
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEight-step total synthesis of (+)-crambescin A†
Zhenhua Gao,
Junchen Li,
Yunyang Song,
Xiaojing Bi,
Xiangyan Meng* and
Yongbiao Guo *
*
State Key Laboratory of NBC Protection for Civilian Research Beijing, 102205, P. R. China. E-mail: van87120@126.com
First published on 26th October 2020
Abstract
(+)-Crambescin A belongs to the polycyclic guanidine natural product family and has been shown to possess various medically important properties. The chiral bicyclic guanidine structure of (+)-crambescin A presents a challenge for chemical synthesis. Here we implement a novel asymmetric Biginelli reaction strategy to achieve the enantiospecific total synthesis of (+)-crambescin A in only 8 steps from the abundant and inexpensive aliphatic aldehyde, urea and methyl 3-oxobutanoate.
Introduction
Crambescins are members of a class of guanidine alkaloids that were isolated from the marine sponges Crambe crambe,1 Monanchora pulchra,2 Pseudaxinella reticulate3 and Monanchora n. sp.4 To date, over 30 structurally related crambescins have been isolated.5,6 Crambescins are classified as three types and are all characterized by at least one chiral cyclic guanidine: type A corresponds to the bicyclic structures; type B to monocyclic ones and type C to spirocyclic frameworks (Fig. 1). Members of the crambescin class exhibit potent biological activity, including inhibition of binding of HIV gp 120 to human CD4,7 suppression of the multiplication of cancer cells,8 blocking of the voltage-gated sodium channels9 and potassium channels,10 and inhibition of the bacteria growth.11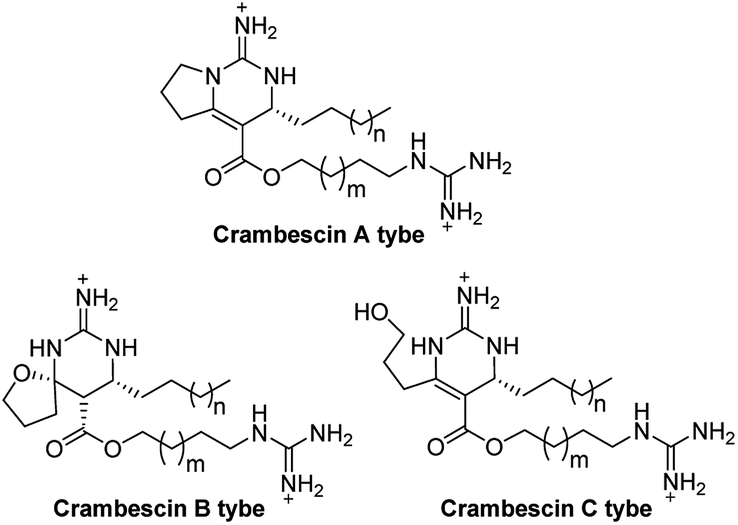 | ||
| Fig. 1 Structures of crambescin A type, B type and C type. | ||
For this family of natural products, a plethora of biological evaluations were done. Synthesis is necessary to secure the access to the molecules as the sponges are not widely accessible and to anticipate an alternative production to preserve fragile marine ecosystems. Only three years after crambescin A–C were isolated, the racemic synthesis of crambescin A–C was reported by Snider.12 More recently, a racemic synthesis of crambescin A was reproted by Thomas and co-workers employing a tethered Biginelli-like reaction between C-2/C-3 activated fatty acids and a central guanidinylated pyrrolinium.13 However, the asymmetric synthesis of crambescin A–C has not been reported. It was not until 2016 that the first asymmetric route to crambescin A–C derivatives (crambescin A–C carboxylic acid) was delineated by Nishikawa and co-workers.9 A cis-enyne (2, Scheme 1) was epoxidized using the Katsuki asymmetric epoxidation to give epoxide (3) and then product recrystallization allowed to reach >99% ee. Epoxide (3) was transformed to the chiral spirocyclic guanidine structure (5) via bromocation-triggered cascade cyclization reaction. Then, subsequent functional group transformations from 6 resulted in the total synthesis of crambescin A carboxylic acid (16 steps from 2, 0.9% total yield), crambescin B carboxylic acid (15 steps from 2, 8.7% total yield) and crambescin C carboxylic acid (15 steps from 2, 4.5% total yield), respectively. Although this achievement is significant, the practicality of this route is questionable because of lower overall yields (0.9–8.7%) and the requirement for a large number of linear synthetic steps (15–16 steps).
