Tungsten–zirconia-supported rhenium catalyst combined with a deoxydehydration catalyst for the one-pot synthesis of 1,4-butanediol from 1,4-anhydroerythritol†
Received
4th March 2020
, Accepted 11th May 2020
First published on 11th May 2020
Abstract
Efficient and reusable catalysts were developed for one-pot reduction of 1,4-anhydroerythritol (1,4-AHERY), which is a promising biomass-derived C4 platform chemical, into 1,4-butanediol (1,4-BuD) with H2. First, various ReOx catalysts on oxide supports were tested for reductive conversion of 2,5-dihydrofuran (2,5-DHF) to 1,4-butanediol (1,4-BuD). ReOx/WO3–ZrO2 showed the best performance, and ReOx catalysts supported on other oxides were much less active in the isomerization of 2,5-DHF to 2,3-DHF which is the first step of the 2,5-DHF conversion to 1,4-BuD. The ReOx/WO3–ZrO2 catalyst was combined with the ReOx–Au/CeO2 catalyst for deoxydehydration (DODH) of 1,4-AHERY into 2,5-DHF to develop a one-pot conversion system of 1,4-AHERY to 1,4-BuD. The highest 1,4-BuD yield of 55% from 1,4-AHERY was obtained. Even though the yield was lower than that obtained over the combination of ReOx/C and ReOx–Au/CeO2 catalysts in our previous study, the regeneration of the combination of ReOx/WO3–ZrO2 and ReOx–Au/CeO2 is possible: calcination at 573 for 3 h of the used catalyst mixture increased the activity to the fresh level. THF was the major by-product in both 1,4-AHERY and 2,5-DHF conversions, which was due to hydrogenation of DHF, disproportionation of DHF and/or dehydration of 1,4-BuD. The W amount in WO3–ZrO2 greatly affected the catalytic performance of ReOx/WO3–ZrO2: too much W above the monolayer level on the ZrO2 support sharply decreased the activity in 2,5-DHF isomerization. On the other hand, WO3–ZrO2 with a tetragonal ZrO2 structure prepared by co-precipitation showed comparable performance to WO3–ZrO2 with a monoclinic ZrO2 structure as the support of the ReOx catalyst, demonstrating that the crystal structure of ZrO2 has little effect on the catalytic performance. The Re species were suggested to be highly dispersed on the WO3 (sub)monolayer on ZrO2 based on the effect of the Re loading amount. The dispersed Re species on monolayer WO3 species on ZrO2 can be the active sites for 2,5-DHF disproportionation to 2,3-DHF.
Introduction
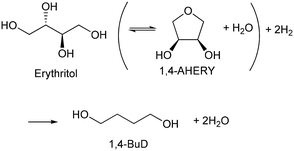
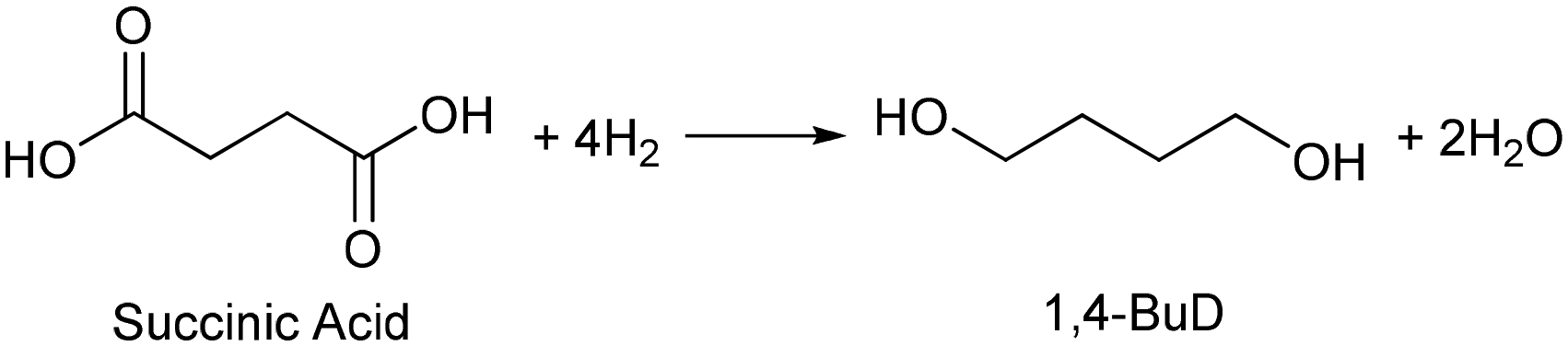
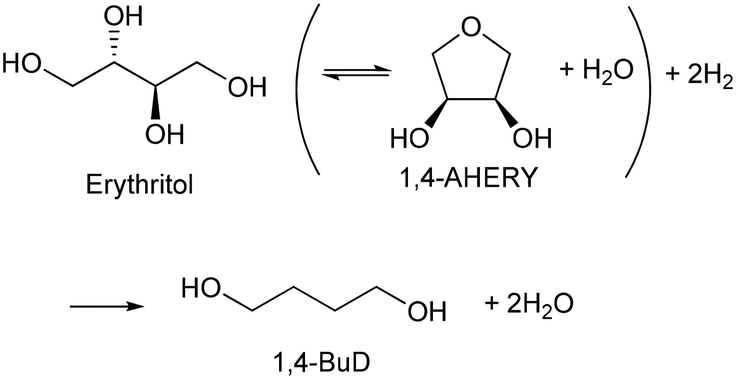
Biomass plays an increasingly significant role as a chemical feedstock and energy resource instead of fossil fuels due to the depletion of oil and natural gas. Although biomass-derived products are more sustainable and renewable, most of them are not convenient and economical as feedstocks for energy generation or the chemical industry due to the high oxygen content.1–5 Catalytic conversions such as selective C–O hydrogenolysis (dissociation of the C–O bond and addition of hydrogen atoms from H2) of biomass-based chemicals can effectively reduce the oxygen numbers and increase their commodity value. Sugar alcohols, such as glycerol, erythritol, xylitol, and sorbitol, are biomass-derived compounds widely used as sweeteners instead of sugar in the food industry. They can be produced on the industrial scale and have large feedstock supply. As platform chemicals, sugar alcohols can be converted to value-added chemicals by selectively removing OH groups,6 for example, glycerol can be converted to 1,3-propanediol or 1,2-propanediol with high selectivity.7–10 However, selective oxygen removal from erythritol, xylitol and sorbitol with a longer chain than glycerol is more difficult because the selectivity to the target product is low due to the large number of isomers of products and the co-production of deeply deoxygenated products, degraded products and/or ring products. Erythritol is manufactured as a low-calorie sweetener and is a potential platform chemical in biomass conversion.11 Erythritol hydrodeoxygenation may produce mixture of butanetriols, butanediols, butanol, and butane.12–14 Among the products from erythritol, 1,4-butanediol (1,4-BuD) is an important non-natural chemical for synthesis of tetrahydrofuran (THF), γ-butyrolactone (GBL) and pharmaceuticals, as well as engineering plastics such as polyesters, polyurethanes, and polyethers.15–17 The current manufacturing method of 1,4-BuD in industry is mainly the refinery of petroleum-based chemicals such as maleic anhydride for industrial scale production.15 Biomass-based production of 1,4-BuD from succinic acid was also reported.15,18,19 However, the production of 1,4-BuD from erythritol and its dehydrated product 1,4-anhydroerythritol (1,4-AHERY) lacks investigation because of the difficulty in selective removal of oxygen. Only 25% yield of 1,4-BuD could be obtained by stepwise hydrogenolysis of erythritol over an Ir–ReOx/SiO2 catalyst12 which is an effective catalyst in glycerol hydrogenolysis to 1,3-propanediol.20–22 Based on the consumption of H2 in production of 1,4-BuD, the transformation from erythritol or 1,4-AHERY consumes half the amount of H2 compared with that from succinic acid (eqn (1) and (2)). Therefore, the production of 1,4-BuD from erythritol is worth improving.| | 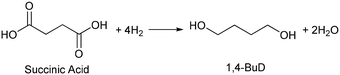 | (1) |
| |  | (2) |
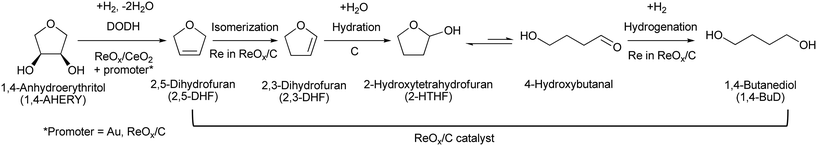
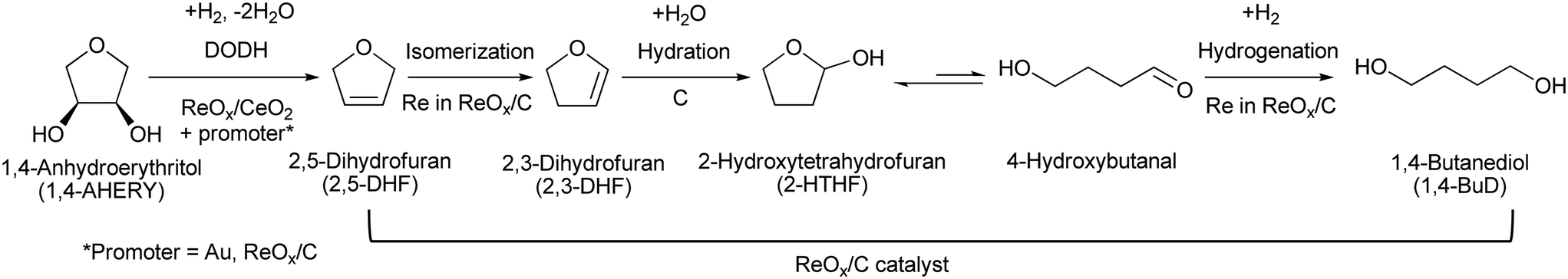
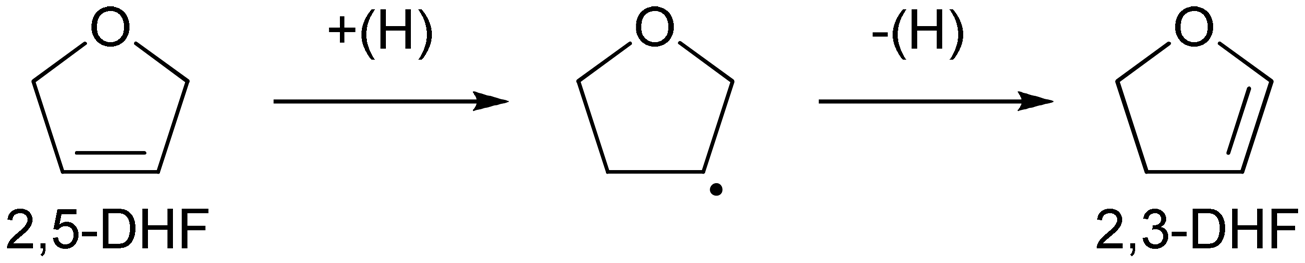
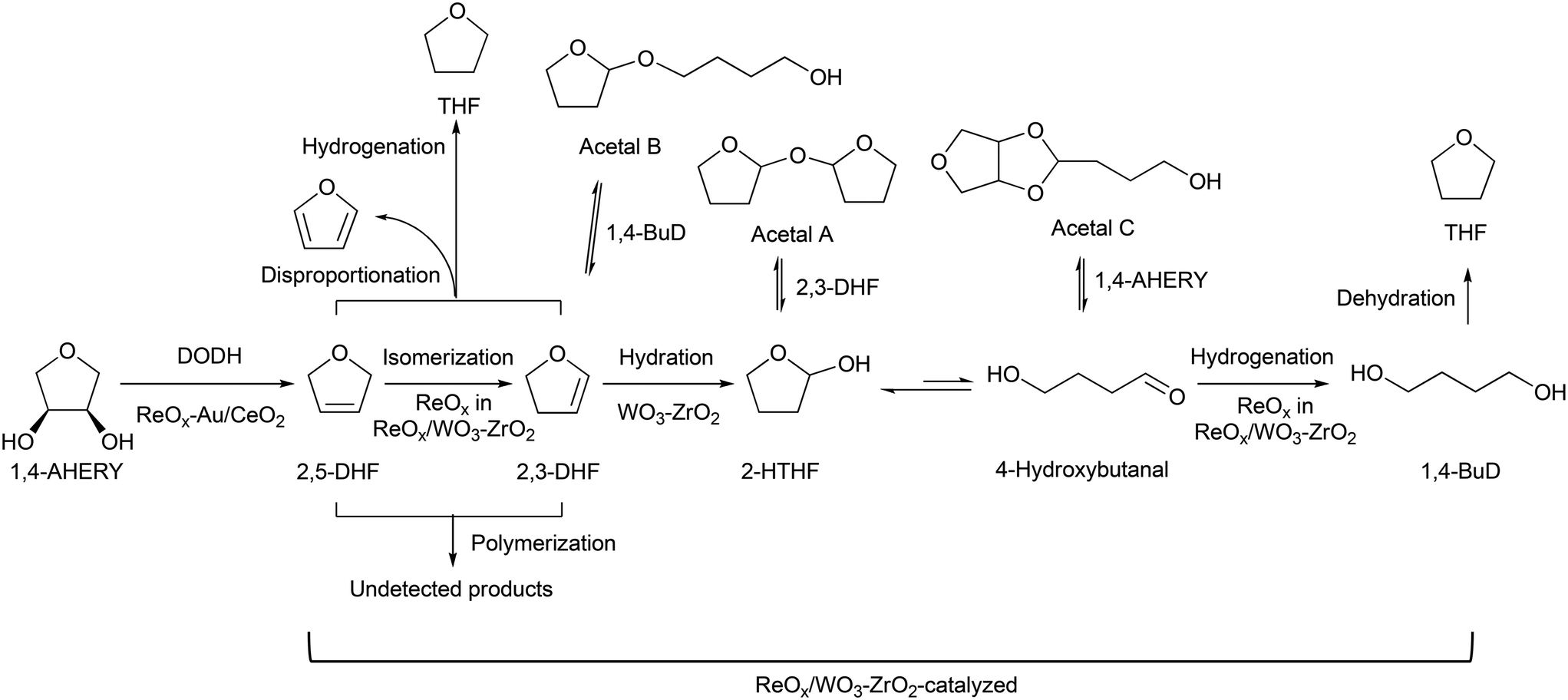
Recently, we have reported that 1,4-BuD could be produced from 1,4-AHERY over the combination of ReOx/C and ReOx–Au/CeO2 catalysts in a one-pot reaction.23 The yield of 1,4-BuD was around 90% which was the highest yield of 1,4-BuD from biomass-derived products so far even including succinic acid. In this co-catalyst system, ReOx–Au/CeO2 firstly catalyzed the reduction of 1,4-AHERY to 2,5-dihydrofuran (2,5-DHF), and ReOx/C catalyzed the further reduction of 2,5-DHF to 1,4-BuD (Scheme 1). The former step is the conversion of cis-vicinal OH groups to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond which is called deoxydehydration (DODH), and this reaction is typically catalyzed by homogeneous Re(VII) catalysts with organic reducing agents such as 3-pentanol and PPh3.24–26 Development of solid catalysts for DODH has been a hot topic in biomass conversion.27–31 Our laboratory has developed a heterogeneous ReOx–Au/CeO2 catalyst for DODH using H2 as the reducing agent.32–35 The highly dispersed monomeric Re oxide species on the crystalline CeO2 surface can be the active sites of DODH.32,33,36–38 Au serves as the activation site of H2, and the activated hydrogen species move on CeO2 probably as pairs of protons and electrons to Re species. Au was selected among various noble metals based on its low CC hydrogenation activity.32 The stabilization of high valent Re species by CeO2 and the capability of hydrogen species to move on the CeO2 surface are the keys of the DODH activity. The latter step catalyzed by ReOx/C consists of the following steps: the isomerization of 2,5-DHF to 2,3-dihydrofuran (2,3-DHF), the hydration of 2,3-DHF to 2-hydroxytetrahydrofuran (2-HTHF), and the reduction of 2-HTHF or its straight chain form 4-hydroxybutanal to 1,4-BuD. In the most recent research, we also found that the ReOx/CeO2 catalyst without an Au promoter still could catalyze the DODH reaction in the presence of ReOx/C, enabling the reduction of 1,4-AHERY to 1,4-butanediol with the mixture of ReOx/CeO2 and ReOx/C.39 The Re species on carbon could activate H2 like an Au promoter for the reduction of Re species on the CeO2 surface which are the active sites in the conversion of 1,4-AHERY. Furthermore, CeO2 without Re loading combined with ReOx/C even could convert 1,4-AHERY to 1,4-BuD, because the Re species on the carbon surface could move to CeO2 surfaces to form ReOx/CeO2. The highest yield of 1,4-BuD reached 85% over the ReOx/CeO2 + ReOx/C system, which was comparable to that in the co-catalyst system with an Au promoter. As above, ReOx/C plays many roles in this system: CC bond movement (isomerization), H2 activation, hydrolysis and CO hydrogenation. Versatile roles of ReOx/C have been also reported in other catalytic reactions with multiple steps such as reductive lignin depolymerization.40,41
