
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borane-induced ring closure reaction of oligomethylene-linked bis-allenes†
Xin
Tao
,
Karel
Škoch
 ,
Constantin G.
Daniliuc
,
Gerald
Kehr
and
Gerhard
Erker
*
,
Constantin G.
Daniliuc
,
Gerald
Kehr
and
Gerhard
Erker
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
First published on 18th December 2019
Abstract
The trimethylene-linked bis-allene 3a reacts with Piers' borane [HB(C6F5)2] by a hydroboration/allylboration sequence to generate the cyclization product 5a. Its pyridine adduct was isolated and characterized by X-ray diffraction. Compound 5a undergoes a typical frustrated Lewis pair 1,2-P/B alkene addition reaction with PPh3 to give the heterobicyclic bridged olefinic zwitterionic product 9a. The tetramethylene-linked bis-allene 3b and its phenylene annulated analogue 3c react with HB(C6F5)2 to give the analogous seven-membered ring products 5b,c under mild conditions. The cyclization product 5a undergoes a series of sequential allylboration reactions with two equivalents of allene followed by ring-closure to give the four-component coupling product 12a. It undergoes FLP addition to an exo-methylene group upon treatment with PPh3. Compound 12a is oxidatively converted to the boron-free alcohol.
Introduction
Allenes are important building blocks in organic synthesis.1 They show interesting and useful stereochemical properties.2 We had recently shown that the borane [HB(C6F5)2]3 catalyses the cyclotrimerization of allene as well as a small series of mono-alkyl-substituted allenes 1 to selectively give the respective 1,3,5-trimethylene cyclohexanes 2 as single isomers under metal-free conditions (Scheme 1).1,4,5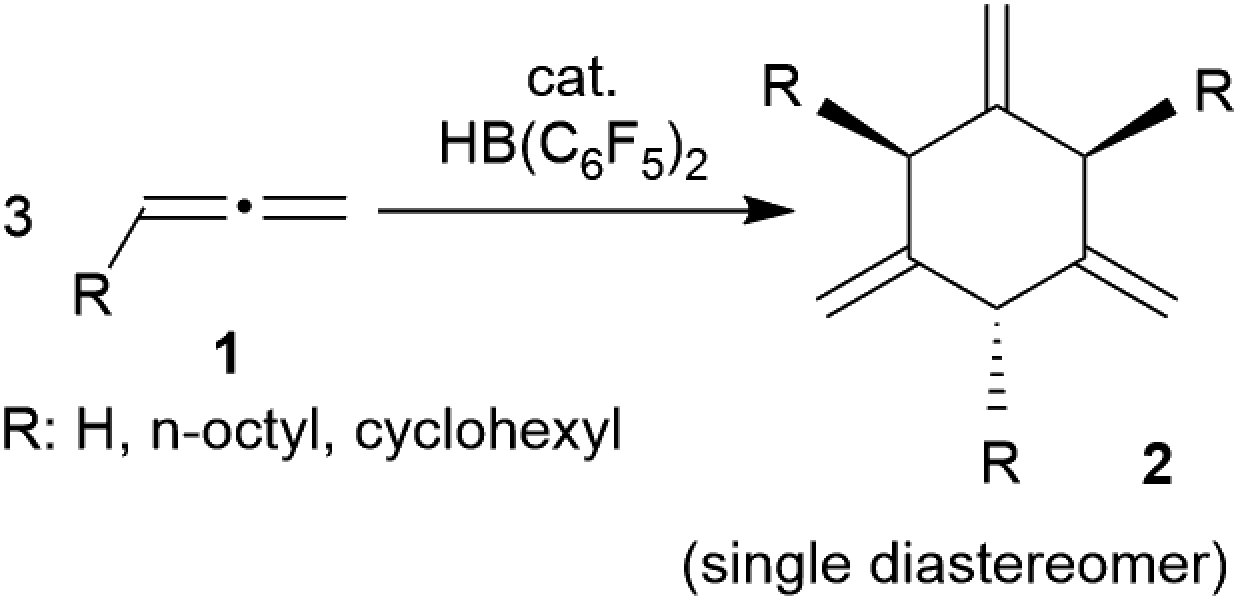 | ||
| Scheme 1 HB(C6F5)2-catalyzed cyclotrimerization of alkyl-substituted allenes. | ||
There is a rich cyclization chemistry of bis-allenes reported in the literature (see Chart 1). Systems I (mostly with X = NTs, less frequently CR2 or O) were reported to rearrange to II thermally induced.6a They added R3Si-SnBu3/or -GeR3 reagents Pd(0) catalysed or radical induced to give the products III.6b,c With H2NR nucleophiles cyclization to medium-sized rings (e.g.IV) was reported.6d The products V and VI of internal [2 + 2] cycloaddition were formed under the influence of Au(I)6e or Pd(0)6a catalysis, respectively. In some cases coupling between two bis-allenes occurred to give hetero-steroidal frameworks.6f,g It should be noted that the vast majority of these reaction is metal catalysed.
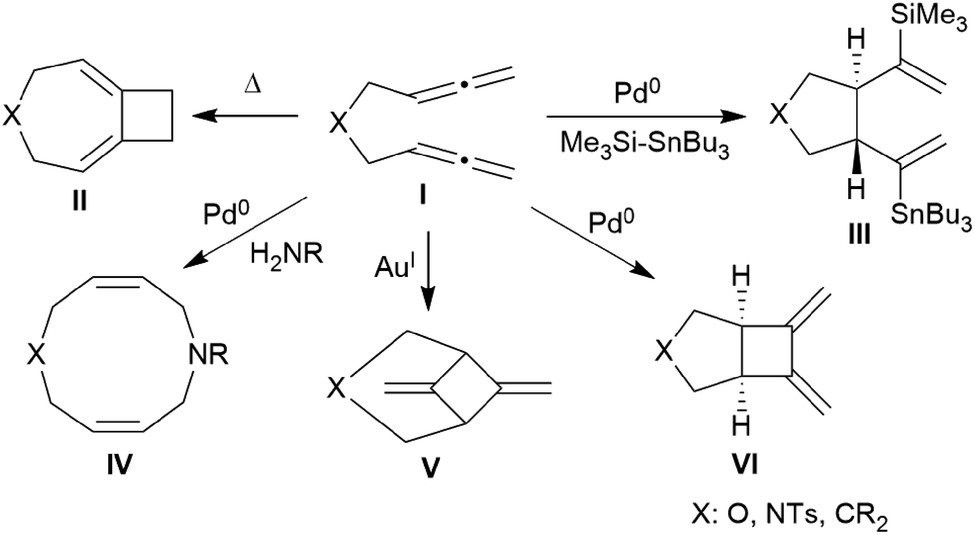 | ||
| Chart 1 Examples of cyclization reactions of bis-allenes. | ||
This posed the question what the favoured reaction pathway would be if we treated e.g. oligomethylene-linked bis-allenes with HB(C6F5)2,7i.e. under metal-free conditions. We have now performed these reactions starting from two examples of that bis-allene family. It turned out that a different cyclization type prevailed under these conditions. The outcome of these reactions will be presented and discussed below.