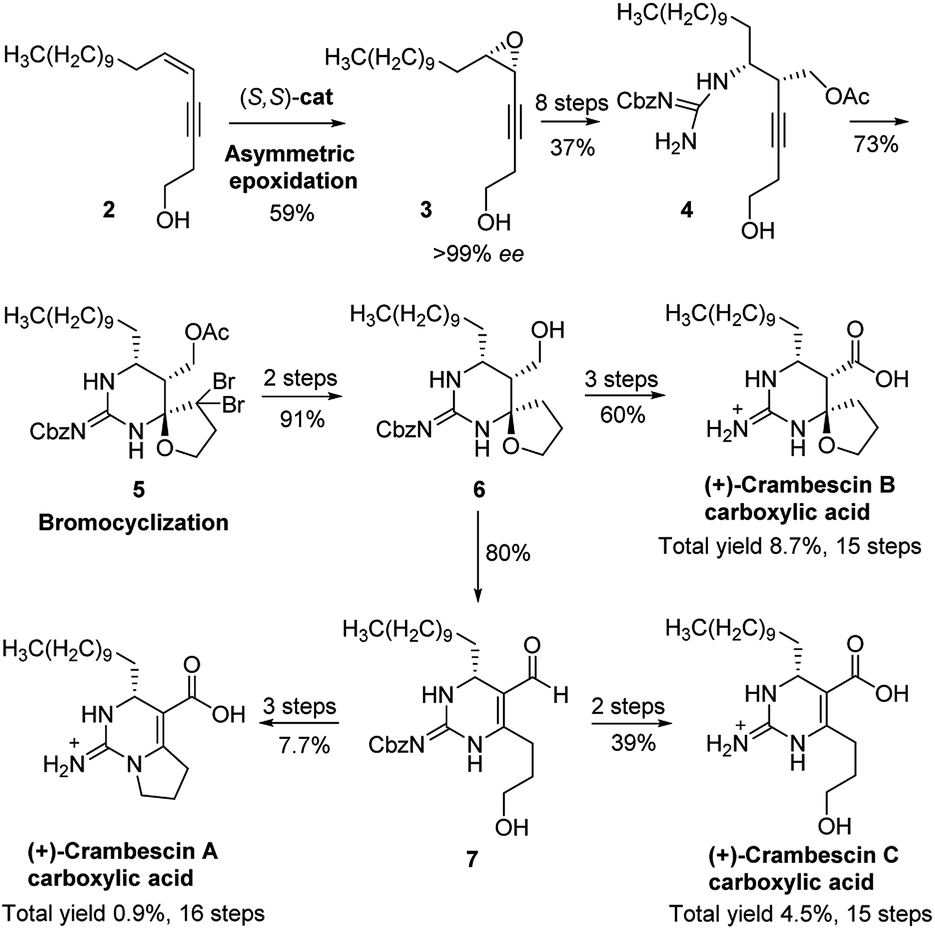 | ||
| Scheme 1 Nishikawa's total synthesis of (+)-crambescin A–C carboxylic acid. | ||
As we are interested in the unique structure of the crambescins, we planned to synthesize the crambescins by a novel strategy. Herein, we report an enantioselective total synthesis of (+)-crambescin A (1) based upon a strategy involving successive Biginelli reaction of aliphatic aldehyde.
As an approach to the synthesis of crambescin A (1), it seemed reasonable to couple the bicyclic guanidine carboxylic acid 8 with the side-chain alcohol 9 by means of an esterification at the final stage of the synthesis (Scheme 2). Compound 8 can be synthesized from the optically active intermediate 10 through four steps, including thionation, S-ethylation, guanidination and Boc protection. The intermediate 10 can be obtained from the key intermediate 11 by employing the C–C and C–N coupling/cyclization cascade reaction. The key intermediate 11 is a kind of 4-alkyl-3,4-dihydropyrimidin-2(1H)-ones (4-alkyl-DHPMs), which can be synthesized by the enantioselective Biginelli reaction of urea, ethyl acetoacetate, and octanal.
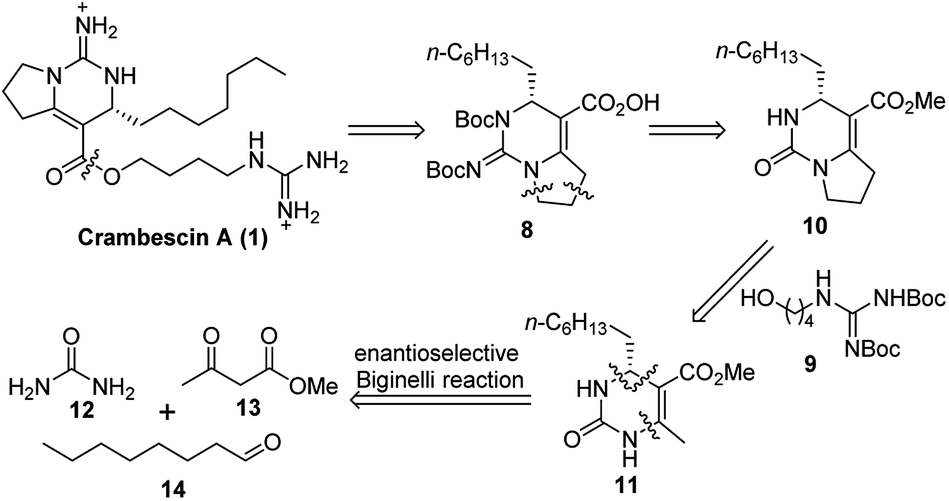 | ||
| Scheme 2 Retrosynthetic analysis of (+)-crambescin A (1). | ||
Results and discussion
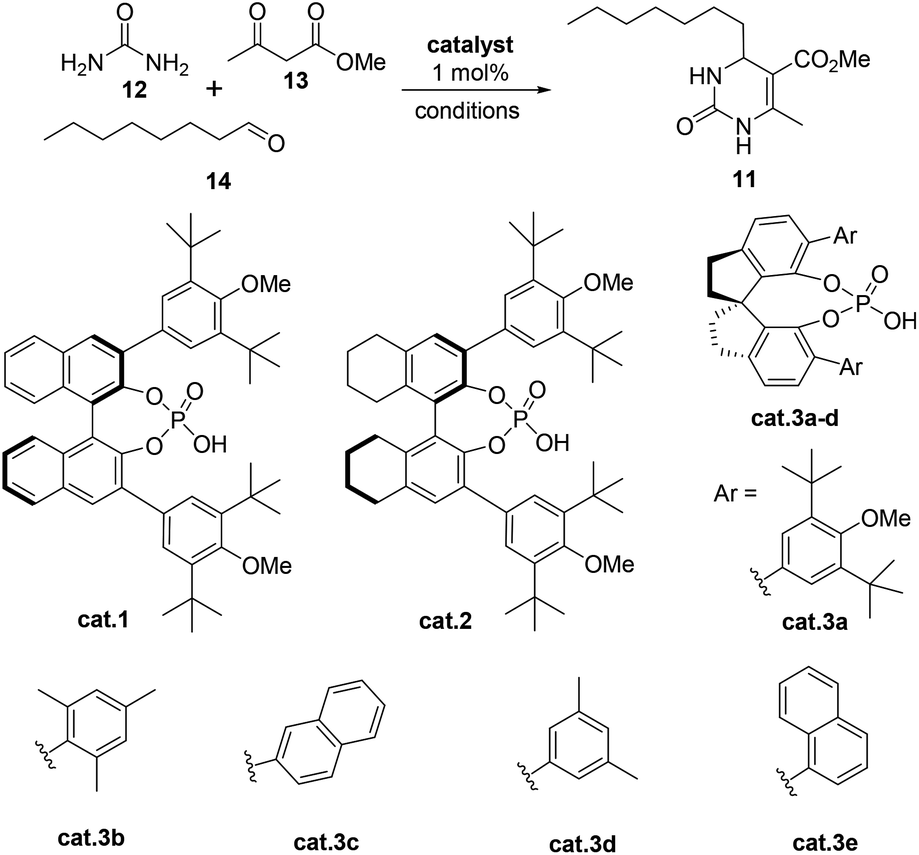
Recently, our group demonstrated that a chiral binaphthol phosphoric acid (cat.1, Table 1, entry 1) can successfully catalyze the asymmetric Biginelli reaction of aliphatic aldehydes, urea and β-ketoesters for the synthesis of optically active 4-alkyl-DHPMs.14,15 However, the reaction time was too long (at 25 °C for 10 days, then at 50 °C for 1 day). Moreover, the enantioselectivity (85% ee, Table 1, entry 1) was not satisfactory. So we commenced our synthetic study by screening more suitable reaction conditions for this particular reaction. Initially, a number of commonly used chiral phosphoric acids (cat.2 and cat.3a–e), were examined in CHCl3 at 25 °C (Table 1). Pleasingly, it was found that all