C double bond which is called deoxydehydration (DODH), and this reaction is typically catalyzed by homogeneous Re(VII) catalysts with organic reducing agents such as 3-pentanol and PPh3.24–26 Development of solid catalysts for DODH has been a hot topic in biomass conversion.27–31 Our laboratory has developed a heterogeneous ReOx–Au/CeO2 catalyst for DODH using H2 as the reducing agent.32–35 The highly dispersed monomeric Re oxide species on the crystalline CeO2 surface can be the active sites of DODH.32,33,36–38 Au serves as the activation site of H2, and the activated hydrogen species move on CeO2 probably as pairs of protons and electrons to Re species. Au was selected among various noble metals based on its low CC hydrogenation activity.32 The stabilization of high valent Re species by CeO2 and the capability of hydrogen species to move on the CeO2 surface are the keys of the DODH activity. The latter step catalyzed by ReOx/C consists of the following steps: the isomerization of 2,5-DHF to 2,3-dihydrofuran (2,3-DHF), the hydration of 2,3-DHF to 2-hydroxytetrahydrofuran (2-HTHF), and the reduction of 2-HTHF or its straight chain form 4-hydroxybutanal to 1,4-BuD. In the most recent research, we also found that the ReOx/CeO2 catalyst without an Au promoter still could catalyze the DODH reaction in the presence of ReOx/C, enabling the reduction of 1,4-AHERY to 1,4-butanediol with the mixture of ReOx/CeO2 and ReOx/C.39 The Re species on carbon could activate H2 like an Au promoter for the reduction of Re species on the CeO2 surface which are the active sites in the conversion of 1,4-AHERY. Furthermore, CeO2 without Re loading combined with ReOx/C even could convert 1,4-AHERY to 1,4-BuD, because the Re species on the carbon surface could move to CeO2 surfaces to form ReOx/CeO2. The highest yield of 1,4-BuD reached 85% over the ReOx/CeO2 + ReOx/C system, which was comparable to that in the co-catalyst system with an Au promoter. As above, ReOx/C plays many roles in this system: CC bond movement (isomerization), H2 activation, hydrolysis and CO hydrogenation. Versatile roles of ReOx/C have been also reported in other catalytic reactions with multiple steps such as reductive lignin depolymerization.40,41
 |
| | Scheme 1 One-pot conversion of 1,4-AHERY to 1,4-BuD over ReOx(–Au)/CeO2 + ReOx/C. | |
The largest drawback of these mixed catalyst systems was catalyst deactivation. The conversion of 1,4-AHERY sharply dropped from 100% to 65% (ReOx–Au/CeO2 + ReOx/C) or 64% (ReOx/CeO2 + ReOx/C) after the first run in reuse tests, indicating the deactivation of ReOx(–Au)/CeO2. The selectivity to 1,4-BuD also decreased, indicating the deactivation of ReOx/C. The activity of noble-metal-modified ReOx/CeO2 catalysts for DODH has been also reported to be deactivated during the reaction; however, they can be regenerated by calcination to recover the activity.32 On the other hand, due to the combustibility of the carbon support, it is not feasible to regenerate the recycled catalyst mixture by calcination. Therefore, in this study, a new Re catalyst with a regenerable support was studied as an alternative to ReOx/C in the co-catalyst system with ReOx–Au/CeO2. Among various oxide-supported ReOx catalysts, ReOx/WO3–ZrO2 is a possible alternative to ReOx/C combined with ReOx–Au/CeO2 in the production of 1,4-BuD from 1,4-AHERY. While the yield of 1,4-BuD over the combination of ReOx/WO3–ZrO2 + ReOx–Au/CeO2 was lower than that over the co-catalyst with a carbon support, the activity of the used catalyst mixture of ReOx/WO3–ZrO2 and ReOx–Au/CeO2 could be regenerated by calcination, which could be comparable to the fresh one.
Experimental section
The ReOx/support catalysts were prepared by impregnating the support materials with NH4ReO4 (Mitsuwa Chemicals Co. Ltd.) aqueous solution at 353 K. The support materials included commercial ZrO2 (Daiichi Kigenso Kagaku Kogyo Co., Ltd., BET surface area: 62 m2 g−1), C (Cabot Co., carbon black BP2000, 1282 m2 g−1), TiO2 (Aerosil Co. Ltd., P25, 47 m2 g−1), SiO2 (Fuji Silysia Co. Ltd., G-6, BET surface area: 535 m2 g−1), Al2O3 (Aerosil Co. Ltd., 87 m2 g−1), MgO (Ube Industries, Ltd., 500A, 34 m2 g−1), HZSM-5 (JRC-Z5-90H(1), Süd-Chemie Catalysts and Catalysis Society of Japan, Si/Al2 = 90, 390 m2 g−1), TiO2–ZrO2 (TiO2 = 30 wt%, Daiichi Kigenso Kagaku Kogyo Co., Ltd., 148 m2 g−1), SiO2–ZrO2 (SiO2 = 10 wt%, Daiichi Kigenso Kagaku Kogyo Co., Ltd., 136 m2 g−1), CeO2–ZrO2 (CeO2 = 50 wt%, Daiichi Kigenso Kagaku Kogyo Co., Ltd., 136 m2 g−1), and WO3–ZrO2 (WO3 = 10 wt%, Daiichi Kigenso Kagaku Kogyo Co., Ltd., 103 m2 g−1). All these supports were calcined at 773 K for 3 h before impregnation. The Re loading amount was 3 wt% and 1 wt% for single-component supports and mixed oxide supports, respectively, unless noted. Other tungsten–zirconia catalysts were also used. cpWO3–ZrO2 (W = 5 wt%) was prepared by a co-precipitation method using zirconium nitrate oxide dihydrate (Kanto Chemical Co. Inc.), ammonium metatungstate hydrate (Strem Chemicals Inc.) and ammonia water (0.1 M). The addition of ammonia water was carried out at 353 K, and the precipitate was filtered, dried in an oven at 373 K for 12 h, and finally calcined at 773 K for 3 h. The BET surface area of cpWO3–ZrO2 was 68 m2 g−1. The ReOx/cpWO3–ZrO2 catalyst (Re 1 wt%) was prepared by impregnation of cpWO3–ZrO2 with NH4ReO4. The ReOx/WO3/support catalysts (Re 1 wt%) were prepared by the sequential impregnation method. The WO3/support was firstly prepared by impregnating the supports, including ZrO2, Al2O3, and TiO2, with ammonium metatungstate hydrate aqueous solution at 353 K. The WO3/support samples were dried (373 K, 12 h), calcined (773 K, 3 h) and then impregnated with NH4ReO4 aqueous solution at 353 K. All the impregnated catalysts were dried in an oven at 373 K for 12 h, and then calcined 773 K for 3 h. The ReOx–Au/CeO2 (1 wt% Re, 0.3 wt% Au) catalyst was prepared by a deposition–precipitation method for Au loading and subsequent impregnation for Re loading according to the previous report.32
The activity test was conducted with an autoclave reactor equipped with an inner glass cylinder. After the reaction, both gas and liquid phases were analyzed by FID-GC. The carbon balance (C.B.) of each analysis result was calculated using eqn (3). The sum of the detected but unidentified products is denoted as “others” in the results. The FID sensitivity of “others” was assumed to be the same as that of 1,4-butanediol. When the C.B. is in the range of 100 ± 10% considering the experimental error, the conversion and selectivity on the carbon basis are calculated using eqn (4) and (5), respectively, and the data of C.B. are not shown in each result. In contrast, when the C.B. is clearly lower than 100% (<90%), the conversion is calculated using eqn (6). The selectivity to “others” is the same as the above case. The data of C.B. are shown in each result when clearly below 100%. The yield was calculated using eqn (7) and (8) for normal cases and cases with low C.B., respectively. The TOF was calculated using eqn (9) with the increase of conversion (Δconversion) from that at 0 h reaction because the reaction proceeded significantly during heating.
| | 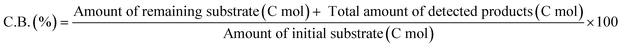 | (3) |
| | 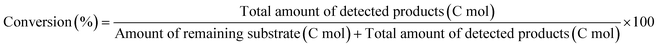 | (4) |
| |  | (5) |
| | 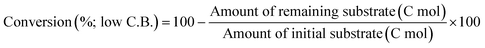 | (6) |
| | 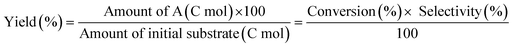 | (7) |
| | 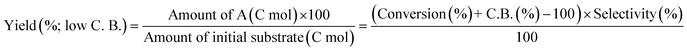 | (8) |
| |  | (9) |
For reuse tests, the catalyst mixture was collected by filtration, washed with 1,4-dioxane and dried for 12 h. The dried catalyst was used directly or after calcination for the next activity test. The calcination conditions were described in each result. The weight loss during the recovery and drying process was about 20%. Typically, multiple runs were carried out simultaneously to collect a sufficient amount of catalyst for the next use, and the number of runs was decreased during reuses (1st use: 4 runs; 2nd use: 3 runs, and so on).