Results and discussion
Borane-induced ring-closure reaction of the bis-allenes
The allenes 3a and 3b (Scheme 2) were prepared by a variant of the Crabbé reaction as described by Ma et al.8 Copper(I) induced treatment of 1,6-heptadiyne with para-formaldehyde as C1-building block and dicyclohexylamine gave the trimethylene-linked bis-allene 3a6h (40% isolated). The analogous reaction starting from 1,7-octadiyne gave the tetramethylene-linked bis-allene 3b6i (63% isolated, see the ESI† for details). The phenylene containing bis-allene 3c was synthesised analogously.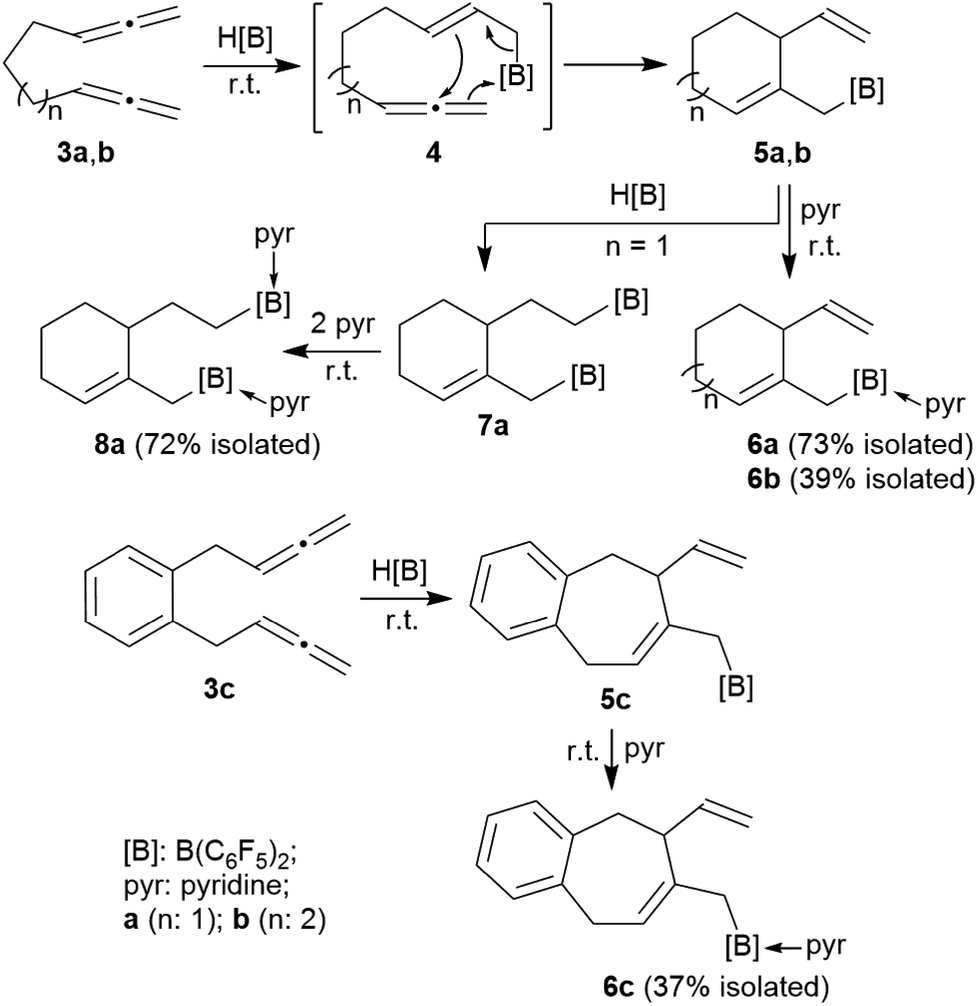 | ||
| Scheme 2 HB(C6F5)2-induced ring closure of the bis-allenes 3. | ||
We reacted compound 3a with one molar equivalent of Piers' borane [HB(C6F5)2]. The rapid reaction (r.t., minutes, in CD2Cl2) gave the cyclized product 5a. It was not isolated but characterized by spectroscopy [1H NMR: δ 5.55 (olefinic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) (C6–H, see Fig. 1 for the unsystematic atom numbering scheme), δ 5.52, 5.03, (–CHCH2 substituent); 11B NMR: δ 69.3; Δδ19Fm,p = 13.1 ppm]. Compound 5a was then generated in situ on a preparative scale and trapped by subsequent addition of pyridine. We isolated the pyridine adduct 6a as a white crystalline solid in 73% yield. The X-ray crystal structure analysis of compound 6a showed the newly formed cyclohexene core that has a vinyl group attached in the allylic position and it bears the –CH2B(C6F5)2(pyridine) substituent adjacent to it at the olefinic ring carbon atom C4 (Fig. 1). In solution (CD2Cl2) we monitored the typical 1H/13C NMR features of the vinyl group at carbon atom C3 and the central doubly substituted cyclohexene core. The 1H NMR features of the coordinated pyridine moiety show up at δ 8.67, 8.11 and 7.65 (11B: δ −0.6). Due to the chiral centre (ring carbon C3) the C6F5 groups at boron are diastereotopic and give rise to a 1
CH–) (C6–H, see Fig. 1 for the unsystematic atom numbering scheme), δ 5.52, 5.03, (–CHCH2 substituent); 11B NMR: δ 69.3; Δδ19Fm,p = 13.1 ppm]. Compound 5a was then generated in situ on a preparative scale and trapped by subsequent addition of pyridine. We isolated the pyridine adduct 6a as a white crystalline solid in 73% yield. The X-ray crystal structure analysis of compound 6a showed the newly formed cyclohexene core that has a vinyl group attached in the allylic position and it bears the –CH2B(C6F5)2(pyridine) substituent adjacent to it at the olefinic ring carbon atom C4 (Fig. 1). In solution (CD2Cl2) we monitored the typical 1H/13C NMR features of the vinyl group at carbon atom C3 and the central doubly substituted cyclohexene core. The 1H NMR features of the coordinated pyridine moiety show up at δ 8.67, 8.11 and 7.65 (11B: δ −0.6). Due to the chiral centre (ring carbon C3) the C6F5 groups at boron are diastereotopic and give rise to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 set of the respective o,p,m-C6F519F NMR signals.
