18), 579sh (3.77).
5h
1H NMR (20% MeOD in CDCl3; δ, ppm; J, Hz): 8.75 (d, 2H, 3J = 4.9, Hβ), 8.66 (d, 2H, 3J = 4.9, Hβ), 8.61 (s, 2H, Hβ), 7.84 (d, 4H, 3J = 8.1, m-CH), 7.78 (d, 4H, 3J = 8.1,CHo-Ar), 7.71 (d, 4H, 3J = 8.1, CHo-Ar), 7.38 (d, 4H, 3J = 8.0, o-CH), 7.10 (d, 8H, 3J = 8.5, CHm-Ar), 4.14–4.08 (m, 8H, CH2O), 1.91 (quint, 4H, 3J = 6.4, CH2), 1.83 (quint, 4H, 3J = 6.3, CH2), 1.60 (sext, 4H, 3J = 7.6, CH2), 1.53 (sext, 4H, 3J = 7.6, CH2), 1.01 (t, 6H, 3J = 7.4, CH3), 0.97 (t, 6H, 3J = 7.4, CH3).13C NMR (20% MeOD in CDCl3; δ, ppm): 159.25, 159.07, 147.94, 147.47, 144.05, 143.62, 142.66, 141.90, 134.55, 134.11, 132.83, 132.71, 132.46, 132.39, 131.95, 131.53, 130.49, 129.33, 120.37, 116.51, 113.09, 113.03, 68.09, 68.04, 31.59, 31.42, 19.44, 19.30, 13.81, 13.77.
MALDI-TOF MS: m/z calcd for C76H68N6NiO8 [M]+ 1250.45, found 1249.90.
UV-vis (CHCl3; λmax, nm (logε)): 345 (4.51), 448 (5.22), 551 (4.27), 581sh (3.86).
6
1H NMR (CDCl3; δ, ppm; J, Hz): 8.63 (s, 4H, Hβ), 8.57 (d, 4H, 3J = 4.8, Hβ), 8.07 (d, 4H, 3J = 4.8, Hβ), 7.85 (br.s, 8H, CHo-Ar), 7.32 (br.d, 8H, 3J = 8.2, CHo-Ar), 7.18 (d, 8H, 3J = 8.3, CHm-Ar), 6.76 (d, 8H, 3J = 8.3, CHm-Ar), 4.19 (t, 8H, 3J = 6.5, CH2O), 3.98 (br.t, 8H, 3J = 6.5, CH2O), 1.92 (quint, 8H, 3J = 6.7, CH2), 1.81 (quint, 8H, 3J = 6.6, CH2), 1.63 (sext, 8H, 3J = 7.4, CH2), 1.52 (sext, 8H, 3J = 7.4, CH2), 1.07 (t, 12H, 3J = 7.4, CH3), 0.95 (t, 12H, 3J = 7.4, CH3).13C NMR (CDCl3; δ, ppm): 159.26, 158.32, 147.60, 145.08, 142.07, 140.76, 134.62, 134.00, 132.76, 132.58, 132.36, 131.98, 131.75, 131.43, 120.38, 115.78, 113.32, 112.95, 68.12, 67.49, 31.77, 31.65, 19.56, 19.54, 14.09, 14.03.
MALDI-TOF MS: m/z calcd for C120H116N10Ni2O8 [M+] 1940.77, found 1940.90.
UV-vis (CHCl3; λmax, nm (logε)): 329 (4.54), 491 (5.12), 424sh, 585 (4.76), 622 (4.25).
