
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimeric boroles: effective sources of monomeric boroles for heterocycle synthesis†
Xiaojun
Su
 ,
J. J.
Baker
and
Caleb D.
Martin
*
,
J. J.
Baker
and
Caleb D.
Martin
*
Department of Chemistry and Biochemistry, Baylor University, One Bear Place #97348, Waco, TX 76798, USA. E-mail: caleb_d_martin@baylor.edu
First published on 29th October 2019
Abstract
Monomeric boroles have been gaining attention as reagents for the synthesis of heterocycles due to their ability to insert atoms into the BC4 ring in a single step. Although unique boron frameworks can be accessed via this methodology, the products feature aryl substitution on the carbon centers as steric bulk is required to preclude borole dimerization. This work demonstrates that insertion chemistry is possible with Diels–Alder dimeric boroles and that such reactivity is not exclusive to monomeric boroles with bulky groups. With 1-phenyl-2,3,4,5-tetramethylborole dimer, the formal 1,1-insertion of a nitrene and sulfur generate the six-membered aromatic 1,2-azaborine and 1,2-thiaborine, respectively. The isolation of the 1,2-thiaborine enabled the synthesis of an η6-chromium complex. Benzophenone and diphenylketene readily insert a CO unit to generate BOC5 seven-membered rings confirming dimeric boroles can serve as monomeric synthons in 1,2-insertion reactions. An epoxide did not furnish the anticipated eight-membered BOC6 ring, instead provided a bicyclic system with a BOC3 ring. The insertion chemistry was demonstrated with two other borole dimers featuring different substitution with diphenylketene as a substrate. This work elevates borole insertion chemistry to a new level to access products that do not require bulky substitution.
Boroles are reactive BC4 heterocycles that feature a three-coordinate boron center linking a 1,3-butadiene backbone first disclosed by Eisch in his seminal report in 1969.1 The four π-electron ring results in an anti-aromatic species high in thermodynamic energy making boroles attractive reagents for more stable species. Within the central ring, the boron center is highly Lewis acidic, the BC4 ring can be reduced, and the diene engages in Diels–Alder reactions.2–4 It has been demonstrated that Lewis acid–base adducts can rearrange if a reactive functionality is pendent on the Lewis base (e.g. imine, nitrile) to access ring expanded products.2c This is particularly significant as boron heterocycles are in small molecule drugs5 and are being investigated in electronic materials.6 Despite this interest, accessing heterocycles containing tricoordinate boron centers is challenging due to the propensity of boron reagents to react with nucleophiles to form four-coordinate species.
Boroles can be accessed through three general routes, one being the direct salt metathesis with a substituted 1,4-dilithio-1,3-butadiene and dihaloborane or organotrifluoroborate.2d,7 Borolide dianions can undergo a two electron oxidation to the neutral boroles and the third method is transmetallation from tin or zirconium precursors.3,8 The transmetallation route is the most popular method due to the ease in manipulating the precursors including compatibility with non-coordinating solvents that enable the isolation of tricoordinate species.8b
Although boroles have been effective reagents for the preparation of ring systems of six to eight atoms,2c,9 a major limitation has been the bulk on the boroles required to kinetically preclude dimerization (e.g.A, Fig. 1).1,10 Boroles bearing a halide on boron (e.g.B) undergo complex decomposition at low temperatures11 while those with organic groups dimerize via [4 + 2] cycloaddition with one equivalent acting as the diene and the other as the dienophile (e.g.C2).3,12 The latter process is dictated by bulk on the carbon centers and is reminiscent of the dimerization of cyclopentadiene. In contrast to cyclopentadiene, the monomers are not isolable, however, reactivity studies suggest that the retro Diels–Alder process can be thermally induced.3,13 Despite these dimers being known since 1985, the only studies have been heating the dimers in the presence of metal precursors to access η5-metal complexes14 or examining their Diels–Alder reactivity.3,13 If dimeric boroles could be utilized as reagents for ring expansion reactions, it would circumvent the requirement of bulky substituted monomeric boroles enabling access to a diverse library of products. We herein investigate the ability of Diels–Alder dimeric boroles to serve as sources of monomers in ring expansion reactions.
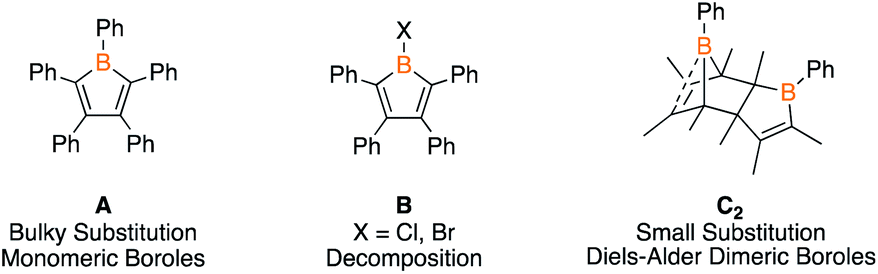 | ||
| Fig. 1 Examples of boroles and a summary of their stability. | ||
A particularly appealing class of molecules are hybrid inorganic/organic analogues of benzene in which a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unit is replaced by boron and lone pair bearing heteroatom.15 Pentaarylboroles have been effective in their preparation by 1,1-insertion reactions but result in products with five bulky groups.16 We envisioned that a dimeric borole, 1-phenyl-2,3,4,5-tetramethylborole dimer C2, may also be an effective synthon of its monomer C to generate six-membered heteroaromatic targets. To target 1,2-azaborines, a 2
C unit is replaced by boron and lone pair bearing heteroatom.15 Pentaarylboroles have been effective in their preparation by 1,1-insertion reactions but result in products with five bulky groups.16 We envisioned that a dimeric borole, 1-phenyl-2,3,4,5-tetramethylborole dimer C2, may also be an effective synthon of its monomer C to generate six-membered heteroaromatic targets. To target 1,2-azaborines, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric mixture of phenyl azide with dimer C2 in toluene was heated to 100 °C and monitored by in situ11B NMR spectroscopy (Scheme 1). The consumption of C2 was observed after 12 h by the disappearance of the peaks at −6.3 and 69.8 ppm coupled with the emergence of a new peak at 35.5 ppm, representative of 1,2-azaborine species.16d–g The identity of the insertion product was confirmed by a single crystal X-ray diffraction study (Fig. 2) and upon workup, the product was isolated as a yellow powder in 70% yield. 1H NMR spectroscopy of the solid redissolved in CDCl3 indicated a simplified methyl region with four signals integrating in equivalent ratios in contrast to eight signals for dimer C2. 1,2-Azaborines are gaining attention for utility in materials and medicinal chemistry and this reaction demonstrates an efficient manner to access these heterocycles in a single step.
