
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric hydrogenation of (Z)-α-dehydroamido boronate esters: direct route to alkyl-substituted α-amidoboronic esters†
Yazhou
Lou
a,
Jun
Wang
a,
Gelin
Gong
a,
Fanfu
Guan
a,
Jiaxiang
Lu
a,
Jialin
Wen
 ab and
Xumu
Zhang
*a
ab and
Xumu
Zhang
*a
aShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, People's Republic of China. E-mail: zhangxm@sustc.edu.cn
bAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen 518000, People's Republic of China
First published on 25th November 2019
Abstract
The direct catalytic asymmetric hydrogenation of (Z)-α-dehydroamino boronate esters was realized. Using this approach, a class of therapeutically relevant alkyl-substituted α-amidoboronic esters was easily synthesized in high yields with generally excellent enantioselectivities (up to 99% yield and 99% ee). The utility of the products has been demonstrated by transformation to their corresponding boronic acid derivatives by a Pd-catalyzed borylation reaction and an efficient synthesis of a potential intermediate of bortezomib. The clean, atom-economic and environment friendly nature of this catalytic asymmetric hydrogenation process would make this approach a new alternative for the production of alkyl-substituted α-amidoboronic esters of great potential in the area of organic synthesis and medicinal chemistry.
Since FDA approval of bortezomib1 for the treatment of multiple myeloma, chiral α-aminoboronic acids have been recognized as key pharmacophores for the design of proteasome inhibitors.2 The incorporation of chiral α-aminoboronic acid motifs at the C-terminal position of a peptide3 to develop potential clinical drug candidates has drawn increasing interest4 (Fig. 1). Meanwhile, chiral α-amidoboronic acids and their derivatives are useful synthetic building blocks for the stereospecific construction of chiral amine compounds.5 The biological and synthetic value of α-amidoboronates has led to considerable efforts for the development of efficient synthetic methods. However, up to now, limited transition-metal-catalyzed asymmetric approaches have been reported. The widely used strategies to synthesize these compounds are stepwise Matteson homologation/N-nucleophilic replacement,6 borylation of imines,7 and alkene functionalization.8 Recently, two other elegant approaches, Ni-catalyzed decarboxylative borylation of α-amino acid derivatives9 and enantiospecific borylation of lithiated α-N-Boc species,10 were reported by the Baran and Negishi groups, respectively. To the best of our knowledge, the majority of the methods relied on either stoichiometric amounts of chiral auxiliaries6,7a,b or substrate-control strategies9 and most of these methods enable the construction of aryl-substituted α-aminoboronates. Enantioselective methods to access unfunctionalized alkyl-substituted α-aminoboronic esters are still rarely developed and so far only two examples have been realized by the Miura8a and Scheidt7f groups, respectively. Considering that most therapeutically relevant α-amidoboronic acid fragments contain an alkyl subunit and the fact that the options for the synthesis of alkyl-substituted α-amidoboronic esters in an enantioselective manner are still rare, the development of other distinct approaches would be highly desirable. Herein, we report a new alternative to access these compounds by catalytic asymmetric hydrogenation of (Z)-α-dehydroamidoboronate esters. With this approach, the desired chiral alkyl-substituted α-amidoboronic esters could be obtained in high yields and generally excellent enantioselectivities (up to 99% yield and 99% ee) with simple purification.