The BET surface area was measured with a Micromeritics Gemini instrument. XRD patterns were obtained with a Rigaku MiniFlex600 diffractometer. H2-TPR profiles were obtained with a home-made apparatus equipped with a fixed-bed quartz reactor, frozen acetone trap and TCD. The sample weight was about 50 mg, and the sample was reduced with 5% H2 in Ar from room temperature to 1173 K at a heating rate of 10 K min−1. Temperature-programmed desorption of NH3 (NH3-TPD) profiles were obtained with a MicrotracBEL BELCAT-II instrument. The sample (50 mg) was first treated with He at 773 K for 1 h, and then NH3/He (5/95) was passed through the sample at 323 K for 30 min. After purging NH3 with He at 323 K, the sample was heated at 10 K min−1 under flowing He. The desorbed NH3 was analyzed with MS at the m/z = 16 signal. XAFS spectra were measured at the BL01B1 station of SPring-8 (Proposal No. 2019A1369). Detectors for Re L3-edge spectra were ion chambers filled with N2/Ar = 85/15 and N2/Ar = 50/50 for I0 and I, respectively. Analysis of data was performed using the REX2000 ver. 2.6 program to obtain the XANES spectra. TG-DTA profiles were obtained with a Rigaku Thermo Plus EVO-II instrument using a 10 mg sample and α-Al2O3 reference under static air.
Results and discussion
Screening of catalysts
In the one-pot reaction of 1,4-anhydroerythritol (1,4-AHERY) to 1,4-butanediol (1,4-BuD) over the mixed catalyst system of ReOx–Au/CeO2 and ReOx/C, the ReOx/C mainly catalyzed the conversion of 2,5-dihydrofuran (2,5-DHF) as an intermediate of 1,4-BuD. Therefore, in order to find a replacement of the ReOx/C catalyst, we tested various Re catalysts in the reaction of 2,5-DHF (Table 1). One equivalent of water was added to the reaction system based on the stoichiometry, while water was supplied by the first DODH step in the reaction of 1,4-AHERY. Various products were detected including 1,4-BuD, THF, 2,3-dihydrofuran (2,3-DHF) and so on. Among products, γ-butyrolactone (GBL) and addition products of 2,3-DHF (2-hydroxytetrahydrofuran (2-HTHF) and acetals) were regarded as possible precursors of 1,4-BuD because the hydrogenation of these compounds can give 1,4-BuD. Although ReOx/TiO2 was reported to catalyze the conversion of 2,5-DHF to 1,4-BuD in a flow reactor with a co-feed of H2 and water with 80% yield,42 under our reaction conditions, ReOx/TiO2 (entry 2) showed a much lower performance (selectivity to 1,4-BuD and its precursors <40%). Nevertheless, ReOx/TiO2 and ReOx/ZrO2 (entries 2 and 3) showed relatively higher conversion and selectivity to 1,4-BuD than Re catalysts on other oxide supports such as ReOx/Al2O3, ReOx/SiO2, ReOx/HZSM5, and ReOx/MgO (entries 4–7). The main products over ReOx/Al2O3 and ReOx/HZSM5 were THF and furan which are the disproportionation products from DHFs (2,5-DHF and 2,3-DHF; 2DHFs → THF + furan). THF was also a main product over ReOx/SiO2 and ReOx/MgO. 2,3-DHF, which is formed by isomerization of 2,5-DHF, was another main product over ReOx/MgO, and it was also formed over ReOx/SiO2 and ReOx/Al2O3. In the reaction of 2,5-DHF to 1,4-BuD, 2,3-DHF formed by isomerization of 2,5-DHF was next converted by hydration which is typically catalyzed by an acid. The lack of acidity of ReOx/MgO is related to the high selectivity to 2,3-DHF. In fact, ReOx/HZSM5 which has high acidity showed 0% selectivity to 2,3-DHF. However, ReOx/HZSM5 showed little activity in isomerization of 2,5-DHF to 2,3-DHF which was the first step of 1,4-BuD formation. ReOx/ZrO2 showed a slightly higher activity of 2,5-DHF conversion than ReOx/TiO2. However, the selectivity to 1,4-BuD over either ReOx/TiO2 or ReOx/ZrO2 was just around 20%, similar to their selectivity to THF.
Table 1 Reaction of 2,5-DHF over various Re catalystsa
| Entry |
Catalyst |
Re loading amount/wt% |
Conv./% |
Selectivity/% |
| 1,4-BuD |
THF |
2,3-DHF |
1-BuOH |
GBL |
Furan |
2-HTHF |
Acetal A |
Acetal B |
Others |
Reaction conditions: 2,5-DHF = 0.15 g, water = 0.04 g, catalyst = 0.15 g (Re = 1 or 3 wt%), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 4 h.
Reported in ref. 23.
TiO2 = 30 wt%.
SiO2 = 10 wt%.
CeO2 = 50 wt%.
WO3 = 10 wt%.
ReOx/WO3–ZrO2 was reduced in solvent at 413 K for 1 h before the reaction.
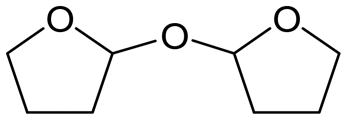
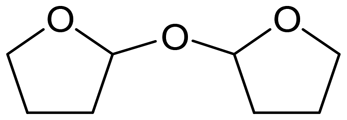
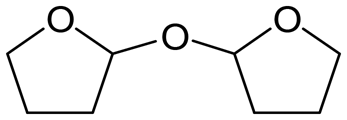
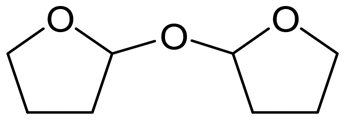
W = 5 wt%; homemade supports prepared by impregnation. BuD: butanediol, DHF: dihydrofuran, THF: tetrahydrofuran, BuOH: butanol, GBL: γ-butyrolactone, HTHF: hydroxytetrahydrofuran, acetal A:  ; acetal B: ; acetal B: 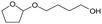 . . |
| 1b |
ReOx/C |
3 |
94 |
60 |
14 |
0 |
2 |
12 |
2 |
0 |
0 |
5 |
5 |
| 2 |
ReOx/TiO2 |
3 |
43 |
21 |
22 |
3 |
18 |
6 |
9 |
2 |
2 |
5 |
11 |
| 3 |
ReOx/ZrO2 |
3 |
62 |
19 |
20 |
6 |
3 |
17 |
7 |
4 |
1 |
16 |
6 |
| 4 |
ReOx/Al2O3 |
3 |
23 |
1 |
27 |
11 |
1 |
2 |
26 |
5 |
4 |
1 |
24 |
| 5 |
ReOx/SiO2 |
3 |
25 |
13 |
48 |
11 |
8 |
0 |
9 |
11 |
0 |
0 |
0 |
| 6 |
ReOx/HZSM5 |
3 |
26 |
2 |
45 |
0 |
7 |
0 |
32 |
1 |
0 |
0 |
12 |
| 7 |
ReOx/MgO |
3 |
25 |
0 |
35 |
46 |
0 |
0 |
11 |
8 |
0 |
0 |
0 |
| 8 |
ReOx/TiO2–ZrO2c |
1 |
57 |
21 |
30 |
10 |
2 |
7 |
4 |
4 |
1 |
17 |
4 |
| 9 |
ReOx/SiO2–ZrO2d |
1 |
52 |
22 |
36 |
1 |
3 |
6 |
6 |
2 |
1 |
17 |
2 |
| 10 |
ReOx/CeO2–ZrO2e |
1 |
17 |
2 |
37 |
28 |
0 |
0 |
0 |
21 |
0 |
0 |
12 |
| 11 |
ReOx/WO3–ZrO2f |
1 |
94 |
43 |
38 |
0 |
4 |
5 |
2 |
1 |
0 |
6 |
1 |
| 12 |
ReOx/WO3–ZrO2f,g |
1 |
63 |
18 |
39 |
1 |
3 |
10 |
3 |
3 |
0 |
12 |
9 |
| 13 |
ReOx/WO3/TiO2h |
1 |
26 |
3 |
31 |
0 |
5 |
0 |
28 |
5 |
0 |
0 |
27 |
| 14 |
ReOx/WO3/Al2O3h |
1 |
22 |
9 |
38 |
1 |
2 |
1 |
21 |
10 |
2 |
6 |
9 |
| 15 |
ReOx/WO3/ZrO2h |
1 |
93 |
40 |
45 |
0 |
5 |
3 |
3 |
0 |
0 |
3 |
1 |
| 16 |
None |
— |
2 |
0 |
38 |
19 |
0 |
0 |
26 |
0 |
11 |
0 |
6 |
All the ReOx catalysts with a single oxide support showed lower activity and 1,4-BuD selectivity than ReOx/C (Table 1, entry 1). Therefore, the Re catalysts with mixed oxide supports were further tested in the reaction of 2,5-DHF. Zirconia was selected as one of the components because of the variety of available mixed oxides and relatively good performance of ReOx/ZrO2. The ReOx/WO3–ZrO2 catalyst (entry 11) using a commercial WO3–ZrO2 support with 10 wt% WO3 loading showed the highest conversion of 2,5-DHF and the highest selectivity to 1,4-BuD among all the catalysts on non-carbon supports. The conversion over ReOx/WO3–ZrO2 was as high as that obtained over ReOx/C, and the selectivity to 1,4-BuD was slightly lower than that of ReOx/C due to the higher selectivity to THF. Other Re catalysts with mixed oxide supports such as ReOx/TiO2–ZrO2, ReOx/SiO2–ZrO2, ReOx/CeO2–ZrO2, ReOx/WO3/TiO2, and ReOx/WO3/Al2O3 (entries 8–10, 13–14) showed lower activity in the conversion of 2,5-DHF and selectivity to 1,4-BuD than ReOx/WO3–ZrO2. The ReOx/WO3–ZrO2 catalyst reduced before use (entry 12) showed a lower activity than the non-pretreated one, and the selectivity to 1,4-BuD also decreased, while the selectivity to other intermediates such as 2-hydroxytetrahydrofuran (addition product of 2,3-DHF and water) and acetals increased. This decrease might be due to the aggregation of catalytically active Re sites.
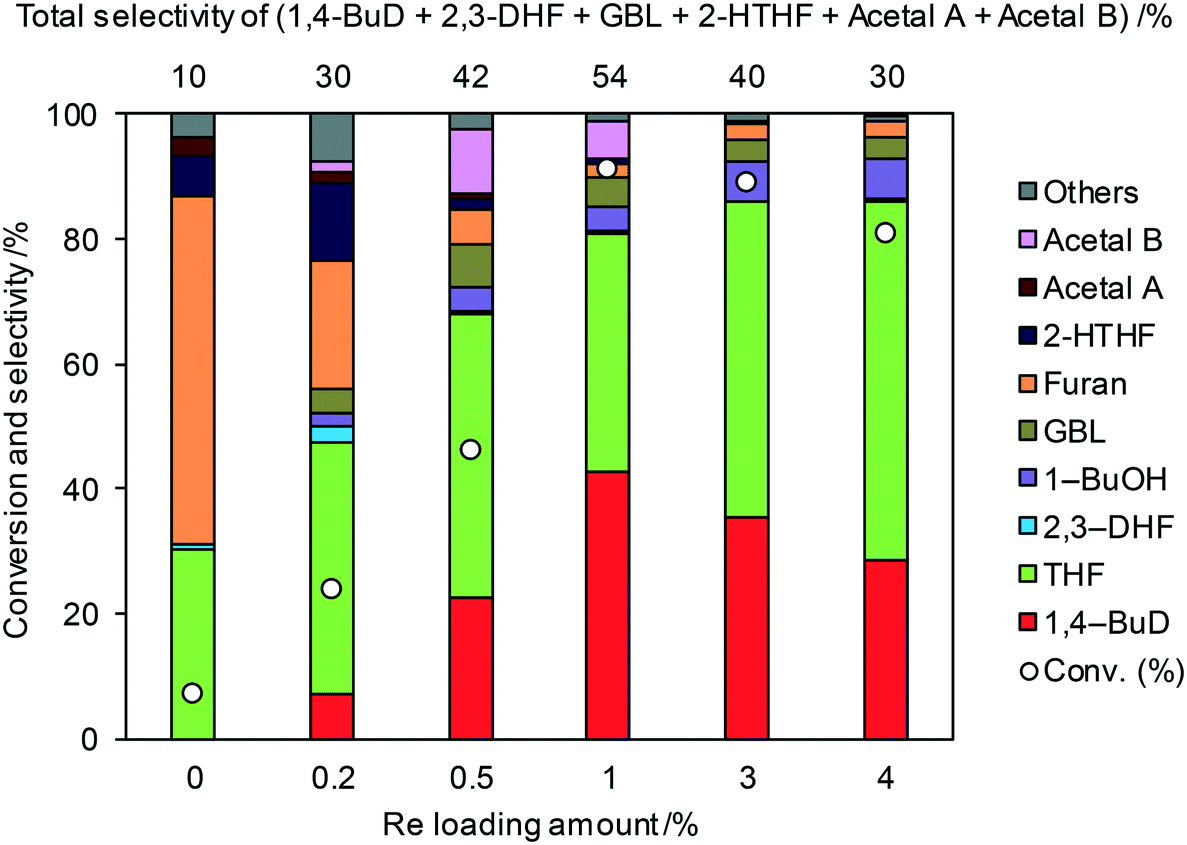
Fig. 1 shows the effect of the Re loading amount of ReOx/WO3–ZrO2 in the reaction of 2,5-DHF. Without Re, the conversion was low and the main products were THF and furan which can be produced by disproportionation. More furan was formed than THF, which suggests that some of furan was formed by dehydrogenation of 2,5-DHF. 1,4-BuD was not formed at all over WO3–ZrO2 alone. The conversion and selectivity to 1,4-BuD dramatically increased with the increase of the Re loading amount from 0 to 1 wt%. The optimized Re loading amount was 1 wt% based on the 2,5-DHF conversion and 1,4-BuD selectivity. However, the formation of THF increased when the Re loading amount was over 1 wt%, while the conversion of 2,5-DHF gradually decreased at the same time. The decrease of conversion can be due to the aggregation of catalytically active Re sites to inactive polymeric Re species (in isomerization of 2,5-DHF to 2,3-DHF). The increase of THF selectivity with the increase of the Re amount can be explained by the hydrogenation ability of polymerized Re species with metallic nature.