:1 stoichiometric mixture of phenyl azide with dimer C2 in toluene was heated to 100 °C and monitored by in situ11B NMR spectroscopy (Scheme 1). The consumption of C2 was observed after 12 h by the disappearance of the peaks at −6.3 and 69.8 ppm coupled with the emergence of a new peak at 35.5 ppm, representative of 1,2-azaborine species.16d–g The identity of the insertion product was confirmed by a single crystal X-ray diffraction study (Fig. 2) and upon workup, the product was isolated as a yellow powder in 70% yield. 1H NMR spectroscopy of the solid redissolved in CDCl3 indicated a simplified methyl region with four signals integrating in equivalent ratios in contrast to eight signals for dimer C2. 1,2-Azaborines are gaining attention for utility in materials and medicinal chemistry and this reaction demonstrates an efficient manner to access these heterocycles in a single step.
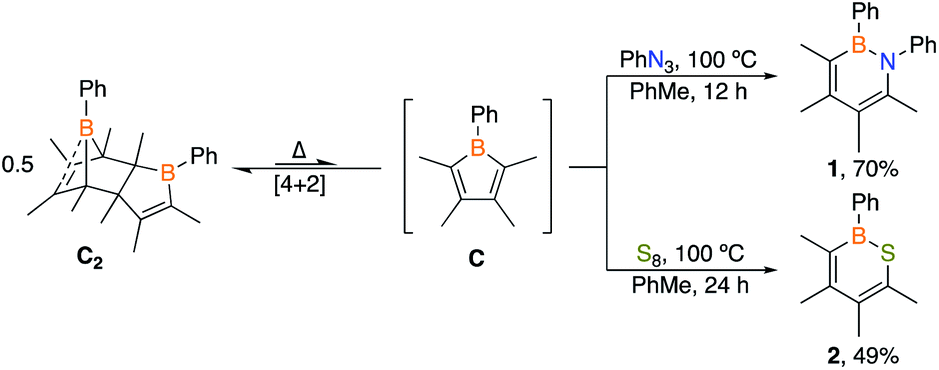 | ||
| Scheme 1 Reactions of borole dimer C2 with phenyl azide and elemental sulfur. | ||
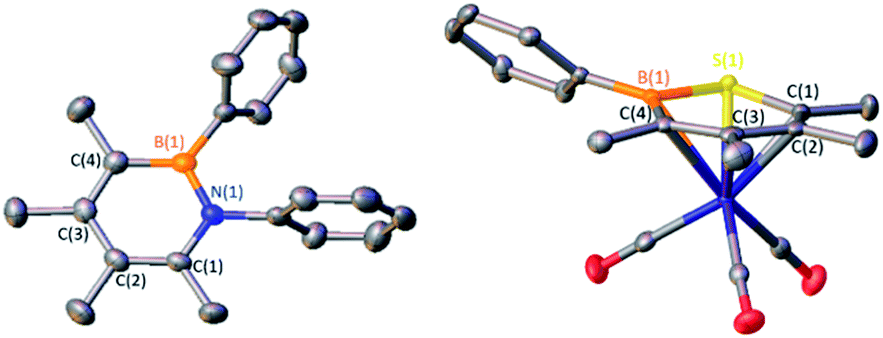 | ||
| Fig. 2 Solid-state structures of 1 (left) and 2·Cr(CO)3 (right). Hydrogen atoms are omitted for clarity and ellipsoids are depicted at the 50% probability level. Selected bond lengths (Å) for 1: B(1)–N(1) 1.443(4), N(1)–C(1) 1.399(4), C(1)–C(2) 1.370(4), C(2)–C(3) 1.442(5), C(3)–C(4) 1.377(5), C(4)–B(1) 1.513(4); for 2·Cr(CO)3: B(1)–S(1) 1.828(2), S(1)–C(1) 1.757(3), C(1)–C(2) 1.397(3), C(2)–C(3) 1.441(3), C(3)–C(4) 1.416(4), B(1)–C(4) 1.515(3), B(1)–Cr 2.388(3), S(1)–Cr 2.4367(6), C(1)–Cr 2.175(2), C(2)–Cr 2.223(3), C(3)–Cr 2.251(3), C(4)–Cr 2.273(2). | ||
To determine if C2 is capable of acting as a source of C in other 1,1-insertion reactions it was reacted with excess elemental sulfur in toluene.17 Monitoring the reaction by in situ11B NMR spectroscopy indicated that upon heating to 100 °C for 24 h a major peak at 49.8 ppm emerged, which lies in the range of reported 1,2-thiaborines (35.8–51.1 ppm).15f,16c,18 Upon work-up, a yellow oil was isolated in 49% yield. The liquid state at room temperature prevented obtaining a solid state structure. Since 1,2-thiaborines are a six-π-electron aromatic system, we postulated that the π-system could coordinate to a transition metal. A suite of 1,2-azaborine metal complexes have been prepared,19 but there is not an example of a 1,2-thiaborine complex in the literature, or any chalcogenaborine metal complexes.15e,15f,20 Our attempts to prepare a 1,2-thiaborine chromium complex from Cr(CO)3(CH3CN)3 and pentaaryl 1,2-thiaborines derived from monomeric pentaarylboroles were unsuccessful, presumably due to the bulky substituents which has also been reported in attempts to coordinate metals to the central ring of hexaphenylbenzene.21 To determine if the smaller 1,2-thiaborine could act as a ligand, the reaction of excess Cr(CO)3(CH3CN)3 with 1,2-thiaborine (2) was conducted in THF at 23 °C and in situ monitoring by 11B NMR spectroscopy revealed an upfield shift from free 2 at 49.8 ppm to 29.1 ppm with the reaction complete after 24 h (Scheme 2). The resonance is consistent with η6-bound 1,2-azaborine metal complexes.19 A single 13C{1H} NMR shift at 229.83 ppm was detected for the three carbonyl groups indicating chemical equivalency on the NMR timescale, attributed to rapid rotation about the BSC4 ring. Upon work up a red solid was isolated in 76% yield and an X-ray diffraction study gratifyingly confirmed the product as the half-sandwich tricarbonylchromium complex 2·Cr(CO)3 (Fig. 2).