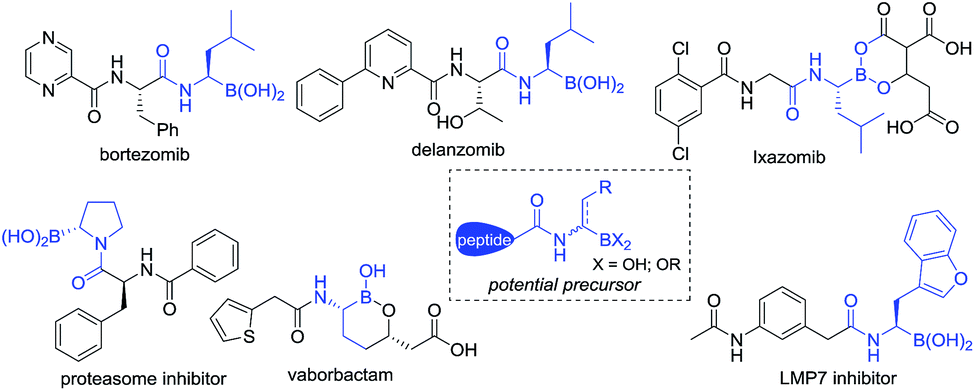 | ||
| Fig. 1 Selected inhibitors containing chiral alkyl-substituted α-amidoboronic acids. | ||
Catalytic asymmetric hydrogenation of olefins is an atom-economic, environmentally friendly and clean process for the synthesis of valuable pharmaceuticals, agricultural compounds and feedstock chemicals.11 Recently, hydrogenation of vinylboronic compounds has emerged for the preparation of chiral boronic compounds in a regiodefined manner.12,13 However, surprisingly α-dehydroamido boronate esters and their derivatives, as elegant precursors to access alkyl-substituted α-amidoboronic compounds, have never been used as substrates in asymmetric hydrogenation and remain a challenging project. To our knowledge, only one efficient hydrogenation approach to (1-halo-1-alkenyl) boronic esters was reported for indirect synthesis of alkyl-substituted α-aminoboronic esters but it was accompanied by inevitable de-halogenated by-products14 (Scheme 1). Given the catalytic efficiency and atom economy of the hydrogenation method, the development of a new direct hydrogenation approach to construct these important chiral alkyl-substituted α-amidoboronic esters would be very appealing.
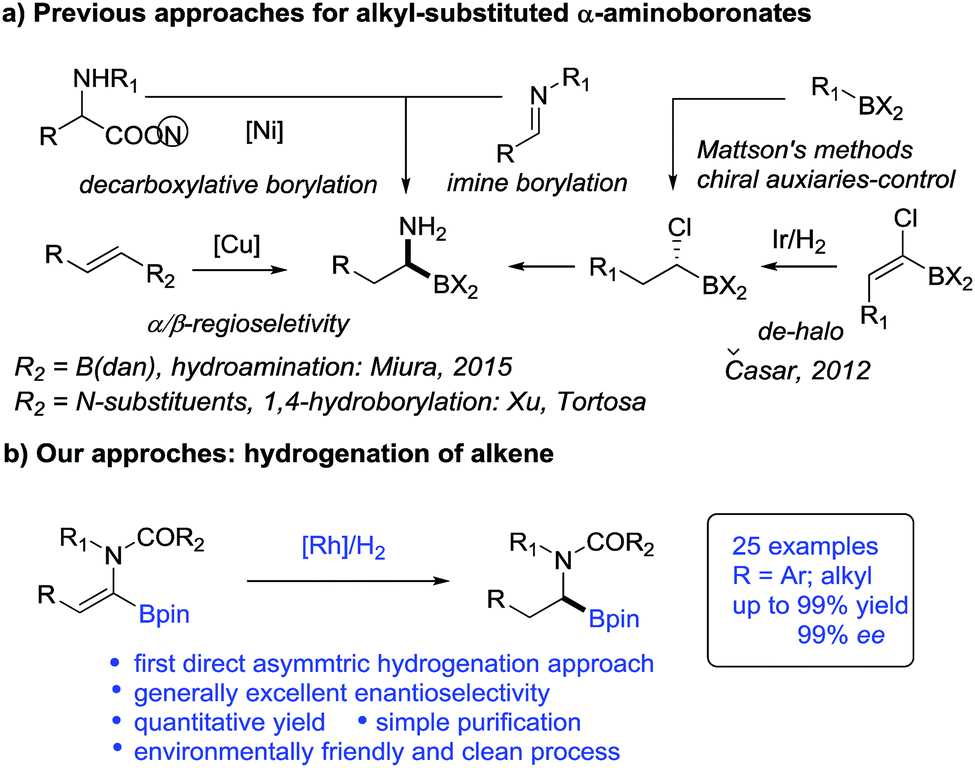 | ||
| Scheme 1 Approaches towards the synthesis of chiral alkyl-substituted α-aminoboronic esters. | ||
The inspiration for our approach to the hydrogenation of α-dehydroamido boronates came from the molecular structures of relevant biologically active inhibitors containing alkyl-substituted α-amidoboronic acid fragments. Due to the limited stability of free α-aminoboronic acids, an electron-withdrawing carboxylic N-substituent is often required.15 Thus, we envisaged that N-carboxyl protected α-dehydroamido boronate esters could serve as a potential precursor for the synthesis of alkyl-substituted α-amidoboronates through Rh-catalyzed asymmetric hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond16 (Fig. 1), a strategically distinct approach to the construction of unfunctionalized alkyl-substituted α-amidoboronic esters. However, challenges still remain, including: (1) how to synthesize α-dehydroamido boronates; (2) the facile transmetalation process of the starting materials leading to deboronated by-products in the hydrogenation process;17 (3) the unknown stability of α-amidoboronic compounds in the presence of a transition-metal catalyst and hydrogen molecules. As part of our continuous efforts to develop efficient hydrogenation approaches to construct valuable motifs,18 here we present the results of the investigation to address the aforementioned challenges.