:1 set of the respective o,p,m-C6F519F NMR signals.
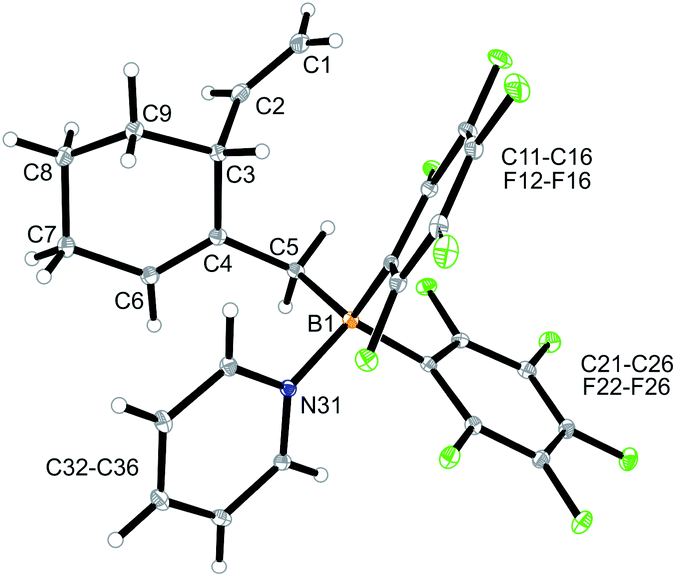 | ||
| Fig. 1 A view of the molecular structure of the borane pyridine adduct 6a. Selected bond lengths (Å) and angles (°): B1–N31 1.613(3), B1–C5 1.651(3), C1–C2 1.322(4), C4–C6 1.337(3), B1–C5–C4 117.7(2). | ||
We assume that the reaction started with hydroboration of the terminal CH2 unit of one allenyl group by the HB(C6F5)2 reagent. This resulted in the formation of the functionalized borane 4 which contained a reactive allylborane unit opposite to a reactive allene unit. This situation was set up for an internal allylboration reaction of the allene, which directly opened a pathway to the observed cyclization product 5a. Addition of the pyridine Lewis base to the strongly Lewis acidic B(C6F5)2 group gave 6a (Scheme 2).
We reacted the tetramethylene-linked bis-allene 3b with HB(C6F5)2 and found that the seven-membered cyclization product 5b was formed analogously. Treatment of the in situ generated borane 5b with pyridine gave the respective pyridine adduct 6b, which we isolated crystalline in 39% yield. Compounds 5b and 6b were characterized by spectroscopy (see the ESI† for details); product 6b was characterized by C, H, N elemental analysis and by an X-ray crystal structure analysis (Fig. 2). It shows the newly formed seven-membered ring in a typical cycloheptene boat-like conformation.9 The –CH2B(C6F5)2 moiety is attached at the sp2-carbon atom C4 (the boron atom bears a pair of C6F5 groups and the coordinated pyridine ligand in a pseudo-tetrahedral geometry). The vinyl substituent is bonded to carbon atom C3.
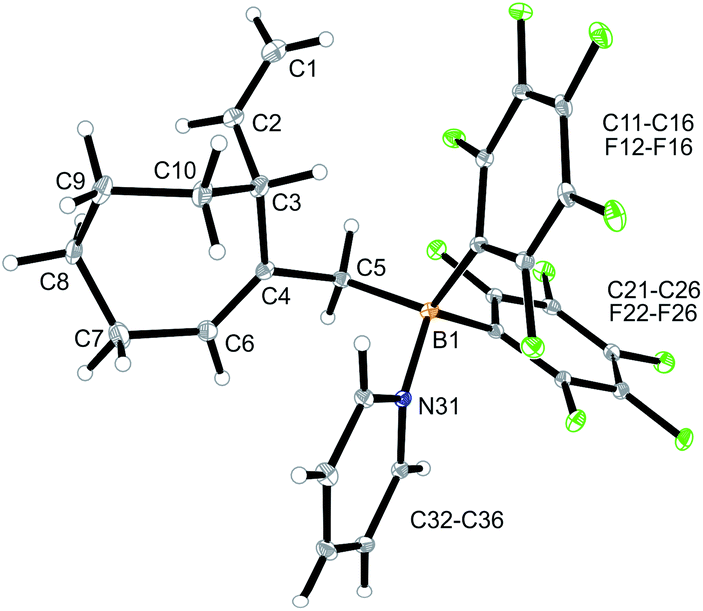 | ||
| Fig. 2 Molecular structure of compound 6b. Selected bond lengths (Å) and angles (°): B1–N31 1.627(5), B1–C5 1.645(6), C1–C2 1.285(8), C4–C6 1.340(6), B1–C5–C4 119.9(3). | ||
The reaction of the bis-allene 3a responded to the stoichiometry of the HB(C6F5)2 reagent since the primary ring-closure product 5a contained a reactive pendant vinyl group. Therefore, the reaction of 3a with two molar equivalents of Piers' borane gave the bis-borane product 7a. Its 11B NMR spectrum showed two signals (δ 73.3 and δ 69.4) that are attributed to the pair of Lewis acidic planar-tricoordinate boron centres. Consequently, we observed two sets of 19F NMR resonances of the pairs of the C6F5 substituents at the two B(C6F5)2 groups. The in situ generated bis-borane 7a was then treated with two molar equivalents of pyridine to give the respective double pyridine adduct 8a (isolated as a white solid in 72% yield). It was characterized by C, H, N elemental analysis, by X-ray diffraction (the structure is shown in the ESI†) and by spectroscopy. Due to the chiral centre (C3) the pair of C6F5 groups at each B(C6F5)2(pyridine) moiety are diastereotopic and, consequently, we observed four sets of o,m,p-C6F519F NMR signals of compound 8a. We observed two sets of pyridine 1H/13C NMR resonances and located the single olefinic 1H NMR signal (C6–H) at δ 4.35 (t, JHH = 3.3 Hz, 1H).