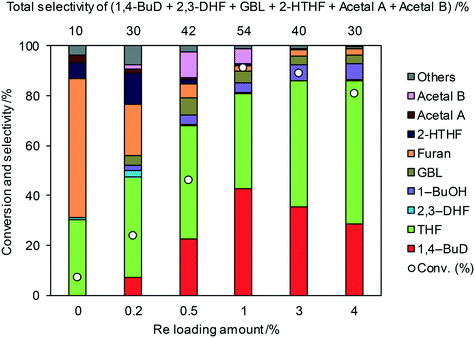 |
| | Fig. 1 Effect of Re loading amount of ReOx/WO3–ZrO2 in the reaction of 2,5-DHF. Reaction conditions: 2,5-DHF = 0.15 g, water = 0.04 g, ReOx/WO3–ZrO2 = 0.15 g (Re = 0–4 wt%, WO3 = 10 wt%), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 4 h. DHF: dihydrofuran, BuD: butanediol, THF: tetrahydrofuran, BuOH: butanol, GBL: γ-butyrolactone, HTHF: hydroxytetrahydrofuran, acetal A: 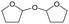 ; acetal B: ; acetal B: 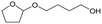 . . | |
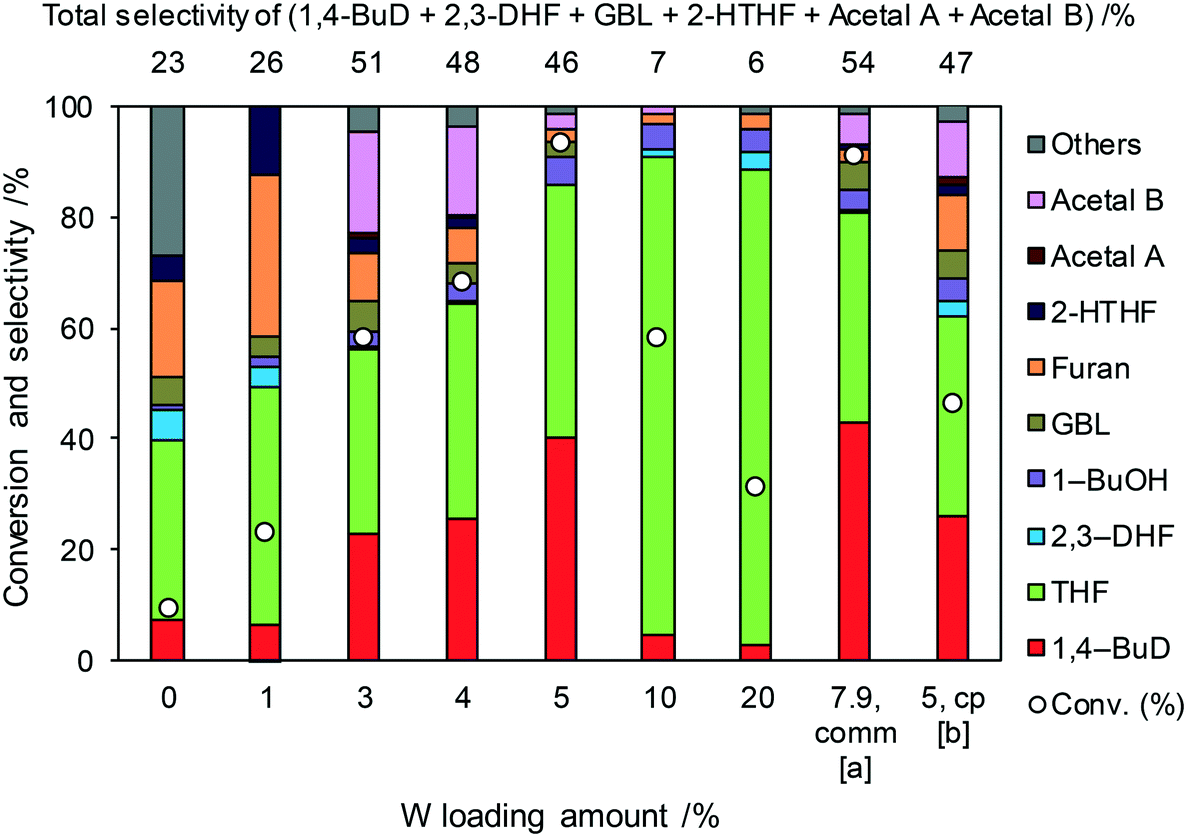
Even though 1,4-BuD was formed over the ReOx/ZrO2 catalyst, adding WO3 to the ZrO2 support improved the conversion and the selectivity to 1,4-BuD dramatically. In addition to the commercial WO3–ZrO2 (10 wt% WO3) support, WO3/ZrO2 supports with various W amounts were prepared by impregnation and tested as ReOx/WO3/ZrO2 catalysts for the reaction of 2,5-DHF (Fig. 2). Up to 5 wt% W, the activity of the ReOx/WO3/ZrO2 catalyst increased with increasing W amount. The selectivity to 1,4-BuD and its precursors (acetals and 2-HTHF) became higher when the W amount was between 3 and 5%. The THF formation sharply increased and the selectivity to 1,4-BuD decreased at a higher W loading amount than 5 wt%. The conversion of 2,5-DHF also decreased dramatically when the W loading amount was higher than 5 wt%. In tungsten–zirconia supported Re catalysts, this homemade WO3/ZrO2 (W = 5 wt%) support (Table 1, entry 15) showed a similar activity to the commercial WO3–ZrO2 (WO3 = 10 wt%) support (entry 11). The best W amount in the homemade support (W = 5 wt%) was similar to the W amount of the commercial support (WO3 = 10 wt%; W = 7.9 wt%) based on the surface area (ZrO2: 62 m2 g−1; WO3–ZrO2: 103 m2 g−1). We also prepared a tungsten–zirconia support by co-precipitation (cpWO3–ZrO2), because the tungsten–zirconia prepared by co-precipitation is known to have a tetragonal ZrO2 structure43 while pure standard ZrO2 has the monoclinic phase. The activity of ReOx/cpWO3–ZrO2 (W = 5 wt%) was lower than those of ReOx/WO3–ZrO2 (WO3 = 10 wt%) with a commercial support and ReOx/WO3/ZrO2 with the same W amount (5 wt%). We used the commercial WO3–ZrO2 (WO3 = 10 wt%) support in the following studies due to the convenience of accessibility, unless noted. The structure–performance relationship will be discussed in a later section based on the characterization data of various tungsten–zirconia supports.
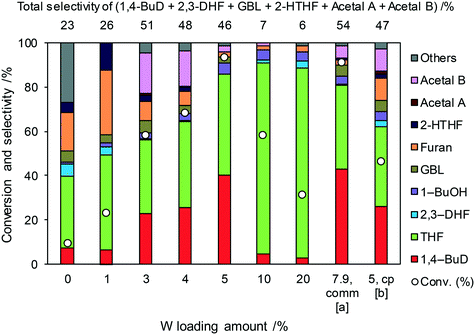 |
| | Fig. 2 Effect of W loading amount of ReOx/impWO3/ZrO2 in the reaction of 2,5-DHF. Reaction conditions: 2,5-DHF = 0.15 g, water = 0.04 g, ReOx/impWO3/ZrO2 = 0.15 g (Re = 1 wt%, W = 1–20 wt%), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 4 h. [a] ReOx/WO3–ZrO2 (Re = 1 wt%, WO3 = 10 wt% (W 7.9 wt%), commercial support). [b] ReOx/cpWO3–ZrO2 (Re = 1 wt%, W = 5 wt%, support prepared by co-precipitation). DHF: dihydrofuran, BuD: butanediol, THF: tetrahydrofuran, BuOH: butanol, GBL: γ-butyrolactone, HTHF: hydroxytetrahydrofuran, acetal A: 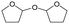 ; acetal B: ; acetal B: 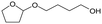 , com: commercial (WO3 = 10 wt%). , com: commercial (WO3 = 10 wt%). | |
Optimization of reaction conditions
The reaction temperature for the conversion of 2,5-DHF over ReOx/WO3–ZrO2 was optimized to 413 K based on the yield of 1,4-BuD (Fig. S1, ESI†). The conversion and selectivity to 1,4-BuD increased with raising the reaction temperature from 393 to 413 K. The selectivity to 1,4-BuD sharply decreased when the temperature was over 413 K; meanwhile the selectivity to THF dramatically increased probably due to the dehydration of 1,4-BuD.
Fig. 3 shows the effect of H2 pressure in the reaction of 2,5-DHF over ReOx/WO3–ZrO2 (the detailed data for the reaction rate determination are shown in Fig. S2 and Table S1, ESI†). The conversion and selectivity to 1,4-BuD increased with the increase in hydrogen pressure from 2 to 8 MPa (Fig. 3(a)). Although the THF selectivity increased at the same time, the reaction rate was much higher under 8 MPa hydrogen pressure. The total selectivity to 1,4-BuD + acetals + 2-HTHF + γ-butyrolactone (GBL) as 1,4-BuD and its precursors was similar to the sum of selectivity to furan + THF under different H2 pressures, even though the 1,4-BuD selectivity was lower and the GBL selectivity was higher under lower H2 pressure. Therefore, GBL may be an intermediate of 1,4-BuD like 2-HTHF and acetals in the conversion of 2,5-DHF.
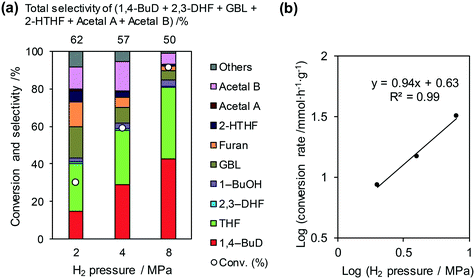 |
| | Fig. 3 Effect of H2 pressure in the reaction of 2,5-DHF over ReOx/WO3–ZrO2. (a) Results at 4 h; (b) initial conversion rate. Reaction conditions: 2,5-DHF = 0.15 g, water = 0.04 g, ReOx/WO3–ZrO2 = 0.15 g (Re = 1 wt%, WO3 = 10 wt%), 1,4-dioxane = 4 g, PH2 = 2–8 MPa, T = 413 K, t = 4 h (a), 0–2 h (b). The detailed data of (b) are shown in Table S1, ESI.† DHF: dihydrofuran, BuD: butanediol, THF: tetrahydrofuran, BuOH: butanol, GBL: γ-butyrolactone, HTHF: hydroxytetrahydrofuran, acetal A:  ; acetal B: ; acetal B:  . . | |
The reaction rate dependence on H2 pressure was also determined based on the reaction results at low conversion levels. The reaction rate of 2,5-DHF increased with increasing H2 pressure over ReOx/WO3–ZrO2. The reaction order with respect to hydrogen pressure was 0.94 (Fig. 3(b)), indicating that the rate-determination step was a reaction involving hydrogen species. Although the conversion of 2,5-DHF to 2,3-DHF does not consume hydrogen, the conversion can proceed by addition of a hydrogen atom to the 4-position of 2,5-DHF and removal of a hydrogen atom at the 2 position (eqn (10)). The rate-determining step might be the activation of H2 or the addition of hydrogen species to the substrate.
