
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational selection of co-catalysts for the deaminative hydrogenation of amides†
Lluís
Artús Suàrez‡
 a,
Upul
Jayarathne‡
b,
David
Balcells
a,
Wesley H.
Bernskoetter
*b,
Nilay
Hazari
c,
Martín
Jaraiz
de and
Ainara
Nova
*af
a,
Upul
Jayarathne‡
b,
David
Balcells
a,
Wesley H.
Bernskoetter
*b,
Nilay
Hazari
c,
Martín
Jaraiz
de and
Ainara
Nova
*af
aHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Oslo, P. O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: ainara.nova@kjemi.uio.no
bDepartment of Chemistry, University of Missouri, Columbia, Missouri 65211, USA
cDepartment of Chemistry, Yale University, P. O. Box 208107, New Haven, Connecticut 06520, USA
dDepartment of Electronics, ETSIT, University of Valladolid, Paseo Belén 15, 47011 Valladolid, Spain
eIU CINQUIMA, University of Valladolid, Paseo de Belén 7, 47011 Valladolid, Spain
fDepartment of Chemistry, UiT-The Arctic University of Norway, N-9037 Tromsø, Norway
First published on 20th January 2020
Abstract
The catalytic hydrogenation of amides is an atom economical method to synthesize amines. Previously, it was serendipitously discovered that the combination of a secondary amide co-catalyst with (iPrPNP)Fe(H)(CO) (iPrPNP = N[CH2CH2(PiPr2)]2−), results in a highly active base metal system for deaminative amide hydrogenation. Here, we use DFT to develop an improved co-catalyst for amide hydrogenation. Initially, we computationally evaluated the ability of a series of co-catalysts to accelerate the turnover-limiting proton transfer during C–N bond cleavage and poison the (iPrPNP)Fe(H)(CO) catalyst through a side reaction. TBD (triazabicyclodecene) was identified as the leading co-catalyst. It was experimentally confirmed that when TBD is combined with (iPrPNP)Fe(H)(CO) a remarkably active system for amide hydrogenation is generated. TBD also enhances the activity of other catalysts for amide hydrogenation and our results provide guidelines for the rational design of future co-catalysts.
Introduction
The selective hydrogenation of carbonyl complexes is one of the most important and widely used catalytic reactions in organic synthesis.1–3 However, the reduction of electron rich carboxylic acid derivatives, such as amides, is still difficult.4,5 The ubiquity of the amide functional group in biological systems, pharmaceuticals, and industrial chemicals6 has spurred considerable effort to create efficient catalytic systems for amide hydrogenation. Nevertheless, amides are still typically reduced using waste generating stoichiometric reagents, such as LiAlH4, and to date only a small number of homogenous catalysts can directly hydrogenate amides to amines.5,7–12 These catalysts, which include both precious and base metal systems, provide proof-of-principle that this atom economic transformation is possible, but can still be improved.1Current mechanistic models for transition metal catalyzed amide reduction, in particular deaminative hydrogenation to produce an amine and an alcohol, propose a sequential reduction of the amide to an intermediate hemiaminal (step 1, Scheme 1), which then undergoes C–N bond cleavage to yield an amine and an aldehyde (step 2). Subsequent hydrogenation of the aldehyde affords the corresponding alcohol (step 3).13,14
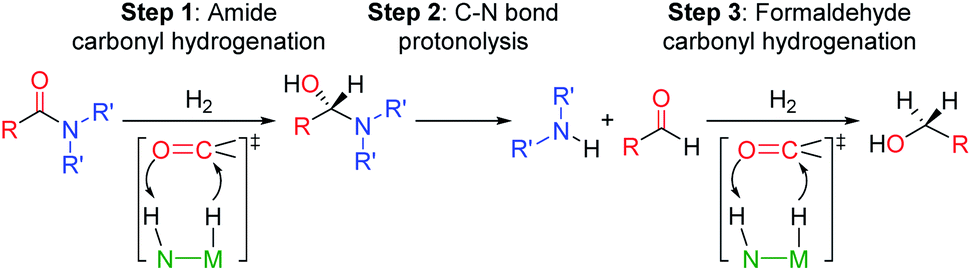 | ||
| Scheme 1 Proposed reaction steps for the deaminative hydrogenation of amides to amines and methanol catalyzed by Noyori type catalysts represented as N(H)–M(H). | ||
Most well-defined catalysts for deaminative hydrogenation rely on a Noyori-type,14,15 bifunctional pathway whereby a metal-hydride and adjacent ligand based proton are delivered to the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety (Scheme 1). Intriguingly, recent mechanistic studies indicate that while the Noyori-type catalyst structure is essential for facilitating the dihydrogen addition steps of the process (1 and 3, in Scheme 1), the proton transfer between the O- and N-ends of the hemiaminal (step 2), which triggers the cleavage of the C–N bond, does not involve necessarily the metal catalyst.13 In addition, step 2 is the turnover-limiting step, indicating that novel methods to facilitate hemiaminal cleavage are required to improve catalytic amide hydrogenation.