of the catalysts could indeed promote the desired reaction giving the 4-alkyl-3,4-dihydropyrimidin-2(1H)-one 6 in variable yields and enantioselectivities with the exception of cat.3c (Table 1, entries 1 to 6). With cat.2 as the best candidate (entry 2), we continued to study the effect of solvents and found that BrCH2CH2Cl was the optimal choice (Table 1, entries 13 vs. 2, 8–12, 14–15). The synthetic application potentiality of this asymmetric Biginelli reaction of aliphatic aldehydes was demonstrated by the gram-scale transformation. A 0.1 mol–scale reaction of 12, 13 and 14 was performed, affording 22.8 g of 11 in 85% yield with 91% ee. There was almost no change in either yield or enantioselectivity. Moreover, ee of 11 could be raised to 99% after a single recrystallization (88% crystallization yield).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (d) | Yieldb (%) | eec (%) |
| a Reactions conditions: 12 (0.2 mmol), 13 (1.5 mmol), 14 (0.24 mmol) and a catalyst (0.002 mmol) in solvent (2 mL) for 5–10 days (25 °C).b Yield was determined by HPLC analysis.c Determined by HPLC (Chiralcel AD-RH).d The reactions was run with 12 (0.1 mol), 13 (0.75 mol), 14 (0.12 mol) and cat.2 (0.001 mol) in BrCH2CH2Cl (100 mL) at 25 °C for 7 days. | |||||
| 1 | cat.1 | CHCl3 | 10 + 1 | 75 | 85 |
| 2 | cat.2 | CHCl3 | 10 | 71 | 89 |
| 3 | cat.3a | CHCl3 | 5 | 26 | 46 |
| 4 | cat.3b | CHCl3 | 5 | 70 | 72 |
| 5 | cat.3c | CHCl3 | 5 | 39 | 0 |
| 6 | cat.3d | CHCl3 | 5 | 49 | 15 |
| 7 | cat.3e | CHCl3 | 5 | 44 | 58 |
| 8 | cat.2 | CHCl3 | 7 | 50 | 59 |
| 9 | cat.2 | CH2Cl2 | 7 | 60 | 90 |
| 10 | cat.2 | CCl4 | 7 | 68 | 78 |
| 11 | cat.2 | Toluene | 7 | 50 | 94 |
| 12 | cat.2 | ClCH2CH2Cl | 7 | 73 | 91 |
| 13 | cat.2 | BrCH2CH2Cl | 7 | 85 | 91 |
| 14 | cat.2 | Benzene | 7 | 54 | 82 |
| 15 | cat.2 | Xylene | 7 | 52 | 81 |
| 16d | cat.2 | BrCH2CH2Cl | 7 | 85 | 91 |