| | 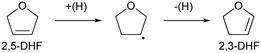 | (10) |
As above, the optimized reaction conditions for 1,4-BuD production from 2,5-DHF were a temperature of 413 K and a H2 pressure of 8 MPa, which were the same reaction conditions used in the reaction of 1,4-AHERY over the combination of ReOx–Au/CeO2 (or ReOx/CeO2) and ReOx/C catalysts as reported in our previous reports.23,39
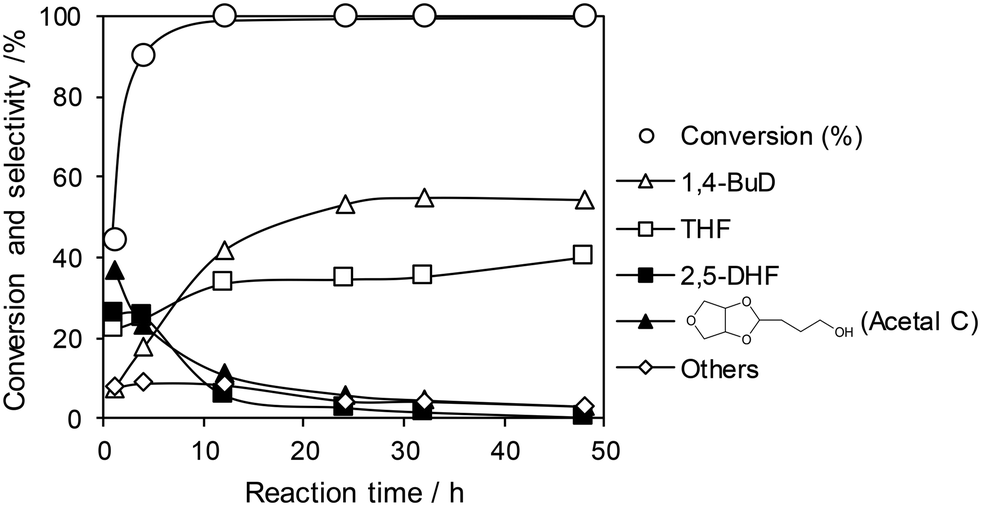
The optimized ReOx/WO3–ZrO2 catalyst was applied to the reaction of 1,4-AHERY combined with the ReOx–Au/CeO2 catalyst for DODH of 1,4-AHERY to 2,5-DHF under the standard reaction conditions. The time course of the reaction of 1,4-AHERY over the mixture of ReOx–Au/CeO2 and ReOx/WO3–ZrO2 is shown in Fig. 4 (the detailed data are shown in Table S2, ESI†). The selectivity to 1,4-BuD and THF increased with the increase of conversion. At the beginning of the reaction, 2,5-DHF as an intermediate and the acetal from 4-hydroxybutanal and 1,4-AHERY were detected, and they were gradually converted with time. Unlike the time course of 1,4-AHERY reduction over the combination of ReOx–Au/CeO2 and ReOx/C where only the acetal was detected as the main intermediate,23 2,5-DHF as an intermediate was detected in a significant amount at a shorter reaction time. This difference means the lower activity of the ReOx/WO3–ZrO2 catalyst in the isomerization of 2,5-DHF to 2,3-DHF than that of ReOx/C. These results are somewhat inconsistent with the similar activity of ReOx/WO3–ZrO2 alone to the ReOx/C catalyst alone in 2,5-DHF reduction (Table 1, entries 1 and 11). The difference can be explained by the movement of Re species from WO3–ZrO2 to the CeO2 support as described below. The DHFs were quickly converted over the ReOx/C catalyst, and the rate-determining step of the ReOx(–Au)/CeO2 + ReOx/C system for 1,4-AHERY reduction was the DODH catalyzed by ReOx(–Au)/CeO2. In the system of ReOx–Au/CeO2 + ReOx/WO3–ZrO2, the rate-determining step in the conversion of 1,4-AHERY was not clear: the DODH step and isomerization step had similar reaction rates. The highest yield of 1,4-BuD was 55% obtained at 32 h, which was significantly lower than the 90% over the co-catalyst of ReOx–Au/CeO2 and ReOx/C, due to the high selectivity to THF in the reaction. Because THF was formed from several pathways (DHF hydrogenation, DHF disproportionation, and 1,4-BuD dehydration), the improvement of the 1,4-BuD yield in the reaction of 1,4-AHERY over the mixture catalyst of ReOx–Au/CeO2 and ReOx/WO3–ZrO2 might be difficult. The TOFRe-total based on the conversion and total Re amount of the mixture of ReOx–Au/CeO2 + ReOx/WO3–ZrO2, which means the TOFRe-total of the first DODH step, is calculated to be 3 × 10 h−1 (conversion of 45% → 90% from 1 to 4 h, Fig. 4 and Table S2†). Although this value could have considerable error because of the limited data points and high conversion levels, this value is comparable to the TOFRe-total of CeO2 + ReOx/C (Re 2 wt%) in 1,4-AHERY reduction to 1,4-BuD (26 h−1)39 at the same temperature and H2 pressure. According to our previous works in DODH using ReOx/CeO2 + promoter catalysts, the DODH activity is controlled by the amount of monomeric ReOx species on CeO2, while Re species on C and other oxide supports are inactive in DODH.39 Therefore, the TOFRe-total value of mixture catalysts is reflected by the ratio of the Re amount on CeO2 to the total Re amount. It was reported that ReOx–Au/CeO2 (1 wt%) + ReOx/C (3 wt%) showed a much lower TOFRe-total (5 h−1) because the number of active Re sites on CeO2 for DODH is fixed by the rapid reduction of both Re species on CeO2 and C to insoluble species. In the case of CeO2 + ReOx/C, the Re species move from the C support to the CeO2 support before reduction to form a large number of active Re sites for DODH. The similar high TOFRe-total value of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 to that of CeO2 + ReOx/C suggests that some of the Re species on WO3–ZrO2 moved to the CeO2 support. On the other hand, the decrease of the Re amount in ReOx/WO3–ZrO2 would lower the activity in 2,5-DHF conversion when it was mixed with ReOx–Au/CeO2.
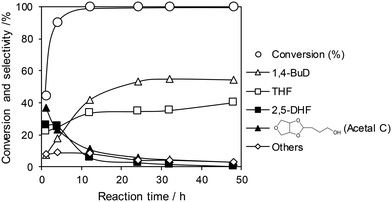 |
| | Fig. 4 Time course of the reaction of 1,4-AHERY over ReOx–Au/CeO2 + ReOx/WO3–ZrO2. Reaction conditions: 1,4-AHERY = 0.3 g, ReOx–Au/CeO2 = 0.15 g (Re = 1 wt%, Au = 0.3 wt%), ReOx/WO3–ZrO2 = 0.15 g (Re = 1 wt%, WO3 = 10 wt%), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 1–48 h. The detailed data are shown in Table S2, ESI.† AHERY: anhydroerythritol, BuD: butanediol, THF: tetrahydrofuran, DHF: dihydrofuran. | |
Reaction mechanism
As described above, 1,4-BuD could be produced from 1,4-AHERY over the physical mixture of ReOx–Au/CeO2 and ReOx/WO3–ZrO2 catalysts (Table 2, entry 1). However, the combination of ReOx/CeO2 and ReOx/WO3–ZrO2 showed very low activity in the conversion of 1,4-AHERY (entry 2). This indicates that the Au promoter is necessary for the activation of H2 in the DODH reaction of 1,4-AHERY to 2,5-DHF, because the single ReOx/WO3–ZrO2 catalyst also showed very low activity to 1,4-AHERY conversion (entry 4). Unlike the mechanism of the mixed catalyst of ReOx/CeO2 and ReOx/C, the Re species on CeO2 could not be reduced by hydrogen species activated on the ReOx/WO3–ZrO2 support. The difference between C and WO3–ZrO2 supports as the promoting ability of ReOx/CeO2 reduction may be related to the electrical conductivity: the C support can more easily transport electrons, which are produced from H2 along with H+, than the oxide support. Therefore, the DODH reaction of 1,4-AHERY to 2,5-DHF was entirely catalyzed by ReOx–Au/CeO2. However, the conversion of 1,4-AHERY over ReOx–Au/CeO2 + ReOx/WO3–ZrO2 (entry 1) was higher than that over ReOx–Au/CeO2 (entry 3). The increase can be explained by the movement of Re species from WO3–ZrO2 to CeO2 as explained in the above section.
Table 2 Reaction of 1,4-AHERY and the intermediates over related catalystsa
| Entry |
Substrate |
Catalyst |
Conv./% (C. B./%) |
Selectivity/% |
| 1,4-BuD |
THF |
2,5-DHF |
2,3-DHF |
1-BuOH |
GBL |
Furan |
2-HTHF |
Acetal A |
Acetal B |
Others |
Reaction conditions: 1,4-AHERY = 0.3 g, catalyst = 0.15 g (or 0.15 g + 0.15 g), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 24 h.
Reported in ref. 23.
1,4-AHERY = 0.5 g.
2,5-DHF = 0.15 g, water 0.04 g, t = 4 h.
2,3-DHF = 0.15 g, water 0.04 g, t = 4 h.
MgO = 0.15 g. C. B.: carbon balance; only described when it was clearly different from 100% (±10%).AHERY: anhydroerythritol, BuD: butanediol, THF: tetrahydrofuran, DHF: dihydrofuran, BuOH: butanol, GBL: γ-butyrolactone, acetal A: 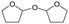 , acetal B: , acetal B: 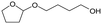 . . |
| 1 |
1,4-AHERY |
ReOx–Au/CeO2 + ReOx/WO3–ZrO2 |
100 |
53 |
35 |
3 |
0 |
2 |
1 |
0 |
0 |
0 |
1 |
6 |
| 2 |
ReOx/CeO2 + ReOx/WO3–ZrO2 |
1 |
0 |
0 |
64 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
36 |
| 3b,c |
ReOx–Au/CeO2 |
64 |
0 |
1 |
89 |
5 |
0 |
0 |
0 |
1 |
0 |
0 |
3 |
| 4 |
ReOx/WO3–ZrO2 |
1 |
0 |
0 |
0 |
0 |
0 |
63 |
0 |
0 |
0 |
0 |
37 |
| 5b,d |
2,5-DHF |
ReOx–Au/CeO2 |
45 |
4 |
34 |
— |
54 |
2 |
0 |
2 |
2 |
0 |
0 |
1 |
| 6d |
ReOx/WO3–ZrO2 |
94 |
43 |
38 |
— |
0 |
4 |
5 |
2 |
1 |
0 |
6 |
1 |
| 7d |
WO3–ZrO2 |
7 |
0 |
30 |
— |
1 |
0 |
0 |
56 |
7 |
3 |
0 |
3 |
| 8d |
ZrO2 |
5 |
0 |
36 |
— |
5 |
1 |
4 |
29 |
8 |
2 |
0 |
16 |
| 9d |
None |
2 |
0 |
38 |
— |
19 |
0 |
0 |
26 |
0 |
11 |
0 |
6 |
| 10b,e |
2,3-DHF |
ReOx–Au/CeO2b |
49 (71) |
8 |
6 |
0 |
— |
0 |
3 |
2 |
41 |
23 |
2 |
13 |
| 11e |
ReOx/WO3–ZrO2 |
100 (20) |
32 |
6 |
0 |
— |
0 |
15 |
0 |
3 |
0 |
15 |
30 |
| 12e |
WO3–ZrO2 |
98 (21) |
9 |
6 |
0 |
— |
0 |
21 |
0 |
18 |
5 |
18 |
23 |
| 13e |
ZrO2 |
76 (62) |
4 |
0 |
0 |
— |
0 |
12 |
0 |
30 |
18 |
16 |
19 |
| 14e,f |
MgO |
32 (79) |
0 |
0 |
3 |
— |
0 |
5 |
0 |
64 |
23 |
0 |
6 |
| 15e |
None |
27 (80) |
0 |
0 |
0 |
— |
0 |
0 |
4 |
76 |
10 |
0 |
10 |
In the conversion of 2,5-DHF, single WO3–ZrO2 (Table 2, entry 7) or ZrO2 (entry 8) without Re loading showed very low activity, and the main products were THF and furan, which were co-produced by disproportionation of DHFs. Generally, furan is easily hydrogenated to THF over a metal catalyst and H2; therefore, furan is an unfavorable by-product. The higher THF selectivity of ReOx/WO3–ZrO2 was due to the activity of the WO3–ZrO2 support in disproportionation of 2,5-DHF to some extent. However, the yield of THF over ReOx/WO3–ZrO2 (entry 6) was higher than that of THF + furan over WO3–ZrO2 (entry 8), suggesting that another route existed for THF formation such as hydrogenation of DHFs. The formation of 2,3-DHF and 1,4-BuD was negligible over the WO3–ZrO2 support without Re, indicating that the Re species have the function of catalyzing the conversion of 2,5-DHF to 2,3-DHF which was the first step in the production of 1,4-BuD from 2,5-DHF. Although ReOx–Au/CeO2 could catalyze a part of the conversion of 2,5-DHF to 2,3-DHF (entry 5), it is difficult to catalyze the hydration of 2,3-DHF to 2-HTHF. The total yield of 1,4-BuD and its precursors over ReOx/WO3–ZrO2 was higher than that over ReOx–Au/CeO2, indicating that the isomerization step of 2,5-DHF to 2,3-DHF in 1,4-AHERY conversion over ReOx–Au/CeO2 + ReOx/WO3–ZrO2 was mainly catalyzed by the Re species on the WO3–ZrO2 support.