O moiety (Scheme 1). Intriguingly, recent mechanistic studies indicate that while the Noyori-type catalyst structure is essential for facilitating the dihydrogen addition steps of the process (1 and 3, in Scheme 1), the proton transfer between the O- and N-ends of the hemiaminal (step 2), which triggers the cleavage of the C–N bond, does not involve necessarily the metal catalyst.13 In addition, step 2 is the turnover-limiting step, indicating that novel methods to facilitate hemiaminal cleavage are required to improve catalytic amide hydrogenation.
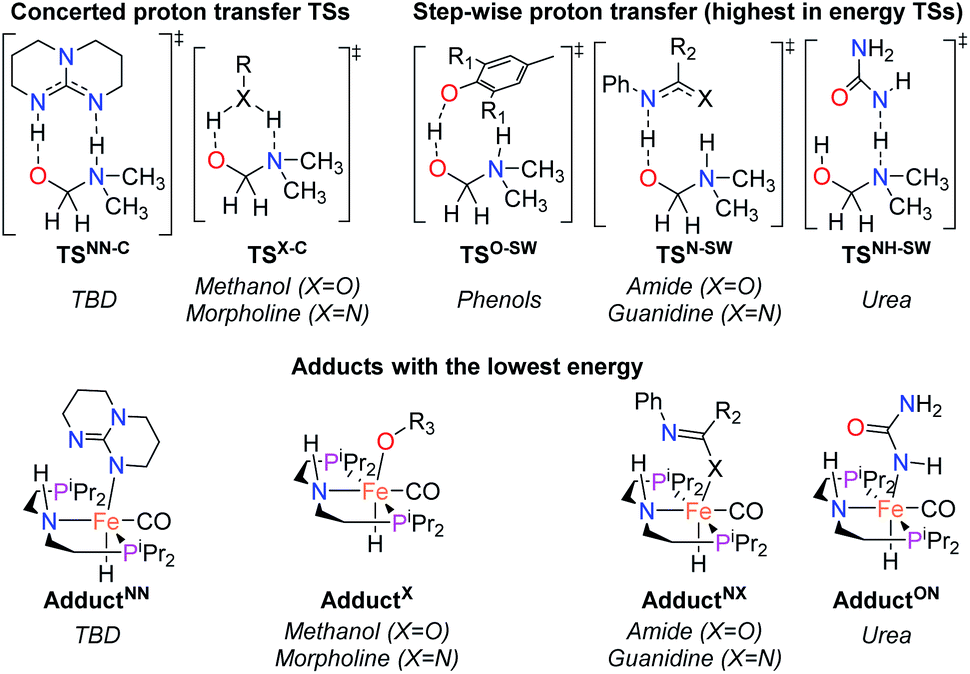
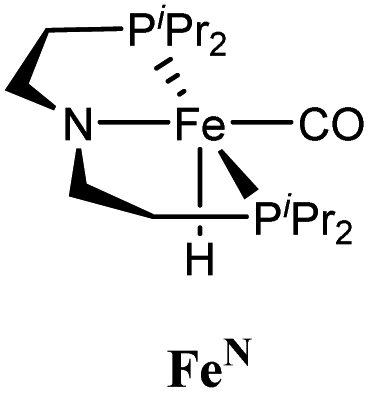
Our laboratories have previously investigated amide hydrogenation catalyzed by the iron(II) complex, (iPrPNP)Fe(H)(CO) (iPrPNP = N[CH2CH2(PiPr2)]2−) (FeN) using both computational and experimental methods (Scheme 2).5,13 In deaminative amide hydrogenation using FeN, a key serendipitous finding was that the reaction is promoted by a co-catalytic amount of a secondary amide (formamide in Scheme 2). This effect was particularly pronounced in the hydrogenation of tertiary alkylic amides, such as DMF, which are important because they are key intermediates in the homogeneous hydrogenation of CO2 to methanol mediated by amines.16,17 The interplay of the two amide equivalents (i.e. one reactant and one co-catalyst) adds complexity to the mechanism. Computational studies indicate that the secondary amide lowers the barrier to the proton transfer that occurs in hemiaminal C–N bond cleavage (ΔG‡HT, in Scheme 2) because the NH moiety acts as a proton-shuttle.13 However, the use of a secondary amide as a co-catalyst has two major pitfalls: (1) secondary amides can form stable adducts with FeN (ΔGadd, in Scheme 2) via 1,2-addition across the iron–amide bond, which lowers the concentration of the active species in catalysis; and (2) the amide co-catalyst can be consumed during the reaction, which undermines its contribution as a co-catalyst and introduces a product separation problem.5 Here, we use a rational approach involving DFT calculations to design co-catalysts tailored for the deaminative hydrogenation of tertiary amides. Our best co-catalyst, triazabicyclodecene (TBD), acts as push–pull proton shuttle for C–N bond cleavage, and leads to significant improvement in iron catalyzed deaminative amide hydrogenation. Importantly, the improvement from TBD also occurs for a number of other transition metal catalysts for deaminative amide hydrogenation, suggesting that the addition of co-catalysts of this type is a general strategy for improving amide reduction.