8g
1H NMR (CDCl3; δ, ppm; J, Hz): 8.61 (d, 2H, 3J = 4.8, Hβ), 8.54 (d, 2H, 3J = 4.8, Hβ), 8.50 (s, 2H, Hβ), 7.71 (dd, 4H, 3JH–P = 12.9, 3JH–H = 7.9, m-CH), 7.51 (dd, 4H, 3JH–H = 8.1, 3JH–P = 3.8 Hz), 7.21 (s, 8H, CHMes), 4.28–4.13 (m, 8H, CH2O), 2.65 (s, 6H, p-CH3), 2.57 (s, 6H, p-CH3), 1.87 (s, 12H, o-CH3), 1.75 (s, 12H, o-CH3), 1.40 (t, 12H, 3J = 7.1, CH3).13C NMR (CDCl3; δ, ppm): 148.26, 146.58, 143.74, 143.33 (d, 3JC–P = 3.3), 142.53, 141.93, 139.08, 138.84, 137.95, 137.37, 137.11, 137.01, 132.72, 132.24, 131.39, 131.32 (d, 3JC–P = 22.1 Hz), 131.24, 130.81 (d, 3JC–P = 15.0), 129.21, 128.02, 127.96, 118.79, 115.27, 62.41 (d, 3JC–P = 5.5), 21.68, 21.66, 21.54, 21.53, 16.54 (d, 3JC–P = 6.3).
31P NMR (CDCl3; δ, ppm): 18.56 (s).
MALDI-TOF MS: m/z calcd for C78H78N6NiO6P2 [M]+ 1314.48, found 1314.24.
UV-vis (CHCl3; λmax, nm (logε)): 343 (4.77), 396 (4.96), 443 (5.49), 547 (4.59), 581 (4.27).
Synthesis of 9
5a (40 mg, 0.03 mmol) was mixed with 4-methoxycarbonylphenyl pinacolborane (39 mg, 0.15 mmol), CsF (20 mg, 0.129 mmol) and Pd(Ph3P)4 (4.6 mg, 4 μmol) under argon. Dry DME (8 mL) was added and the mixture was refluxed overnight. The complete conversion of the starting compound was detected by TLC (silica gel, hexane/DCM = 1:2), and afterwards the reaction mixture was evaporated to dryness. The residue was applied to a column packed with silica gel in hexane. The column was eluted with hexane/DCM mixtures (0 → 100% of DCM) and the fraction containing 9 was evaporated to provide 37 mg (84%) of the product.
1H NMR (CDCl3; δ, ppm; J, Hz): 8.86 (d, 2H, 3J = 4.9, Hβ), 8.77 (d, 2H, 3J = 4.9, Hβ), 8.73 (s, 2H, Hβ), 8.12 (d, 4H, 3J = 8.0, CHAr), 7.89 (d, 4H, 3J = 8.2, CHo-Ar), 7.81 (d, 4H, 3J = 8.2, CHo-Ar), 7.69 (d, 4H, 3J = 8.1, CHAr), 7.57 (d, 4H, 3J = 8.1, CHAr), 7.52 (d, 4H, 3J = 8.1, CHAr), 7.20 (d, 4H, 3J = 6.3, CHm-Ar), 7.19 (d, 4H, 3J = 6.6, CHm-Ar), 4.19 (t, 4H, 3J = 6.5, CH2O), 4.16 (t, 4H, 3J = 6.5, CH2O), 3.97 (s, 6H, COOCH3), 1.98–1.89 (m, 8H, CH2), 1.68–1.55 (m, 8H, CH2), 1.09 (t, 6H, 3J = 7.4, CH3), 1.03 (t, 6H, 3J = 7.4, CH3).
13C NMR (CDCl3; δ, ppm): 167.10, 159.34, 159.26, 148.26, 147.53, 145.09, 144.28, 142.77, 141.98, 139.95, 139.54, 134.75, 134.33, 133.18, 132.95, 132.74, 132.66, 132.03, 131.65, 131.35, 130.25, 129.22, 126.96, 126.69, 120.52, 116.56, 113.22, 113.16, 68.11, 68.07, 52.28, 31.92, 31.65, 19.76, 19.53, 14.14, 14.09.
MALDI-TOF MS: m/z calcd for C90H80N6NiO8 [M]+ 1430.54, found 1430.66.
UV-vis (CHCl3; λmax, nm (logε)): 308 (4.56), 345 (4.53), 396 (4.56), 446 (5.26), 550 (4.25), 580 (3.81).