We performed the reaction of the phenylene bridged bis-allene system 3c in a similar way. The reaction of 3c with one molar equiv. of HB(C6F5)2 was carried out in CD2Cl2 at r.t. and the almost instantaneously in situ generated product was characterized by NMR spectroscopy [1H: δ 5.48/5.13/5.08 (vinyl substituent), 5.72 (ring-CH), 11B: δ 71.9, 19F: Δδ19Fm,p = 13.2]. The latter heteroatom NMR signals are typical for the presence of strongly Lewis acidic tricoordinate boron with this substituent pattern. Compound 5c was treated with pyridine and the respective pyridine/borane Lewis adduct 6c was isolated in 37% yield after workup involving pentane extraction. The NMR spectra now show a 11B NMR resonance in the typical range of tetracoordinate boron (δ −0.5) and the C6F5 substituents at boron are diastereotopic (see the ESI† for further details).
Subsequent FLP ring-closure reactions
The compounds 5 each contain a sterically encumbered, strongly Lewis acidic borane functionality and in its vicinity an accessible reactive vinyl group. We used this for carrying out a typical frustrated Lewis pair10 reaction, namely a 1,2-borane/phosphane addition to the CC double bond.11
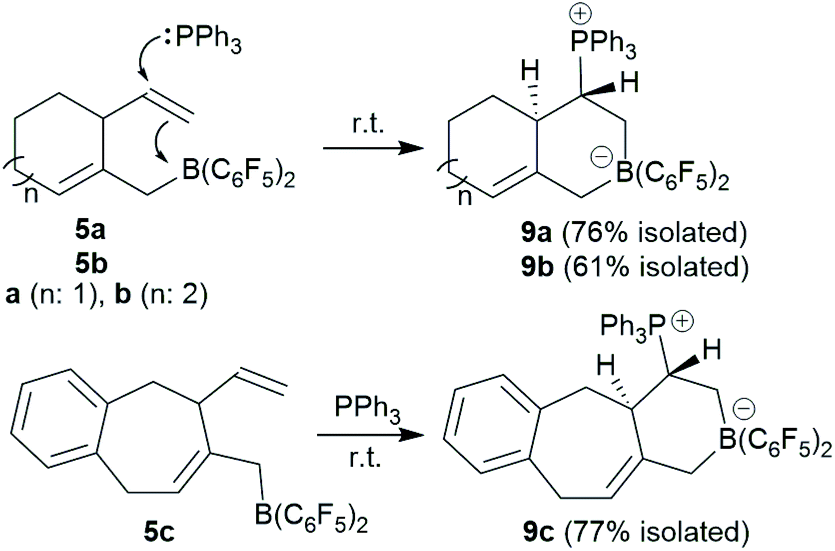
We reacted the in situ generated cyclization product 5a with triphenylphosphane at room temperature. The reaction was practically instantaneous under these typical conditions and we isolated the P/B addition product 9a to the internal vinyl group as a white powder in 76% yield (Scheme 3). Single crystals of compound 9a that were suited for the X-ray crystal structure analysis were obtained at room temperature from a solution in dichloromethane that was layered with pentane. It showed that 1,2-phosphane/borane addition to the vinyl substituent had occurred. The internal –B(C6F5)2 Lewis acid had been added to the CH2 terminus of the alkene and the external PPh3 nucleophile to the –CH carbon atom. The resulting zwitterionic heterobicyclo[4.4.0]decene type system features a bridgehead CC double bond (C4–C6). There is a borate system inside the heterocyclic six-membered ring that was formed in the FLP addition reaction. Consequently, the –PPh3+ phosphonium substituent is found attached at the same ring at carbon atom C2. We have isolated a single diastereoisomer of 9a from this reaction; it features the hydrogen atoms at carbon atoms C2 and C3 oriented trans to each other at the heterobicyclic framework (Fig. 3). In solution compound 9a shows the NMR heteronuclear resonances at δ −12.3 (11B) and δ 27.9 (31P). A diastereotopic pair of C6F5 substituents is bonded at boron, giving rise to two separate sets of 19F NMR resonances. The 1H NMR [P]–CH– signal is found at δ 3.64 and the single olefinic CH– signal at δ 5.17.
