
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile assembly of 1,5-diazocan-2-ones via cyclization of tethered sulfonamides to cyclopropenes†
Jonathon P. Mathenya,
Pavel M. Yamanushkina,
Peter A. Petillob and
Michael Rubin *ac
*ac
aDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Fax: +1-785-864-5396; Tel: +1-785-864-5071
bDesign-Zyme LLC, 4950 Research Park Way, Lawrence, KS 66047, USA
cDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation. E-mail: alexaks05@rambler.ru
First published on 15th December 2020
Abstract
The sulfonamide moiety was evaluated as an activating and stabilizing functional group in the metal-templated strain release-driven intramolecular nucleophilic addition of amines to cyclopropenes to generate 1,5-diazocan-2-ones.
Introduction
Compounds containing the 1,5-diazocin-2-one moiety are scarce in nature,1 with the best known examples occurring in the Homalium alkaloid family (1a–d, Fig. 1). The four known naturally-occurring Homalium alkaloids isolated from the leaves of Homalium pronyense Guillaum. are biogenically-derived from spermine and the appropriate α,β-unsaturated fatty or cinnamic acids.1,2 These unique bis-azalactams have been the targets of a number of synthetic efforts over the past three decades.1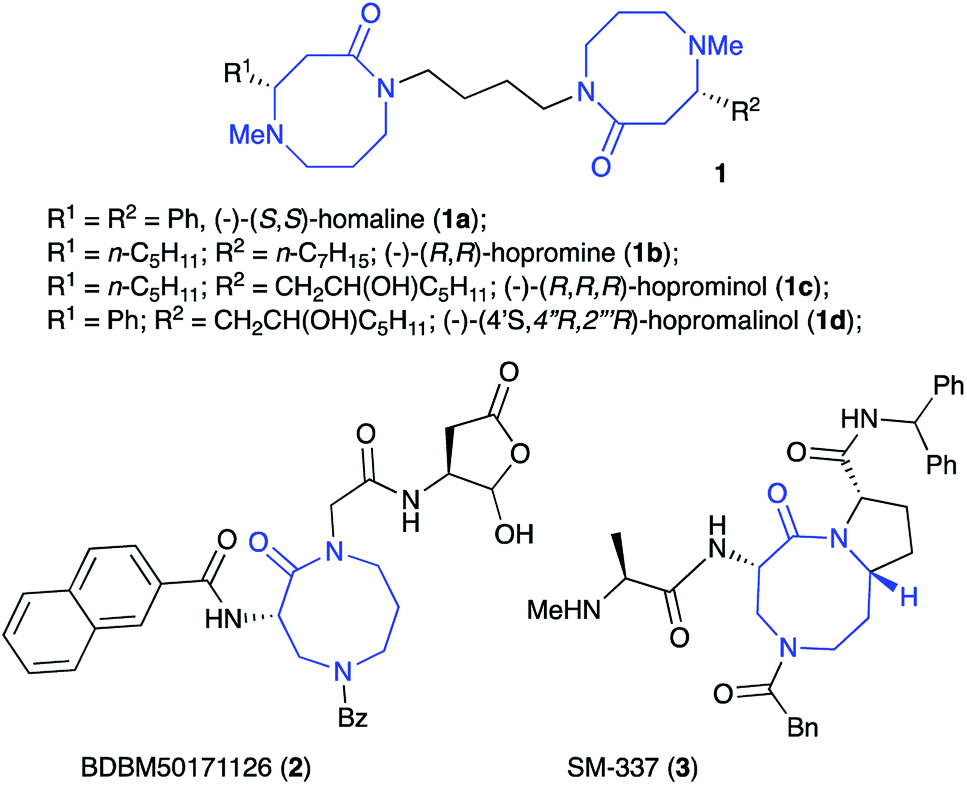 | ||
| Fig. 1 Biologically active 1,5-diazocan-2-ones. | ||
The 1,5-diazocin-2-one core has recently been exploited in new therapeutic agents (Fig. 1). For example, diazocan peptomimetic BDBM50171126 (2) exhibits high levels of activity as a selective caspase-1 inhibitor. Compounds in this class has shown promising anti-inflammatory and analgesic activity in animal models for the treatment of rheumatoid arthritis.3,4 Compound SM-337 (3) belongs to a family of conformationally-constrained mimetics of the endogenous IAP antagonist Smac. Over the past decade, Smac mimetics have garnered increasing attention showing great potential as a new class of antitumor drugs.5,6
It is known that medium-sized 8-membered rings are difficult to assemble via conventional methods of cyclization,7 largely due to the enthalpic cost incurred in the transition state as well as the decreased entropy of the cyclic products relative to their linear precursors.8 Several alternative synthetic approaches to the eight-membered 1,5-diazocin-2-one core have been developed, including Beckman rearrangement,9 fragmentation of 1,5-diazabicyclo[3.3.1]nonan-2-ones,10 reductive N–N scission of tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazol-1-ones,11–14 as well as various ring closures, exploiting intramolecular versions of reductive amination,15–18 amine acylation,7,19–22 strain release-driven transamidation,23–25 and SNAr reactions.26 Herein, we disclose a new application of metal-templated intramolecular 8-exo-trig cyclization involving nucleophilic addition of sulfonamides tethered to a cyclopropene moiety.
Results and discussion
We have recently reported on the key role of potassium cations in the chelation-controlled cyclization of cyclopropenes 4 linked to nucleophilic alkoxide moieties.27–29 This innovative approach allows for a highly efficient and diastereoselective assembly of cyclic ethers 5 with ring sizes 7 to 10 (Scheme 1).27–29 These bicyclic structures produced new, highly selective anti-mycobacterial agents.29 Closely related diazepinones and diazecanones 7 demonstrated antitumor activity. These species could be accessed via the potassium-templated cyclization of cyclopropenes 6 with tethered carbamates (Scheme 1).30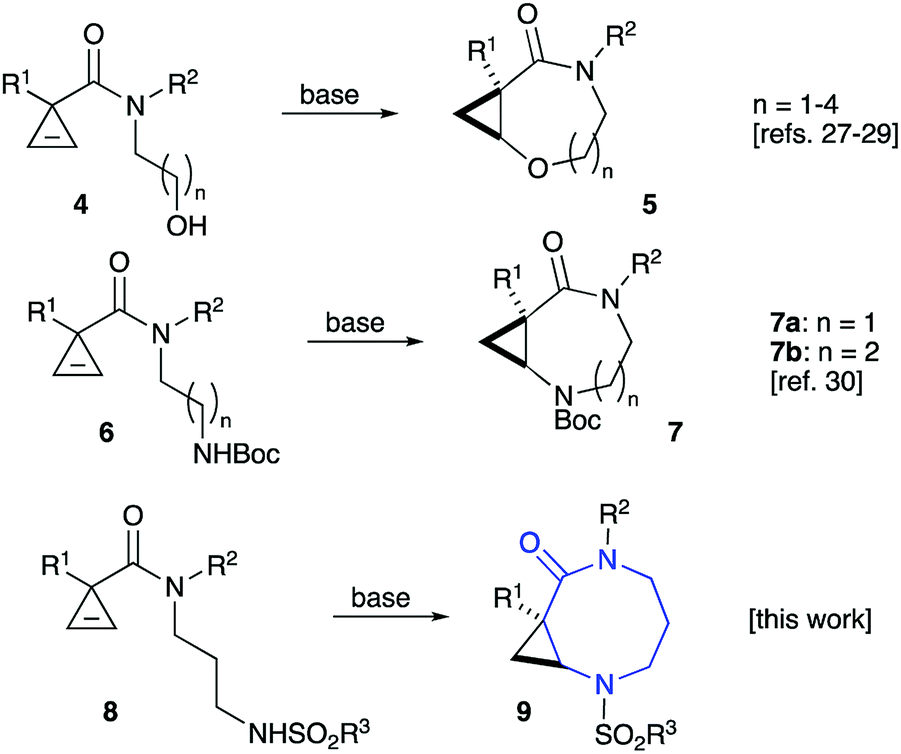 | ||
| Scheme 1 | ||
In order to carry out more focused and complete SAR studies, we wanted to gain access to analogs within scaffold 7 by replacing the Boc protecting group with a range of substituents. This study is focused on the synthesis of sulfonamides 9, which give rise to three points of diversity. It should be pointed out, that a scope of substituents R1 and R2 as well as ring size in such bicyclic scaffolds was already previously investigated.29,30 The current study is focused on evaluating the activating properties of different sulfonyl protecting groups. We also briefly examined the use of these sulfonamides as chiral auxiliaries.
The diversity-oriented approach to 9 was to rely on the late-stage functionalization of cyclic secondary amine 10, which was to be accessible via simple acid-assisted deprotection of routinely available carbamates 7b (Scheme 2). Removal of the Boc group moderating the electronic density at the N-2 resulted in intermediate cyclopropane 10 undergoing facile ring cleavage and subsequent decomposition via cyclic imine 11 (Scheme 2). The intermolecular version of this small ring cleavage reaction allowed for the expeditious access of GABA amides.31 Based on the decomposition of 10 via 11 to other species, the access of sulfonamides 9, ultimately required the installation of the sulfonyl group prior to the cyclization step.