With large quantities of optically active 4-alkyl-DHPM 11 in hand, we next examined the conversion of 11 into the bicyclic dihydropyrimidinone 10 through the C–C and C–N coupling/cyclization cascade reaction. Initially, we envisaged that C–C and C–N coupling/cyclization cascade reaction of 11 with 1,2-dibromoethane by the Singh16,17 method would afford 10. However, the yield of bicyclic intermediate 10 was very low (10%). Thus, several bases and electrophiles for this conversion were further investigated, and the results are summarized in Table 2. The best yield (42%, 71% conv.) was obtained when 5.0 equivalents of n-BuLi and 5.0 equivalents of 1-bromo-2-chloroethane were used. Control experiments performed by chiral HPLC indicated that no erosion of the ee occurred during the cyclization process.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) | Conversion (%) |
| a Reagents and conditions: 11 (0.5 mmol), base (1.75–3.0 mmol), dry THF (5 mL), −20 °C – r.t., 1 h, then 1,2-dibromoethane or 1-bromo-2-chloroethane (2.5 mmol), r.t. 3 h.b Isolated yield. | ||||
| 1 | n-BuLi (3.5) | 1,2-Dibromoethane | 10 | — |
| 2 | n-BuLi (3.5) | 1-Bromo-2-chloroethane | 34 | 65 |
| 3 | n-BuLi (4.0) | 1-Bromo-2-chloroethane | 38 | 61 |
| 4 | n-BuLi (5.0) | 1-Bromo-2-chloroethane | 42 | 71 |
| 5 | n-BuLi (6.0) | 1-Bromo-2-chloroethane | 40 | 64 |
| 6 | LDA (5.0) | 1-Bromo-2-chloroethane | 38 | 58 |
| 7 | LiHMDS (5.0) | 1-Bromo-2-chloroethane | Trace | — |
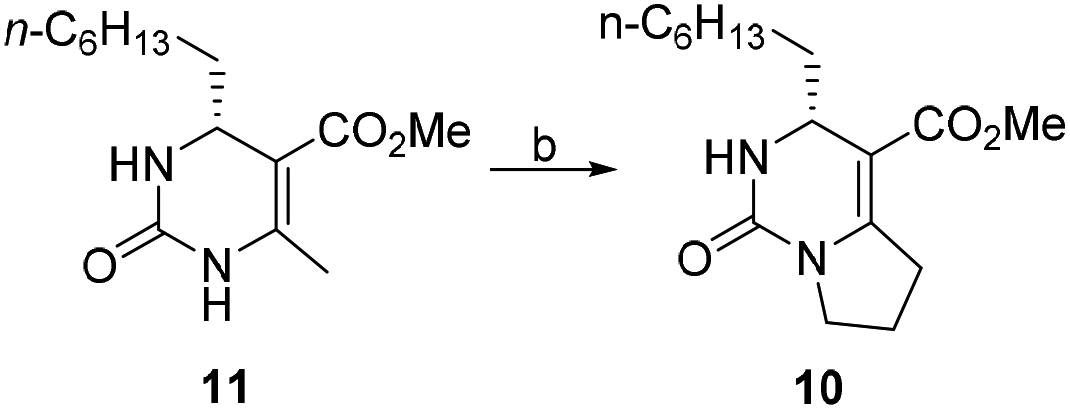
Compound 10 could be treated directly with ethylation reagent (Et3O+BF4−), leading to ethoxypyrimidine 15, which was then converted to bicyclic guanidine 16 via guanidination reaction with ammonium propionate. However, the total yield (53%) of two steps was not satisfactory owing to formation of the side product 10 in the guanidination step. So we decided to prevent its formation by substituting sulfur atom for oxygen atom to avoid the side reaction. Dihydropyrimidinthione 17 was generated in quantitative yield by treatment of 10 with Lawesson's reagent. By following the same procedure of 10, we were pleased that the yield of 16 could be increased to 81% from 10 (Scheme 3).