C bond16 (Fig. 1), a strategically distinct approach to the construction of unfunctionalized alkyl-substituted α-amidoboronic esters. However, challenges still remain, including: (1) how to synthesize α-dehydroamido boronates; (2) the facile transmetalation process of the starting materials leading to deboronated by-products in the hydrogenation process;17 (3) the unknown stability of α-amidoboronic compounds in the presence of a transition-metal catalyst and hydrogen molecules. As part of our continuous efforts to develop efficient hydrogenation approaches to construct valuable motifs,18 here we present the results of the investigation to address the aforementioned challenges.
The desired aryl-substituted (Z)-α-dehydroamido boronates could be obtained by Cu-catalyzed regioselective hydroborylation of ynamide according to a previous report.19 However, different α/β-regioselectivity was observed for the preparation of alkyl-substituted (Z)-α-dehydroamido boronate esters and a new synthetic route was developed (Scheme 2, see the ESI† for details). Of note, (Z)-α-dehydroamido boronate esters should be purified with deactivated silica gel,7c or else protodeborylation would occur readily with flash chromatography.
 | ||
| Scheme 2 Synthetic route to (Z)-α-dehydroamino boronates. | ||
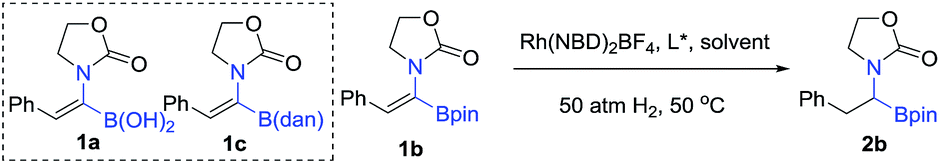
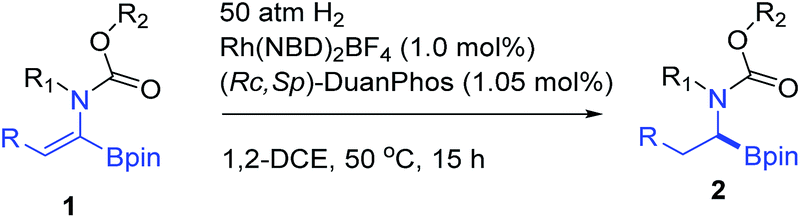
In order to check the feasibility of our hypothesis, three substrates were prepared with Rh(NBD)2BF4 and examined and our group prepared (Rc,Sp)-DuanPhos under 50 atm hydrogen pressure (Table 1). Gratifyingly, substrate 1b reacted smoothly to provide the desired product 2b in high yield and enantioselectivity (>99% conv., 98% ee, entry 2) whilst the reaction with substrate 1a yielded a mixture of deborylation products and 1c did not work at all (entries 1 and 3). Of note, we did not observe deborylation products with 1b under the current reaction conditions and we did not select (Z)-α-dehydroamido boronic acid 1a as the model substrate because of its poor solubility in most solvents. Then, a