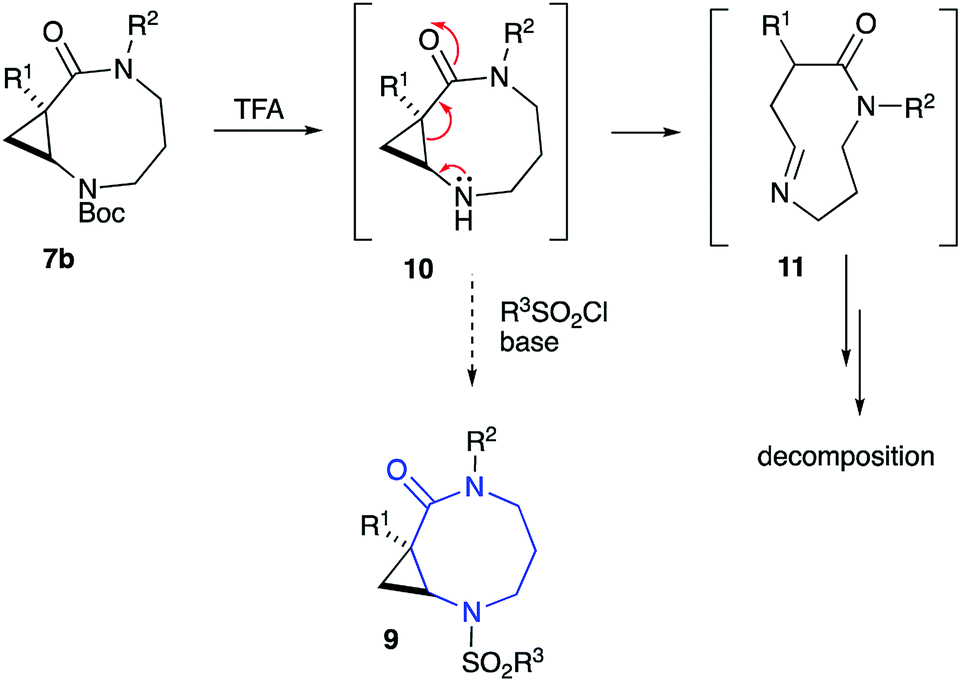 | ||
| Scheme 2 | ||
Primary amine hydrochloride 15 was envisioned to serve as a common precursor to the linear sulfonamides series 8. To this end, readily available cyclopropene-3-carboxylic acid 1232,33 was employed in acylation of tert-butyl (3-(benzylamino)propyl)carbamate hydrochloride (13)34 to afford amide 14. The carbamate protecting group in the latter was removed via treatment with anhydrous HCl in dichloromethane to provide the desired salt 15 in good overall yield (Scheme 3). Next, the series of sulfonamides 8a–h was prepared by treating 15 with the corresponding sulfonyl chlorides in the presence of base. These reactions proceeded uneventfully, affording moderate to good yields (Scheme 3). It should be pointed out that the signals of the two different rotamers present in NMR spectra of all amides 8 complicated spectral analysis. However, all these materials were chromatographically pure and perfectly suitable for further transformation. Subsequently, sulfonamides 8 were treated with freshly ground powdered KOH in anhydrous THF at 50 °C, as these reaction conditions were previously shown to be optimal for the cyclization of carbamates.30 Gratifyingly, most of sulfonamides tested (8a–f) underwent the reaction smoothly affording the corresponding products 9a–f in high yields. The reaction seems to be very tolerant to steric hindrance at the sulfonyl group. Indeed, only minor reduction in yield was observed in the formation of product 9c bearing the bulky 2,5-xylyl group. An attempt to employ sulfonamides bearing electron-withdrawing substituents revealed a somewhat more serious limitation of this methodology. As expected, the N–H bond in these sulfonamides is much more acidic, but the corresponding conjugate base is significantly less nucleophilic. Such negative trend between acidity of the nucleophilic reagents and their effective nucleophilicity was previously demonstrated for a related base-assisted reaction of cyclopropenes with phenols.35,36 Evidently, the same tendency exists for the reaction of sulfonamides. Indeed, a notable reduction of the yield of bicyclic product 9f was observed in cyclization of N-brosylate 8f, whereas nosylate 8g reacted very sluggishly, and the corresponding product 9g was formed only in marginal yield (Scheme 4).
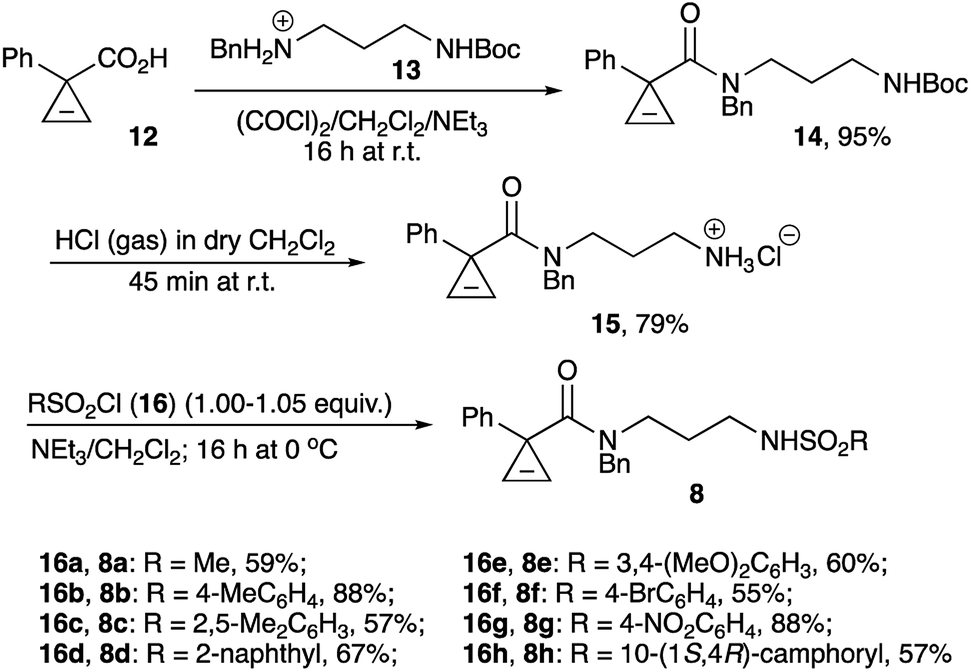 | ||
| Scheme 3 | ||
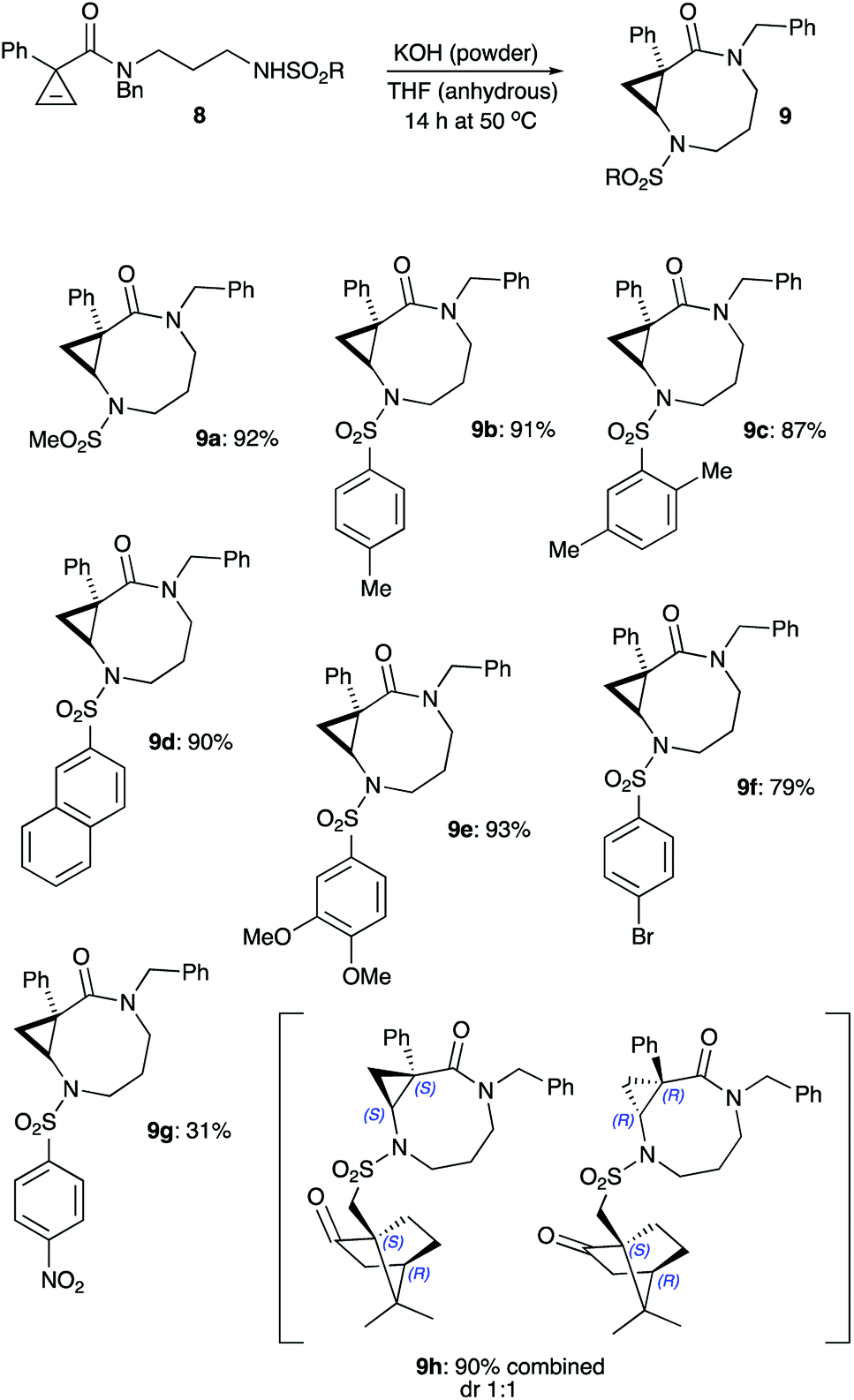 | ||
| Scheme 4 | ||
Also, the possibility to carry out diastereoselective cyclization employing the 10-(4S,1R)-camphorsulfonyl group as a chiral auxiliary was evaluated. To this end, camphorsulfonamide 8h was cyclized under the standard reaction conditions. The reaction proceeded smoothly, affording a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomeric products 9h. Evidently, asymmetric induction in this case was highly inefficient.