The reaction of 2,3-DHF was investigated next (Table 2, entries 10–15). Similar to the reaction of 2,5-DHF, although ReOx–Au/CeO2 could partially catalyze the hydration of 2,3-DHF to 2-HTHF and the acetals (entry 10), the conversion activity was lower than that of ReOx/WO3–ZrO2 (entry 11) or WO3–ZrO2 (entry 12), suggesting that the hydration of 2,3-DHF was mainly catalyzed by WO3–ZrO2. Acidity is probably necessary in the hydration of 2,3-DHF to 2-HTHF and its derivatives. Basic supports such as MgO (entry 14) showed much lower conversion than non-basic supports like ZrO2 (entry 13) and WO3–ZrO2 (entry 12), and the results were similar to the case without a catalyst (entry 15). Furthermore, the addition of WO3 to ZrO2 increased the conversion probably due to the stronger acidity of WO3–ZrO2 than ZrO2. The carbon balance over ReOx/WO3–ZrO2, WO3–ZrO2, and ZrO2 (entries 11–13) was low, which might be due to the formation of polymers from 2,3-DHF as well as the evaporation of 2,3-DHF (boiling point: 328 K). A similar low carbon balance in the reaction of 2,3-DHF has been also reported over the ReOx/C catalyst.23 The low concentration of 2,3-DHF in the reaction of 1,4-AHERY and the presence of basic ReOx–Au/CeO2 can suppress the loss of carbon balance in the reaction of 1,4-AHERY over ReOx–Au/CeO2 + ReOx/WO3–ZrO2. The selectivity to 1,4-BuD was dramatically increased when the Re species were loaded on WO3–ZrO2. This indicates that the hydrogenation of 2-HTHF-derived products to 1,4-BuD was mainly catalyzed by the Re species on WO3–ZrO2, although the W species in WO3–ZrO2 have some activity in hydrogenation (or transfer hydrogenation) of 2-HTHF-derived products to 1,4-BuD.
Based on these results, we propose a reaction mechanism of 1,4-AHERY to 1,4-BuD over the combination of ReOx–Au/CeO2 and ReOx/WO3–ZrO2 (Scheme 2) similar to the one we proposed for the mixture of ReOx–Au/CeO2 and ReOx/C.23,39 It is composed of ReOx–Au/CeO2-catalyzed DODH of 1,4-AHERY to 2,5-DHF and ReOx/WO3–ZrO2-catalyzed reduction of 2,5-DHF to 1,4-BuD. In the conversion of 2,5-DHF, the Re species of ReOx/WO3–ZrO2 firstly catalyzed the isomerization of 2,5-DHF to 2,3-DHF, and then WO3–ZrO2 catalyzed the hydration of 2,3-DHF to 2-HTHF-derivatives; finally the Re species on ReOx/WO3–ZrO2 catalyzed the hydrogenation of the 2-HTHF-derived intermediates to 1,4-BuD.
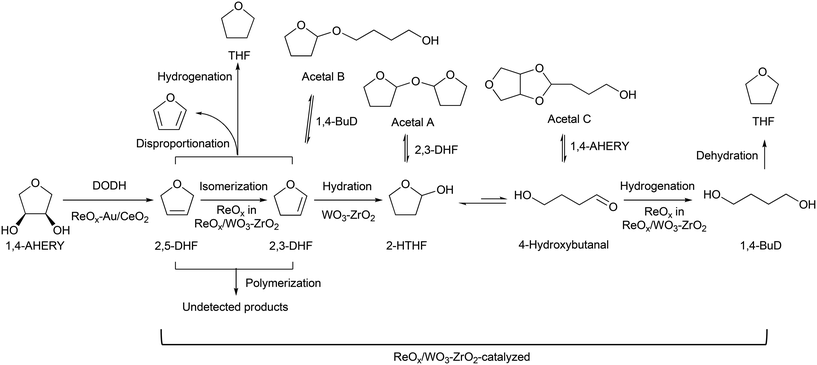 |
| | Scheme 2 Reaction routes of 1,4-AHERY reduction to 1,4-BuD over ReOx–Au/CeO2 + ReOx/WO3–ZrO2. | |
Catalyst stability
We were unable to reuse the mixed catalyst of ReOx–Au/CeO2 + ReOx/C due to the deactivation after catalytic use, as well as the difficulty in regeneration because of the combustibility of the carbon support,23,39 while ReOx–Au/CeO2 for DODH can be totally regenerated by calcination without a change of catalyst structure including the Au particle size.32 Therefore, the reusability of the ReOx/WO3–ZrO2 catalyst combined with ReOx–Au/CeO2 was the most important part in this study. Table 3 shows the results of reuse tests of the mixed catalyst of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 in the reaction of 1,4-AHERY to 1,4-BuD. When the catalyst mixture was reused without treatment, the selectivity to 1,4-BuD of the ReOx–Au/CeO2 + ReOx/WO3–ZrO2 mixture dramatically decreased from 53% in the first run to 25% in the second run, and the conversion also decreased from 100% to 97% (entries 1 and 2). The product composition was similar to the reaction result of half the reaction time (12 h, Fig. 4), indicating that the catalytic activity of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 decreased after the reaction. Due to the oxide support, the ReOx/WO3–ZrO2 catalyst could be calcined to remove the deposits on the catalyst surface. According to the TG-DTA profile of the ReOx–Au/CeO2 + ReOx/WO3–ZrO2 mixture after use (Fig. S3, ESI†), the deposits on the catalyst surface could be removed in the calcination temperature range from 450 K to 650 K. Therefore, the calcination temperature was set at 573 K (entries 3–6) or 773 K (entries 7 and 8). The best results were obtained with the calcination at 573 K for a longer time (3 h; entries 5 and 6). The conversion and selectivity to 1,4-BuD were almost the same in the reaction of 1,4-AHERY over the recycled catalyst mixture of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 calcined at 573 K for 1 or 3 h in the second run (entries 3 and 5, respectively). However, the conversion of 1,4-AHERY obviously dropped in the third run over the recycled catalyst mixture calcined at 573 K for 1 h, and the selectivity to 1,4-BuD also decreased with increasing THF selectivity (entry 4). On the other hand, the conversion was still 100% in the third run over the recycled catalyst mixture calcined at 573 K for 3 h, and the selectivity to 1,4-BuD was similar to that of the fresh one (entry 6). Therefore, the co-catalyst system of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 showed potential to be reused by calcination at 573 K for 3 h to regenerate the catalytic activity. The conversion and 1,4-BuD selectivity sharply decreased in the reuse tests over the recycled catalyst mixture calcined at the higher temperature of 773 K for 3 h (entries 7 and 8), indicating that the higher calcination temperature also caused the deactivation of the catalyst mixture. The replenishment of the fresh ReOx–Au/CeO2 catalyst to the recycled mixture of ReOx–Au/CeO2 + ReOx/WO3–ZrO2 without calcination could recover the activity to the fresh level (entry 9). On the other hand, the replenishment of fresh ReOx/WO3–ZrO2 to the used catalyst mixture showed the conversion of 1,4-AHERY at almost the expected level by the decreased amount of ReOx–Au/CeO2 (ca. 20% weight loss during the collection; conversion decreased by about 30%) (entry 10). These results demonstrated that the decrease of performance was mainly caused by the deactivation of the ReOx–Au/CeO2 catalyst.
Table 3 Reuse of the ReOx–Au/CeO2 + ReOx/WO3–ZrO2 catalyst mixture in the reaction of 1,4-AHERYa
| Entry |
Calcination conditions |
Usage times |
Conv./% |
Selectivity/% |
| 1,4-BuD |
THF |
GBL |
1-BuOH |
2,5-DHF |
2,3-DHF |
Acetal A |
Acetal B |
Acetal C |
Others |
Reaction conditions: 1,4-AHERY = 0.3 g, ReOx–Au/CeO2 = 0.15 g (Re = 1 wt%, Au = 0.3 wt%), ReOx/WO3–ZrO2 = 0.15 g (Re = 1 wt%, WO3 = 10 wt%), 1,4-dioxane = 4 g, PH2 = 8 MPa, T = 413 K, t = 24 h.
Recycled ReOx–Au/CeO2 + ReOx/WO3–ZrO2 mixture = 0.3 g.
Recycled ReOx–Au/CeO2 + ReOx/WO3–ZrO2 mixture = 0.24 g, and fresh ReOx–Au/CeO2 = 0.06 g (Re = 1 wt%, Au = 0.3 wt%) was added.
Recycled ReOx–Au/CeO2 + ReOx/WO3–ZrO2 mixture = 0.24 g, and fresh ReOx/WO3–ZrO2 = 0.06 g (Re = 1 wt%, WO3 = 10 wt%) was added. AHERY: anhydroerythritol, BuD: butanediol, THF: tetrahydrofuran, DHF: dihydrofuran, BuOH: butanol, GBL: γ-butyrolactone, HTHF: hydroxytetrahydrofuran, acetal A: 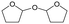 , acetal B: , acetal B: 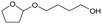 , acetal C: , acetal C: 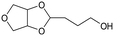 . . |
| 1 |
— |
1 |
100 |
53 |
31 |
1 |
2 |
5 |
0 |
0 |
1 |
6 |
0 |
| 2b |
None |
2 |
97 |
25 |
23 |
4 |
1 |
12 |
2 |
0 |
20 |
8 |
5 |
| 3b |
573 K, 1 h |
2 |
100 |
46 |
38 |
0 |
2 |
1 |
0 |
0 |
1 |
11 |
1 |
| 4b |
573 K, 1 h |
3 |
84 |
35 |
44 |
0 |
2 |
2 |
0 |
0 |
1 |
13 |
3 |
| 5b |
573 K, 3 h |
2 |
100 |
49 |
39 |
0 |
2 |
5 |
0 |
0 |
0 |
4 |
1 |
| 6b |
573 K, 3 h |
3 |
100 |
43 |
34 |
1 |
2 |
13 |
1 |
0 |
3 |
2 |
1 |
| 7b |
773 K, 3 h |
2 |
80 |
38 |
41 |
0 |
2 |
1 |
1 |
0 |
1 |
14 |
2 |
| 8b |
773 K, 3 h |
3 |
68 |
26 |
18 |
6 |
1 |
26 |
1 |
0 |
8 |
6 |
9 |
| 9c |
None |
2 |
100 |
51 |
35 |
1 |
2 |
0 |
0 |
0 |
1 |
10 |
1 |
| 10d |
None |
2 |
68 |
32 |
36 |
0 |
2 |
2 |
0 |
0 |
3 |
23 |
2 |
Characterization
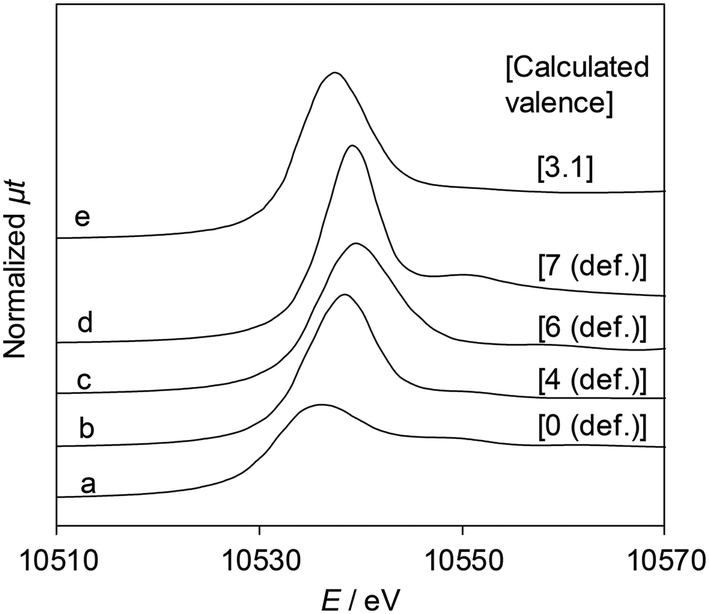
Fig. 5 shows the Re L3-edge XANES spectra of ReOx/WO3–ZrO2 and reference compounds. The detailed linear relationship between white line areas and average valence44 is shown in Table S4 and Fig. S4, ESI.† The average Re valence of ReOx/WO3–ZrO2 after use in 2,5-DHF reduction was +3.1. On the other hand, ReOx/WO3–ZrO2 showed activity in hydrogenation of 4-hydroxybutanal to 1,4-BuD, suggesting that some of the Re species were reduced to the metallic state. We have reported that the Re/SiO2 catalyst with a similar average Re valence (2.6) determined by the same XANES method contains Re0 sites interacting with Ren+, and the Re/SiO2 catalyst has activity in carboxylic acid hydrogenation.45
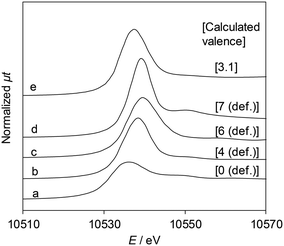 |
| | Fig. 5 Re L3-edge XANES spectra and the Re valence calculated using the white line area. (a) Re powder, (b) ReO2, (c) ReO3, (d) Re2O7, and (e) ReOx/WO3–ZrO2 after the reaction (reaction conditions: 2,5-DHF 0.15 g, water 0.03 g, catalyst 0.15 g, 1,4-dioxane 4 g, H2 8 MPa, 413 K, 4 h). The relationship between the white line area and Re valence is shown in Table S4 and Fig. S4, ESI.† | |
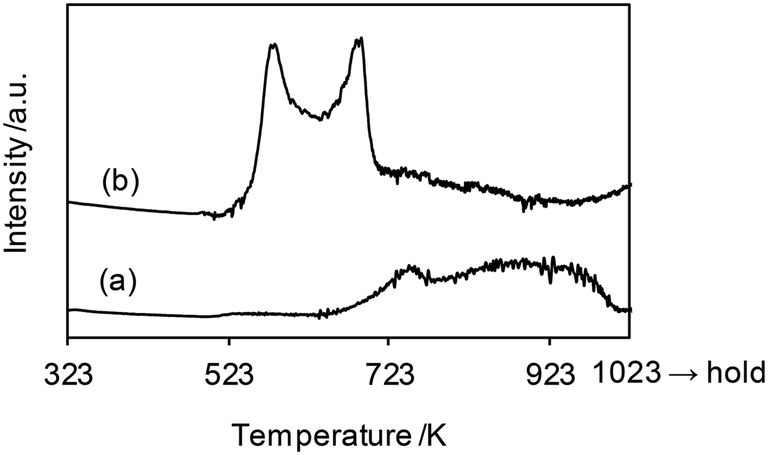
The H2-TPR profiles of WO3–ZrO2 and ReOx/WO3–ZrO2 are shown in Fig. 6, and Table 4 summarizes the H2 consumption amount in H2-TPR. WO3–ZrO2 showed a broad signal at 623–1000 K, which can be assigned to the reduction of WO3 in WO3–ZrO2. The H2 consumption of this broad signal was 0.0082 mmol (50 mg sample), which corresponded to the valence change of W by 0.74. On the other hand, ReOx/WO3–ZrO2 had strong two bands in the range 500–723 K and a broad band up to 923 K. The broad reduction signal of W was shifted from that in the case without ReOx to lower temperature, probably by the supply of hydrogen species activated on the reduced Re site. We assume that the band for W reduction with the same H2 consumption amount (0.0082 mmol) as WO3–ZrO2 was overlapped with Re reduction bands. With this assumption, the H2 consumption for W reduction in ReOx/WO3–ZrO2 between 500 and 723 K was estimated to be 0.0032 mmol (= 0.0082–0.0050 mmol), and the H2 consumption amount for Re reduction was calculated to be 0.0052 mmol (= 0.084–0.0032 mmol). This consumption amount corresponds to the Re valence change from +7 to +3.1, which agreed well with the XANES results. Although the temperature range of this signal (500–723 K) was higher than the reaction temperature (413 K), the reduction of Re species could take place in the reaction temperature due to the higher H2 pressure.45 Considering the higher temperature range and small H2 consumption amount for the reduction of W, only a small portion of W species in ReOx/WO3–ZrO2 was probably reduced to the +5 valence state during the catalysis, while most of the W species retained the +6 valence state.