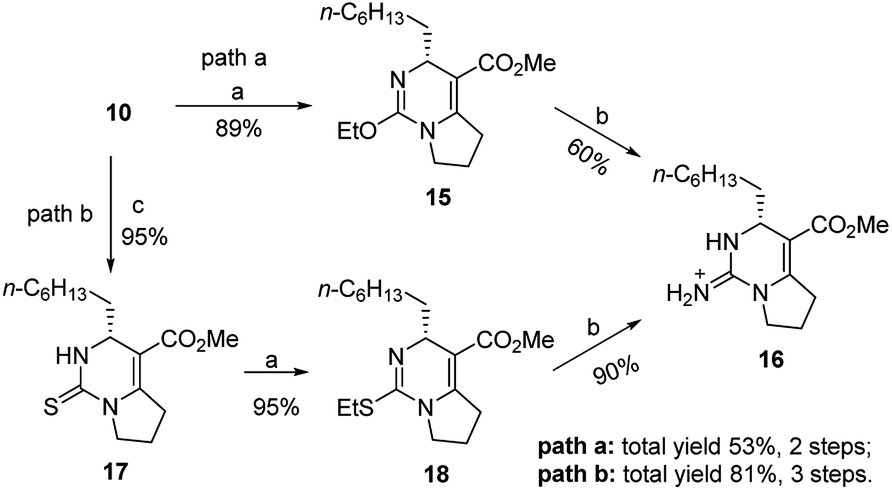 | ||
| Scheme 3 Reagents and conditions: (a) Et3O+BF4−, NaHCO3, dry CH2Cl2, r.t., 1 h; (b) NH4+EtCO2−, 80 °C, 3 h; (c) Lawesson's reagent, xylene, 120 °C, 2 h, 95%. | ||
The last three steps completed the total synthesis of crambescin A (Scheme 4). Treatment of the bicyclic guanidine 16 with (Boc)2O in the presence of NaH and DMAP gave the fully protected bicyclic guanidine 19. Then, demethylation of 19 with EtSLi and subsequent condensation with the guanidine alcohol 9 provided the fully protected crambescin A 20. Finally, CF3CO2H promoted removal of the four Boc groups, furnishing the targeted crambescin A (only 8 steps, 10.7% total yield). All of the analytical data including 1H, 13C, 2D NMR, and [α]D value of the synthetic 1 were identical to those of the natural product (Tables S1 and S2†).11
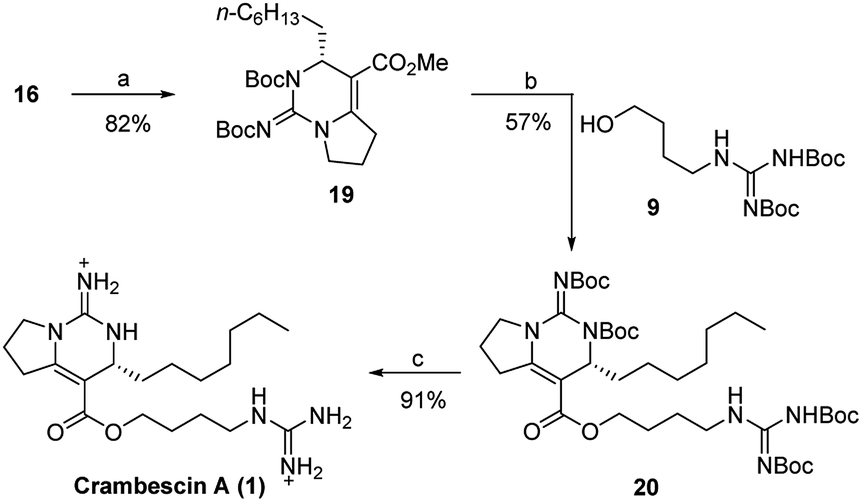 | ||
| Scheme 4 Reagents and conditions: (a) NaH, dry THF, 0 °C, then DMAP, (Boc)2O, r.t.; (b) EtSLi, HMPA, r.t., then BOPCl, NEt3, dry CH2Cl2, r.t.; (c) CF3CO2H, dry CH2Cl2. | ||
Conclusions
The first asymmetric total synthesis of (+)-crambescin A (1) has been accomplished via a short and practical route from urea, ethyl acetoacetate, and octanal as the starting material. The key transformations include a chiral phosphoric acid-catalyzed Biginelli reaction and a C–C and C–N coupling/cyclization cascade reaction under strong alkali condition. This study should open up a new efficient route for synthesizing crambescins.Experimental section
General information
Reagents and solvents were purchased from common commercial suppliers and were used without further purification. Column chromatography was generally performed on silica gel (200–300 mesh). Melting points were determined with a Büchi B-545 melting-point apparatus. 600 MHz 1H NMR and 150 MHz 13C NMR spectra were recorded on Varian VMS-600 spectrometers, respectively. The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in hertz (Hz). High-resolution mass spectra (HRMS) were obtained on Agilent 6502 Q-TOF HPLC and mass spectrometry.Synthesis of 4-alkyl-DHMPs 11
A mixture of urea 12 (0.1 mol), catalyst cat.2 (0.001 mol), 14 (0.12 mol), 13 (0.75 mol) and BrCH2CH2Cl (100 mL) was stirred at 25 °C for 7 days. Then the resulting solution was concentrated under reduced pressure, and the orange residue was purified by column chromatography ([eluting with hexane/i-PrOH 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)] to afford 11 (22.8 g, 85%) as a white crystalline solid. Then the optically pure 11 were obtained in 88% yield with 99% ee by recrystallization from MeCN. m.p. 128–130 °C; 1H NMR (600 MHz, CDCl3): δ (ppm) 8.27 (s, 1H), 5.87 (s, 1H), 4.28 (dt, J = 7.8 Hz, 3.6 Hz, 1H), 3.71 (s, 3H), 2.27 (s, 3H), 2.27 (s, 3H) 1.57–1.48 (m, 2H), 1.38–1.25 (m, 10H), 0.87 (t, J = 6.6 Hz, 3H). 13 C NMR (150 MHz, CDCl3): δ (ppm) 166.4, 154.6, 147.0, 101.4, 51.6, 51.1, 36.9, 31.8, 29.3, 29.2, 24.4, 22.6, 18.6, 14.1. HRMS (m/z) calcd for C14H25N2O3 [M + H]+ 269.1860, found 269.1862; [α]D25 = +140 (c = 0.30, CH2Cl2); The ee was determined by HPLC analysis (Chiralpak OD-RH, CH3CN/H2O = 60/40, v/v, 1.0 mL min−1, 280 nm, tR (minor) = 3.4 min, tR (major) = 5.1 min), 99% ee.