:1 mixture of diastereomeric products 9h. Evidently, asymmetric induction in this case was highly inefficient.
Conclusion
The utilization of various sulfonyls as activating groups in the cation-templated 8-exo-trig nucleophilic additions of amines across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of cyclopropenes was assessed. We demonstrated that most of the sulfonyls provide excellent yields of the corresponding eight-membered cyclic products. Electron-deficient sulfonamides, such as nosylate, afforded reduced yields, showing a consistent limitation of the featured methodology. The possibility to carry out diastereoselective cyclization employing 10-(4S,1R)-camphorsulfonyl as a chiral auxiliary was evaluated. This reaction, however, afforded a 1:1 mixture of diastereomeric cyclic product, demonstrating the total inefficiency of such an approach.
C bond of cyclopropenes was assessed. We demonstrated that most of the sulfonyls provide excellent yields of the corresponding eight-membered cyclic products. Electron-deficient sulfonamides, such as nosylate, afforded reduced yields, showing a consistent limitation of the featured methodology. The possibility to carry out diastereoselective cyclization employing 10-(4S,1R)-camphorsulfonyl as a chiral auxiliary was evaluated. This reaction, however, afforded a 1:1 mixture of diastereomeric cyclic product, demonstrating the total inefficiency of such an approach.
Experimental part
General information
NMR spectra were recorded on a Bruker Avance DRX-500 (500 MHz) with a dual carbon/proton cryoprobe (CPDUL). 13C NMR spectra were registered with broadband decoupling. The (+) and (−) designations represent positive and negative intensities of signals in 13C DEPT-135 experiments. IR spectra were measured on a ThermoFisher Nicolet™ iS™ 5 FT-IR Spectrometer. HRMS was carried out on an LCT Premier (Micromass Technologies) instrument employing ESI TOF detection techniques. Glassware used in moisture-free syntheses was flame-dried under vacuum prior to use. Column chromatography was carried out on silica gel (Sorbent Technologies, 40–63 μm). Pre-coated silica gel plates (Sorbent Technologies Silica XG 200 μm) were used for TLC analysis. Anhydrous CH2Cl2, THF, and triethylamine were each prepared by refluxing commercially available solvent over CaH2 followed by distillation under a stream of dry nitrogen at ambient pressure and stored over 3 Å molecular sieves under dry nitrogen. Anhydrous DMSO and DMF were prepared by stirring commercial solvents over CaH2 at 100 °C and 80 °C, respectively, followed by distillation under reduced pressure. Thus, obtained dry solvents were stored over 3 Å molecular sieves under dry nitrogen. All other reagents, unless otherwise specified, were used in their commercially-available forms and purities. All manipulations of powdered KOH were conducted under inert atmosphere (<8 ppm residual oxygen and moisture) using a combination of glovebox and standard Schlenk techniques.tert-Butyl (3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propyl)carbamate (14)
A flame-dried round bottom flask was charged with 1-phenylcycloprop-2-ene-1-carboxylic acid (12) (760 mg, 4.74 mmol, 1.00 equiv.), anhydrous dichloromethane (21 mL), and DMF (3 drops) under nitrogen atmosphere. Oxalyl chloride (623 μL, 903 mg, 7.12 mmol, 1.50 equiv.) was then added dropwise and the mixture was stirred at room temperature for 2 h. Volatiles were removed under reduced pressure to provide the crude acyl chloride. To a flame-dried flask containing tert-butyl (3-(benzylamino)propyl)carbamate hydrochloride (13) (1.65 g, 5.48 mmol, 1.16 equiv.), triethylamine (2.00 mL, 1.44 g, 14.2 mmol, 3.00 equiv.), and anhydrous dichloromethane (9.2 mL) was added dropwise a solution of the crude acyl chloride in anhydrous dichloromethane (6.1 mL). The reaction mixture was stirred for 16 hours at RT. The mixture was diluted with dichloromethane (40 mL) and washed with 1 N HCl (3 × 30 mL). The combined aqueous layers were back-extracted with dichloromethane (1 × 30 mL). The combined organic layers were then washed with brine (1 × 20 mL), dried with MgSO4, filtered, and concentrated under reduced pressure. The product was purified by column chromatography on silica gel eluting with CH2Cl2/EtOAc (7:1) to afford the title compound as a pale yellow oil (1.83 g, 4.50 mmol, 95%); Rf 0.36 (CH2Cl2/EtOAc, 7:1); NMR spectra indicate the presence of two rotamers (ratio of 2.6:1): 1H NMR (500 MHz, CDCl3) δ ppm [7.35 (s) & 7.33–7.12 (m) & 7.13 (d, J = 7.1 Hz) & 7.09 (s) & 6.94 (d, J = 6.9 Hz), Σ12H], [5.45 (t, J = 6.3 Hz) & 4.61 (s) & 4.49 (s) & 4.27 (m), Σ3H], [3.37 (t, J = 6.7 Hz) & 3.23 (m), Σ2H], [3.13 (q, J = 6.3 Hz) & 2.79 (q, J = 6.5 Hz), Σ2H], [1.66 (p, J = 6.5 Hz) & 1.41 (m), Σ11H]; 13C NMR (126 MHz CDCl3) δ ppm 175.1, 174.4, 156.2, 155.9, 143.3, 143.0, 137.6, 137.0, 128.9 (+), 128.8 (+), 128.7 (+), 128.6 (+), 128.3 (+), 127.7 (+), 127.5 (+), 126.9 (+), 126.8 (+), 126.7 (+), 126.4 (+), 126.0 (+), 110.4 (+), 109.9 (+), 79.4, 79.1, 51.3 (−), 47.6 (−), 44.6 (−), 42.1 (−), 38.0 (−), 37.7 (−), 32.3, 32.1, 28.6 (+), 28.5 (+), 28.5 (−), 27.6 (−); FT-IR (NaCl, cm−1): 3334, 3085, 3061, 3028, 2976, 2931, 1708, 1623, 1514, 1495, 1452, 1425, 1365, 1273, 1250, 1171, 997, 737, 700, 654, 605; HRMS (TOF ES): found 429.2149, calculated for C25H30N2O3Na [M + Na]+ 429.2154 (1.2 ppm).
3-(N-Benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15)
Gaseous hydrogen chloride was bubbled through a solution of (3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propyl)carbamate (14) (589 mg, 1.45 mmol) in dichloromethane (25 mL) while stirring at rt. The reaction was allowed to proceed until TLC analysis indicated consumption of the protected amine (45 min). Volatiles were removed under reduced pressure. The resultant solid was triturated with diethyl ether and collected via vacuum filtration to afford the title compound as a white crystalline solid (390 mg, 1.14 mmol, 79%); mp 89.1 °C (decomposed); NMR spectra indicate the presence of two rotamers (ratio of 1.4:1): 1H NMR (500 MHz, DMSO-d6) δ [7.93 (s) & 7.89 (s) & 7.62 (s), Σ5H], [7.38–7.17 (m) & 7.12–7.05 (m) & 7.01–6.98 (m), Σ10H], [4.54 (s) & 4.52 (s), Σ2H], [3.26 (t, J = 7.9 Hz) & 3.21 (t, J = 7.2 Hz), Σ2H], [2.71 (q, J = 6.6 Hz) & 2.54 (q, J = 6.4 Hz), Σ2H], [1.78 (p, J = 7.4 Hz) & 1.72–1.64 (m), Σ2H]; FT-IR (NaCl, cm−1): 3122, 3084, 2817, 2788, 2712, 1590, 1531, 1441, 1430, 1367, 1242, 738, 709, 696, 662, 537; HRMS (TOF ES): found 307.1827, calculated for C20H23N2O (M+) 307.1810 (5.5 ppm).