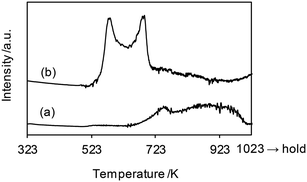 |
| | Fig. 6 H2-TPR profiles of (a) WO3–ZrO2 (WO3 = 10 wt%) and (b) ReOx/WO3–ZrO2 (Re = 1 wt%, WO3 = 10 wt%). Measurement conditions: sample weight 50 mg, H2/Ar = 5/95, 10 K min−1. The H2 consumption amount is summarized in Table 4. | |
Table 4 Summary of TPR results (Fig. 6)
| Sample |
Sample weight/mg |
Re amount/mmol |
W amount/mmol |
H2 consumption amount/mmol (temperature range/K) |
| (a) WO3–ZrO2 |
50 |
— |
0.022 |
0.0082 (500–1000) |
| (b) ReOx/WO3–ZrO2 |
50 |
0.0027 |
0.022 |
0.0084 (500–723) |
| 0.0050 (723–923) |
An NH3-TPD experiment was carried out to determine the acidity of catalysts. The NH3-TPD profiles of WO3–ZrO2, carbon and Re catalysts on these supports are shown in Fig. S5, ESI.† The desorbed NH3 amount from WO3–ZrO2 was larger than that from the carbon support, indicating that the acidity of WO3–ZrO2 is stronger than that of carbon. However, the performance of ReOx/C combined with ReOx–Au/CeO2 in the reaction of 1,4-AHERY is much higher than that of ReOx/WO3–ZrO2. As mentioned above, while acidity is probably necessary in the hydration of 2,3-DHF to 2-HTHF-derivatives, it might not be a significant factor on the overall reaction of 1,4-AHERY to 1,4-BuD since DODH of 1,4-AHERY and 2,5-DHF isomerization to 2,3-DHF are slower steps than hydration of 2,3-DHF. In addition, mixing a basic material (CeO2 in ReOx–Au/CeO2) with ReOx/WO3–ZrO2 in the reaction media can affect the acidity. Also considering the low carbon balance in the reaction tests of 2,3-DHF (Table 2), we do not give a decisive conclusion for the acidity–performance relationship from these NH3-TPR data.
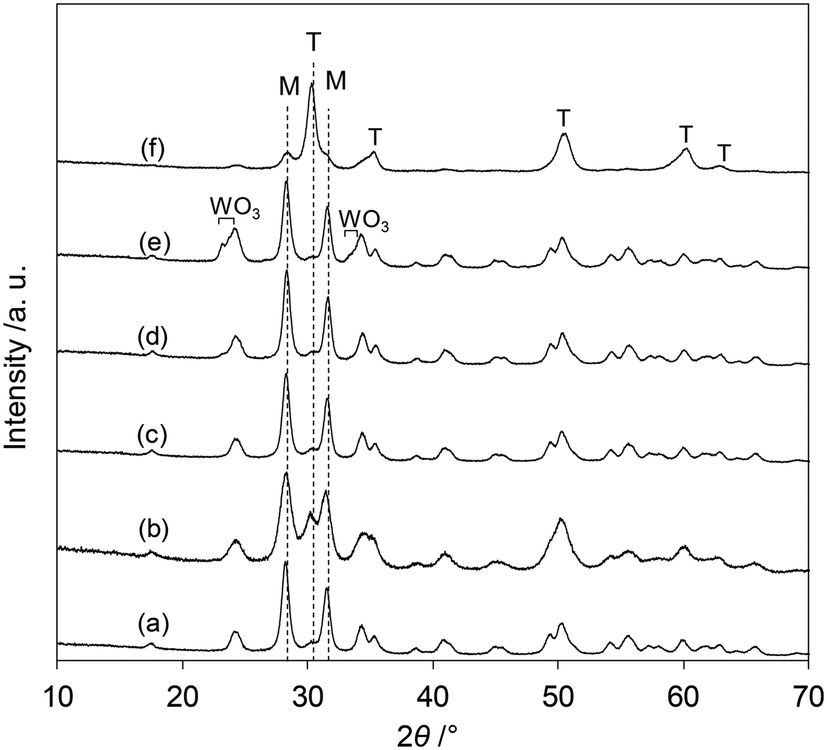
The XRD patterns of the tungsten–zirconia-supported Re catalysts are shown in Fig. 7. The Re species did not show peaks due to the low loading amount and/or low crystallinity on the support surface for all the catalysts. The catalyst with commercial WO3–ZrO2 which has been mainly used as a support in this study (Fig. 7(b)) had mainly a monoclinic structure of ZrO2 (characteristic peaks at around 28 and 32°) and a small amount of tetragonal ZrO2 structure (characteristic peak at 30°). In order to determine which phase is more active, the crystalline structures of the series of catalysts with home-made supports were determined and the catalytic performances were correlated with the structures. The commercial ZrO2 had a monoclinic crystalline structure (Fig. 7(a)). WO3/ZrO2 prepared by impregnation of ZrO2 showed also a monoclinic ZrO2 structure while there were small peaks of WO3 when the loading of W was high (≥10 wt%) (Fig. 7(c–e)). The ZrO2 of cpWO3–ZrO2 prepared by the co-precipitation method formed a tetragonal crystalline structure (t-ZrO2) (Fig. 7(f)), as reported in the literature.43 As shown in Fig. 1, all the ReOx/tungsten–zirconia catalysts except ReOx/WO3/ZrO2 with high W loading (≥10 wt%) showed activity in 2,5-DHF reduction to 1,4-BuD. The activity difference can be simply explained by the surface concentration of W species, because WO3–ZrO2 prepared by impregnation should have a higher surface concentration of W than cpWO3–ZrO2 prepared by co-precipitation with a similar surface area (actual surface area of supports: 62 and 68 m2 g−1, respectively) and the same W amount (5 wt%). The performance of ReOx/cpWO3–ZrO2 corresponded to that of ReOx/WO3/ZrO2 with 2–3 wt% W loading. Therefore, the crystalline structure of ZrO2 does not have a critical role in the catalysts. On the other hand, ReOx/WO3/ZrO2 with high W loading had very low activity (Fig. 2). The presence of WO3 crystallites seemed to deactivate the Re species. The similar crystal structure of WO3 and ReO3 (partially reduced rhenium oxide) might be related to the low activity, by incorporation of Re species into the WO3 crystal. In the literature, WO3 species on ZrO2 with lower loading can form monolayer or sub-monolayer species composed of a two-dimensional plane of corner-shared WO6 octahedra,46 and the loading amount per surface area (5 wt% on 62 m2 g−1 support; 2.7 W atoms per nm2) was slightly lower than the monolayer coverage (5 W atoms per nm2).47 The similar catalytic performance of ReOx/WO3–ZrO2 (commercial) to ReOx/WO3/ZrO2 (5 wt% W) and similar W loading amount per surface area (7.9 wt% in 103 m2 g−1 WO3–ZrO2) suggest that the good catalytic performance of ReOx/WO3–ZrO2 is also derived from surface submonolayer WO3 on ZrO2. Also considering the similar structure of WO3 and ReO3, Re species during reduction can spread over the WO3 (sub)monolayer and high dispersion of Re species can be obtained. Considering that both 2,5-DHF isomerization and 4-hydroxybutanal hydrogenation, which were catalyzed by the Re species in ReOx/WO3–ZrO2, involve hydrogen species, the highly dispersed Re0 species on the WO3–ZrO2 support can be the active sites for these steps.
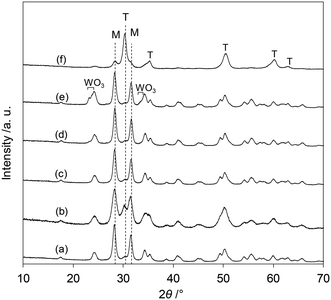 |
| | Fig. 7 XRD patterns of tungsten–zirconia-supported Re catalysts after calcination. (a) ReOx/ZrO2 (Re = 3 wt%), (b) ReOx/WO3–ZrO2 (Re = 1 wt%, WO3 = 10 wt%, commercial support), (c) ReOx/WO3/ZrO2 (Re = 1 wt%, W = 5 wt%), (d) ReOx/WO3/ZrO2 (Re = 1 wt%, W = 10 wt%), (e) ReOx/WO3/ZrO2 (Re = 1 wt%, W = 20 wt%), and (f) ReOx/cpWO3–ZrO2 (Re = 1 wt%, W = 5 wt%; WO3–ZrO2 was prepared by the co-precipitation method). T: tetragonal ZrO2, M: monoclinic ZrO2. | |
Conclusions
In order to solve the problem of the reusability of the mixture of ReOx–Au/CeO2 and carbon-supported Re catalysts to produce 1,4-butanediol (1,4-BuD) from 1,4-anhydroerythritol (1,4-AHERY) in a one-pot reaction, oxide supports were used to replace the combustible carbon support. We found that the tungsten–zirconia-supported Re catalyst combined with ReOx–Au/CeO2 could catalyze the one-pot reduction of 1,4-AHERY to 1,4-BuD. Even though the 55% yield of 1,4-BuD was lower than that over the combination of ReOx/C and ReOx(–Au)/CeO2 catalysts, the reuse of the recovered mixture of ReOx/WO3–ZrO2 and ReOx–Au/CeO2 catalysts was possible by calcination at 573 K for 3 h. Similar to the mixed catalyst system of ReOx/C + ReOx–Au/CeO2, ReOx–Au/CeO2 catalyzed the deoxydehydration (DODH) reaction of 1,4-AHERY to 2,5-dihydrofuran (2,5-DHF), and ReOx/WO3–ZrO2 catalyzed the subsequent reactions of 2,5-DHF to 1,4-BuD. The lower yield of 1,4-BuD was accompanied by larger formation of THF, which can be due to the disproportionation of 2,5-DHF to THF and furan over the WO3–ZrO2 support, the hydrogenation of 2,5-DHF over ReOx/WO3–ZrO2, and cyclization of 1,4-BuD. The performance of ReOx/WO3–ZrO2 in the reaction of 2,5-DHF strongly depended on the W loading amount, while the type of crystal structure of ZrO2 (monoclinic or tetragonal) had little effect. Too much WOx above the level for monolayer coverage of the ZrO2 support sharply decreased the activity of Re species in isomerization of 2,5-DHF to 2,3-DHF which is the first step and the most important step in the conversion of 2,5-DHF to 1,4-BuD. The Re species were suggested to be highly dispersed on the WO3 (sub)monolayer on ZrO2, and they can be the active sites for 2,5-DHF disproportionation to 2,3-DHF.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by JSPS KAKENHI 18H05247. Part of this work was carried out on commission by the Ministry of the Environment, Japan, as “Demonstration project for plastics resource circulation system for decarbonized society”.