 | ||
| Scheme 3 FLP reaction of the boranes 5 with PPh3. | ||
 | ||
| Fig. 3 Molecular structure of the P/B FLP addition product 9a. Selected bond lengths (Å) and angles (°): B1–C1 1.647(2), B1–C5 1.640(2), P1–C2 1.856(2), C1–C2 1.549(2), C4–C6 1.333(2), C1–B1–C5 105.7(1), B1–C1–C2 114.6(1), C1–C2–C3 112.4(1), C2–C3–C4 109.2(1), C3–C4–C5 114.9(1), C4–C5–B1 108.9(1), P1–C2–C1 109.3(1), P1–C2–C3 111.9(1), P1–C2–C3–C4 174.9(1), P1–C2–C1–B1 175.9(1). | ||
The seven-membered ring compounds 5b and 5c react analogously with PPh3. Compound 5b was in situ generated and triphenylphosphane was added. We isolated the product 9b in 61% yield by crystallization. It was characterized by C, H-elemental analysis, by spectroscopy and by X-ray diffraction. The molecular structure is similar to that of 9a. It also contains a trans-relationship of the C3–H and C2–H hydrogen atoms of the heterobicyclic ring system. The hetero-NMR signals occur at δ −13.3 (11B) and δ 30.5 (31P). Compound 9c was prepared analogously from 5c (see Scheme 3). It was isolated as a white solid in 77% yield after workup and characterized by NMR spectroscopy and by an X-ray crystal structure analysis. For further details of the characterization of the compounds 9b and 9c including their depicted molecular structures see the ESI.†
Subsequent cyclooligomerization reaction with allene
Internal allene allylboration12 represents the important step of the ring-closure reaction sequence starting from 3 to form the products 5. The compounds 5 themselves each contain an allylborane functionality which might show the respective reactivity towards added allene reagents. Therefore, we exposed the cyclization product 5a to an excess of the parent allene H2CCCH2. An NMR experiment revealed a close to complete conversion to the new product 12a within 24 h at room temperature. We carried out this reaction on a preparative scale under analogous conditions. Workup involving crystallization from pentane at −35 °C (3 d) gave the crystalline product 12a, which we isolated in 37% yield. Compound 12a was characterized by C, H elemental analysis, by spectroscopy and by an X-ray crystal structure analysis. This showed (Fig. 4) that the endocyclic allylborane moiety of the starting material 5a had reacted with two molar equivalents of H2CCCH2 to give the functionalized decalin derivative 12a. Apparently, compound 5a had undergone an allylboration reaction with allene to generate the intermediate 10a. This itself represents an “elongated” allylborane system, that subsequently took up another allene equivalent to give 11a. The intermediate 11a could in principle have reacted with further allene, but instead its allylborane “found” the remaining exo-methylene group at the six-membered core with which it underwent a favoured intramolecular allylboration reaction4 to directly give the observed product 12a (Scheme 4).
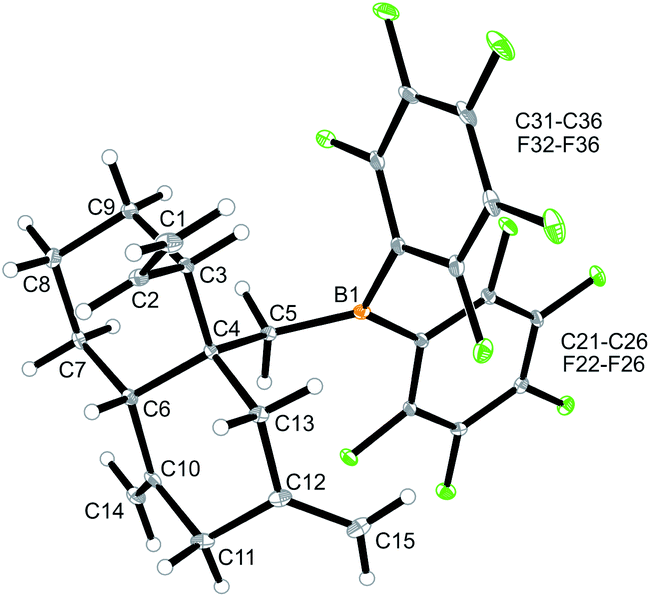 | ||
| Fig. 4 Molecular structure of compound 12a. Selected bond lengths (Å) and angles (°): B1–C5 1.554(3), C1–C2 1.313(3), C4–C5 1.557(3), C4–C6 1.566(3), C10–C14 1.324(3), C12–C15 1.324(3), B1–C5–C4 123.2(2), C5–C4–C6 109.0(2). | ||
 | ||
| Scheme 4 Formation of compound 12a and allene by sequential allylboration reactions. | ||
The X-ray crystal structure analysis of compound 12a shows the newly formed trans-decalin framework that was formed by the consecutive C–C coupling between 5a and two molar equivalents of allene. The ring carbon atom C3 bears the remaining vinyl substituent; carbon atoms C10 and C12 are both part of the pair of exo-methylene groups 1,3-positioned in the second ring. The –CH2B(C6F5)2 group is attached at the bridgehead carbon (C4 in Fig. 4).
In solution (CD2Cl2) compound 12a shows the typical NMR features of the vinyl substituent. The pair of CH2 exo-methylene groups shows a total of four 1H NMR resonances (δ 4.85/4.61 and δ 4.68/4.42). The –CH2[B] substituent shows the 1H NMR signals of a pair of diastereotopic hydrogen atoms (AB system at δ 2.30/2.15; 13C: δ 36.7). The corresponding 11B NMR feature is at δ 76.4, i.e. in a typical range of a strongly Lewis acidic tri-coordinated boron atom in this substituent situation.13 Consequently, we observed three 19F NMR signals of the pair of the C6F5 substituents at boron with a large Δδ19Fm,p = 11.5 ppm chemical shift separation.
Some typical reactions of the borane 12a
Compound 12a is a reactive borane and it contains CC double bond functionalities. Therefore, it should be suitable to undergo typical FLP addition to one of the olefinic units in the presence of an external phosphane nucleophile.11 We, consequently, reacted the in situ generated borane 12a with triphenylphosphane in dichloromethane. The reaction with PPh3 was instantaneous. The volatiles were removed and the residue was washed with pentane to give the P/B addition product 13a, which we isolated as a white solid in 56% yield (see Scheme 5). Compound 13a was characterized by C, H-elemental analysis, by spectroscopy and by X-ray diffraction (Fig. 5). It showed that a P/B FLP addition had taken place at the proximal CCH2 moiety (C12C15 in compound 12a, see Fig. 4) by using the adjacent pendent internal borane and the external phosphane. Compound 13a is the isomer that was formed by borane addition to the CCH2 terminus and, consequently, phosphane addition to the sp2-ring carbon atom. This resulted in the formation of a heterocyclic six-membered ring-system that had become 1,3-attached at the “lower” six-membered decalin ring of compound 12a. The phosphonium PPh3+ moiety is found attached at the new bridgehead atom C12 (Fig. 5).