N-Benzyl-N-(3-((3,4-dimethoxyphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8e): typical procedure 1
An oven-dried 5 mL V-Vial equipped with a magnetic spin vane was charged with 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (94 mg, 0.274 mmol, 1.00 equiv.), dichloromethane (1.5 mL), and triethylamine (115 μL, 83 mg, 0.822 mmol, 3.00 equiv.). The solution was cooled to 0 °C and 3,4-dimethoxybenzenesulfonyl chloride (16e) (68.0 mg, 0.287 mmol, 1.05 equiv.) was added in a single portion. The reaction mixture was stirred at 0 °C for 2 hours. The reaction mixture was then stirred for an additional 16 hours at RT. The reaction mixture was diluted with dichloromethane (20 mL) and washed successively with 1 M HCl (2 × 6 mL), 5% NaHCO3 (2 × 6 mL), water (2 × 6 mL), and brine (1 × 8 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The product was purified by column chromatography on silica gel eluting with CH2Cl2/EtOAc (3:1) to afford the title compound as a thick, colorless oil (83 mg, 0.164 mmol, 60%); Rf = 0.26 (CH2Cl2/EtOAc, 3:1); NMR spectra indicate the presence of two rotamers (ratio of 8:1): 1H NMR (500 MHz, CDCl3) δ ppm [7.52 (dd, J = 8.4, 2.1 Hz) & 7.39 (d, J = 2.1 Hz) & 7.38–7.17 (m) & 7.09 (s) & 6.97 (dd, J = 6.9, 2.8 Hz) & 6.93–6.88 (m), Σ15H], [6.08 (t, J = 5.5 Hz) & 4.59 (s) & 4.44 (s), 3.94 (s) & 3.93 (s) & 3.90 (s) & 3.60 (t, J = 5.9 Hz), Σ9H], [3.37 (t, J = 6.2 Hz) & 3.33–3.23 (m), Σ2H], [2.92 (q, J = 5.8 Hz) & 2.55 (q, J = 6.3 Hz), Σ2H], [1.62 (p, J = 6.2 Hz) & 1.37–1.32 (m), Σ2H]; 13C NMR (126 MHz CDCl3) δ ppm 175.6, 152.4, 149.2, 142.7, 136.4, 132.3, 129.0 (+), 128.8 (+), 128.7 (+), 128.2 (+), 127.9 (+), 126.9 (+), 126.9 (+), 126.7 (+), 126.0 (+), 121.1 (+), 110.6 (+), 110.1 (+), 109.9 (+), 100.1, 56.4 (+), 56.3 (+), 51.2 (−), 47.7 (−), 44.4 (−), 41.4 (−), 40.1 (−), 32.0, 29.9 (+), 27.3 (+). FT-IR (NaCl, cm−1): 3269, 3148, 3103, 3061, 2934, 2856, 1613, 1509, 1443, 1325, 1262, 1237, 1182, 1153, 1095, 1021, 765, 701, 578; HRMS (TOF ES): found 529.1759, calculated for C28H30N2O5SNa [M + Na]+ 529.1773 (2.6 ppm).
N-Benzyl-N-(3-(methylsulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8a)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (167 mg, 0.487 mmol, 1.00 equiv.), triethylamine (204 μL, 148 mg, 1.46 mmol, 3 equiv.), and methanesulfonyl chloride (16a) (40 μL, 57 mg, 0.511 mmol, 1.05 equiv.) to yield the title compound as a thick, colorless oil (110 mg, 0.286 mmol, 59%); Rf = 0.29 (CH2Cl2/EtOAc, 2:3); NMR spectra indicate the presence of two rotamers (ratio of 6.9:1): 1H NMR (500 MHz, CDCl3) δ [7.42 (s) & 7.39–7.22 (m), Σ8H], [7.17–7.12 (m) & 6.99–6.94 (m), Σ3H], [5.85 (s) & 4.65 (s) & 4.55 (s), Σ3H], [3.54 (t, J = 6.1 Hz) & 3.47 (t, J = 6.2 Hz) & 3.38–3.32 (m) & 3.15 (t, J = 6.1 Hz), Σ4H], [3.07–3.01 (m) & 2.97 (s) & 2.80 (s) & 2.77–2.71 (m), Σ3H], [1.73 (p, J = 6.1 Hz) & 1.46 (p, J = 6.8 Hz), Σ2H]; 13C NMR (126 MHz, CDCl3) δ 175.6, 174.4, 143.2, 142.7, 137.4, 136.3, 129.2 (+), 128.9 (+), 128.8 (+), 128.7 (+), 128.6 (+), 128.1 (+), 127.8 (+), 126.9 (+), 126.8 (+), 126.6 (+), 126.3 (+), 125.9 (+), 110.3 (+), 110.0 (+), 51.4 (−), 47.6 (−), 44.3 (−), 41.6 (−), 40.5 (−), 40.4 (+), 40.2 (−), 40.0 (+), 32.2, 31.9, 28.4 (−), 28.0 (−); FT-IR (NaCl, cm−1): 3259, 3062, 3030, 2932, 1721, 1623, 1453, 1319, 1150, 1079, 975, 735, 702, 521; HRMS (TOF ES): found 407.1414, calculated for C21H24N2O3SNa [M + Na]+ 407.1405 (2.2 ppm).
N-Benzyl-N-(3-((4-methylphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8b)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (163 mg, 0.475 mmol, 1.00 equiv.), triethylamine (199 μL, 144 mg, 1.43 mmol, 3.00 equiv.), and 4-methylbenzenesulfonyl chloride (16b) (95 mg, 0.499 mmol, 1.05 equiv.) to yield the title compound as a thick, colorless oil (193 mg, 0.419 mmol, 88%). Rf = 0.32 (CH2Cl2/EtOAc, 6:1); NMR spectra indicate the presence of two rotamers (ratio of 5.6:1): 1H NMR (500 MHz, CDCl3) δ ppm [7.79 (d, J = 8.2 Hz) & 7.61 (d, J = 8.0 Hz), Σ2H], [4.78 (s) & 7.32–7.16 (m) & 7.11 (s) & 7.01–6.95 (m) & 6.90 (d, J = 6.9 Hz) & 6.06 (t, J = 6.6 Hz), Σ14H], [6.06 (t, J = 6.6 Hz) & 4.57 (s) & 4.43 (s) & 3.74 (t, J = 6.2 Hz), Σ3H], [3.36 (t, J = 6.2 Hz) & 3.26 (m), Σ2H], [2.91 (q, J = 6.3 Hz) & 2.54 (q, J = 6.2 Hz), Σ2H], [2.42 (s), 2.40 (s), 1.60 (p, J = 6.1 Hz) & 1.34 (p, J = 6.7 Hz), Σ2H]; 13C NMR (126 MHz CDCl3) δ ppm 175.6, 143.1, 142.8, 129.9 (+), 129.8 (+), 129.0 (+), 128.8 (+), 128.7 (+), 128.7 (+), 128.2 (+), 127.9 (+), 127.5 (+), 127.3 (+), 127.1 (+), 127.0 (+), 126.8 (+), 126.7 (+), 126.6 (+), 126.0 (+), 110.4 (+), 110.2 (+), 51.2 (−), 47.6 (−), 44.4 (−), 41.4 (−), 40.5 (−), 40.1 (−), 32.0 (−), 28.0 (−), 27.3 (−), 21.6 (+); FT-IR (KBr, cm−1): 3147, 3103, 3061, 3028, 2925, 2869, 1612, 1445, 1426, 1207, 1152, 1093, 815, 738, 700, 658, 551; HRMS (TOF ES): found 483.1736, calculated for C27H28N2O3SNa [M + Na]+ 483.1718 (3.7 ppm).
N-Benzyl-N-(3-((2,5-dimethylphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8c)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (115 mg, 0.274 mmol, 1.00 equiv.), triethylamine (114 μL, 83 mg, 0.821 mmol, 3.00 equiv.), and 2,5-dimethylbenzenesulfonyl chloride (16c) (59 mg, 0.287 mmol, 1.05 equiv.) to yield the title compound as a thick, colorless oil (73.9 mg, 0.156 mmol, 57%); Rf = 0.35 (6:1, CH2Cl2/EtOAc); NMR spectra indicate the presence of two rotamers (ratio of 5.9:1): 1H NMR (500 MHz, CDCl3) δ [7.79 (d, J = 1.9 Hz) & 7.67 (s), Σ1H], [7.36 (s) & 7.32–7.15 (m) & 7.13 (s) & 7.07–7.03 (m) & 6.93–6.89 (m), Σ14H], [6.09 (t, J = 6.8 Hz) & 4.58 (s) & 4.44 (s) & 3.77 (t, J = 6.3 Hz), Σ3H], [3.38 (t, J = 6.2 Hz) & 3.30–3.23 (m), Σ2H], [2.94 (q, J = 6.3 Hz) & 2.67 (s) & 2.51 (q, J = 6.5 Hz) & 2.47 (s) & 2.36 (s), Σ8H], [1.57 (p, J = 6.1 Hz) & 1.33 (p, J = 6.7 Hz), Σ2H]; 13C NMR (126 MHz, CDCl3) δ 175.6, 174.5, 142.8, 138.4, 136.5, 136.3, 136.0, 135.5, 134.2, 133.7 (+), 133.2 (+), 132.6 (2C, (+)), 130.1 (+), 129.8 (+), 129.6, 129.0 (+), 128.8 (2C, (+)), 128.7 (+), 128.2 (+), 127.9 (+), 127.5 (+), 127.0 (+), 126.9 (+), 126.8 (+), 126.7 (+), 126.1 (+), 110.4 (+), 110.2 (+), 51.2 (−), 47.6 (−), 44.4 (−), 41.4 (−), 40.3 (−), 39.9 (−), 32.4, 32.1, 28.0 (−), 27.6 (−), 21.0 (+), 20.0 (+), 19.9 (+); FT-IR (NaCl, cm−1): 3142, 3100, 3053, 3028, 2927, 2869, 1614, 1451, 1427, 1207, 1151, 1095, 816, 738, 701, 682, 655, 594; HRMS (TOF ES): found 497.1872, calculated for C28H30N2O3SNa [M + Na]+ 497.1875 (0.6 ppm).