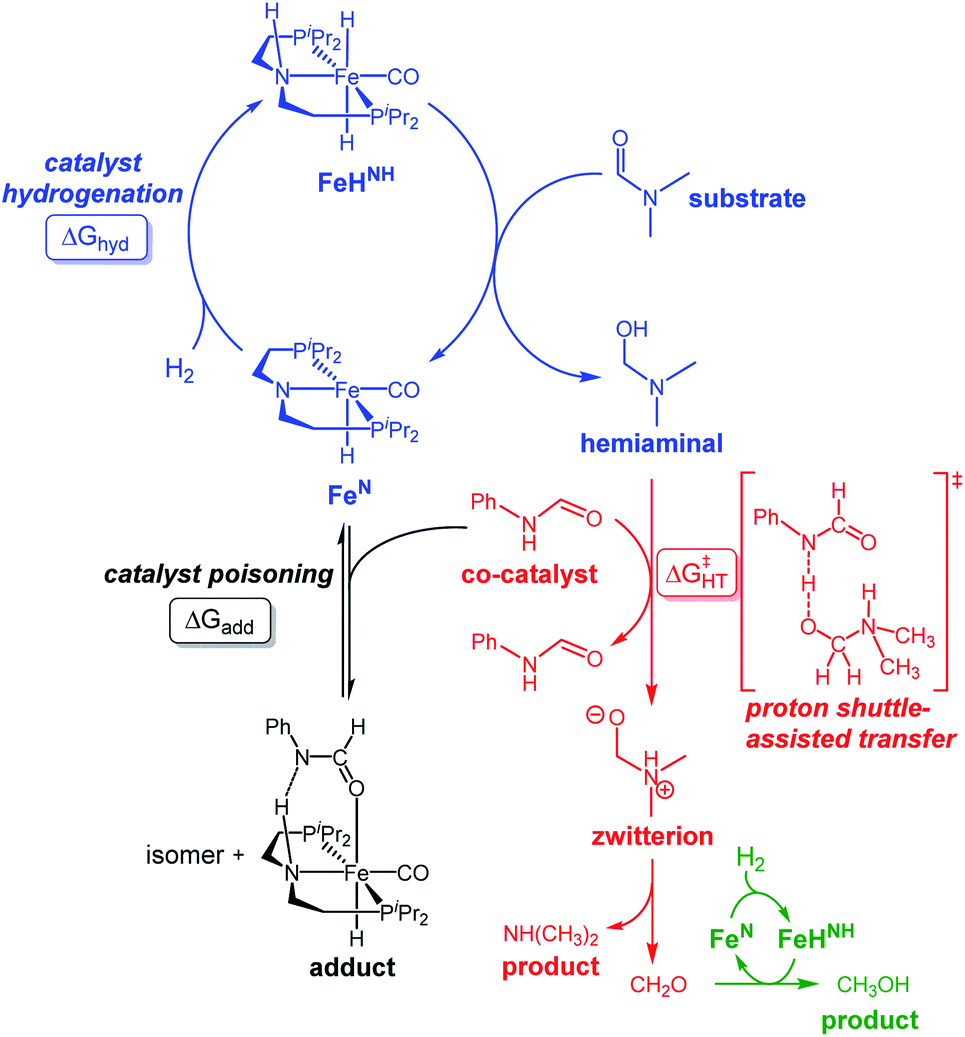 | ||
| Scheme 2 Reaction mechanism for the deaminative hydrogenation of amides by Noyori-type catalysts. Isomer = Fe N-bound form of the adduct. Color code: hemiaminal formation (blue), C–N cleavage by proton transfer (red), formaldehyde hydrogenation (green) and adduct formation (black). | ||
Results and discussion
Computational co-catalyst design
On the basis of the mechanism shown in Scheme 2, DFT calculations were performed on a series of potential organic co-catalysts for the hydrogenation of DMF using FeN (Table 1; see ESI† for computational details). The co-catalysts assessed included molecules with either single site hydrogen bond donors (entries 4–7 and 10) or with both hydrogen bond donor and acceptor sites which could act as push–pull proton shuttles (entries 1–3, 8 and 11). Various aryl and alkyl substituents (i.e. H, Me, iPr, tBu, Ph) were introduced into the pool of co-catalysts, to sample a wide range of stereoelectronic effects. In molecules with CO or CN functional groups, only electron-rich systems were chosen to minimize the hydrogenation of the co-catalyst. Although there are co-catalysts that are hydrogenated when used as reactants (e.g. formanilide and acetamide),5 when they are used in catalytic concentrations their consumption is slower than that of the reactants, enabling their co-catalytic effect.13 The ability of each potential co-catalyst to assist with the hemiaminal proton transfer5,18,19 involved in the C–N bond cleavage (step 2 in Scheme 1) was quantified by computing the transition state(s) associated with this process (ΔG‡HT; Scheme 2 and Fig. 1), which can be either concerted (with TBD, methanol and morpholine) or step-wise (all other co-catalysts). In the latter, the N-protonation of the hemiaminal is followed by its O-deprotonation, which is rate-limiting for all co-catalysts except urea. The thermodynamic preference of the co-catalyst to trap the iron complex was quantified by computing the free energy for the formation of the off-cycle adducts from FeN (ΔGadd; Scheme 2 and Fig. 1). In this framework, all co-catalysts were screened with the aim of finding an optimal balance between a low ΔG‡HT and a thermoneutral or endergonic value of ΔGadd. Catalyst hydrogenation (ΔGhyd; Scheme 2) competes with adduct formation and, thus, there is an interplay between the free energies of both reactions. In the case of FeN, ΔGhyd (which will depend on the nature of the catalyst) was calculated to be −10.2 kcal mol−1 under the experimental