 | ||
| Scheme 5 Reaction of compound 12a with PPh3 and oxidative replacement of the boryl group. | ||
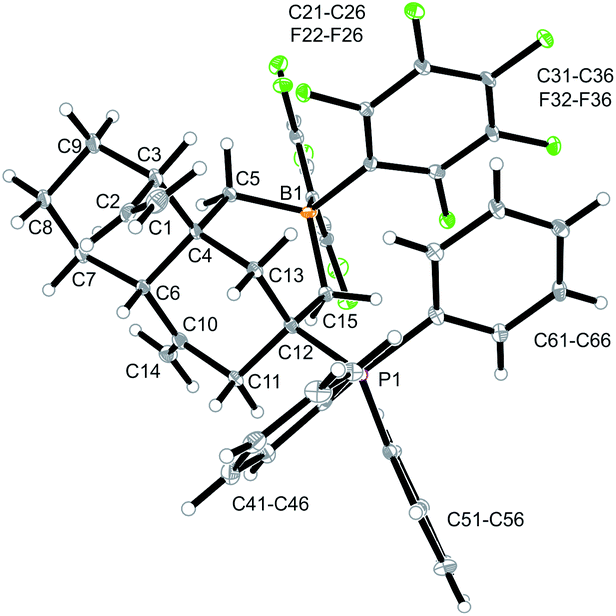 | ||
| Fig. 5 A view of the P/B FLP alkene addition product 13a. Selected bond lengths (Å) and angles (°): B1–C5 1.640(6), B1–C15 1.667(6), P1–C12 1.858(4), C1–C2 1.302(6), C4–C6 1.565(5), C10–C14 1.328(6), C12–C15 1.555(5), C5–B1–C15 110.3(3), C12–C15–B1 114.3(3), P1–C12–C15–B1 −129.8(3). | ||
Compound 13a shows a typical borate 11B NMR resonance at δ −15.1 in solution (CD2Cl2, 273 K) and a phosphonium 31P NMR signal at δ 31.1. It shows the 19F NMR features of a pair of diastereotopic C6F5 groups at the boron atom (for further details of the NMR characterization of compound 13a see the ESI†).
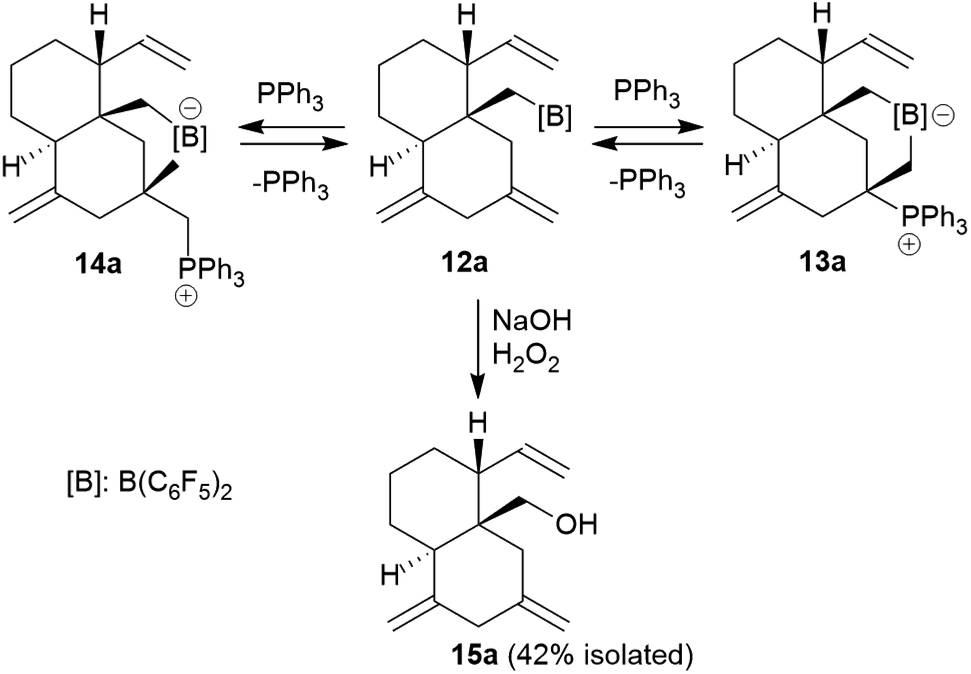
Compound 13a is the P/B FLP addition product that has been formed under kinetic control. When we stored the CD2Cl2 solution of compound 13a for 7 days at room temperature the resulting NMR spectra showed the formation of an equilibrium mixture of 13a (ca. 20 mol%), the starting material 12a (plus PPh3, ca. 8 mol%) and the new compound 14a (ca. 65 mol%) (plus some minor contaminants). The major product 14a, apparently formed under thermodynamic control, was prepared similarly on a preparative scale (24 h, r.t., CH2Cl2 layered with pentane) and crystallized from the mixture. Crystalline compound 14a was isolated in 60% yield and the product was characterized by C, H-elemental analysis, by spectroscopy and by X-ray diffraction.
The X-ray crystal structure analysis of 14a shows the presence of a five-membered boratacycle that is 1,3-annulated to the “lower” six-membered ring of the trans-decalin framework. It was apparently formed by a 1,2-P/B FLP addition to the C12C15 carbon–carbon double bond of the starting material 12a, similar as we had seen it in the formation of its isomer 13a, only that in this case PPh3 addition had taken place at the CH2 terminus of the exo-methylene group concurrent with borane addition to its adjacent doubly substituted sp2-carbon atom. The structure of the resulting P/B zwitterion 14a is depicted in Fig. 6.