N-Benzyl-N-(3-(naphthalene-2-sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8d)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (115 mg, 0.335 mmol, 1.00 equiv.), triethylamine (140 μL, 102 mg, 1.00 mmol, 3.00 equiv.), and naphthalene-2-sulfonyl chloride (16d) (84 mg, 0.369 mmol, 1.05 equiv.) to yield the title compound as a thick, colorless oil (112 mg, 226 mmol, 67%). Rf = 0.33 (CH2Cl2/EtOAc, 6:1); NMR spectra indicate the presence of two rotamers (ratio of 5.6:1): 1H NMR (500 MHz, CDCl3) δ [8.46 (s) & 8.32 (s), Σ2H], [7.97–7.86 (m) & 7.71–7.54 (m), Σ6H], [7.34–7.21 (m) & 7.19–6.99 (m) & 6.95–6.92 (m) & 6.87 (dd, J = 7.4, 2.1 Hz), Σ12H], [6.30 (t, J = 6.7 Hz) 4.56 (s) & 4.40 (s) & 3.83 (t, J = 6.5 Hz), Σ3H], [3.37 (t, J = 6.2 Hz) & 3.28–3.22 (m), Σ2H], [2.95 (q, J = 6.0 Hz) & 2.57 (q, J = 6.4 Hz), Σ2H], [1.59 (p, J = 6.1 Hz) & 1.34 (p, J = 6.7 Hz), Σ2H]; 13C NMR (126 MHz, CDCl3) δ 175.7, 142.6, 137.5, 136.4, 134.9, 132.4, 129.7 (+), 129.5 (+), 129.4 (+), 129.3 (+), 129.2 (+), 129.1 (+), 129.0 (+), 128.9 (+), 128.7 (2C, (+)), 128.6 (+), 128.5 (+), 128.3 (+), 128.3 (+), 128.2 (+), 128.0 (+), 127.9 (+), 127.5 (+), 127.1 (+), 127.0 (+), 126.8 (+), 126.7 (+), 126.4 (+), 126.0 (+), 122.8 (+), 122.3 (+), 110.4 (+), 110.2 (+), 51.2 (−), 47.6 (−), 44.4 (−), 41.4 (−), 40.6 (−), 40.1 (−), 32.4, 32.0, 28.0 (−), 27.3 (−); FT-IR (NaCl, cm−1): 3276, 3149, 3105, 3058, 3029, 2935, 2872, 1611, 1494, 1425, 1328, 1267, 1157, 1131, 1076, 818, 735, 700, 616, 550, 478; HRMS (TOF ES): found 519.1734, calculated for C30H28N2O3SNa [M + Na]+ 519.1718 (3.1 ppm).
N-Benzyl-N-(3-((4-bromophenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8f)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (122 mg, 0.356 mmol, 1.00 equiv.), triethylamine (149 μL, 108 mg, 1.07 mmol, 3.00 equiv.), and 4-bromobenzenesulfonyl chloride (16f) (95 mg, 0.374 mmol, 1.05 equiv.) to yield the title compound as a colorless oil (102 mg, 0.194 mmol, 55%); Rf = 0.28 (hexanes/EtOAc/MeOH, 7:2:1); NMR spectra indicate the presence of two rotamers (ratio of 7.9:1): 1H NMR (500 MHz, CDCl3) δ ppm [7.77–7.73 (m) & 7.63–7.55 (m), Σ4H], [7.36 (s) & 7.32–7.14 (m) & 7.11 (s) & 7.01–6.96 (m) & 6.91–6.87 (m), Σ12H], [6.37 (t, J = 6.6 Hz) & 4.57 (s) & 4.45 (s) & 4.19 (t, J = 6.4 Hz), Σ3H], [3.36 (t, J = 6.2 Hz) & 3.30–3.23 (m), Σ2H], [2.90 (q, J = 6.1 Hz) & 2.53 (q, J = 6.3 Hz), Σ2H], [1.60 (p, J = 6.1 Hz) & 1.37 (p, J = 6.6 Hz), Σ2H]; 13C NMR (126 MHz CDCl3) δ ppm 175.9, 174.9, 143.1, 142.5, 139.6, 138.8, 137.2, 136.1, 132.5 (+), 132.4 (+), 129.0 (+), 128.9 (+), 128.8 (+), 128.8 (+), 128.7 (+), 128.6 (+), 128.2 (+), 128.0 (+), 127.6 (+), 127.0 (+), 126.8 (+), 126.6 (+), 125.9 (+), 110.3 (+), 110.1 (+), 51.4 (−), 47.7 (−), 44.4 (−), 41.6 (−), 40.5 (−), 40.1 (−), 32.2, 31.9, 27.9 (−), 27.3 (−); FT-IR (NaCl, cm−1): 3262, 3149, 3105, 3062, 3029, 2933, 2872, 1612, 1576, 1494, 1426, 1357, 1163, 1010, 823, 736, 700, 654, 605, 562; HRMS (TOF ES): found 547.0645, calculated for C26H25BrN2O3SNa [M + Na]+ 547.0667 (4.0 ppm).
N-Benzyl-N-(3-((4-nitrophenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8g)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (94 mg, 0.274 mmol, 1.00 equiv.), triethylamine (115 μL, 83 mg, 0.822 mmol, 3.00 equiv.), and 4-nitrobenzenesulfonyl chloride (16g) (64 mg, 0.288 mmol, 1.05 equiv.) to yield the title compound as a thick, colorless oil (112 mg, 0.228 mmol, 83%); Rf = 0.29 (CH2Cl2/EtOAc, 6:1); NMR spectra indicate the presence of two rotamers (ratio of 14.5:1): 1H NMR (500 MHz, CDCl3) δ [8.29 (d, J = 8.9 Hz) & 8.08 (d, J = 8.8 Hz) & 7.89 (d, J = 8.5 Hz), Σ4H], [7.38 (s) & 7.32–7.26 (m) & 7.25–7.17 (m) & 7.12 (s) & 6.98 (dd, J = 7.5, 2.1 Hz) & 6.91–6.88 (m) & 6.82 (t, J = 6.5 Hz) & Σ15H], [4.59 (s) & 4.47 (s), Σ2H], [3.39 (t, J = 6.1 Hz) & 3.32–3.27 (m), Σ2H], [2.96 (q, J = 6.1 Hz) & 2.64–2.54 (m), Σ2H], [1.61 (p, J = 6.1 Hz) & 1.44–1.37 (m), Σ2H]; 13C NMR (126 MHz, CDCl3) δ 176.0, 150.0, 146.6, 142.5, 136.0, 129.1 (+), 128.8 (+), 128.5 (+), 128.1 (+), 127.0 (+), 127.0 (+), 125.8 (+), 124.3 (+), 110.1 (+), 51.5 (−), 41.4 (−), 40.2 (−), 31.9, 27.4 (−); FT-IR (NaCl, cm−1): 3145, 3103, 3064, 3029, 2933, 2866, 1608, 1529, 1445, 1426, 1207, 1152, 1093, 855, 737, 700, 610, 554; HRMS (TOF ES): found 514.1413, calculated for C26H25N3O5SNa [M + Na]+ 514.1418 (1.0 ppm).