conditions.13 The value of ΔGadd − ΔGhyd (ΔGP, in Table 1) is therefore a measure of how adduct formation may limit the reaction by catalyst poisoning (i.e. a more positive value is indicative of less deactivation). For example, the production of methanol may be expected to inhibit catalysis by adduct formation with FeN (entry 7; ΔGadd = −6.9 kcal mol−1). However, catalyst hydrogenation is even more favorable (ΔGhyd = −10.2 kcal mol−1) making the FeHNH the likely preferred species (ΔGP = 3.3 kcal mol−1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Co-catalyst | ΔG‡HTa | ΔGaddb | ΔGPc | TONd | Conv.d |
| a ΔG‡HT (in kcal mol−1) corresponds to the calculated energy of the proton-transfer transition state with the highest energy for DMF assisted by the co-catalysts (Scheme 2, Fig. 1). b ΔGadd (in kcal mol−1) corresponds to the calculated energy for the formation of the adduct (isomer with the lowest energy) formed by [FeN] with the co-catalysts (Scheme 2, Fig. 1). c ΔGP = ΔGadd − ΔGhyd (−10.2 kcal mol−1 for all co-catalysts). d Experimental reaction conditions: 30 atm H2, 5 μmol of [FeN] (0.07 mol%), (1.75 mol%) of each additive and 7 mmol of 4-formylmorpholine in 5 mL of THF at 100 °C for 2 h. TON and conv. were determined by GC-FID analysis of the products and remaining starting material. Each entry is the average of two or more trials. | ||||||
| 1 |
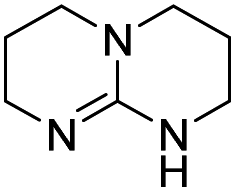
|
21.3 | −1.5 | 8.7 | 830 | 59% |
| 2 |
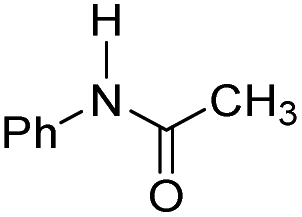
|
25.3 | −8.3 | 1.9 | 780 | 55% |
| 3 |
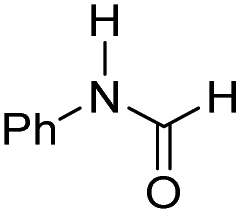
|
22.6 | −12.2 | −2.0 | 630 | 45% |
| 4 |
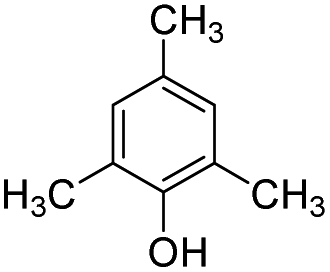
|
24.3 | −11.5 | −1.3 | — | — |
| 5 |
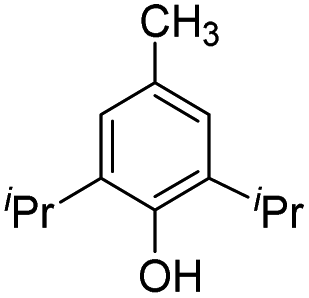
|
21.8 | −11.4 | −1.2 | — | — |
| 6 |
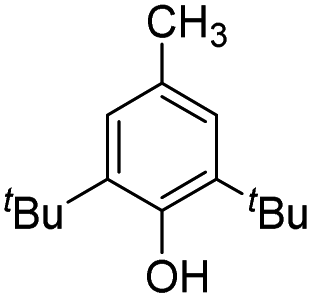
|
25.5 | 4.4 | 14.6 | 560 | 40% |
| 7 | CH3OH | 29.6 | −6.9 | 3.3 | 510 | 37% |
| 8 |
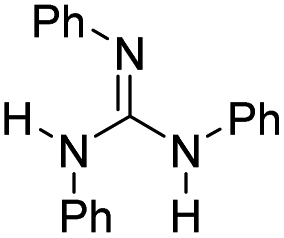
|
22.3 | −9.3 | 1.4 | 440 | 31% |
| 9 | No additive | — | — | — | 320 | 22% |
| 10 |
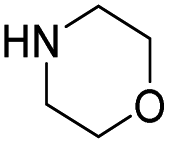
|
34.6 | 8.0 | 18.2 | 320 | 22% |
| 11 |
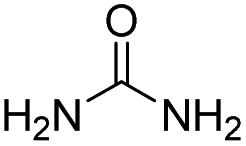
|
35.3 | −8.8 | 1.4 | 90 | 6% |
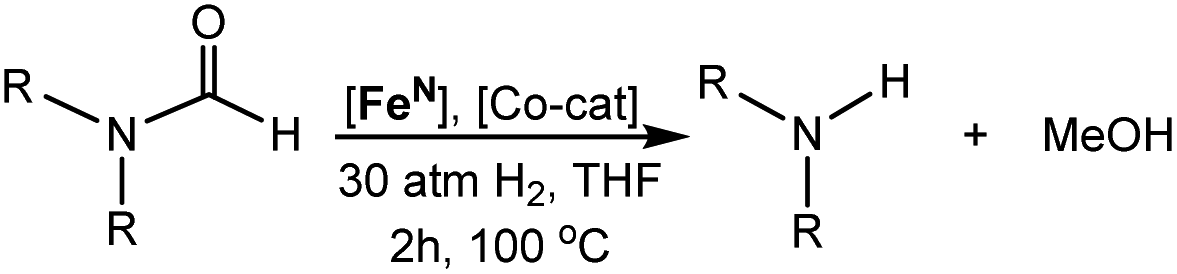
 | ||
| Fig. 1 TSs and adducts obtained from DFT calculations to compute the assisted proton transfer barrier (ΔG‡HT) and adduct formation free energy (ΔGadd) for the co-catalysts shown in Table 1. | ||