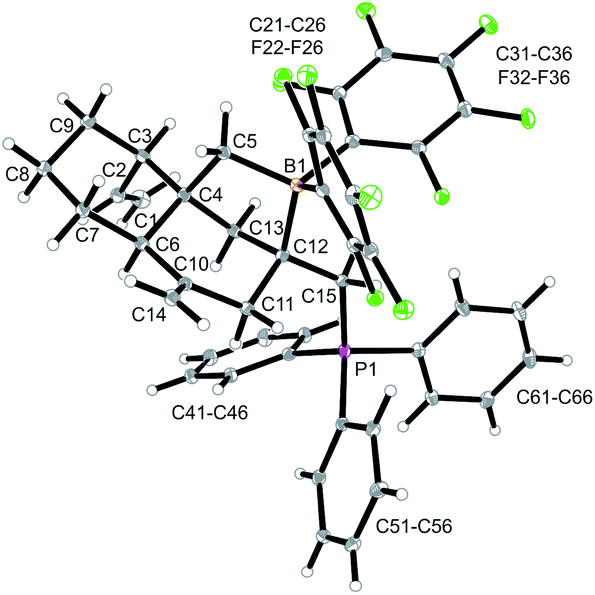 | ||
| Fig. 6 A projection of the molecular structure of compound 14a. Selected bond lengths (Å) and angles (°): B1–C5 1.657(4), B1–C12 1.695(3), P1–C15 1.823(2), C1–C2 1.301(13), C4–C6 1.556(3), C10–C14 1.325(4), C12–C15 1.540(3), C5–B1–C12 99.9(2), C12–C15–B1 112.1(2), B1–C12–C15–P1 179.1(2). | ||
The 1H NMR spectrum of compound 14a (in CD2Cl2, at 299 K) shows the P-coupled system of the exocyclic –CH2–[P] moiety at δ 4.18/2.70 and the resonances of the endocyclic –CH2–[B] group at δ 1.32/0.35. The heteroatom NMR signals occur at δ −6.8 (11B) and 19.9 (31P), respectively and we observed two sets of 19F NMR signals of the pair of diastereotopic C6F5 groups at boron (for further details see the ESI†).
We eventually converted the borane-induced multi-component cyclization product 12a to a boron-free derivative.14 This was carried out in the usual way of oxidative deborylation as it is done in conventional hydroboration chemistry.15 Treatment of the strongly electrophilic –B(C6F5)2 borane 12a with NaOH/H2O2 gave the alcohol 15a that we isolated as a white solid in 42% yield after workup (see the ESI† for its characterization by NMR spectroscopy).
We briefly investigated the reaction of the bis-allenic ether 166c,h,j with HB(C6F5)2. The reaction was carried out in CD2Cl2 solution at r.t. The products of the reaction were not isolated but directly identified in situ generated from the solution. We subsequently added a total of three molar equivalents of HB(C6F5) to eventually achieve a complete conversion of compound 16 with a clean product formation. The NMR analysis (for details see the ESI†) revealed that the by far predominant reaction was ether cleavage. This gave the (C6F5)2B–O–CH2–CHCCH2 cleavage product 17 (see Scheme 6) and butadiene (18) as primary products. The latter was then subsequently converted by added HB(C6F5)2 to the bis-hydroboration product 19. The boryl ether 17 also was not stable under the reaction conditions, probably due to subsequent ether cleavage with additional HB(C6F5)2 (see the ESI† for details). We also investigated briefly the reaction of the respective bis-allenic N-tosyl amine with HB(C6F5)2, but that gave a complicated mixture of as yet unidentified products.
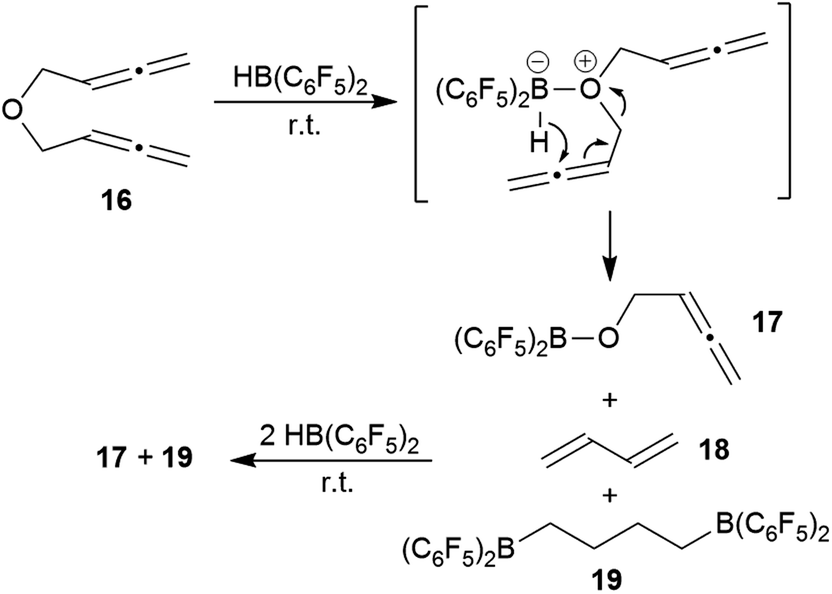 | ||
| Scheme 6 Reaction of the bis-allenic ether 16 with HB(C6F5)2. | ||
Conclusions
With this study we have found a new variant of our borane induced carbon–carbon coupling reactions between allene building blocks. In this case the reaction starts as it is commonly observed in our systems by 1,2-[B]–H addition16 to a terminal alleneCH2 group by the strongly electrophilic HB(C6F5)2 hydroboration reagent to probably generate a reactive allylborane intermediate in situ, which is set for undergoing rapid intramolecular ring-closure with the pendant second allenyl moiety to generate the products 5a to 5c, respectively. These are then obviously protected by their special geometry from undergoing further intermolecular allylborane coupling under the applied reaction conditions, so that the reaction stopped at the functionalized six-or seven-membered ring products. The compounds 5 are, however, in principle still active allylboration reagents. This we could show by the rapid reaction of the example 5a with the parent allene H2CCCH2. Two equivalents of allene were consumed in a sequence of consecutive intramolecular allylboration reactions, followed by a final intramolecular allylboration ring-closure reaction to give the four-component coupling product 12a. This in turn was oxidatively converted to the boron-free product 15a. These metal-free reactions are markedly different from the common metal catalysed bis-allenic cyclization reactions reported in the literature (see Chart 1 and the respective references). We will see how the products of our metal free cyclization reactions and their follow-up products (and related systems) might become easily available useful reagents for further external C–C coupling reactions using either of the newly generated functionalities.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS; (b) Modern Allene Chemistry, ed. N. Krause, and A. S. K. Hashmi, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (c) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef; (d) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (e) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (f) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC.
- C. Gropp, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2016, 55, 14444 CrossRef CAS PubMed.