variety of chiral diphosphine ligands were investigated along with Rh(NBD)2BF4 and the results are shown in Table 1. In most cases, the reaction proceeded smoothly to furnish the desired products and the best results were obtained when (Rc,Sp)-DuanPhos was used as the ligand (entries 2 and 4–12). Poor results were obtained with axially bidentate phosphine ligands (entries 5, 6 and 9). (R,R)-QuinoxP* and (R,R)-Ph-BPE also gave good conversion with a slightly decreased ee whilst (R,R)-iPr-DuPhos exhibited poor results (entries 4, 7 and 10). Subsequent solvent screening revealed that the desired products could be obtained in most of the solvents and 1,2-DCE was the best solvent. (Entry 13, see the ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Sub | Ligand | Conv.b (%) | eec (%) |
| a Unless otherwise mentioned, the reactions were performed with 1 (0.1 mmol), Rh(NBD)2BF4 (10 mol%), and a ligand (11 mol%) in 1.0 mL THF at 50 °C for 15 h. b Determined by crude 1H NMR. c Determined with chiral HPLC. d The reaction was performed in iPrOH. e Rh(NBD)2BF4 (1.0 mol%) and ligand (1.05 mol%) were used. f Isolated yield in parentheses. g 1,2-DCE was used as the solvent. Pin = 2,3-dimethyl-2,3-butanediol; dan = 1,8-diaminonaphthalene. | ||||
| 1d | 1a | (Rc,Sp)-DuanPhos | 89 | n.d. |
| 2e | 1b | (Rc,Sp)-DuanPhos | >99 | 98 |
| 3 | 1c | (Rc,Sp)-DuanPhos | n.r. | n.d. |
| 4 | 1b | (R,R)-QuinoxP* | >99 | 97 |
| 5 | 1b | (S)-SegPhos | >99 | 17 |
| 6 | 1b | (S)-BINAP | >99 | 10 |
| 7 | 1b | (R,R)-iPr-DuPhos | >99 | 3 |
| 8 | 1b | (R,S)-Cy-JosiPhos | >99 | 14 |
| 9 | 1b | (R)-BIPHEP | >99 | −30 |
| 10 | 1b | (R,R)-Ph-BPE | >99 | −86 |
| 11 | 1b | (S,S)-f-Binaphane | >99 | 61 |
| 12 | 1b | (2S,4S)-BDPP | >99 | 59 |
| 13e,f,g | 1b | (Rc,Sp)-DuanPhos | >99(99) | 99 |
|
||||
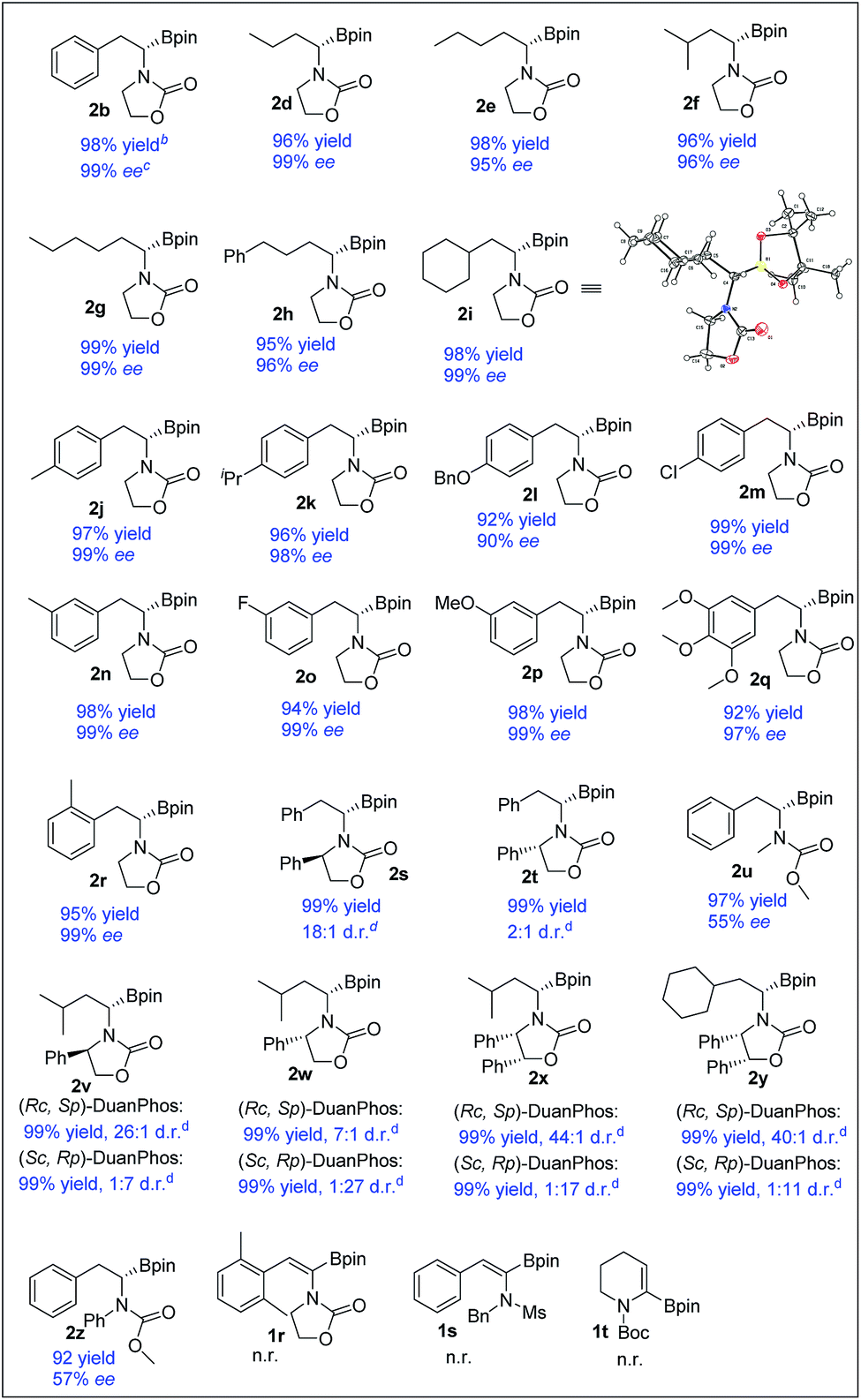