The DFT calculations using DMF as a model substrate yielded optimal results for TBD (triazabicyclodecene) as a co-catalyst (Table 1, entry 1). The basic and rigid character of the guanidine scaffold provides a low proton transfer barrier (ΔG‡HT = 21.3 kcal mol−1), facilitating the C–N bond cleavage of the hemiaminal intermediate. Additionally, TBD yielded a ΔGadd close to zero (−1.5 kcal mol−1) and the second largest ΔGadd − ΔGhyd (8.7 kcal mol−1), suggesting that the formation of the adduct does not compete with the hydrogenation of the amide. 1,2,3-Triphenylguanidine (entry 8) yielded a similar ΔG‡HT barrier, but with a more negative ΔGadd value (−9.3 kcal mol−1), likely due to its lower basicity compared to TBD. Acetanilide (entry 2) also afforded promising results, in this case showing that replacement of H by Me in the originally reported formanilide co-catalyst (entry 3) changes ΔGadd − ΔGhyd from negative to positive, meaning lower competition of the adduct formation towards amide hydrogenation. Among single site hydrogen bond donors, phenols (entries 4–6) exhibited some promise as a proton shuttle, although sterically large substituents were required to alleviate formation of iron adducts (AdductO, Fig. 1). Interestingly, morpholine and urea yield the largest energy barrier of all of the co-catalysts (34.6 and 35.3 kcal mol−1, respectively). This result suggests that a purely basic co-catalyst, although beneficial to prevent adduct formation, does not assist with the hemiaminal proton transfer. Overall, the computational results indicate that the best co-catalysts are those which provide spatially separated hydrogen bond donor and acceptor sites which can act as push–pull proton shuttles, together with a basic character and/or steric bulky groups to prevent the formation of adducts.
The high co-catalytic activity predicted for TBD (entry 1) was further analyzed by performing microkinetic modelling20–22 using the complex reaction network we previously found for amide hydrogenation (see ESI†).13 Under the conditions typically used experimentally (1.4 M of DMF, 0.02 M of TBD, 1 mM of FeN and fixed concentration of 0.162 M of H2, at 100 °C),5 the microkinetic model yielded a high conversion of 27% over a short reaction time of 2 hours. This conversion is substantially higher than the conversion with formanilide as co-catalyst (12%). The same trend was observed by using 4-formylmorpholine as the substrate (see ESI†),23 a benchmark tertiary amide used in our prior studies on FeN-catalyzed catalyzed deaminative hydrogenation. In this case, the conversions with TBD and formanilide were 56% and 46%, respectively.