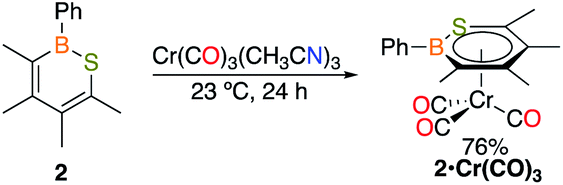 | ||
| Scheme 2 Reaction of 1,2-thiaborine 2 with Cr(CH3CN)3(CO)3. | ||
The solid state structure of 2·Cr(CO)3 confirms η6-coordination to chromium with a planar ring (max. deviation from planarity = 0.04 Å), although the chromium bonds to boron [Cr–B = 2.388(3) Å] and sulfur [Cr–S = 2.4367(6) Å] are longer than those to the four carbon atoms [B–C range 2.175(2)-2.273(2) Å]. The bond lengths within the 1,2-thiaborine ring of 2·Cr(CO)3 are marginally longer in comparison to the only non-disordered uncomplexed 1,2-thiaborine, which is expected upon coordination.16c The carbonyl stretching frequencies are a gauge of the donor properties of aromatic ligands.22 FT-IR spectroscopy revealed C–O stretching frequencies of 1964, 1908 and 1873 cm−1 for 2·Cr(CO)3 which are lower than those of the chromium complex with 1,2-dihydro-1-methyl-2-phenyl-1,2-azaborine (1979, 1916, 1900 cm−1).19a This indicates that 1,2-thiaborine 2 induces more π-backbonding to the CO ligands by enriching the electron density at chromium more than the 1,2-azaborine.
The resiliency of the π-coordination of the 1,2-thiaborine to chromium was investigated. No decomposition or migration to the B-phenyl group was observed at room temperature. Heating a THF-d8 solution of 2·Cr(CO)3 resulted in free 2 by 1H NMR spectroscopy and an insoluble grey precipitate, indicating slow decomposition of 2·Cr(CO)3 with no evidence of migration of the chromium center. Adding excess benzene as an extraneous π-donor into a solution of 2·Cr(CO)3 in THF-d8 did not result in any reaction at room temperature. During the course of heating, in situ1H NMR spectroscopy revealed that 2·Cr(CO)3 decomposed by generating (benzene)chromium tricarbonyl, free 2, and a similar grey precipitate. Attempts to access η1-coordination chemistry via the sulfur atom in 2 were unsuccessful due to the relatively weak sulfur donor (see ESI†).
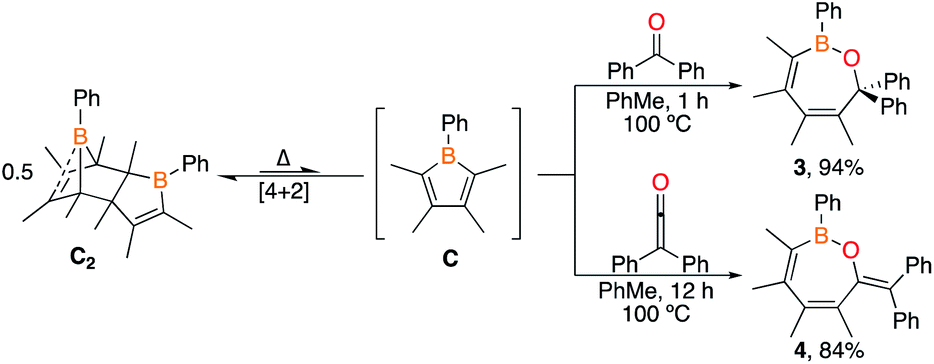
To examine the ability of borole synthon C2 to undergo 1,2-insertions to access seven-membered rings, the 1:2 stoichiometric reactions of C2 with benzophenone and diphenylketene in toluene-d8 at room temperature were investigated. No reaction occurred at room temperature but upon heating the solutions to 100 °C (1 h for benzophenone and 12 h for diphenylketene), 1H NMR spectroscopy revealed consumption of dimer C2, corroborated by 11B NMR spectroscopy with new resonances at 43.6 ppm for benzophenone and 43.2 ppm for diphenylketene (Scheme 3). Both are consistent with a three-coordinate boron bound to an oxygen.9e,16b Single crystals grown for X-ray diffraction studies identified the products as the seven-membered BOC5 rings 3 and 4 in which the 1,2-dipolar CO unit is inserted into the endocyclic B–C bond of the monomeric borole (Fig. 3). Both species adopt boat-like conformations.