:1)] to afford 11 (22.8 g, 85%) as a white crystalline solid. Then the optically pure 11 were obtained in 88% yield with 99% ee by recrystallization from MeCN. m.p. 128–130 °C; 1H NMR (600 MHz, CDCl3): δ (ppm) 8.27 (s, 1H), 5.87 (s, 1H), 4.28 (dt, J = 7.8 Hz, 3.6 Hz, 1H), 3.71 (s, 3H), 2.27 (s, 3H), 2.27 (s, 3H) 1.57–1.48 (m, 2H), 1.38–1.25 (m, 10H), 0.87 (t, J = 6.6 Hz, 3H). 13 C NMR (150 MHz, CDCl3): δ (ppm) 166.4, 154.6, 147.0, 101.4, 51.6, 51.1, 36.9, 31.8, 29.3, 29.2, 24.4, 22.6, 18.6, 14.1. HRMS (m/z) calcd for C14H25N2O3 [M + H]+ 269.1860, found 269.1862; [α]D25 = +140 (c = 0.30, CH2Cl2); The ee was determined by HPLC analysis (Chiralpak OD-RH, CH3CN/H2O = 60/40, v/v, 1.0 mL min−1, 280 nm, tR (minor) = 3.4 min, tR (major) = 5.1 min), 99% ee.
Synthesis of 10
After stirring a solution of 11 (2 mmol) in dry THF (20 mL) under nitrogen atmosphere at −20 °C, 2.4 M nBuLi (4.2 mL, 10 mmol) was added dropwise at −20 °C. After the addition, reaction mixture was warmed to room temperature and stirred for additional 1 h. Then 1-bromo-2-chloroethane (12 mmol) was added dropwise using cannula at room temperature. Then the mixture was stirred at room temperature for 3 h. After which a saturated aqueous solution of NH4Cl was introduced to terminate the reaction. The reaction was extracted with diethyl ether (3 × 25 mL) treated in sequence with brine and water, dried over anhydrous Na2SO4 and filtered. After the excess solvent was evaporated, the resulting orange gum was purified by column chromatography on a silica gel column [eluting with hexane/ethyl acetate (3:1)] to obtain 10 (247 mg, 42%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ (ppm) 5.48 (s, 1H), 4.27 (dt, J = 7.2 Hz, 3.6 Hz, 1H), 3.71 (s, 3H), 3.72–3.65 (m, 2H), 3.20 (ddd, J = 18.0 Hz, 8.0 Hz, 4.2 Hz, 1H), 3.00 (dt, J = 18.0 Hz, 8.0 Hz, 1H), 2.06–1.91 (m, 2H), 1.55–1.48 (m, 2H), 1.38–1.24 (m, 10H), 0.86 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 166.2, 153.9, 153.1, 98.4, 51.9, 51.0, 46.6, 37.1, 31.9, 31.8, 29.2, 29.2, 24.4, 22.6, 21.4, 14.1. HRMS (m/z) calcd for C16H27N2O3 [M + H]+ 295.2016, found 295.2029; [α]D25 = +146 (c = 0.2, CH2Cl2); the ee was determined by HPLC analysis (Chiralpak OD-H, CH3CN/H2O = 60/40, v/v, 1.0 mL min−1, 280 nm, tR (minor) = 5.5 min, tR (major) = 6.9 min), 99% ee.
Synthesis of 17
A mixture of 10 (0.8 mmol) and Lawesson's reagent (0.96 mmol) in xylene was heated to 120 °C for 2 h. After the excess solvent was evaporated, the resulting orange gum was purified by column chromatography on a silica gel column [eluting with hexane/EtOAc (4:1)] to obtain 17 (236 mg, 95%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ (ppm) 6.96 (s, 1H), 4.28 (dt, J = 7.5, 3.6 Hz, 1H), 4.01 (ddd, J = 9.1, 5.5, 3.4 Hz, 2H), 3.73 (s, 3H), 3.30 (ddd, J = 18.2, 8.5, 3.6 Hz, 1H), 2.99 (dt, J = 18.4, 9.3 Hz, 1H), 2.06 (ddt, J = 12.9, 10.8, 4.0 Hz, 1H), 2.00–1.89 (m, 1H), 1.53 (dddd, J = 24.4, 14.1, 8.5, 3.9 Hz, 2H), 1.39–1.25 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 176.3, 165.9, 150.4, 100.4, 52.0, 51.9, 51.4, 36.9, 32.0, 31.8, 29.2, 29.1, 24.1, 22.6, 21.1, 14.1. HRMS (ESI) m/z calcd for C16H27N2O2S [M + H]+: 311.1793; found 311.1788. [α]D25 = +240 (c = 0.30, CH2Cl2).