Experimental co-catalyst and catalyst testing
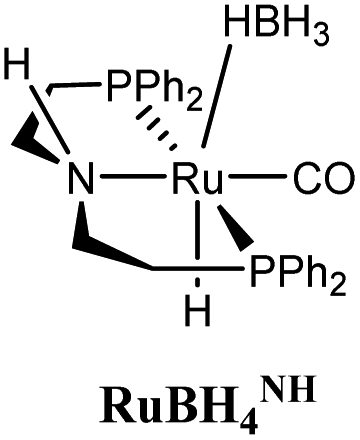
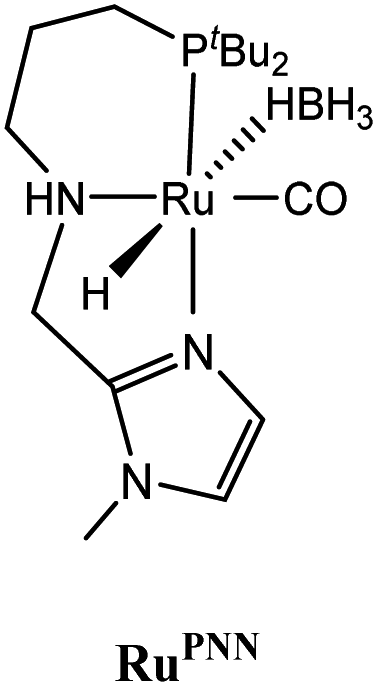
The computational predictions of co-catalyst efficacy were examined experimentally using 4-formylmorpholine (Table 1). Each potential co-catalyst was tested in catalytic trials with a 1.75 mol% loading, along with 0.07 mol% FeN under previously optimized conditions.5 Due to the high activity of the catalyst, the variable amount of time required to manipulate the pressure vessel between trials, and the need to equilibrate the vessel at the reaction temperature, it was not possible to acquire reliable initial rate measurements (conversions < 10%). Instead, reaction times were limited to 2 hours to minimize conversion and provide kinetically relevant comparisons. The reaction progress was monitored by amide conversion because of chromatographic issues in quantifying the morpholine product. However, no signals other than starting materials, morpholine and methanol were observed by GC-FID. As predicted by DFT, TBD proved a remarkable co-catalyst, affording a greater than two fold enhancement in TON (compare entries 1 and 9) over the short reaction time. Examination of the influence of TBD loading from 0 to 250 μmol (see ESI; Fig. S7†) indicated a strong correlation between TON and [TBD], saturating at approximately 200 μmol. The computational results also successfully predicted the relative ability of the other co-catalysts. For additives in which ΔGP is positive, the best co-catalysts should be those which lower the ΔG‡HT involved in the hemiaminal C–N bond cleavage, as illustrated in entries 1, 2, 6 and 7. In cases where iron deactivation is problematic due to a large negative ΔGadd, then the key barrier to amide hydrogenation is approximated by the total energy difference between ΔGadd − ΔGhyd and ΔG‡HT, which explains the superior performance of acetanilide over formanilide (entries 2 and 3). The only discernable variation from this trend is the unexpectedly poor performance of 1,2,3-triphenylguanidine (entry 8), which may react in a different manner as indicated by an immediate color change upon treatment with FeN. The use of urea in the reaction appeared to inhibit the reaction (lower conversion than without additive), which is likely due to other irreversible reactions with the iron species or difficulties in drying the very hydroscopic parent amide. Still, our rational co-catalyst design has led to the identification of a remarkably active catalytic system for selective amide hydrogenation.The few homogenous transition metal catalysts reported for deaminative hydrogenation are all proposed to follow similar pathways (Scheme 1), with Noyori-type bifunctional mechanisms being prominent.14,15 Given the importance of non-metal mediated hemiaminal cleavage in our computed mechanism, we hypothesized that the co-catalytic enhancements observed here with FeN should be generalizable to other systems. Indeed, highly active ruthenium catalysts recently reported by Beller and Sanford24–26 also exhibit substantial enhancement in activity upon co-catalytic addition of TBD or formanilide (Table 2). The (PhPNHP)Ru(H)(CO)(BH4) (RuBH4NH) precatalyst (PhPNHP = HN[CH2CH2(PPh2)]2) exhibited a near 4-fold increase in TON for 4-formylmorpholine hydrogenation over a short 2 hour reaction time in the presence of TBD, making it one of the most active systems for hydrogenation of this benchmark substrate. In this case, formanilide inhibits the reaction by forming a stable ruthenium adduct (Fig. S6†). In contrast, with the (PNN)Ru(H)(CO)(BH4) (RuPNN) (PNN = 3-(di-tert-butylphosphino)-N-[(1-methyl-1H-imidazol-2-l)methyl]propylamine) precatalyst, the relative difference in performance between TBD and formanilide is not as large, likely because the steric bulk of the tert-butyl substituents on the phosphine donors lowers the stability of a formanilide adduct.