 | ||
| Scheme 3 Reaction of dimer C2 with benzophenone and diphenylketene. | ||
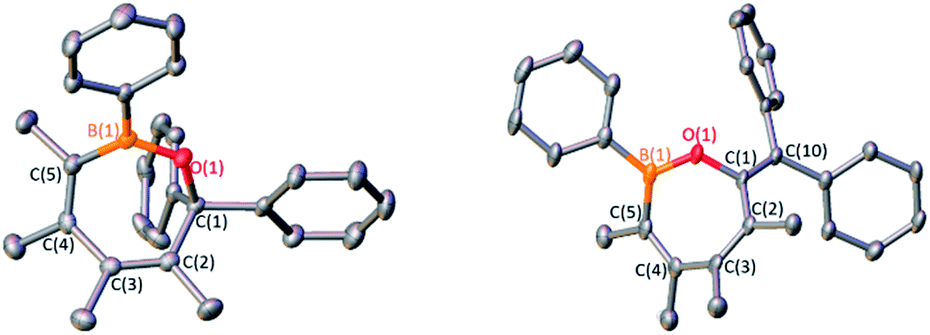 | ||
| Fig. 3 (a) Solid-state structure of 3 (left) and 4 (right) hydrogen atoms are omitted for clarity and ellipsoids are depicted at the 50% probability level. Selected bond lengths (Å) for 3: B(1)–O(1) 1.366(5), O(1)–C(1) 1.452(5), C(1)–C(2) 1.542(5), C(2)–C(3) 1.348(5), C(3)–C(4) 1.486(5), C(4)–C(5) 1.347(5), C(5)–B(1) 1.563(5); for 4: B(1)–O(1) 1.368(2), O(1)–C(1) 1.392(3), C(1)–C(2) 1.484(2), C(2)–C(3) 1.346(2), C(3)–C(4) 1.489(2), C(4)–C(5) 1.353(3), C(5)–B(1) 1.558(3), C(1)–C(10) 1.345(3). | ||
1,1-Diphenylethylene oxide reacts with pentaphenylborole to generate a rare eight-membered boron heterocycle from the insertion of the C2O unit.9a In the reaction of two equivalents of 1,1-diphenylethylene oxide with C2 at 100 °C in toluene, in situ11B NMR spectroscopy revealed the complete consumption of C2 after 14 h and a new signal at 59.4 ppm, differing from the eight-membered boracycle in the corresponding reaction with pentaphenylborole (46.3 ppm).9a Single crystal X-ray diffraction identified the product as a bicyclic compound composed of fused BC4 and BOC3 rings (5, Fig. 4) with a boron and carbon atom at the ring junctions. The boron center is essentially trigonal planar with the sum of angles about boron being 358.0(6)°. The presence of the phenyl group on the carbon adjacent to boron, C(6), suggests a rearrangement occurred. The B(1)–O(1) bond [1.353(3) Å] is consistent with a B–O single bond.9a,23 The carbon–carbon bond lengths in the BC4 ring derived from the borole alternate [C(3)–C(4) = 1.530(3), C(4)–C(5) = 1.342(4), C(5)–C(6) = 1.546(3) Å] which is consistent with 1-bora-cyclopent-3-ene ring systems.9d,24 BOC3 rings with a fused ring have been gaining attention with the effective drugs Kerydin® (onychomycosis) and Eucrisa® (eczema) containing such bicyclic systems.5d,5e
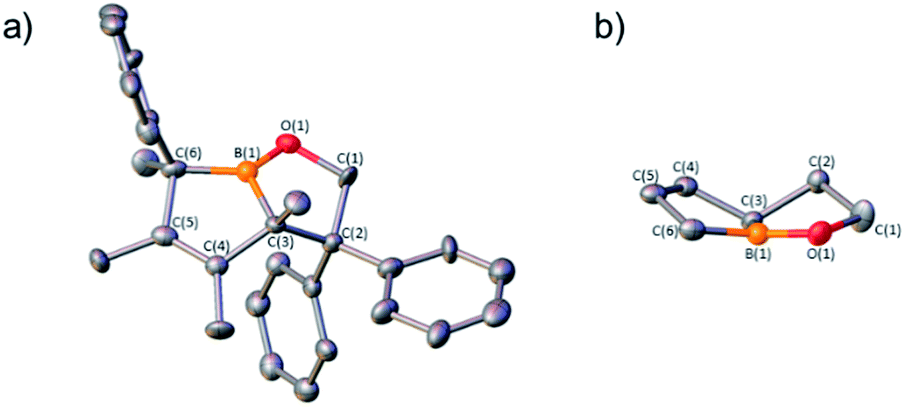 | ||
| Fig. 4 (a) Solid-state structure of 5. Hydrogen atoms are omitted for clarity and ellipsoids are depicted at the 50% probability level. Selected bond lengths (Å): B(1)–O(1) 1.353(3), O(1)–C(1) 1.460(3), C(1)–C(2) 1.567(3), C(2)–C(3) 1.609(3), C(3)–C(4) 1.530(3), C(4)–C(5) 1.342(4), C(5)–C(6) 1.546(3), C(6)–B(1) 1.582(4), B(1)–C(3) 1.565(4); (b) View of the fused bicyclic core along the boron plane. | ||
A proposed mechanism can be drawn that proceeds through an initial Lewis acid–base adduct Int1 (Scheme 4). The epoxide ring opens via the oxygen–carbon bond of the diphenyl carbon to furnish the resonance stabilized carbocation Int2. The diene attacks the carbocation to forge the bicyclic framework and the phenyl group migrates to the adjacent carbon to give 5. The migration occurs on the same face rationalizing the anti-stereochemistry of the methyl groups observed in the solid state