Synthesis of 10
5a (40 mg, 0.03 mmol), Pd(OAc)2 (7.7 mg, 0.03 mmol) and xantphos (35 mg, 0.06 mmol) were mixed under a CO atmosphere, an then BuOH (5 mL) and Et3N (125 μL, 0.9 mmol) were added. The mixture was heated at 100 °C and bubbled with CO for 3 h. Afterwards, the mixture was cooled to ambient temperature and evaporated to dryness. The residue was applied to a column packed with silica gel in hexane and eluted with hexane/DCM (0 → 100% of DCM) and then with DCM/MeOH (0 → 100% of MeOH) mixtures. The evaporation of the fractions containing 10 provided 22 mg (54%) of the target compound.1H NMR (CDCl3; δ, ppm; J, Hz): 8.87 (d, 2H, 3J = 4.9, Hβ), 8.77 (d, 2H, 3J = 4.9, Hβ), 8.73 (s, 2H, Hβ), 7.95 (d, 4H, 3J = 8.0, m-CH), 7.89 (d, 4H, 3J = 8.3, CHo-Ar), 7.82 (d, 4H, 3J = 8.2, CHo-Ar), 7.50 (d, 4H, 3J = 8.1, o-CH), 7.24–7.18 (m, 8H, CHm-Ar), 4.38 (t, 4H, 3J = 6.7, CH2O), 4.27–4.17 (m, 8H, CH2O), 2.08–2.01 (m, 4H, CH2), 1.97–1.91 (m, 4H, CH2), 1.86–1.79 (m, 4H, CH2), 1.72 (sext, 4H, 3J = 7.4, CH2), 1.64 (sext, 4H, 3J = 7.6, CH2), 1.54 (sext, 4H, 3J = 7.9, CH2), 1.14 (t, 6H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3), 1.04 (t, 6H, 3J = 7.4, CH3).
13C NMR (CDCl3; δ, ppm): 166.59, 159.46, 159.29, 148.02, 147.65, 144.21, 143.75, 142.83, 142.07, 134.75, 134.29, 133.04, 132.88, 132.59, 132.54, 132.16, 131.73, 130.71, 130.56, 129.23, 120.57, 116.69, 113.24, 113.21, 68.15, 68.14, 65.11, 31.84, 31.66, 31.02, 29.85, 19.69, 19.54, 19.46, 14.19, 14.10, 13.95.
MALDI-TOF MS: m/z cacld for C84H84N6NiO8 [M]+ 1362.57, found 1362.68.
UV-vis (CHCl3; λmax, nm (logε)): 349 (4.50), 402sh (4.60), 447 (5.21), 550 (4.26), 581 (3.83).
Synthesis of 5g by the phosphonylation of 5a
5a (19 mg, 0.014 mmol), Pd(OAc)2 (1.9 mg, 8 μmol) and dppf (4.7 mg, 8 μmol) were mixed under argon. Toluene (2 mL), EtOH (0.5 mL), diethylphosphite (110 μL, 0.86 mmol) and Et3N (29 μL, 0.21 mmol) were subsequently added. The resulting solution was refluxed for 20 h. Afterwards, the reaction mixture was evaporated, and the residue was applied to a column packed with silica gel in hexane. The column was eluted with DCM/MeOH mixtures (0 → 20% of MeOH) and the fraction containing 5g was evaporated to provide 19 mg (95%) of the product.Synthesis of 11
5a (40 mg, 0.03 mmol), PdCl2dppf (2 mg, 0.003 mmol), KOAc (18 mg, 0.18 mmol) and dipinacoldiborane (23 mg, 0.09 mmol) were mixed under argon, and dioxane (6 mL) was added. The obtained suspension was stirred and heated to 100 °C. The starting material completely dissolved at ca. 80 °C. The mixture was heated for 18 h and then cooled to ambient temperature and evaporated to dryness. The residue was applied