Synthesis of 18
To a mixture of 17 (0.7 mmol) and NaHCO3 (2.8 mmol) in dry CH2Cl2 (15 mL) was added dropwise Et3O+BF4− (0.2 M CH2Cl2 solution, 7 mL, 1.4 mmol) under nitrogen atmosphere at room temperature. The resulting solution was stirred at room temperature for 0.5 h and then water (30 mL) was added at room temperature. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2SO4 and filtered. After the excess solvent was evaporated, the resulting orange gum was purified by column chromatography on a silica gel column [eluting with hexane/ethyl acetate (6:1)] to obtain 18 (225 mg, 95%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ = 4.62 (s, 1H), 3.72–3.69 (m, 1H), 3.68 (s, 3H), 3.51 (dt, J = 16.8, 8.5 Hz, 1H), 3.21 (ddd, J = 17.9, 8.4, 1.9 Hz, 1H), 3.14 (s, 1H), 2.96 (s, 1H), 2.85 (dt, J = 15.9, 9.3 Hz, 1H), 2.07–2.03 (m, 1H), 1.98–1.91 (m, 1H), 1.47–1.39 (m, 3H),1.34–1.25 (m, 12H), 0.86 (t, J = 7.2 Hz, 3H).13C NMR (150 MHz, CDCl3): δ = 167.4, 153.9, 150.5, 97.2, 55.9, 50.8, 48.4, 37.2, 31.9, 30.9, 29.7, 29.6, 29.3, 25.4, 24.7, 22.6, 22.0, 14.3, 14.1. HRMS: m/z calcd for C18H31N2O2S [M + H]+: 339.2106; found: 339.2114.
Synthesis of 16
A mixture of 19 (0.5 mmol) and ammonium propionate (1.0 g) was stirred at 80 °C under nitrogen atmosphere for 3 h and then the mixture was concentrated under reduced pressure. The resulting orange gum was purified by column chromatography on a silica gel column [eluting with EtOAc/MeOH/TFA (100:10:1)] to obtain 16 (TFA salt, 183 mg, 90%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ (ppm) 10.12 (s, 1H), 4.43 (s, 1H), 3.87(t, J = 8.4 Hz 1H), 3.75 (s, 3H), 3.69 (dd, J = 11.4 Hz, 8.4 Hz, 1H), 3.33 (ddd, J = 18.0 Hz, 8.4 Hz, 3.0 Hz, 1H), 2.94 (dt, J = 18.6 Hz, 9.6 Hz, 1H), 2.25–2.19 (m, 1H), 2.13–2.06 (m, 1H), 1.58–1.48 (m, 2H), 1.40–1.25 (m, 10H), 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 165.1, 152.0, 150.4, 102.3, 51.6, 50.1, 47.4, 36.7, 31.7, 30.7, 29.1, 29.0, 23.9, 22.6, 21.8, 14.0. HRMS (m/z) calcd for C16H28N3O2 [M + H]+ 294.2176, found 294.2177; [α]D25 = +50 (c = 0.30, MeOH, TFA salt).
Synthesis of 20
To a solution of 16 (0.4 mmol) in THF (5 mL) at 0 °C was added NaH (4 mmol). After stirring for 15 min, DMAP (0.2 mmol) and Boc2O (2.4 mmol) were added and the solution was allowed to warm to RT. After stirring for 12 h, the solution was quenched with saturated NH4Cl (5 mL) and then the solution was extracted with diethyl ether (3 × 30 mL) treated in sequence with brine and water, dried over anhydrous Na2SO4 and filtered. After the excess solvent was evaporated, the resulting orange gum was purified by column chromatography on a silica gel column [eluting with hexane/ethyl acetate (3:1)] to obtain crude 19 (162 mg, 82%, crude yield) as a colorless oil. Then, to crude 19 (0.328 mol) was added a solution of EtSLi in HMPA (3 mL of a 1.6 M solution). After stirring at 25 °C for 2 h, the reaction was quenched with a saturated solution of NH4Cl (0.3 mL) and diluted with Et2O (50 mL). After washing H2O (5 × 30 mL), the combined aqueous layer was acidified to pH 3 with 0.1 N HCl and extracted with Et2O (2 × 50 mL). The combined organic layers were then washed with 0.1 N HCl (3 × 30 mL), dried over anhydrous Na2SO4 and filtered. After the excess solvent was evaporated, the crude carboxylic acid was obtained. A mixture of crude carboxylic acid, guanidine alcohol 9 (0.5 mmol), BOPCl (0.5 mmol), Et3N (15 mmol) and dry CH2Cl2 (5 mL) was stirred at 25 °C for 15 h. Then the resulting solution was concentrated under reduced pressure, and the orange residue was purified by column chromatography [eluting with hexane/EtOAc (4:1)] to afford 20 (180 mg, 57%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ (ppm) 11.50 (s, 1H), 8.37 (s, 1H), 5.21 (dd, J = 8.4 Hz, 4.2 Hz, 1H), 4.20–4.12 (m, 2H), 3.95 (dd, J = 12.0 Hz, 7.8 Hz, 1H), 3.73 (ddd, J = 12.0 Hz, 8.4 Hz, 4.2 Hz, 1H), 3.49–3.44 (m, 2H), 3.22 (ddd, J = 18.0 Hz, 8.4 Hz, 4.2 Hz, 1H), 2.91 (dt, J = 18.0 Hz, 9.0 Hz, 1H), 2.08–2.04 (m, 1H), 2.01–1.95 (m, 1H), 1.75–1.66 (m, 4H), 1.63–1.57 (m, 1H), 1.51 (s, 9H), 1.50 (s, 9H), 1.49 (s, 9H), 1.48 (s, 9H), 1.40–1.36 (m, 1H), 1.30–1.23 (m, 10H), 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 165.1, 158.9, 156.1, 153.3, 152.0, 150.8, 144.8, 103.0, 83.1(2), 79.7(2), 63.6, 52.9, 48.9, 33.6, 31.9, 31.2, 29.2, 29.1, 28.3(3), 28.2(3), 28.1(3), 28.0(3), 26.2, 25.8, 24.8, 22.6, 21.4, 14.1. HRMS (m/z) calcd for C40H69N6O10 [M + H]+ 793.5070, found 793.5084.