With the optimized reaction conditions in hand, a series of (Z)-α-dehydroamido boronate esters were tested and the results are summarized in Table 2. All the substrates reacted smoothly to give the corresponding alkyl-substituted α-amidoboronates in high yields with good to excellent enantioselectivities (2b, 2d–2r, and 2u, 99% yield, 57–99% ee). Alkyl-substituted (Z)-α-dehydroamido boronate esters were well tolerated in the current reaction, providing the corresponding α-amidoboronates in high yields and excellent enantioselectivities (2d–2i, 99% yield, 96–99% ee). Aryl-substituted (Z)-α-dehydroamido boronate esters with electron-donating (2j–l, 2n and 2p–r) and withdrawing (2m and 2o) substituents could also give the desired products in excellent yield with excellent enantioselectivities (90–99% ee). The ortho-methyl-substituted substrate 1r reacted smoothly to give the desired product with excellent enantioselectivity, but the 2,6-dimethyl-substituted substrate 1z could not react at all. Functional groups such as ether, halo and benzyl were well tolerated in the current reaction (2k, 2l, 2m and 2o–q). Replacement of the N-substituents with acyclic carbamate was also tolerated but with a decreased ee (2u and 2z). Substrates containing a chiral oxazolidin-2-one unit bearing bulky Ph-substituents around the nitrogen and oxygen were also competent, yielding the desired products with good to excellent diastereoselectivities (2s, 2t, and 2v–y). Of note, the substrate 1s bearing an N-Ms substituent and the cyclic substrate 1t did not work in the current reaction. The absolute configuration of generated α-amidoboronates was assigned as (S) by X-ray crystallographic analysis of 2i (Scheme 3).20
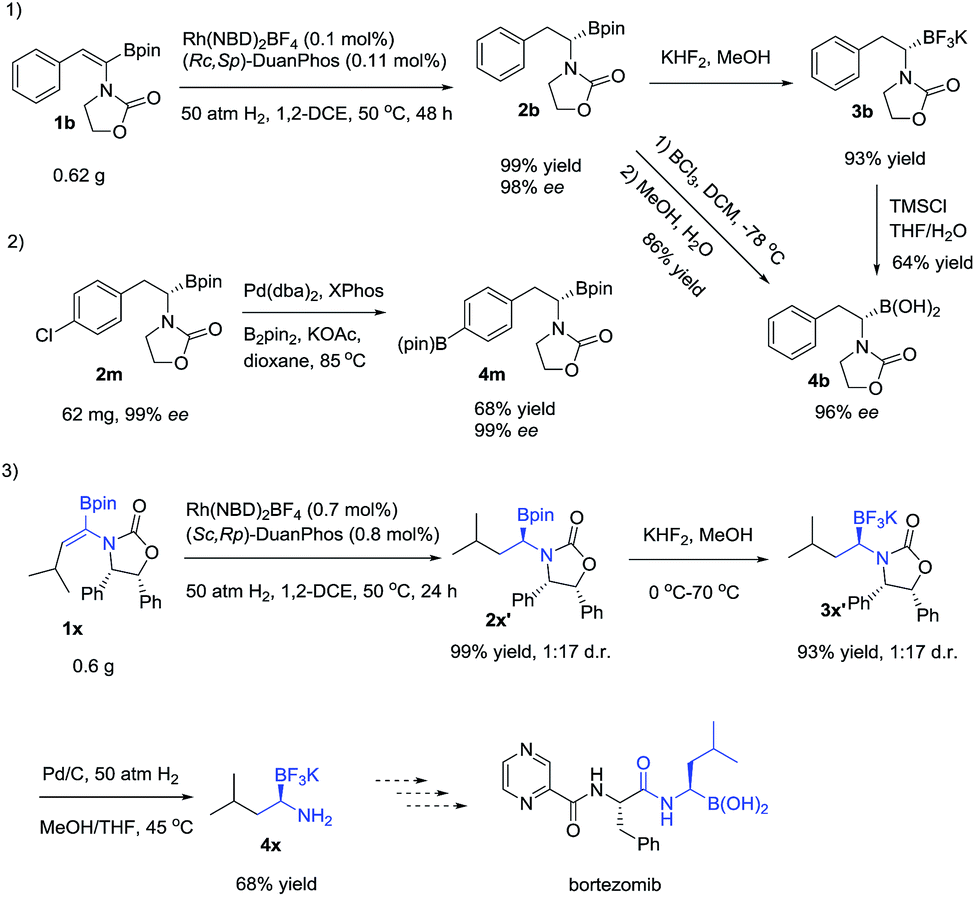 | ||
| Scheme 3 Scale-up synthesis and synthetic utility. | ||
To demonstrate the utility of the products, a scale-up reaction (0.62 g) was successfully performed with 0.1 mol% catalytic loading, giving 2b in 99% yield and 98% ee, and 2b could be easily transformed to a more stable α-amidoborate 3b with KHF2,6d,21 followed by hydrolysis with TMSCl to yield α-amido boronic acid 4b in 46% yield,22 which could also be obtained from 2b by treating it with BCl3 in 84% yield, without loss of the optical purity.8b2m could easily be transformed to 4m in 68% yield by a