structure. The centrosymmetric P21/n space group indicates that both enantiomers are generated. This differs from the pentaphenylborole product in which the endocyclic B–C bond of the borole attacks the carbocation in Int2 to generate an eight-membered ring.9a,25 Although this bicyclic framework has not been observed previously in a product from a borole reaction, it has been proposed as an intermediate in insertion reactions and may provide insight into other reaction mechanisms.16e,26
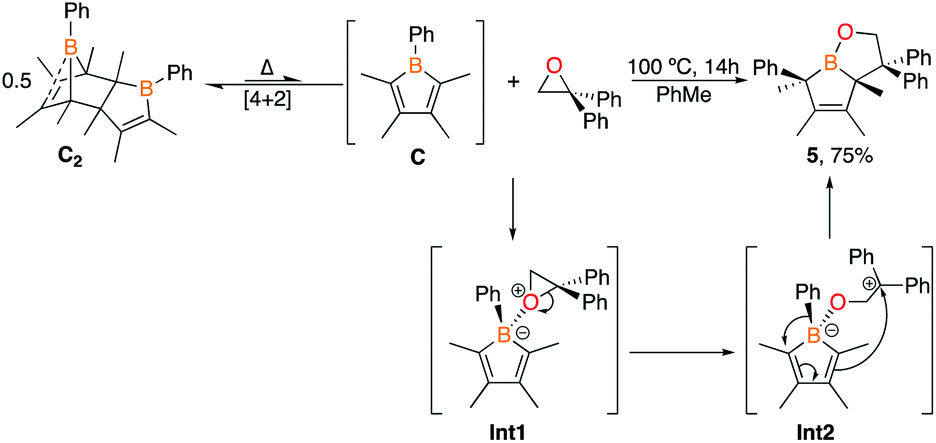 | ||
| Scheme 4 Reaction of 1,1-diphenylethylene oxide with C2 and proposed mechanism. | ||
To determine if other Diels–Alder dimeric boroles are capable of generating insertion products, two other Diels–Alder borole dimers, 1-phenyl-2,3,4,5-tetraethylborole dimer (D2) and 1-biphenyl-2,3,4,5-tetramethylborole dimer (E2), were prepared via transmetallation of zirconium precursors (Scheme 5). The identity of the tetraethyl dimer D2 was established by obtaining an X-ray diffraction structure that is similar to C2 with the notable feature of a CC bond coordinating to the bridgehead boron (Fig. 5). A 13C DEPT experiment showed eight distinct resonances for both methylene and methyl groups indicating the presence of eight chemically inequivalent ethyl groups, consistent with the structure. The identity of 1-biphenyl dimer E2 was confirmed by multinuclear NMR spectroscopy and elemental analysis. The anticipated eight methyl signals were observed for E2 by 1H NMR and 13C NMR spectroscopy. Obtaining 11B NMR spectra for D2 and E2 revealed two resonances, one for the tricoordinate boron center and another for the pseudo-five coordinate bridgehead boron akin to C2 (D2 = 68.5, −2.7 ppm; E2 = 70.4–5.8 ppm; cf.C2 = 69.8, −6.3 ppm).
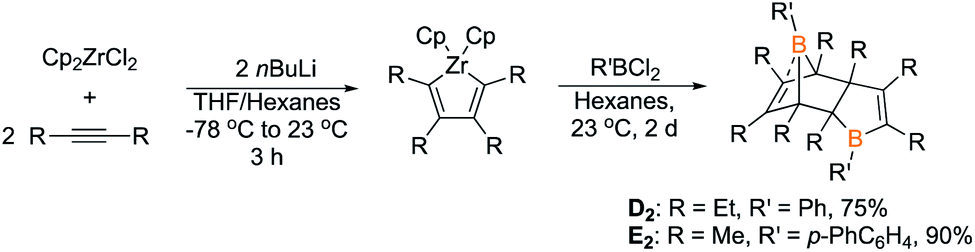 | ||
| Scheme 5 Synthesis of borole dimers D2 and E2. | ||
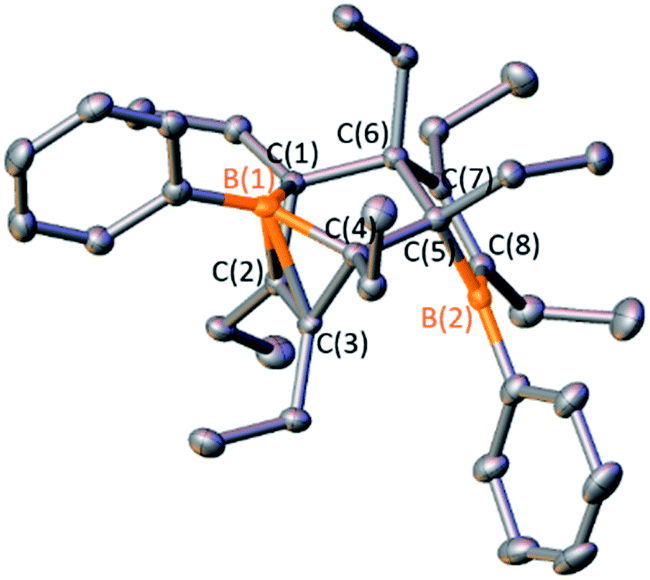 | ||
| Fig. 5 Solid-state structure of D2. Hydrogen atoms are omitted for clarity and ellipsoids are depicted at the 50% probability level. Selected bond lengths (Å) for D2: B(1)–C(1) 1.619(4), B(1)–C(4) 1.611(4), C(1)–C(2) 1.514(4), C(2)–C(3) 1.380(4), C(3)–C(4) 1.524(4), C(4)–C(5) 1.585(3), C(5)–C(6) 1.573(4), C(6)–C(7) 1.527(4), C(7)–C(8) 1.357(4), B(2)–C(5) 1.599(5), B(2)–C(8) 1.540(4). | ||
The 1,2-insertion reactions of borole dimers D2 and E2 with two equivalents of diphenylketene were investigated (Scheme 6). Upon heating the toluene-d8 solutions to 100 °C (1 h for D2 and 8 h for E2), 1H and 11B NMR spectroscopy revealed the consumption of the borole dimers. The 11B signals at 43.7 and 44.9 ppm for the reactions with D2 and E2, respectively, are reminiscent of 4 (43.2 ppm). The reactions were scaled up and X-ray diffraction studies confirmed the products from D2 and E2 as the seven-membered BOC5 rings 6 and 7, respectively (Fig. 6). The experiments confirm that insertion reactions are effective with other dimeric boroles although the reaction times differ. The reactions with the dimers featuring methyl groups on carbon were completed in 12 h (C2) and 8 h (E2) while the reaction with the dimer bearing ethyl groups on the carbon centers (D2) was complete within an hour.17