Synthesis of (+)-crambescin A
A mixture of urea 20 (0.2 mol),CF3CO2H (2.5 mol) and dry CH2Cl2 (7.5 mL) was stirred at 25 °C for 1 h. Then the resulting solution was concentrated under reduced pressure, and the orange residue was purified by column chromatography [eluting with EtOAc/MeOH/TFA (100:10:1)] to afford (+)-crambescin A (113 mg, 91%, TFA salt) as a colorless oil. 1H NMR (600 MHz, CD3OD): δ (ppm) 4.40 (dd, J = 6.6 Hz, 4.8 Hz, 1H), 4.22 (td, J = 14.4 Hz, 6.6 Hz, 1H), 3.82 (td, J = 8.4 Hz, 1.8 Hz, 1H), 3.67 (dd, J = 16.2 Hz, 9.0 Hz, 1H), 3.35–3.31 (m, 1H), 3.23 (t, J = 7.2 Hz, 2H), 2.99(dt, J = 18.0 Hz, 9.0 Hz, 1H), 2.26–2.18 (m, 1H), 2.15–2.06 (m, 1H), 1.79–1.74 (m, 2H), 1.70–1.66 (m, 2H), 1.62–1.54 (m, 2H), 1.44–1.30 (m, 10H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (150 MHz, CD3OD): δ (ppm) 165.9, 158.5, 152.9, 152.6, 103.0, 64.9, 51.1, 48.7, 41.8, 37.2, 32.7, 31.7, 30.1, 30.0, 26.8, 26.4, 25.0, 23.5, 22.7, 14.2. HRMS (m/z) calcd for C20H37N6O2 [M + H]+ 393.2973, found 393.2987. [α]D25 = +13 (c = 0.1, MeOH, TFA salt), Lit:11 [α]D = +10 (c = 0.1, MeOH, TFA salt).
Conflicts of interest
There are no conflicts to declare.References
- R. G. S. Berlinck, J. C. Braekman and D. Daloze, Tetrahedron lett, 1990, 31, 6531–6534 CrossRef CAS.
- T. N. Makarieva, E. K. Ogurtsova and V. A. Denisenko, Org. Lett., 2014, 16, 4292–4295 CrossRef CAS.
- M. T. Jamison and T. F. Molinski, J. Nat. Prod., 2015, 78, 557–561 CrossRef CAS.
- A. El-Demerdash, C. Moriou and M. T. Martin, J. Nat. Prod., 2016, 79, 1929∼1937 CrossRef.
- R. G. S. Berlinck and S. Romminger, Nat. Prod. Rep., 2016, 33, 456–490 RSC.
- R. G. S. Berlinck, A. F. Bertonha, M. Takaki and J. P. G. Rodriguez, Nat. Prod. Rep., 2017, 34, 1264–1301 RSC.
- A. D. Patil, N. V. Kumar and W. C. Kokke, J. Org. Chem., 1995, 60, 1182–1188 CrossRef CAS.
- M. Roel, J. A. Rubiolo and E. Ternon, Mar. Drugs, 2015, 13, 4633–4653 CrossRef CAS.
- A. Nakazaki, Y. Nakane, Y. Ishikawa, M. Yostsu-Yamashia and T. Nishikawa, Org. Biomol. Chem., 2016, 14, 5304–5309 RSC.
- V. Martín, C. Vale and S. Bondu S, Chem. Res. Toxicol., 2013, 26, 169–178 Search PubMed.
- M. T. Jamison and T. F. Molinski, J. Nat. Prod., 2015, 78, 557–561 CrossRef CAS.
- B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828–3839 CrossRef CAS.
- S. B. L. Silva, F. Oberhänsli, M. A. Tribalat, G. Genta-Jouve, J. L. Teyssié, M. Y. Dechraoui-Bottein, J. F. Gallard, L. Evanno, E. Poupon and O. P. Thomas, Angew. Chem., Int. Ed., 2019, 58, 520–525 CrossRef CAS.
- Y. Guo, Z. Gao, X. Meng, Y. Huang, H. Zhong, H. Yu, X. Ding, H. Tang and C. Zou, Synlett, 2017, 28, 2041–2045 CrossRef CAS.
- Y. Guo, Z. Gao, C. Fan, J. Chen, J. Li, Y. Huang and C. Zou, Synthesis, 2018, 50, 2394–2406 CrossRef CAS.
- K. Singh and S. Singh, J. Org. Chem., 2005, 70, 6114–6117 CrossRef CAS.
- K. Singh and S. Singh, Tetrahedron, 2008, 64, 11718–11723 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08726b |
| This journal is © The Royal Society of Chemistry 2020 |