- (a) D. J. Parks, R. E. H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef CAS; (b) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS; (c) M. Hoshi, K. Shirakawa and M. Okimoto, Tetrahedron Lett., 2007, 48, 8475 CrossRef CAS; (d) A. Schnurr, K. Samigullin, J. M. Breunig, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2011, 30, 2838 CrossRef CAS; (e) J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757 CrossRef CAS PubMed.
- (a) X. Tao, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2017, 56, 1376 CrossRef CAS PubMed; (b) See also: X. Tao, C. Wölke, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2019, 10, 2478 RSC.
- Arylallenes react differently, see: X. Tao, C. G. Daniliuc, D. Dittrich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2018, 57, 13922 CrossRef CAS PubMed.
- (a) X. Jiang, X. Cheng and S. Ma, Angew. Chem., Int. Ed., 2006, 45, 8009 CrossRef CAS PubMed; (b) S.-K. Kang, T.-G. Baik, A. N. Kulak, Y.-H. Ha, Y. Lim and J. Park, J. Am. Chem. Soc., 2000, 122, 11529 CrossRef CAS; (c) Y.-T. Hong, S.-K. Yoon, S.-K. Kang and C.-M. Yu, Eur. J. Org. Chem., 2004, 4628 CrossRef CAS; (d) J. Cheng, X. Jiang and S. Ma, Org. Lett., 2011, 13, 5200 CrossRef CAS PubMed; (e) S. M. Kim, J. H. Park, Y. K. Kang and Y. K. Chung, Angew. Chem., Int. Ed., 2009, 48, 4532 CrossRef CAS PubMed; (f) S. Ma, P. Lu, L. Lu, H. Hou, J. Wei, Q. He, Z. Gu, X. Jiang and X. Jin, Angew. Chem., Int. Ed., 2005, 44, 5275 CrossRef CAS PubMed; (g) S. Ma and L. Lu, Chem.–Asian J., 2007, 2, 199 CrossRef CAS PubMed; (h) X. Lian and S. Ma, Chem.–Eur. J., 2010, 16, 7960 CrossRef CAS PubMed; (i) V. A. D'yakonov, G. N. Kadikova, L. M. Khalilov and U. M. Dzhemilev, Russ. J. Org. Chem., 2013, 49, 1139 CrossRef; (j) S.-K. Kang, Y.-H. Ha, D.-H. Kim, Y. Lim and J. Jung, Chem. Commun., 2001, 1306 RSC.
- For examples of thermally induced reactions of bis-allenes, see: (a) L. Skattebol and S. Solomon, J. Am. Chem. Soc., 1965, 87, 4506 CrossRef; (b) W. R. Roth, M. Heiber and G. Erker, Angew. Chem., Int. Ed. Engl., 1973, 12, 504 CrossRef; (c) S. Sakai, J. Phys. Chem. A, 2006, 110, 9443 CrossRef CAS PubMed.
- (a) J. Kuang and S. Ma, J. Org. Chem., 2009, 74, 1763 CrossRef CAS PubMed ; for the original Crabbé reaction see e.g.; (b) P. Crabbé, H. Fillion, D. André and J.-L. Luche, J. Chem. Soc., Chem. Commun., 1979, 859 RSC ; see also; (c) S. Kitagaki, M. Komitzu and C. Mukai, Synlett, 2011, 1129 CrossRef CAS.
- Basic Organic Stereochemistry, ed. E. L. Eliel, S. H. Wilen and M. P. Doyle , John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- (a) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968 CrossRef CAS PubMed; (b) J. B. Sortais, T. Voss, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2009, 7417 RSC; (c) T. Voss, J.-B. Sortais, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2011, 30, 584 CrossRef CAS; (d) X. X. Zhao and D. W. Stephan, J. Am. Chem. Soc., 2011, 133, 12448 CrossRef CAS PubMed; (e) G. Ménard, L. Tran, J. S. J. McCahill, A. J. Lough and D. W. Stephan, Organometallics, 2013, 32, 6759 CrossRef.
- (a) B. M. Mikhailov and Y. N. Bubnov, Izv. Akad. Nauk SSSR, Ser. Khim., 1964, 13, 1874 Search PubMed; (b) Organoboron Compounds in Organic Synthesis, ed. B. M. Mikhailov and Y. N. Bubnov, Harvood, Chur, 1984 Search PubMed; (c) Y. N. Bubnov, Pure Appl. Chem., 1987, 59, 895 CAS; (d) R. W. Hoffmann, Pure Appl. Chem., 1988, 60, 123 CAS; (e) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (f) S. Y. Erdyakov, M. E. Gurskii, A. V. Ignatenko and Y. N. Bubnov, Mendeleev Commun., 2004, 14, 242 CrossRef; (g) M. E. Gurskii, S. Y. Erdyakov, T. V. Potapova and Y. N. Bubnov, Russ. Chem. Bull., 2008, 57, 802 CrossRef CAS.
- For a comparison, the 11B NMR signal of B(C6F5)3 is δ 59.5. (a) G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245 CrossRef; (b) A. G. Massey and A. J. Park, J. Organomet. Chem., 1966, 5, 218 CrossRef CAS.
- We had previously shown that alkenyl-B(C6F5)2 derivatives undergo typical borane reactions, e.g. Pd-catalyzed cross-coupling with iodobenzene: (a) C. Chen, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2010, 132, 13594 CrossRef CAS PubMed; (b) C. Chen, T. Voss, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2011, 13, 62 CrossRef CAS PubMed.
- (a) H. C. Brown and B. C. Rao, J. Am. Chem. Soc., 1956, 78, 5694 CrossRef CAS; (b) H. C. Brown and B. C. Rao, J. Org. Chem., 1957, 22, 1136 CrossRef CAS; (c) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1959, 81, 247 CrossRef CAS.
- For an example of a rare alternative 1,1-hydroboration reaction, see: A. Ueno, J. Yu, X. Tao, C. G. Daniliuc, G. Kehr and G. Erker, Organometallics, 2018, 37, 2665 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, further spectral and crystallographic data. CCDC deposition numbers are 1922906–1922913 and 1957134. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03870a |
| This journal is © The Royal Society of Chemistry 2020 |