to a column packed with silica gel in hexane and eluted with hexane/DCM (0 → 100% of DCM) and then DCM/MeOH mixtures (0 → 3% of MeOH) mixtures. The fractions containing 11 were combined, evaporated and repeatedly purified by means of gel-permeation chromatography at Bio-Beads SX-1 with CHCl3/MeOH eluent (2.5% of MeOH) to provide 17 mg (40%) of 11 after evaporation.1H NMR (CDCl3; δ, ppm; J, Hz): 8.86 (d, 2H, 3J = 4.9, Hβ), 8.76 (d, 2H, 3J = 4.9, Hβ), 8.71 (s, 2H, Hβ), 7.89 (d, 4H, 3J = 8.2, CHo-Ar), 7.81 (d, 4H, 3J = 8.2, CHo-Ar), 7.69 (d, 4H, 3J = 7.7, m-CH), 7.43 (d, 4H, 3J = 7.7, o-CH), 7.21 (d, 4H, 3J = 8.4, CHm-Ar), 7.20 (d, 4H, 3J = 8.1, CHm-Ar), 4.24 (t, 4H, 3J = 6.4, CH2O), 4.20 (t, 4H, 3J = 6.5, CH2O), 2.05 (quint, 4H, 3J = 6.4, CH2), 1.94 (quint, 4H, 3J = 6.3, CH2), 1.76 (sext, 4H, 3J = 7.6, CH2), 1.64 (sext, 4H, 3J = 7.6, CH2), 1.39 (s, 24H, CH3Pin), 1.16 (t, 6H, 3J = 7.4, CH3), 1.09 (t, 6H, 3J = 7.4, CH3).
13C NMR (CDCl3; δ, ppm): 159.43, 159.23, 149.09, 147.59, 144.26, 142.64, 142.26, 141.85, 134.74, 134.30, 133.31, 132.88, 132.74, 132.57, 131.93, 131.65, 129.99, 120.35, 116.58, 113.20, 83.91, 68.15, 68.13, 31.93, 31.67, 25.12, 19.70, 19.55, 14.35, 14.10.
MALDI-TOF MS: m/z calcd for C86H90B2N6NiO8 [M]+ 1414.64, found 1413.78.
UV-vis (CHCl3; λmax, nm (logε)): 300 (3.36), 342 (3.43), 394sh (3.50), 443 (4.29), 548 (3.24), 579sh (2.82).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Russian Foundation for Basic Research and Moscow city Government according to the research project no. 19-33-70036 and Council of the President of Russian Federation for support of young scientists (grant MK-1454.2019.3, synthesis of compounds 4f,g and 5f,g) for financial support. The measurements were made at the Shared Facility Centers of the Institute of Physical Chemistry and Electrochemistry RAS.Notes and references
- J. Liu, Q. Liang, R. Zhao, S. Lei and W. Hu, Mater. Chem. Front., 2020, 4, 354–368 RSC.
- D. Ma, J. Li, A. Liu and C. Chen, Materials, 2020, 13, 1734 CrossRef CAS.
- A. G. Navrotskaya, D. D. Aleksandrova, E. F. Krivoshapkina, M. Sillanpää and P. V. Krivoshapkin, Front. Chem., 2020, 8, 1–9 CrossRef.
- O. A. T. Dias, S. Konar, A. L. Leão, W. Yang, J. Tjong and M. Sain, Front. Chem., 2020, 8, 420 CrossRef CAS.
- S. W. Shin, J. S. Yuk, S. H. Chun, Y. T. Lim and S. H. Um, Nano Convergence, 2020, 7, 2 CrossRef CAS.
- C.-X. Yao, N. Zhao, J.-C. Liu, L.-J. Chen, J.-M. Liu, G.-Z. Fang and S. Wang, Polymers, 2020, 12, 691 CrossRef CAS.
- A. Kayan, Adv. Compos. Hybrid Mater., 2019, 2, 34–45 CrossRef CAS.
- N. Aslankoohi, D. Mondal, A. S. Rizkalla and K. Mequanint, Polymers, 2019, 11, 1437 CrossRef CAS.