N-Benzyl-N-(3-((((1S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptan-1-yl)methyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8h)
The compound was prepared according to typical procedure 1 employing 3-(N-benzyl-1-phenylcycloprop-2-ene-1-carboxamido)propan-1-aminium chloride (15) (250 mg, 0.729 mmol, 1 equiv.), triethylamine (305 μL, 221 mg, 2.19 mmol, 3.00 equiv.), and (1S)-(+)-10-camphorsulfonyl chloride (16h) (201 mg, 0.802 mmol, 1.10 equiv.) to yield the title compound as a thick, pale yellow oil (215 mg, 0.413 mmol, 57%); Rf = 0.31 (CH2Cl2/EtOAc, 3:1); NMR spectra indicate the presence of two rotamers (ratio of 3:1): 1H NMR (500 MHz, CDCl3) δ [7.41 (d, J = 4.9 Hz, 1H), 7.36–7.23 (m), 7.20 (dd, J = 9.2, 5.0 Hz), 7.17–7.09 (m), 6.98–6.94 (m), Σ12H], [5.82 (t, J = 6.5 Hz), 4.74 (t, J = 6.2 Hz), 4.69–4.57 (m), 4.52 (m), Σ3H], [δ 3.52–3.27 (m), 3.23 (d, J = 15.2 Hz), 3.18 (q, J = 6.4 Hz), 2.91 (d, J = 15.0 Hz), 2.86–2.73 (m), 2.40 (p, J = 3.3, 2.9 Hz), 2.36 (t, J = 3.8 Hz), 2.22–2.12 (m), 2.11 (t, J = 4.6 Hz), 2.04 (tq, J = 12.2, 4.2 Hz), 1.92 (d, J = 18.6 Hz), 1.84 (ddd, J = 14.3, 9.4, 4.8 Hz), 1.76 (tt, J = 10.6, 5.2 Hz), 1.46 (dddd, J = 29.4, 13.0, 7.8, 3.7 Hz), Σ15H], [1.07 (s), 1.00 (s), Σ3H], [0.89 (s), 0.88 (s), Σ3H]; 13C NMR (126 MHz, CDCl3) δ 217.3, 216.5, 175.3, 174.6, 143.3, 143.0, 137.6, 136.8, 129.0 (+), 128.8 (+), 128.7 (+), 128.7 (+), 128.3 (+), 127.8 (+), 127.5 (+), 127.0 (+), 126.8 (+), 126.7 (+), 126.6 (+), 126.1 (+), 110.8 (+), 110.2 (+), 110.0 (+), 109.9 (+), 59.3, 59.0, 51.5 (−), 49.3 (−), 49.2 (−), 49.0, 48.6, 47.7 (−), 44.6 (−), 43.1 (−), 43.0 (−), 42.9 (+), 42.0 (−), 41.2 (−), 40.9 (−), 32.3, 32.1, 29.8 (−), 28.6 (−), 28.2 (−), 27.2 (−), 26.7 (−), 25.9 (−), 20.0 (+), 20.0 (+), 19.9 (+), 19.6 (+). FT-IR (NaCl, cm−1): 3281, 3205, 3148, 3103, 3060, 3028, 2958, 2887, 1743, 1645, 1618, 1446, 1424, 1329, 1146, 1067, 736, 700, 607, 567; HRMS (TOF ES): found 543.2292, calculated for C30H36N2O4SNa [M + Na]+ 543.2294 (0.4 ppm).
(1S*,8S*)-6-Benzyl-2-((3,4-dimethoxyphenyl)sulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9e): typical procedure 2
A 5 mL oven-dried V-Vial equipped with a magnetic spin vane was charged with N-benzyl-N-(3-((4-methylphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8e) (19.5 mg, 0.038 mmol, 1.00 equiv.), freshly-ground potassium hydroxide (4.3 mg, 0.077 mmol, 2.00 equiv.), and dry THF (800 μL). The reaction mixture was stirred for 14 h at 50 °C. Then, the reaction mixture was cooled to room temperature and passed through a short plug of silica eluting with EtOAc. The filtrate was concentrated in vacuum. The resulting crude product was purified by column chromatography on silica gel eluting with hexanes/EtOAc (2:3) to afford the title compound as a colorless solid (18.1 mg, 0.036 mmol, 93%); Rf 0.35 (hexanes/EtOAc, 2:3); mp 194–198 °C; 1H NMR (500 MHz, CDCl3) δ ppm 7.47 (dd, J = 8.4, 2.1 Hz, 1H), 7.34–7.17 (m, 9H), 7.12 (dd, 7.2, 1.7 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 5.31 (d, J = 14.8 Hz, 1H), 4.16 (dt, J = 14.0, 3.6 Hz, 1H), 4.04 (d, J = 14.9 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 3.68 (dd, J = 15.7, 10.9 Hz, 1H), 3.05 (dd, J = 15.6, 6.3 Hz, 1H), 2.77–2.65 (m, 3H), 1.95 (dtd, J = 15.8, 11.5, 4.6 Hz, 1H), 1.60–1.50 (m, 1H), 1.33 (p, J = 5.6 Hz, 1H); 13C NMR (126 MHz CDCl3) δ ppm 169.3, 153.9, 149.3, 138.7, 137.5, 129.5, 129.0 (+), 128.7 (+), 128.4 (+), 127.6 (+), 127.2 (+), 125.5 (+), 121.7 (+), 110.9 (+), 110.4 (+), 56.5 (+), 56.4 (+), 53.5 (−), 49.3 (−), 46.6 (+), 46.0 (−), 28.3 (−), 23.9 (−); FT-IR (NaCl, cm−1): 3060, 3027, 2965, 2934, 2848, 1641, 1587, 1509, 1441, 1346, 1263, 1140, 1020, 733, 702, 573; HRMS (TOF ES): found 529.1778, calculated for C28H30N2O5SNa [M + Na]+ 529.1773 (0.9 ppm).
(1S*,8S*)-6-Benzyl-2-(methylsulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9a)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-(methylsulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8a) (32.7 mg, 0.085 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (9.5 mg, 0.170 mmol, 2.00 equiv.) to yield the title compound as a colorless crystalline solid (30.2 mg, 0.079 mmol, 92%). Rf 0.23 (hexane/EtOAc/MeOH, 6:3:1, 0.2% TFA); mp 129–131 °C (decomposed); 1H NMR (500 MHz, CDCl3) δ 7.35–7.23 (m, 8H), 7.19–7.15 (m, 2H), 5.28 (d, J = 15.4 Hz, 1H), 4.19 (dt, J = 14.2, 3.8 Hz, 1H), 4.11 (d, J = 14.7 Hz, 1H), 3.79 (dd, J = 15.5, 11.0 Hz, 1H), 3.17–3.11 (m, 2H), 3.05 (ddd, J = 15.0, 12.4, 3.0 Hz, 1H), 2.93 (s, 3H), 2.59 (dd, J = 7.1, 5.3 Hz, 1H), 2.00–1.87 (m, 1H), 1.67–1.57 (m, 1H), 1.49 (t, J = 7.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 170.0, 138.2, 137.0, 129.2 (+), 128.9 (+), 128.6 (+), 127.9 (+), 127.4 (+), 125.4 (+), 52.9 (−), 49.6 (−), 46.6 (+), 46.3 (−), 38.2 (+), 36.6, 28.2 (−), 23.4 (−); FT-IR (NaCl, cm−1): 3083, 3060, 2923, 2907, 2850, 1701, 1638, 1446, 1350, 1167, 823, 745, 700, 614, 580, 542; HRMS (TOF ES): found 407.1388, calculated for C21H24N2O3SNa [M + Na]+ 407.1405 (4.2 ppm).
(1S*,8S*)-6-Benzyl-8-phenyl-2-tosyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9b)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-((2,5-dimethylphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8b) (25.0 mg, 0.054 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide freshly-ground potassium hydroxide (6.1 mg, 0.109 mmol, 2.00 equiv.) to yield the title compound as a white solid (22.8 mg, 0.049 mmol, 91%); Rf 0.36 (hexanes/EtOAc, 3:2); mp 172–174 °C; 1H NMR (500 MHz, CDCl3) δ ppm 7.74 (d, J = 8.3 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.33–7.16 (m, 8H), 7.12 (dd, J = 7.1, 1.8 Hz, 1H), 5.31 (d, J = 14.8 Hz, 1H), 4.16 (dt, J = 13.9, 3.4 Hz, 1H), 4.06 (d, J = 14.8 Hz, 1H), 3.67 (dd, J = 15.6, 11.0 Hz, 1H), 3.06 (dd, J = 15.6, 6.3 Hz, 1H), 2.74–2.62 (m, 3H), 2.46 (s, 2H), 1.97 (dtd, J = 15.7, 11.9, 4.6 Hz, 1H), 1.55 (ddt, J = 14.8, 5.6, 2.4 Hz, 1H), 1.36 (dd, J = 7.1, 6.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 169.3, 144.1, 138.8, 137.6, 134.8, 130.1 (+), 129.0 (+), 128.8 (+), 128.5 (+), 127.9 (+), 127.7 (+), 127.2 (+), 125.6 (+), 53.6 (−), 49.4 (−), 46.8 (+), 46.1, 36.7, 28.4 (−), 23.9 (−), 21.8 (+); FT-IR (NaCl, cm−1): 3061, 3030, 2961, 2924, 2855, 1641, 1598, 1495, 1479, 1442, 1425, 1380, 1344, 1165, 1129, 1090, 816, 734, 712, 699, 563, 551, 541; HRMS (TOF ES): found 483.1721, calculated for C27H28N2O3SNa [M + Na]+ 483.1718 (0.6 ppm).