|
|
|||
|---|---|---|---|
| Catalyst | Co-catalyst | TONb | Conv.b |
| a Reaction conditions: 30 atm H2, 5 μmol of [Fe or Ru] (0.07 mol%), 125 μmol of co-catalyst, and 7 mmol of 4-formylmorpholine in 5 mL of THF at 100 °C for 2 h. For [Ru] co-catalysts 10 μmol of NEt3 was added to activate the catalyst. b Determined by GC-FID analysis of the products and remaining starting material. Each entry is the average of two or more trials. c Formanilide reacts irreversibly with this Ru catalyst to form an adduct, see ESI for details. | |||
|
None | 320 | 23% |
| TBD | 830 | 59% | |
| HCONHPh | 630 | 45% | |
|
None | 310 | 22% |
| TBD | 1200 | 86% | |
| HCONHPh | 0c | 0c | |
|
None | 440 | 31% |
| TBD | 1170 | 84% | |
| HCONHPh | 1040 | 74% | |
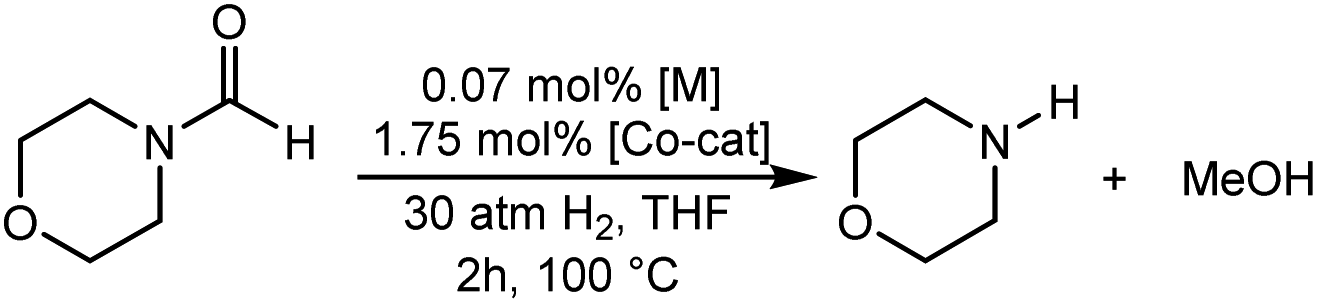
The co-catalytic effect of TBD with FeN across different classes of amides was also investigated experimentally (Table 3). Examples of dialkyl and diaryl formamides (entries 1 and 2) exhibited significant enhancement in TON in the presence of TBD compared to the reaction without co-catalyst. N-Phenylacetamide (entry 3), a substrate that previously proved challenging for FeN, was also hydrogenated with greater productivity in the presence of TBD. However, no enhancement was observed upon TBD treatment of the corresponding benzamide (entry 4). This may be due to steric limitation at the carbonyl moiety created by the larger phenyl substituent. In this case, substituting TBD for a smaller co-catalyst provided a modest increase in TON. These results suggest the co-catalytic effect of TBD and related shuttles may be effective with more diverse amides. Admittedly, the enhancement observed with diphenylformanilide was initially unexpected because a mechanism involving the iron-catalyst instead of formanilide, was previously proposed for the hemiaminal C–N bond cleavage using aryl amide substrates.13 However, the calculated ΔG‡HT using the diphenylformanilide hemianimal intermediate and TBD is lower (ΔG‡ = 21.8 kcal mol−1) than the barrier for the iron-assisted mechanism (ΔG‡ = 28.6 kcal mol−1, see Fig. 2). This result is in agreement with the enhanced reactivity observed for the hydrogenation of diphenylformanilide using TBD as co-catalyst (see Fig. 2).