- M. J. F. Calvete, G. Piccirillo, C. S. Vinagreiro and M. M. Pereira, Coord. Chem. Rev., 2019, 395, 63–85 CrossRef CAS.
- Z. Sun, G. Cui, H. Li, Y. Liu, Y. Tian and S. Yan, J. Mater. Chem. B, 2016, 4, 5194–5216 RSC.
- Ł. Klapiszewski, K. Siwińska-Stefańska and D. Kołodyńska, Chem. Eng. J., 2017, 330, 518–530 CrossRef.
- J. Zhang, M. Li, L. Cheng and T. Li, Polym. Chem., 2017, 8, 6527–6533 RSC.
- A. Wang, J. Zhang, W. Zhao, W. Zhu and Q. Zhong, J. Alloys Compd., 2018, 748, 929–937 CrossRef CAS.
- Y. Qiu, Y. Zhang, L. Jin, L. Pan, G. Du, D. Ye and D. Wang, Org. Chem. Front., 2019, 6, 3420–3427 RSC.
- J. G. Mahy, C. A. Paez, C. Carcel, C. Bied, A. S. Tatton, C. Damblon, B. Heinrichs, M. Wong Chi Man and S. D. Lambert, J. Photochem. Photobiol., A, 2019, 373, 66–76 CrossRef CAS.
- L. Wang, P. Jin, S. Duan, J. Huang, H. She, Q. Wang and T. An, Environ. Sci.: Nano, 2019, 6, 2652–2661 RSC.
- V. V. Arslanov, M. A. Kalinina, E. V. Ermakova, O. A. Raitman, Y. G. Gorbunova, O. E. Aksyutin, A. G. Ishkov, V. A. Grachev and A. Y. Tsivadze, Russ. Chem. Rev., 2019, 88, 775–799 CrossRef CAS.
- S. Supriya, V. S. Shetti and G. Hegde, New J. Chem., 2018, 42, 12328–12348 RSC.
- L.-Y. Huang, J.-F. Huang, Y. Lei, S. Qin and J.-M. Liu, Catalysts, 2020, 10, 656 CrossRef CAS.
- J. Min Park, J. H. Lee and W.-D. Jang, Coord. Chem. Rev., 2020, 407, 213157 CrossRef.
- W. Gao, J. Tian, Y. Fang, T. Liu, X. Zhang, X. Xu and X. Zhang, Chemosphere, 2020, 243, 125334 CrossRef CAS.
- T. O. Shekunova, L. A. Lapkina, A. B. Shcherbakov, I. N. Meshkov, V. K. Ivanov, A. Y. Tsivadze and Y. G. Gorbunova, J. Photochem. Photobiol., A, 2019, 382, 111925 CrossRef CAS.
- I. A. Abdulaeva, K. P. Birin, A. Bessmertnykh-Lemeune, A. Y. Tsivadze and Y. G. Gorbunova, Coord. Chem. Rev., 2020, 407, 213108 CrossRef CAS.
- A. V. Shokurov, D. S. Kutsybala, A. G. Martynov, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun, Y. G. Gorbunova, A. Y. Tsivadze, A. V. Zaytseva, D. Novikov, V. V. Arslanov and S. L. Selektor, Langmuir, 2020, 36, 1423–1429 CrossRef CAS.
- I. Jiménez-Munguía, A. K. Fedorov, I. A. Abdulaeva, K. P. Birin, Y. A. Ermakov, O. V. Batishchev, Y. G. Gorbunova and V. S. Sokolov, Biomolecules, 2019, 9, 853 CrossRef.
- Y. Y. Enakieva, A. A. Sinelshchikova, M. S. Grigoriev, V. V. Chernyshev, K. A. Kovalenko, I. A. Stenina, A. B. Yaroslavtsev, Y. G. Gorbunova and A. Y. Tsivadze, Chem.–Eur. J., 2019, 25, 10552–10556 CrossRef CAS.
- K. P. Birin, A. I. Poddubnaya, E. V. Isanbaeva, Y. G. Gorbunova and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2017, 21, 406–415 CrossRef CAS.