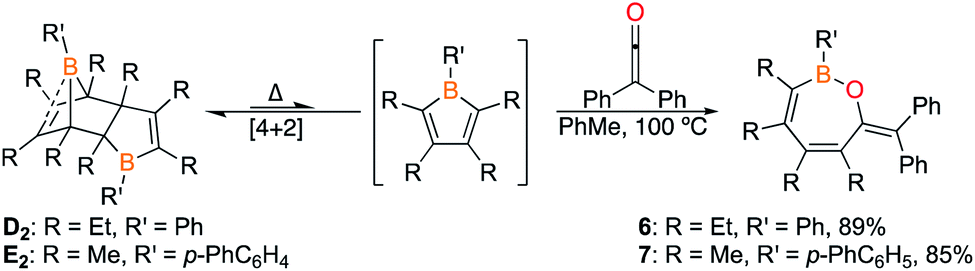 | ||
| Scheme 6 Reaction of diphenylketene with D2 and E2. Reaction times: 1 h for 6 and 8 h for 7. | ||
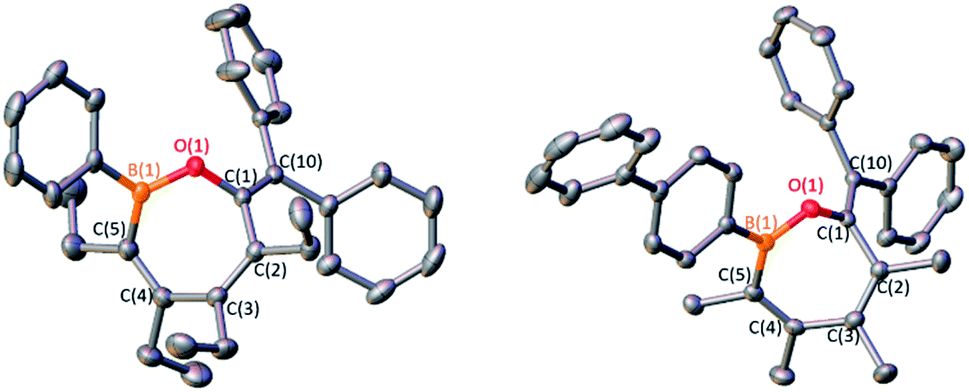 | ||
| Fig. 6 (a) Solid-state structure of 6 (left) and 7 (right) hydrogen atoms are omitted for clarity and ellipsoids are depicted at the 50% probability level. Selected bond lengths (Å) for 6: B(1)–O(1) 1.368(2), O(1)–C(1) 1.398(2), C(1)–C(2) 1.489(2), C(2)–C(3) 1.351(2), C(3)–C(4) 1.500(2), C(4)–C(5) 1.351(2), C(5)–B(1) 1.561(1), C(1)–C(10) 1.345(2); for 7: B(1)–O(1) 1.372(2), O(1)–C(1) 1.398(2), C(1)–C(2) 1.483(2), C(2)–C(3) 1.341(2), C(3)–C(4) 1.482(2), C(4)–C(5) 1.355(2), C(5)–B(1) 1.563(2), C(1)–C(10) 1.340(2). | ||
Conclusions
In conclusion, we report the first examples of ring expansion reactions with borole dimers as precursors, specifically the 1,1-insertion with elemental sulfur and an azide as well as the 1,2-insertion with benzophenone and diphenylketene to access six- and seven-membered boracycles, respectively. The sulfur insertion product, a 1,2-thiaborine, could be coordinated to chromium in an η6 fashion. It is notable that this complex could not be accessed from the peraryl-substituted monomeric borole products. An attempt to access an eight-membered ring was unsuccessful with 1,1-diphenylethylene oxide, instead forging a new BOC3 ring that is a component in effective pharmacophores. The reactions all required heat to crack the dimer but were all complete within 24 h at 100 °C. Dimers with different substitution on boron and carbon are also capable of insertion reactions, exemplified in the reactions with diphenylketene. These studies solidify that bottleable monomeric boroles are not essential for insertion chemistry and that dimers have great potential to act as borole synthons to furnish a wealth of heterocycles with less restrictions on the substitution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Welch Foundation (Grant No. AA-1846) and the National Science Foundation for a CAREER Award (Award No. 1753025) for their generous support of this work. We thank John R. Tidwell for assistance in X-ray diffraction analysis.Notes and references
- J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef CAS.
- (a) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC; (b) H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS; (c) J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC; (d) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS.
- P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS.