(1S*,8S*)-6-Benzyl-2-((2,5-dimethylphenyl)sulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9c)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-((2,5-dimethylphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8c) (28.3 mg, 0.060 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (6.7 mg, 119 mmol, 2.00 equiv.) to yield the title compound as a colorless crystalline solid (24.7 mg, 0.052 mmol, 87%); Rf 0.33 (hexanes/EtOAc/MeOH, 7:2:1); mp 182–185 °C; 1H NMR (500 MHz, CDCl3) δ ppm 7.86 (d, J = 2.0 Hz, 1H), 7.35–7.19 (m, 10H), 7.16 (dd, J = 7.4, 1.8 Hz, 2H), 5.43 (d, J = 14.7 Hz, 1H), 4.37 (dt, J = 14.6, 3.8 Hz, 1H), 3.90 (d, J = 14.7 Hz, 1H), 3.74 (dd, J = 15.7, 11.0 Hz, 1H), 3.16 (dd, J = 8.0, 5.4 Hz, 1H), 3.11–2.99 (m, 2H), 2.59 (s, 3H), 2.41 (s, 3H), 1.98–1.83 (m, 2H), 1.68–1.58 (m, 2H), 1.19 (dd, J = 8.0, 6.7 Hz, 1H); 13C NMR (126 MHz CDCl3) δ ppm 169.4, 138.6, 137.3, 136.6, 136.2, 135.3, 134.4 (+), 133.0 (+), 131.1 (+), 129.0 (+), 128.8 (+), 128.5 (+), 127.7 (+), 127.1 (+), 125.5 (+), 51.7 (−), 48.9 (−), 46.3 (+), 45.7 (−), 36.5, 28.0 (−), 23.3 (−), 21.0 (+), 19.9 (+); FT-IR (NaCl, cm−1): 3060, 3029, 2923, 1641, 1494, 1441, 1323, 1156, 821, 735, 713, 701, 588; HRMS (TOF ES): found 497.1876, calculated for C28H30N2O3SNa [M + Na]+ 497.1875 (0.2 ppm).
(1S*,8S*)-6-Benzyl-2-(naphthalen-1-ylsulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9d)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-(naphthalene-2-sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8d) (26.7 mg, 0.054 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (6.0 mg, 0.108 mmol, 2.00 equiv.) to yield the title compound as a colorless crystalline solid (23.9 mg, 0.048 mmol, 90%); Rf 0.38 (hexanes/EtOAc/MeOH, 7:2:1); mp 185–190 °C (decomposed); 1H NMR (500 MHz, CDCl3) δ 8.34 (d, J = 1.8 Hz, 1H), 7.92 (t, J = 7.8 Hz, 2H), 7.88–7.84 (m, 1H), 7.75 (dd, J = 8.7, 1.8 Hz, 1H), 7.57 (dddd, J = 19.9, 8.0, 6.9, 1.3 Hz, 2H), 7.24–7.05 (m, 8H), 7.01–6.97 (m, 2H), 5.22 (d, J = 14.8 Hz, 1H), 4.16 (dt, J = 14.0, 3.8 Hz, 1H), 3.97 (d, J = 14.8 Hz, 1H), 3.58 (dd, J = 15.7, 11.0 Hz, 1H), 2.96 (dd, J = 15.6, 6.3 Hz, 1H), 2.75–2.54 (m, 3H), 1.99–1.78 (m, 1H), 1.51–1.44 (m, 1H), 1.31 (dd, J = 7.9, 6.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 169.2, 138.6, 137.5, 135.1, 134.8, 132.4, 129.6 (+), 129.5 (+), 129.2 (+), 129.2 (+), 129.0 (+), 128.8 (+), 128.5 (+), 128.2 (+), 127.9 (+), 127.6 (+), 127.2 (+), 125.5 (+), 122.9 (+), 53.7 (−), 49.3 (−), 46.7 (+), 46.0 (−), 36.8, 28.4 (−), 23.9 (−); FT-IR (NaCl, cm−1): 3060, 3041, 2965, 2855, 1641, 1598, 1447, 1442, 1425, 1360, 1344, 1165, 1090, 816, 734, 712, 699, 655, 563, 551, 541; HRMS (TOF ES): found 519.1748, calculated for C30H28N2O3SNa [M + Na]+ 519.1718 (5.8 ppm).
(1S*,8S*)-6-Benzyl-2-((4-bromophenyl)sulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9f)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-((4-bromophenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8f) (27.9 mg, 0.053 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (6.0 mg, 0.11 mmol, 2.00 equiv.) to yield the title compound as a colorless crystalline solid (22.0 mg, 0.042 mmol, 79%); Rf 0.25 (hexanes/EtOAc/MeOH, 7:2:1); mp 151 °C (decomposed); 1H NMR (500 MHz, CDCl3) δ 7.77–7.67 (m, 4H), 7.35–7.18 (m, 8H), 7.14–7.10 (m, 2H), 5.29 (d, J = 14.8 Hz, 1H), 4.18–4.11 (m, 1H), 4.09 (d, J = 14.8 Hz, 1H), 3.69 (dd, J = 15.7, 11.0 Hz, 1H), 3.08 (dd, J = 15.6, 6.3 Hz, 1H), 2.78–2.71 (m, 1H), 2.71–2.65 (m, 2H), 2.03–1.93 (m, 1H), 1.61–1.54 (m, 1H), 1.37 (dd, J = 6.9, 5.7 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 169.3, 138.3, 137.3, 136.8, 132.7 (+), 129.3 (+), 129.1 (+), 128.8 (+), 128.5 (+), 128.4, 127.7 (+), 127.3 (+), 125.5 (+), 53.6 (−), 49.4 (−), 46.5 (−), 46.0 (−), 36.8, 29.9, 28.3 (−), 23.7 (−); FT-IR (NaCl, cm−1): 3087, 3061, 2920, 2850, 1703, 1640, 1445, 1349, 1167, 822, 745, 700, 609, 561; HRMS (TOF ES): found 547.0660, calculated for C26H25BrN2O3SNa [M + Na]+ 547.0667 (1.3 ppm).
(1S*,8S*)-6-Benzyl-2-((4-nitrophenyl)sulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9g)
The compound was prepared according typical procedure 2 employing N-benzyl-N-(3-((4-nitrophenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8g) (26.2 mg, 0.053 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (6.0 mg, 0.107 mmol, 2.00 equiv.). The reaction mixture was stirred for 24 hours at 50 °C. The target compound was obtained as a white crystalline solid (8.2 mg, 0.016 mmol, 31%); Rf 0.26 (hexanes/EtOAc/MeOH, 6:3:1) mp 145 °C (decomposed); 1H NMR (500 MHz, CDCl3) δ 8.43 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 7.36–7.18 (m, 8H), 7.17–7.08 (m, 2H), 5.25 (d, J = 14.8 Hz, 1H), 4.22–4.16 (m, 1H), 4.12 (d, J = 14.8 Hz, 1H), 3.72 (dd, J = 15.7, 11.0 Hz, 1H), 3.10 (dd, J = 15.7, 6.2 Hz, 1H), 2.91–2.77 (m, 1H), 2.77–2.63 (m, 2H), 2.02–1.91 (m, 1H), 1.63–1.57 (m, 1H), 1.39 (t, J = 6.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 169.0, 150.6, 143.9, 138.2, 137.3, 129.1 (+), 129.0 (+), 128.8 (+), 128.5 (+), 127.7 (+), 127.5 (+), 125.5 (+), 124.7 (+), 53.6 (−), 49.6 (−), 46.3, 46.0 (−), 37.0, 28.4 (−), 23.5 (−); FT-IR (NaCl, cm−1): 3052, 2926, 2852, 1701, 1640, 1530, 1446, 1377, 1350, 1163, 853, 737, 700, 609, 591; HRMS (TOF ES): found 514.1422, calculated for C26H25N3O5SNa [M + Na]+ 514.1413 (1.8 ppm).