References
- F. M. Girio, C. Fonseca, F. Carvalheiro, L. C. Duarte, S. Marques and R. Bogel-Lukasik, Bioresour. Technol., 2010, 101, 4775–4800 CrossRef CAS PubMed.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- H. Shi, ChemCatChem, 2019, 11, 1824–1877 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. K. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179–190 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, J. Mater. Chem. A, 2014, 2, 6688–6702 RSC.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Appl. Catal., B, 2016, 193, 75–92 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, Res. Chem. Intermed., 2018, 44, 3879–3903 CrossRef CAS.
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520–2530 CrossRef CAS.
- Y. Amada, H. Watanabe, Y. Hirai, Y. Kajikawa, Y. Nakagawa and K. Tomishige, ChemSusChem, 2012, 5, 1991–1999 CrossRef CAS.
- A. Said, D. D. S. Perez, N. Perret, C. Pinel and M. Besson, ChemCatChem, 2017, 9, 2768–2783 CrossRef CAS.
- T. Arai, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2016, 9, 1680–1688 CrossRef CAS PubMed.
- Z. Huang, K. J. Barnett, J. P. Chada, Z. J. Brentzel, Z. Xu, J. A. Dumesic and G. W. Huber, ACS Catal., 2017, 7, 8429–8440 CrossRef CAS.
- J. B. McKinlay, C. Vieille and J. G. Zeikus, Appl. Microbiol. Biotechnol., 2007, 76, 727–740 CrossRef CAS PubMed.
- D. R. Vardon, A. E. Settle, V. Vorotnikov, M. J. Menart, T. R. Eaton, K. A. Unocic, K. X. Steirer, K. N. Wood, N. S. Cleveland, K. E. Moyer, W. E. Michener and G. T. Beckham, ACS Catal., 2017, 7, 6207–6219 CrossRef CAS.
- D. Sun, S. Sato, W. Ueda, A. Primo, H. Garciac and A. Corma, Green Chem., 2016, 18, 2579–2597 RSC.
- M. Tamura, Y. Nakagawa and K. Tomishige, Asian J. Org. Chem., 2020, 9, 126–143 CrossRef CAS.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., B, 2019, 256, 117775 CrossRef.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- T. Wang, S. Liu, M. Tamura, Y. Nakagawa, N. Hiyoshi and K. Tomishige, Green Chem., 2018, 20, 2547–2557 RSC.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed.
- A. R. Petersen and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef CAS PubMed.
- S. Raju, M. Moret and R. J. M. K. Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- L. Sandbrink, E. Klindtworth, H.-U. Islam, A. M. Beale and R. Palkovits, ACS Catal., 2016, 6, 677–680 CrossRef CAS.
- L. Sandbrink, K. Beckerle, I. Meiners, R. Liffmann, K. Rahimi, J. Okuda and R. Palkovits, ChemSusChem, 2017, 10, 1375–1379 CrossRef CAS PubMed.
- Y. Kon, M. Araque, T. Nakashima, S. Paul, F. Dumeignil and B. Katryniok, ChemistrySelect, 2017, 2, 9864–9868 CrossRef CAS.
- B. E. Sharkey and F. C. Jentoft, ACS Catal., 2019, 9, 11317–11328 CrossRef CAS.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS.
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
- J. Cao, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Catal., 2019, 9, 3725–3729 CrossRef CAS.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 CrossRef CAS.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed.
- N. Ota, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa and K. Tomishige, Angew. Chem., Int. Ed., 2018, 57, 8058–8062 CrossRef CAS PubMed.
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS.
- B. Zhang, Z. Qi, X. Li, J. Ji, W. Luo, C. Li, A. Wang and T. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 208–215 CrossRef CAS.
- B. Zhang, Z. Qi, X. Li, J. Ji, L. Zhang, H. Wang, X. Liu and C. Li, Green Chem., 2019, 21, 5556–5564 RSC.
-
R. Pinkos, R. H. Fischer, B. Breitscheidel and P. Polanek, CA Pat., 2168458C, 2004 Search PubMed.
- S. Ramu, N. Lingaiah, B. L. A. P. Devi, R. B. N. Prasad, I. Suryanarayana and P. S. S. Prasad, Appl. Catal., A, 2004, 276, 163–168 CrossRef CAS.
- M. Rønning, T. Gjervan, R. Prestivik, D. G. Nicholson and A. Holmen, J. Catal., 2001, 204, 292–304 CrossRef.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2015, 5, 7034–7047 CrossRef CAS.
- E. I. Ross-Medgaarden, W. V. Knowles, T. Kim, M. S. Wong, W. Zhou, C. J. Kiely and I. E. Wachs, J. Catal., 2008, 256, 108–125 CrossRef CAS.
- T. Yamamoto, A. Teramachi, A. Orita, A. Kurimoto, T. Motoi and T. Tanaka, J. Phys. Chem. C, 2016, 120, 19705–19713 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00085j |
| ‡ Current address: Research Center for Artificial Photosynthesis, The Advanced Research Institute for Natural Science and Technology, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi, Osaka, 558-8585, Japan. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Masazumi
Tamura‡
*ab,
Masazumi
Tamura‡
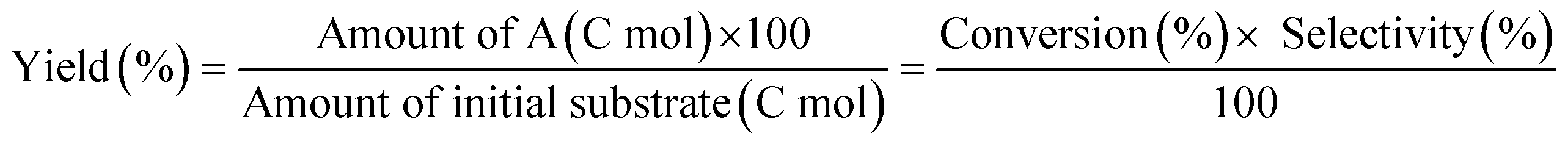
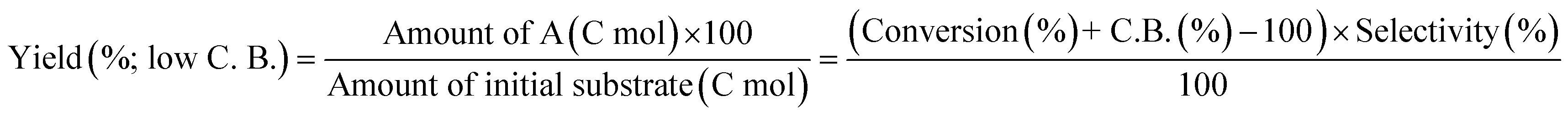
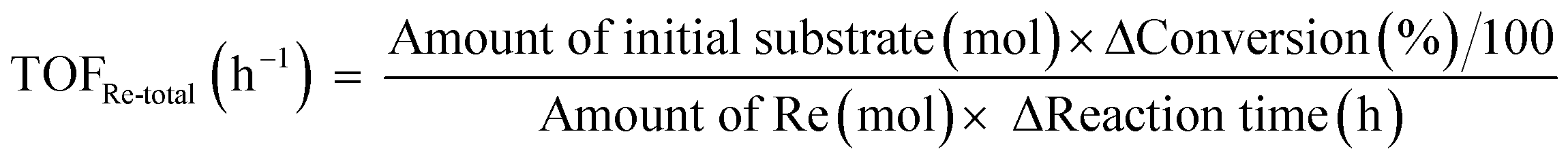
 ; acetal B:
; acetal B: 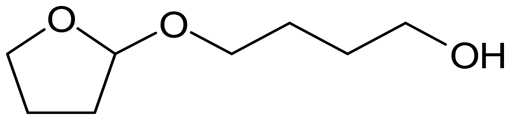 .
.
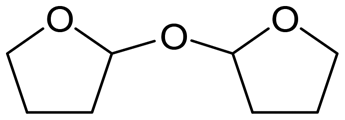 ; acetal B:
; acetal B: 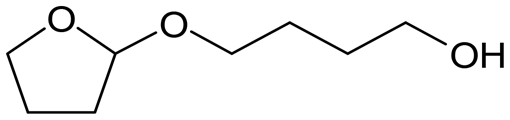 .
.
 ; acetal B:
; acetal B: 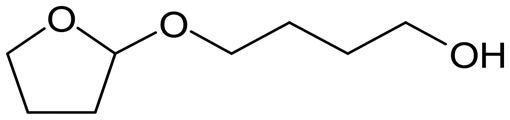 , com: commercial (WO3 = 10 wt%).
, com: commercial (WO3 = 10 wt%).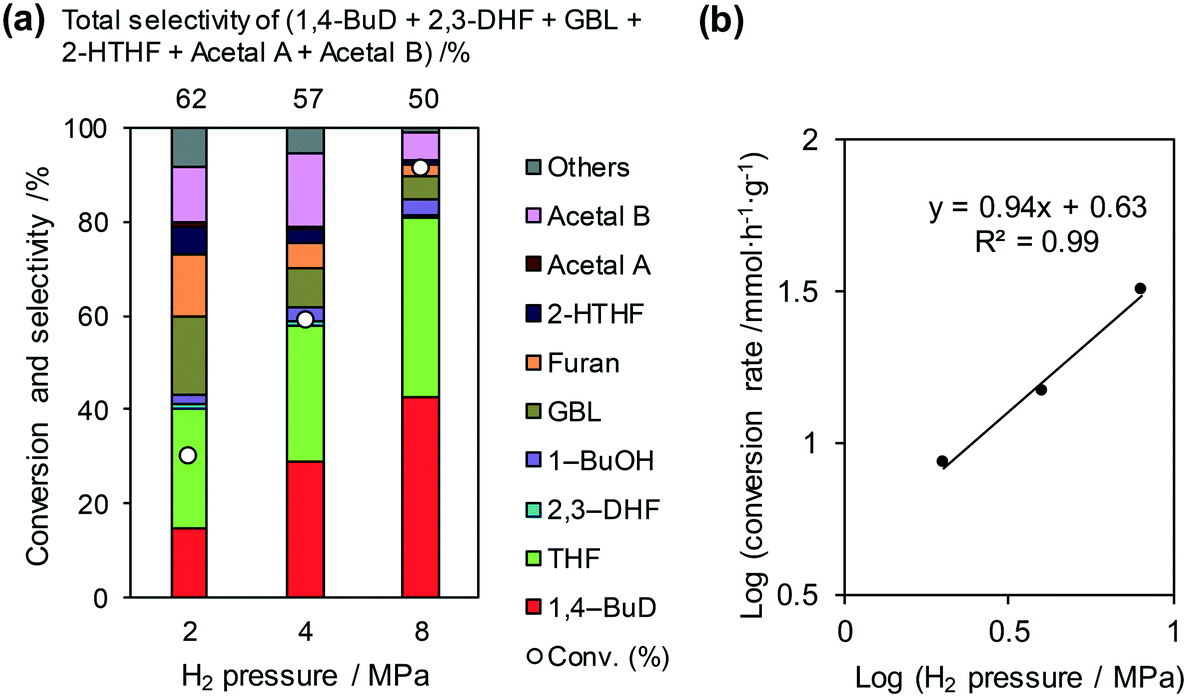
 ; acetal B:
; acetal B: 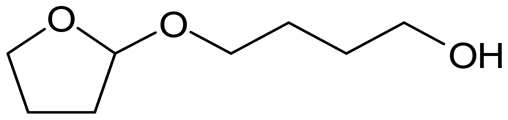 .
.
 , acetal B:
, acetal B: 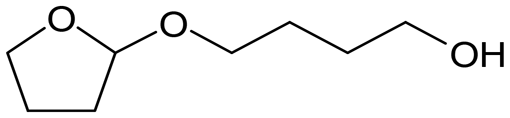 .
.
 , acetal B:
, acetal B: 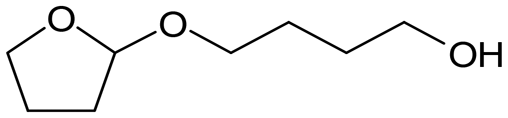 , acetal C:
, acetal C: 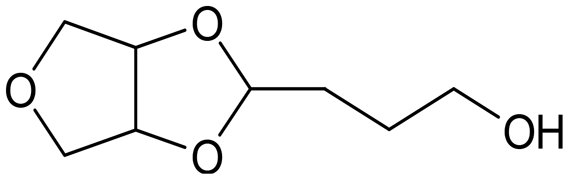 .
.