- (a) G. E. Herberich, B. Buller, B. Hessner and W. Oschmann, J. Organomet. Chem., 1980, 195, 253–259 CrossRef CAS; (b) C. W. So, D. Watanabe, A. Wakamiya and S. Yamaguchi, Organometallics, 2008, 27, 3496–3501 CrossRef CAS; (c) C. Sindlinger and P. N. Ruth, Angew. Chem., Int. Ed., 2019, 58, 15051–15056 CrossRef CAS.
- (a) C. Baldock, J. B. Rafferty, S. E. Sedelnikova, P. J. Baker, A. R. Stuitje, A. R. Slabas, T. R. Hawkes and D. W. Rice, Science, 1996, 274, 2107–2110 CrossRef CAS; (b) L. J. Liu, A. J. V. Marwitz, B. W. Matthews and S. Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 6817–6819 CrossRef CAS; (c) D. H. Knack, J. L. Marshall, G. P. Harlow, A. Dudzik, M. Szaleniec, S. Y. Liu and J. Heider, Angew. Chem., Int. Ed., 2013, 52, 2599–2601 CrossRef CAS; (d) S. Jinna and J. Finch, Drug Des. Dev. Ther., 2015, 9, 6185–6190 CAS; (e) A. S. Paller, W. L. Tom, M. G. Lebwohl, R. L. Blumenthal, M. Boguniewicz, R. S. Call, L. F. Eichenfield, D. W. Forsha, W. C. Rees, E. L. Simpson, M. C. Spellman, L. F. S. Gold, A. L. Zaenglein, M. H. Hughes, L. T. Zane and A. A. Hebert, J. Am. Acad. Dermatol., 2016, 75, 494–503 CrossRef CAS.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef; (b) E. Von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (c) A. Lorbach, A. Hubner and M. Wagner, Dalton Trans., 2012, 41, 6048–6063 RSC; (d) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (e) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS; (f) D. T. Yang, S. K. Mellerup, J. B. Peng, X. Wang, Q. S. Li and S. N. Wang, J. Am. Chem. Soc., 2016, 138, 11513–11516 CrossRef CAS; (g) F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS; (h) F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- (a) Z. F. Xi, Acc. Chem. Res., 2010, 43, 1342–1351 CrossRef CAS; (b) Z. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC; (c) T. Heitkemper and C. P. Sindlinger, Chem.–Eur. J., 2019, 25, 6628–6637 CrossRef CAS.
- (a) G. E. Herberich, B. Buller, B. Hessner and W. Oschmann, J. Organomet. Chem., 1980, 195, 253–259 CrossRef CAS; (b) X. Y. Yan and C. J. Xi, Acc. Chem. Res., 2015, 48, 935–946 CrossRef CAS.
- (a) S. Yruegas, C. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2017, 36, 2581–2587 CrossRef CAS; (b) H. Braunschweig, F. Hupp, I. Krummenacher, L. Mailänder and F. Rauch, Chem.–Eur. J., 2015, 21, 17844–17849 CrossRef CAS; (c) A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814–1818 RSC; (d) K. Huang, S. A. Couchman, D. J. Wilson, J. L. Dutton and C. D. Martin, Inorg. Chem., 2015, 54, 8957–8968 CrossRef CAS; (e) K. Huang and C. D. Martin, Inorg. Chem., 2015, 54, 1869–1875 CrossRef CAS; (f) Y. T. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- (a) T. Heitkemper and C. P. Sindlinger, Chem.–Eur. J., 2019, 25, 6628–6637 CrossRef CAS; (b) Z. L. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC.
- (a) H. Braunschweig, C. W. Chiu, A. Damme, K. Ferkinghoff, K. Kraft, K. Radacki and J. Wahler, Organometallics, 2011, 30, 3210–3216 CrossRef CAS; (b) H. Braunschweig, C. W. Chiu, J. Wahler, K. Radacki and T. Kupfer, Chem.–Eur. J., 2010, 16, 12229–12233 CrossRef CAS; (c) Z. L. Zhang, Z. Wang, M. Haehnel, A. Eichhorn, R. M. Edkins, A. Steffen, A. Krueger, Z. Y. Lin and T. B. Marder, Chem. Commun., 2016, 52, 9707–9710 RSC; (d) Z. Wang, Y. Zhou, K. H. Lee, W. H. Lam, R. D. Dewhurst, H. Braunschweig, T. B. Marder and Z. Y. Lin, Chem.–Eur. J., 2017, 23, 11587–11597 CrossRef CAS.
- (a) P. J. Fagan, W. A. Nugent and J. C. Calabrese, J. Am. Chem. Soc., 1994, 116, 1880–1889 CrossRef CAS; (b) G. E. Herberich and H. Ohst, Chem. Ber., 1985, 118, 4303–4313 CrossRef CAS.
- J. J. Baker, K. H. M. Al Furaiji, O. T. Liyanage, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem.–Eur. J., 2019, 25, 1581–1587 CrossRef CAS.
- (a) G. E. Herberich, B. Hessner, H. Ohst and I. A. Raap, J. Organomet. Chem., 1988, 348, 305–316 CrossRef CAS; (b) G. E. Herberich, M. Negele and H. Ohst, Chem. Ber., 1991, 124, 25–29 CrossRef CAS.