Pd-catalyzed borylation reaction. Meanwhile, after hydrogenation of 1x to 2x′ and transformation of 2x′ to its trifluoroborate derivative 3x′, removal of the benzyl group of 3x′ with Pd/C under hydrogenation conditions23 yielded the primary α-aminoborate 4x in 62% yield in three steps, which could serve as a potential precursor15 to synthesize bortezomib.
Conclusions
In summary, we have presented a strategically distinct alternative for the direct synthesis of chiral alkyl-substituted α-amidoboronates by Rh-catalyzed asymmetric hydrogenation. A series of (Z)-α-dehydroamido boronate esters could be hydrogenated to the corresponding alkyl-substituted α-amidoboronic esters in excellent yields with generally excellent enantioselectivities. The boronic acid derivatives and the potential precursor of bortezomib could be facilely obtained with this approach. Additional asymmetric transformations of the alkyl-substituted α-amidoboronates are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr. Xiaoyong Chang (Department of Chemistry, SUSTech) for the crystallographic analysis and the helpful suggestions from Prof. Jian Liao (Chinese Academy of Sciences, Chengdu Institute of Biology). Prof. Xumu Zhang is grateful for the financial support from the Science, Technology and Innovation Committee of Shenzhen (No. JSGG20160608140847864 and KQTD20150717103157174), Shenzhen Nobel Prize Scientists Laboratory Project (C17783101), and SZDRC Discipline Construction Program.Notes and references
- (a) R. C. Kane, P. F. Bross, A. T. Farrell and R. Pazdur, Oncologist, 2003, 8, 508 CrossRef; (b) J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304 CrossRef CAS; (c) R. C. Kane, R. Dagher, A. Farrell, C.-W. Ko, R. Sridhara, R. Justice and R. Pazdur, Clin. Cancer Res., 2007, 13, 5291 CrossRef CAS PubMed.
- Selected reviews, see: (a) L. R. Dick and P. E. Fleming, Drug Discovery Today, 2010, 15, 243 CrossRef CAS PubMed; (b) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279 RSC; (c) W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346 CrossRef CAS PubMed.