(1S*,8S*)-6-Benzyl-2-((((1S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptan-1-yl)methyl)sulfonyl)-8-phenyl-2,6-diazabicyclo[6.1.0]nonan-7-one (9h)
The compound was prepared according to typical procedure 2 employing N-benzyl-N-(3-((3,4-dimethoxyphenyl)sulfonamido)propyl)-1-phenylcycloprop-2-ene-1-carboxamide (8h) (32.9 mg, 0.063 mmol, 1.00 equiv.) and freshly-ground potassium hydroxide (7.1 mg, 0.126 mmol, 2.00 equiv.) to afford the title compound as an inseparable mixture of diastereomers (∼1:1) as a colorless solid (29.5 mg, 0.057 mmol, 90%); Rf 0.25 (hexanes/EtOAc/MeOH, 6:3:1); mp 92–103 °C; 1H NMR (500 MHz, CDCl3) δ 7.27–7.10 (m, 20H), 5.21 (dd, J = 22.0, 14.7 Hz, 2H), 4.10 (ddt, J = 19.2, 16.6, 3.6 Hz, 2H), 4.03 (d, J = 14.8 Hz, 1H), 3.97 (d, J = 14.8 Hz, 1H), 3.70 (ddd, J = 15.5, 10.9, 4.2 Hz, 2H), 3.41 (d, J = 14.6 Hz, 1H), 3.33 (d, J = 14.5 Hz, 1H), 3.27 (dd, J = 8.2, 5.3 Hz, 1H), 3.17 (dd, J = 8.3, 5.4 Hz, 1H), 3.09 (ddd, J = 15.0, 12.4, 3.1 Hz, 1H), 3.06–2.97 (m, 3H), 2.90 (d, J = 14.6 Hz, 1H), 2.76 (d, J = 14.6 Hz, 1H), 2.67 (dd, J = 6.7, 5.3 Hz, 1H), 2.55 (dd, J = 7.0, 5.4 Hz, 1H), 2.47 (dddd, J = 21.6, 14.8, 11.8, 4.0 Hz, 2H), 2.35 (q, J = 4.0 Hz, 1H), 2.32 (q, J = 4.0 Hz, 1H), 2.06 (q, J = 4.3 Hz, 2H), 2.00 (tq, J = 12.2, 4.4 Hz, 2H), 1.91 (d, J = 5.2 Hz, 1H), 1.87 (d, J = 5.3 Hz, 1H), 1.62 (dddd, J = 14.0, 9.3, 7.1, 4.7 Hz, 4H), 1.50 (dddd, J = 11.3, 8.6, 6.4, 3.1 Hz, 2H), 1.42–1.33 (m, 4H), 1.09 (s, 3H), 1.07 (s, 3H), 0.85 (d, J = 2.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 215.8, 215.8, 169.5, 169.3, 138.8, 138.8, 137.6, 137.5, 129.0 (+), 128.7 (2C, (+)), 128.5 (+), 127.6 (2C, (+)), 127.1 (+), 125.5 (+), 125.4 (+), 58.8, 58.7, 53.7 (−), 53.1 (−), 49.4 (−), 49.1 (−), 48.4, 48.1, 46.7 (−), 46.6 (+), 46.3 (+), 46.2 (−), 46.0 (−), 43.0 (+), 42.9 (+), 42.8 (−), 36.7, 36.5, 28.6 (−), 28.3 (−), 27.1 (2C, (−)), 25.5 (2C, (−)), 23.6 (−), 23.3 (−), 20.2 (+), 20.1 (+), 20.0 (+); FT-IR (NaCl, cm−1): 3060, 3028, 2961, 1745, 1640, 1496, 1480, 1342, 1052, 757, 699, 562, 526. HRMS (TOF ES): found 543.2291, calculated for C30H36N2O4SNa [M + Na]+ 543.2294 (0.6 ppm).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the grant from the Ministry of Education and Science of the Russian Federation (grant #0795-2020-0031).Notes and references
- S. G. Davies and J. E. Thomson, Alkaloids: Chem. Biol. Perspect., 2015, 74, 121–158 Search PubMed.
- L. Crombie, R. C. F. Jones and D. Haigh, Tetrahedron Lett., 1986, 27, 5147–5150 Search PubMed.
- K. A. Oppong, C. D. Ellis, M. C. Laufersweiler, S. V. O'Neil, Y. Wang, D. L. Soper, M. W. Baize, J. A. Wos, B. De, G. K. Bosch, A. N. Fancher, W. Lu, M. K. Suchanek, R. L. Wang and T. P. Demuth, Bioorg. Med. Chem. Lett., 2005, 15, 4291–4294 Search PubMed.
- K. Rudolphi, N. Gerwin, N. Verzijl, d. K. P. van and d. B. W. van, Osteoarthritis Cartilage, 2003, 11, 738–746 Search PubMed.
- Y. Peng, H. Sun, Z. Nikolovska-Coleska, S. Qiu, C.-Y. Yang, J. Lu, Q. Cai, H. Yi, S. Kang, D. Yang and S. Wang, J. Med. Chem., 2008, 51, 8158–8162 Search PubMed.
- R. Sheng, H. Sun, L. Liu, J. Lu, D. McEachern, G. Wang, J. Wen, P. Min, Z. Du, H. Lu, S. Kang, M. Guo, D. Yang and S. Wang, J. Med. Chem., 2013, 56, 3969–3979 Search PubMed.
- G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 Search PubMed.
- L. Mandolini, Adv. Phys. Org. Chem., 1986, 22, 1–111 Search PubMed.
- P. Pigeon and B. Decroix, J. Heterocycl. Chem., 1997, 34, 375–380 Search PubMed.
- M. Denzer and H. Ott, J. Org. Chem., 1969, 34, 183–187 Search PubMed.
- J. A. Martinez-Perez, M. A. Pickel, E. Caroff and W.-D. Waggon, Synlett, 1999, 1875–1878 Search PubMed.
- R. G. Sherrill, Tetrahedron Lett., 2007, 48, 7053–7056 Search PubMed.
- M. Yoshida, N. Sassa, T. Kato, S. Fujinami, T. Soeta, K. Inomata and Y. Ukaji, Chem.–Eur. J., 2014, 20, 2058–2064 Search PubMed.
- T. M. T. Tong, T. Soeta, T. Suga, K. Kawamoto, Y. Hayashi and Y. Ukaji, J. Org. Chem., 2017, 82, 1969–1976 Search PubMed.
- Y. Peng, H. Sun and S. Wang, Tetrahedron Lett., 2006, 47, 4769–4770 Search PubMed.
- Q. Cai, H. Sun, Y. Peng, J. Lu, Z. Nikolovska-Coleska, D. McEachern, L. Liu, S. Qiu, C.-Y. Yang, R. Miller, H. Yi, T. Zhang, D. Sun, S. Kang, M. Guo, L. Leopold, D. Yang and S. Wang, J. Med. Chem., 2011, 54, 2714–2726 Search PubMed.
- Y. Peng, H. Sun, J. Lu, L. Liu, Q. Cai, R. Shen, C.-Y. Yang, H. Yi and S. Wang, J. Med. Chem., 2012, 55, 106–114 Search PubMed.
- H. Sun, J. Lu, L. Liu, C.-Y. Yang and S. Wang, ACS Chem. Biol., 2014, 9, 994–1002 Search PubMed.
- Y. Hirokawa, H. Yamazaki and S. Kato, J. Heterocycl. Chem., 2002, 39, 727–731 Search PubMed.
- C. Ensch and M. Hesse, Helv. Chim. Acta, 2002, 85, 1659–1673 Search PubMed.
- E. Aiello, G. Dattolo, G. Cirrincione, A. M. Almerico and I. D'Asdia, J. Heterocycl. Chem., 1981, 18, 1153–1155 Search PubMed.
- I. Stansfield, C. Ercolani, A. Mackay, I. Conte, M. Pompei, U. Koch, N. Gennari, C. Giuliano, M. Rowley and F. Narjes, Bioorg. Med. Chem. Lett., 2009, 19, 627–632 Search PubMed.
- L. Crombie, R. C. F. Jones and D. Haigh, Tetrahedron Lett., 1986, 27, 5151–5154 Search PubMed.
- L. Crombie, D. Haigh, R. C. F. Jones and A. R. Mat-Zin, J. Chem. Soc., Perkin Trans. 1, 1993, 2047–2054 Search PubMed.
- L. Crombie, D. Haigh, R. C. F. Jones and A. R. Mat-Zin, J. Chem. Soc., Perkin Trans. 1, 1993, 2055–2068 Search PubMed.
- S. Seto, A. Tanioka, M. Ikeda and S. Izawa, Bioorg. Med. Chem., 2005, 13, 5717–5732 Search PubMed.
- B. K. Alnasleh, M. Rubina and M. Rubin, Chem. Commun., 2016, 52, 7494–7496 Search PubMed.
- P. Ryabchuk, J. P. Matheny, M. Rubina and M. Rubin, Org. Lett., 2016, 18, 6272–6275 Search PubMed.
- V. A. Maslivetc, D. N. Turner, K. N. McNair, L. Frolova, S. Rogelj, A. A. Maslivetc, N. A. Aksenov, M. Rubina and M. Rubin, J. Org. Chem., 2018, 83, 5650–5664 Search PubMed.
- V. A. Maslivetc, L. V. Frolova, S. Rogelj, A. A. Maslivetc, M. Rubina and M. Rubin, J. Org. Chem., 2018, 83, 13743–13753 Search PubMed.
- V. A. Maslivetc, M. Rubina and M. Rubin, Org. Biomol. Chem., 2015, 13, 8993–8995 Search PubMed.
- A. Edwards, M. Rubina and M. Rubin, Chem.–Eur. J., 2018, 24, 1394–1403 Search PubMed.
- L.-a. Liao, N. Yan and J. M. Fox, Org. Lett., 2004, 6, 4937–4939 Search PubMed.
- P. Thanigaimalai, K.-C. Lee, S.-C. Bang, J.-H. Lee, C.-Y. Yun, E. Roh, B.-Y. Hwang, Y. Kim and S.-H. Jung, Bioorg. Med. Chem., 2010, 18, 1135–1142 Search PubMed.
- J. E. Banning, A. R. Prosser, B. K. Alnasleh, J. Smarker, M. Rubina and M. Rubin, J. Org. Chem., 2011, 76, 3968–3986 Search PubMed.
- J. E. Banning, A. R. Prosser and M. Rubin, Org. Lett., 2010, 12, 1488–1491 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09014j |
| This journal is © The Royal Society of Chemistry 2020 |