- I. A. Abdulaeva, D. A. Polivanovskaia, K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Mendeleev Commun., 2020, 30, 162–164 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, M. S. Batov, A. G. Martynov, I. V. Nefedova, Y. G. Gorbunova, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Inorg. Chem., 2019, 58, 5058–5068 CrossRef CAS.
- A. G. Martynov, E. A. Safonova, A. Y. Tsivadze and Y. G. Gorbunova, Coord. Chem. Rev., 2019, 387, 325–347 CrossRef CAS.
- M. V. Volostnykh, S. M. Borisov, M. A. Konovalov, A. A. Sinelshchikova, Y. G. Gorbunova, A. Y. Tsivadze, M. Meyer, C. Stern and A. Bessmertnykh-Lemeune, Dalton Trans., 2019, 48, 8882–8898 RSC.
- K. P. Birin, Y. G. Gorbunova, A. Y. Tsivadze, A. G. Bessmertnykh-Lemeune and R. Guilard, Eur. J. Org. Chem., 2015, 2015, 5610–5619 CrossRef CAS.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, RSC Adv., 2015, 5, 67242–67246 RSC.
- I. A. Abdulaeva, K. P. Birin, J. Michalak, A. Romieu, C. Stern, A. Bessmertnykh-Lemeune, R. Guilard, Y. G. Gorbunova and A. Y. Tsivadze, New J. Chem., 2016, 40, 5758–5774 RSC.
- K. P. Birin, A. I. Poddubnaya, I. A. Abdulaeva, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2018, 156, 243–249 CrossRef CAS.
- K. P. Birin, I. A. Abdulaeva, A. I. Poddubnaya, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2020, 181, 108550 CrossRef CAS.
- I. A. Abdulaeva, K. P. Birin, A. A. Sinelshchikova, M. S. Grigoriev, K. A. Lyssenko, Y. G. Gorbunova, A. Y. Tsivadze and A. Bessmertnykh-Lemeune, CrystEngComm, 2019, 21, 1488–1498 RSC.
- I. A. Abdulaeva, K. P. Birin, Y. G. Gorbunova, A. Y. Tsivadze and A. Bessmertnykh-Lemeune, J. Porphyrins Phthalocyanines, 2018, 22, 619–631 CrossRef CAS.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2011, 40, 11539 RSC.
- K. P. Birin, A. I. Poddubnaya, Y. G. Gorbunova and A. Y. Tsivadze, Macroheterocycles, 2017, 10, 514–515 CrossRef CAS.
- M. J. Crossley, L. G. King, I. A. Newsom and C. S. Sheehan, J. Chem. Soc., Perkin Trans. 1, 1996, 2675–2684 RSC.
- K. P. Birin, I. A. Abdulaeva, D. A. Polivanovskaia, A. A. Sinelschikova, L. I. Demina, A. E. Baranchikov, Y. G. Gorbunova and A. Y. Tsivadze, Russ. J. Inorg. Chem., 2021 DOI:10.31857/S0044457X21020021.
- E. V. Vinogradova, Y. Y. Enakieva, S. E. Nefedov, K. P. Birin, A. Y. Tsivadze, Y. G. Gorbunova, A. G. Bessmertnykh Lemeune, C. Stern and R. Guilard, Chem.–Eur. J., 2012, 18, 15092–15104 CrossRef CAS.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 1981, 56–57 CrossRef.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Elsevier/Butterworth-Heinemann, 2009 Search PubMed.
- M. Lo, J.-F. Lefebvre, D. Leclercq, A. van der Lee and S. Richeter, Org. Lett., 2011, 13, 3110–3113 CrossRef CAS.
- M. Morisue, N. Haruta, D. Kalita and Y. Kobuke, Chem.–Eur. J., 2006, 12, 8123–8135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete NMR characteristics of the compounds. See DOI: 10.1039/d0ra08603g |
| This journal is © The Royal Society of Chemistry 2020 |