- (a) M. D. Valery, Q. Abed Al Aziz and S. Morris, Mini-Rev. Med. Chem., 2004, 4, 1001 CrossRef PubMed; (b) D. S. Matteson, Med. Res. Rev., 2008, 28, 233 CrossRef CAS; (c) L. J. Milo, J. H. Lai, W. Wu, Y. Liu, H. Maw, Y. Li, Z. Jin, Y. Shu, S. E. Poplawski, Y. Wu, D. G. Sanford, J. L. Sudmeier and W. W. J. Bachovchin, Med. Chem., 2011, 54, 4365 CrossRef CAS PubMed; (d) N. Micale, K. Scarbaci, V. Troiano, R. Ettari, S. Grasso and M. Zappalá, Med. Res. Rev., 2014, 34, 1001 CrossRef CAS PubMed.
- Selected reviews and examples, see: (a) S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275 CrossRef CAS PubMed; (b) L. Han, Y. Wen, R. Li, B. Xu, Z. Ge, X. Wang, T. Cheng, J. Cui and R. Li, Bioorg. Med. Chem., 2017, 25, 4031 CrossRef CAS; (c) K. Markus, H. Oliver, H. Philipp and B. Michaul, PCT Int. Appl., WO 2016050359 A1, 2016; (d) B. D. Dorsey, M. Iqbal, S. Chatterjee, E. Menta, R. Bernardini, A. Bernareggi, P. G. Cassarà, G. D. Arasmo, E. Ferretti, S. D. Munari, A. Oliva, G. Pezzoni, C. Allievi, I. Strepponi, B. Ruggeri, M. A. Ator, M. Williams and J. P. Mallamo, J. Med. Chem., 2008, 51, 1068 CrossRef CAS; (e) B. A. Teicher and J. E. Tomaszewski, Biochem. Pharmacol., 2015, 96, 1 CrossRef CAS PubMed; (f) A. Rentsch, D. Landsberg, T. Brodmann, L. Bülow, A. Girbig and M. Kalesse, Angew. Chem., Int. Ed., 2013, 52, 5450 CrossRef CAS; (g) D. Chauhan, Z. Tian, B. Zhou, D. Kuhn, R. Orlowski, N. Raje, P. Richardson and K. C. Anderson, Clin. Cancer Res., 2011, 17, 5311 CrossRef CAS.
- (a) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738 CrossRef CAS PubMed; (b) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef CAS PubMed; (c) T. Ohmura, T. Awano and M. Suginome, Chem. Lett., 2009, 38, 664 CrossRef CAS; (d) T. Ohmura, K. Miwa, T. Awano and M. Suginome, Chem.—Asian J., 2018, 13, 2414 CrossRef CAS PubMed.
- Reviews and selected examples, see: (a) D. S. Matteson, Chem. Rev., 1989, 89, 1535 CrossRef CAS; (b) D. S. Matteson, Acc. Chem. Res., 1988, 21, 294 CrossRef CAS; (c) D. S. Matteson and M. Debesh, J. Am. Chem. Soc., 1980, 102, 7588 CrossRef CAS; (d) D. S. Matteson and K. M. Sadhu, J. Am. Chem. Soc., 1981, 103, 5241 CrossRef CAS.
- (a) M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910 CrossRef CAS PubMed; (b) A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671 CrossRef CAS PubMed; (c) K. Hong and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 9252 CrossRef CAS PubMed; (d) S. Zhang, Y. Zhao, P. Tian and G. Lin, Synlett, 2013, 4, 437 Search PubMed; (e) D. Wang, P. Cao, B. Wang, T. Jia, Y. Lou, M. Wang and J. Liao, Org. Lett., 2015, 17, 2420 CrossRef CAS; (f) C. B. Schwamb, K. P. Fitzpatrick, A. C. Brueckner, H. C. Richardson, P. H. Cheong and K. A. Scheidt, J. Am. Chem. Soc., 2018, 140, 10644 CrossRef CAS.
- (a) D. Nishikawa, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 15620 CrossRef CAS PubMed; (b) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef CAS PubMed; (c) L. Chen, X. Zou, H. Zhao and S. Xu, Org. Lett., 2017, 19, 3676 CrossRef CAS PubMed; (d) A. López, T. B. Clark, A. Parra and M. Tortosa, Org. Lett., 2017, 19, 6272 CrossRef PubMed; (e) L. Chen, J.-J. Shen and S. Xu, Chem. Sci., 2018, 9, 5855 RSC.
- C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, 1045 CAS.
- Q. Qi, X. Yang, X. Fu, S. Xu and E. Negishi, Angew. Chem., Int. Ed., 2018, 57, 15138 CrossRef CAS PubMed.