- (a) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (b) P. G. Campbell, A. J. V. Marwitz and S. Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS; (c) E. R. Abbey, A. N. Lamm, A. W. Baggett, L. N. Zakharov and S. Y. Liu, J. Am. Chem. Soc., 2013, 135, 12908–12913 CrossRef CAS PubMed; (d) H. Braunschweig, K. Geetharani, J. O. C. Jimenez-Halla and M. Schafer, Angew. Chem., Int. Ed., 2014, 53, 3500–3504 CrossRef CAS PubMed; (e) J. H. Chen, Z. Bajko, J. W. Kampf and A. J. Ashe III, Organometallics, 2007, 26, 1563–1564 CrossRef CAS; (f) A. D. Rohr, M. M. B. Holl, J. W. Kampf and A. J. Ashe III, Organometallics, 2011, 30, 3698–3700 CrossRef CAS; (g) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS; (h) P. A. Brown, C. D. Martin and K. L. Shuford, Phys. Chem. Chem. Phys., 2019, 21, 18458–18466 RSC; (i) B. L. Wang, Y. X. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7 Search PubMed; (j) B. L. Wang and R. Kinjo, Chem. Sci., 2019, 10, 2088–2092 RSC; (k) A. W. Baggett, F. Guo, B. Li, S. Y. Liu and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS.
- (a) J. H. Barnard, P. A. Brown, K. L. Shuford and C. D. Martin, Angew. Chem., Int. Ed., 2015, 54, 12083–12086 CrossRef CAS; (b) S. Yruegas, D. C. Patterson and C. D. Martin, Chem. Commun., 2016, 52, 6658–6661 RSC; (c) S. Yruegas and C. D. Martin, Chem.–Eur. J., 2016, 22, 18358–18361 CrossRef CAS; (d) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS; (e) S. A. Couchman, T. K. Thompson, D. J. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC; (f) H. Braunschweig, M. A. Celik, F. Hupp, I. Krummenacher and L. Mailänder, Angew. Chem., Int. Ed., 2015, 54, 6347–6351 CrossRef CAS PubMed; (g) H. Braunschweig, M. A. Celik, T. Dellermann, G. Frenking, K. Hammond, F. Hupp, H. Kelch, I. Krummenacher, F. Lindl and L. Mailänder, Chem.–Eur. J., 2017, 23, 8006–8013 CrossRef CAS.
- In an attempt to observe monomeric species, conducting elevated temperature 1H and 11B NMR spectroscopic experiments on C2 and E2 in toluene-d8 to 80°C did not show any change in speciation (Fig. S102, S103 and S106, S107†). Given the observed reaction times, this equilibrium is presumably shifted towards the dimer. NMR spectroscopic studies on D2 revealed new 1H resonances in the aryl region that emerged at 45 °C and temperatures above. Although no corresponding new 11B NMR resonance could be detected, it is possible that the new species detected by 1H NMR spectroscopy could be monomeric borole D (Fig. S104†).
- (a) P. J. Grisdale and J. L. Williams, J. Org. Chem., 1969, 34, 1675–1677 CrossRef CAS; (b) F. A. Davis and M. J. S. Dewar, J. Am. Chem. Soc., 1968, 90, 3511–3515 CrossRef CAS.
- (a) A. J. Ashe III, X. D. Fang, X. G. Fang and J. W. Kampf, Organometallics, 2001, 20, 5413–5418 CrossRef; (b) J. Pan, J. W. Kampf and A. J. Ashe III, Organometallics, 2006, 25, 197–202 CrossRef CAS; (c) J. Pan, J. Wang, M. M. B. Holl, J. W. Kampf and A. J. Ashe III, Organometallics, 2006, 25, 3463–3467 CrossRef CAS; (d) J. Pan, J. W. Kampf and A. J. Ashe III, Organometallics, 2009, 28, 506–511 CrossRef CAS.
- F. A. Tsao and D. W. Stephan, Chem. Commun., 2017, 53, 6311–6314 RSC.
- B. Mailvaganam, B. G. Sayer and M. J. Mcglinchey, J. Organomet. Chem., 1990, 395, 177–185 CrossRef CAS.
- G. Klopman and K. Noack, Inorg. Chem., 1968, 7, 579–584 CrossRef CAS.
- (a) Y. K. Loh, C. C. Chong, R. Ganguly, Y. X. Li, D. Vidovic and R. Kinjo, Chem. Commun., 2014, 50, 8561–8564 RSC; (b) L. E. Laperriere, S. Yruegas and C. D. Martin, Tetrahedron, 2019, 75, 937–943 CrossRef CAS; (c) J. N. Bentley and C. B. Caputo, Tetrahedron, 2019, 75, 31–35 CrossRef CAS.
- (a) H. Braunschweig, A. Damme, C. Hörl, T. Kupfer and J. Wahler, Organometallics, 2013, 32, 6800–6803 CrossRef CAS; (b) S. Yruegas, K. Huang, D. J. Wilson, J. L. Dutton and C. D. Martin, Dalton Trans., 2016, 45, 9902–9911 RSC; (c) B. C. Caputo, Z. J. Manning, J. H. Barnard and C. D. Martin, Polyhedron, 2016, 114, 273–277 CrossRef CAS; (d) V. A. K. Adiraju and C. D. Martin, Dalton Trans., 2017, 46, 10324–10331 RSC; (e) H. Braunschweig, M. Domling, S. Kachel, H. Kelch, T. Kramer, I. Krummenacher, C. Lenczyk, S. J. Lin, Z. Y. Lin, C. Possiel and K. Radacki, Chem.–Eur. J., 2017, 23, 16167–16170 CrossRef CAS PubMed; (f) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen and M. Parvez, J. Am. Chem. Soc., 2010, 132, 9604–9606 CrossRef CAS PubMed; (g) A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers and H. M. Tuononen, J. Am. Chem. Soc., 2013, 135, 941–947 CrossRef CAS PubMed.
- It is unclear with the experimental data whether the product is dictated by kinetics or thermodynamics.
- H. Braunschweig, I. Krummenacher, L. Mailänder and F. Rauch, Chem. Commun., 2015, 51, 14513–14515 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1942996–1943000 and 1960237–1960239. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04053f |
| This journal is © The Royal Society of Chemistry 2020 |