|
|
|||
|---|---|---|---|
| Entry | Substrate | [TBD] | TONb |
| a Reaction conditions: 60 atm H2, 5 μmol of [Fe] (0.07 mol%), x μmol of TBD, and 7 mmol of substrate in 5 mL of THF at 120 °C for 16 h. b TON was determined by GC-FID and NMR analysis of the products and remaining starting material. Each entry is the average of three or more trials. c TBD was substituted by N-phenylacetamide (Table 1; entry 2). | |||
| 1 |
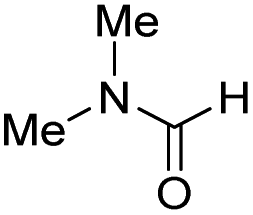
|
0 | 50 |
| 1.75 | 300 | ||
| 2 |
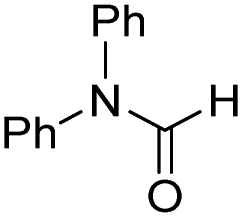
|
0 | 1150 |
| 0.45 | 5180 | ||
| 3 |
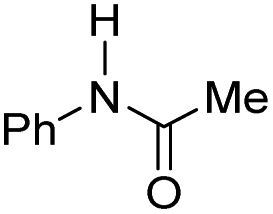
|
0 | 140 |
| 1.75 | 230 | ||
| 4 |
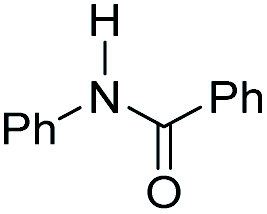
|
0 | 120 |
| 1.75 | 120 | ||
| 1.75c | 250c | ||

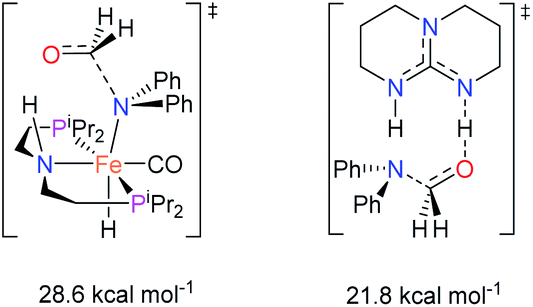 | ||
| Fig. 2 Gibbs energies associated with the C–N bond cleavage TSs for diphenylformamide assisted by FeN and the TBD co-catalyst. | ||
Conclusions
In conclusion, this work establishes the basis for co-catalyst optimization in amide deaminative hydrogenation reactions using Noyori-type catalysts. Key factors in the co-catalyst design include a push–pull motif of hydrogen bonding sites to assist the C–N bond cleavage of the hemiaminal and controlled acidity and steric hindrance to prevent catalyst poisoning. Notably, these design principles yielded co-catalysts enhancing the activity of systems based on different transition metals. The generality of the co-catalyst effect and its mechanistic understanding provide new opportunities for the catalytic hydrogenation of challenging electron-rich carbonyl compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ll. A., D. B. and A. N. thank the support from the Research Council of Norway (FRINATEK Grant No. 250044 and Center of Excellence Grand No. 262695), the Norwegian Metacenter for Computational Science (NOTUR, nn4654k) and NordForsk, (Grand No. 85378). U. J., W. B. and N. H. acknowledge support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program (DE-SC0018222).Notes and references
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC.
- G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- U. Jayarathne, Y. Zhang, N. Hazari and W. H. Bernskoetter, Organometallics, 2017, 36, 409–416 CrossRef CAS.
- G. Arthur, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, Hoboken, 1st edn, 2000 Search PubMed.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- D. G. Gusev, ACS Catal., 2017, 7, 6656–6662 CrossRef CAS.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- J. M. John and S. H. Bergens, Angew. Chem., Int. Ed., 2011, 50, 10377–10380 CrossRef CAS PubMed.
- M. Beller, W. Baumann, E. Alberico, H.-J. J. Drexler, J. R. Cabrero-Antonino, H. Junge, K. Junge, E. Alberico, H.-J. J. Drexler, W. Baumann, K. Junge, H. Junge and M. Beller, ACS Catal., 2016, 6, 47–54 CrossRef.
- J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285–5288 RSC.
- L. Artús Suàrez, Z. Culakova, D. Balcells, W. H. Bernskoetter, O. Eisenstein, K. I. K. I. Goldberg, N. Hazari, M. Tilset and A. Nova, ACS Catal., 2018, 8, 8751–8762 CrossRef.
- P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2018, 119, 2681–2751 CrossRef PubMed.
- S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS.
- C. Tang, K. Sordakis, H. Junge, M. Beller, L. K. Vogt, G. Laurenczy and P. J. Dyson, Chem. Rev., 2017, 118, 372–433 Search PubMed.
- A. Wakatsuki, Y. Kabe, M. Matsumoto, N. Watanabe and H. K. Ijuin, Tetrahedron Lett., 2018, 59, 971–977 CrossRef.
- B. Yuan, R. He, W. Shen, Y. Xu, X. Liu and M. Li, ChemistrySelect, 2016, 1, 2971–2978 CrossRef CAS.
- The microkinetic model was performed using COPASI 4.22 software.
- S. Hoops, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- M. Besora and F. Maseras, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 1372, 1–13 Search PubMed.
- Microkinetic models show that 4-formylmorpholine reacts in a similar manner than DMF (see ESI†).
- C. Bornschein, K. P. J. Gustafson, O. Verho, M. Beller and J. E. Bäckvall, Chem.–Eur. J., 2016, 22, 11583–11586 CrossRef CAS PubMed.
- R. Adam, E. Alberico, W. Baumann, H. J. Drexler, R. Jackstell, H. Junge and M. Beller, Chem.–Eur. J., 2016, 22, 4991–5002 CrossRef CAS PubMed.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details (including procedure for co-catalyst screening and synthesis of (PhPNHP)RuH(CO)(HCONPh)) and computational details (including information on the microkinetic models with DMF and 4-formylmorpholine, results obtained with diphenylformanilide, and optimized coordinates). CCDC 1943231. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03812d |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2020 |