- Selected reviews and examples, see: (a) F. Glorius, Org. Biomol. Chem., 2005, 3, 4171 RSC; (b) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem.–Eur. J., 2015, 21, 12226 CrossRef CAS PubMed; (c) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979 CrossRef CAS PubMed; (d) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (e) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357 CrossRef CAS PubMed; (f) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS PubMed; (g) Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS PubMed; (h) W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2019, 51, 1858 CrossRef PubMed.
- Rh-catalysed asymmetric hydrogenation of vinylboronic compounds, see: (a) J. B. Morgan and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 15338 CrossRef CAS PubMed; (b) W. J. Moran and J. P. Morken, Org. Lett., 2006, 8, 2413 CrossRef CAS PubMed; (c) G. Liu, A. Li, X. Qin, Z. Han, X.-Q. Dong and X. Zhang, Adv. Synth. Catal., 2019, 361, 2844 CrossRef CAS. Ni-catalysed example: Z. Han, G. Liu, X. Zhang, A. Li, X.-Q. Dong and X. Zhang, Org. Lett., 2019, 21, 3923 CrossRef CAS PubMed.
- Ir-catalyzed asymmetric hydrogenation of vinylboronic compounds, see: (a) A. Ganić and A. Pfaltz, Chem.–Eur. J., 2012, 18, 6724 CrossRef PubMed; (b) A. Paptchikhine, P. Cheruku, M. Engman and P. G. Andersson, Chem. Commun., 2009, 45, 5996 RSC.
- (a) I. G. Smilović, E. Casas-Arcé, S. J. Roseblade, U. Nettekoven, A. Zanotti-Gerosa, M. Kovačevič and Z. Časar, Angew. Chem., Int. Ed., 2012, 51, 1014 CrossRef PubMed; (b) S. J. Roseblade, I. G. Smilović and Z. Časar, Tetrahedron, 2014, 70, 2654 CrossRef CAS; (c) S. J. Roseblade, E. Casas-Arcé, U. Nettekoven, I. G. Smilović, A. Zanotti-Gerosa and Z. Časar, Synthesis, 2013, 45, 2824 CrossRef CAS.
- S. Touchet, F. Carreaux, G. A. Molander, B. Carboni and A. Bouillon, Adv. Synth. Catal., 2011, 353, 3391 CrossRef CAS PubMed.
- C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746 CrossRef CAS.
- (a) G. He, Q. Zhang, H. Huang, S. Chen, Q. Wang, D. Zhang, R. Zhang and H. Zhu, Eur. J. Org. Chem., 2013, 6979 CrossRef CAS; (b) E. M. Simmons, B. Mudryk, A. G. Lee, Y. Qiu, T. M. Razler and Y. Hsiao, Org. Process Res. Dev., 2017, 21, 1659 CrossRef CAS.
- Selected reviews and examples, see: (a) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed; (b) G.-Q. Chen, B.-J. Lin, J.-M. Huang, L.-Y. Zhao, Q.-S. Chen, S.-P. Jia, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 8064 CrossRef CAS PubMed; (c) X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024 CrossRef CAS PubMed.
- Here, an incorrect Cu-catalyzed regioselective hydroborylation of alkyl-substituted ynamides was observed in the reference: G. He, S. Chen, Q. Wang, H. Huang, Q. Zhang, D. Zhang, R. Zhang and H. Zhu, Org. Biomol. Chem., 2014, 12, 5945 RSC . Thus, we developed a new synthetic route using alkyl-substituted (Z)-α-dehydroamido boronate esters and on the gram-scale the reaction worked smoothly to yield the desired products.
- CCDC 1897699 ((S)-2i) contains the supplementary crystallographic data for this paper.†.
- V. Bagutski, A. Ros and V. K. Aggarwal, Tetrahedron, 2009, 65, 9956 CrossRef CAS.
- S. R. Inglis, E. C. Y. Woon, A. L. Thompson and C. J. Schofield, J. Org. Chem., 2010, 75, 468 CrossRef CAS PubMed.
- (a) O. Tamura, M. Hashimoto, Y. Kobayashi, T. Katoh, K. Nakatani, M. Kamada, I. Hayakawa, T. Akiba and S. Terashima, Tetrahedron Lett., 1992, 33, 3487 CrossRef CAS; (b) P. B. D. Ranslow, L. S. Hegedus and C. D. L. Rios, J. Org. Chem., 2004, 69, 105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04534a |
| This journal is © The Royal Society of Chemistry 2020 |