
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated alkaline hydrogen evolution on M(OH)x/M-MoPOx (M = Ni, Co, Fe, Mn) electrocatalysts by coupling water dissociation and hydrogen ad-desorption steps†
Lishan
Peng‡
 a,
Mansheng
Liao‡
a,
Xingqun
Zheng‡
a,
Yao
Nie
b,
Ling
Zhang
a,
Minjie
Wang
a,
Rui
Xiang
a,
Jian
Wang
a,
Li
Li
*a and
Zidong
Wei
*a
a,
Mansheng
Liao‡
a,
Xingqun
Zheng‡
a,
Yao
Nie
b,
Ling
Zhang
a,
Minjie
Wang
a,
Rui
Xiang
a,
Jian
Wang
a,
Li
Li
*a and
Zidong
Wei
*a
aThe State Key Laboratory of Power Transmission Equipment & System Security and New Technology, Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China. E-mail: zdwei@cqu.edu.cn; liliracial@cqu.edu.cn
bCollege of Chemistry, Chongqing Normal University, Chongqing 400047, China
First published on 27th January 2020
Abstract
Developing efficient and cheap electrocatalysts for the alkaline hydrogen evolution reaction is still a big challenge due to the sluggish water dissociation kinetics as well as poor M–Had energetics. Herein, hydroxide modification and element incorporation have been demonstrated to realize a synergistic modulation on a new class of M(OH)x/M-MoPOx catalysts for accelerating water dissociation and hydrogen ad-desorption steps in the HER. Theoretical and experimental results disclosed that in situ modification with hydroxide endowed M(OH)x/M-MoPOx with a strong ability to dissociate water, and meanwhile, oxygen incorporation effectively optimized the M–Had energetics of the NiMoP catalyst. Moreover, the interaction between M(OH)x and M-MoPOx components in M(OH)x/M-MoPOx further enhances their ability to catalyze the two elementary steps in alkaline hydrogen evolution, providing a wide avenue for efficiently catalyzing hydrogen evolution. In general, the optimized Ni(OH)2/NiMoPOx catalyst exhibits excellent alkaline HER activity and durability, superior to the state-of-the-art Pt/C catalyst when the overpotential exceeds 65 mV.
Introduction
Hydrogen has been extensively researched as an alternative fuel source to fossil fuels. Water electrolysis is an appealing, but challenging method for large-scale production of H2. In a water-alkali electrolyser, there are two half-cell reactions occurring at the cathode and anode, i.e. the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). The kinetics of these two electrochemical reactions under typical operation conditions are inherently slow, which necessitates the use of catalysts with high activity and stability to reduce electricity consumption.1–4 Nowadays, the most effective HER catalysts are Pt-based metals, but their industrial application is limited by their scarcity and high cost. Although some studies have been conducted to replace Pt with Ru compounds (that are relatively low in cost), the intrinsic limitation of noble metals has still not been overcome completely.5,6 Thus, it is especially important to use highly active HER electrocatalysts derived from low-cost materials.In recent few years, abundant non-noble metal-based materials have been developed as promising electrocatalysts for hydrogen evolution under alkaline conditions.7,8 Among these transition metal catalysts, molybdenum-based compounds,9 such as alloys,10 carbides,11–15 chalcogenides,16–18 and phosphides,19–21 have recently attracted significant research interest due to their desired chemical and physical properties. For example, as a prototypical model, MoS2 is the first reported transition metal-based HER catalyst, in which only the surface edges are catalytically active.22–24 Unlike MoS2, which only shows HER activity at surface edges, all the sites of bulk MoP show high HER activity. Calculations of P sites on MoP indicate that P acts like a ‘hydrogen deliverer’ as hydrogen adsorbs on P at low coverage whilst it desorbs at high coverage.25,26 Accordingly, molybdenum phosphides usually exhibit higher HER current density than MoS2, as more active sites are available for the intensive electrocatalytic reaction. However, even though these MoP-based catalysts have abundant active sites for catalyzing protons to hydrogen, their HER activities are still unsatisfactory, especially in alkaline solution.
Under alkaline conditions, the HER process inevitably undergoes the first water dissociation step (Volmer step), because there are few protons existing in alkaline solutions.27,28 In 2012, Markovic et al. probed the surface HER reactivity of the materials with near-optimal M–Had energetics (such as Pt) and found that it can be enhanced by modifying them with more efficient active sites (such as 3d metal hydroxides) for water molecule dissociation.29 Recently, our group has found that there is a strong interaction occurring at the interface of Ni@Ni(OH)2–Pd, which is effective for catalysing hydrogen generation in alkaline solution compared with the pure Pd catalyst.30 With this in mind, we speculate that two types of active sites are required for efficient alkaline hydrogen evolution, i.e., one with optimized M–Had energetics (ΔG(H)) and another with the capability of accelerating the dissociation of water. However, all the above-mentioned HER catalysts contain noble metals (e.g. Pt and Pd) which seem to act as indispensable parts for the high HER activity. Pure metal oxides or hydroxides are generally considered as HER inactive materials, although they facilitate the water dissociation step.31,32 Such strong dependency on noble metals is undesirable for future massive applications. Therefore, achieving a satisfactory synergism in accelerating both water dissociation and hydrogen ad-desorption (i.e. optimal M–Had energetics) steps on a non-noble material is urgent but challenging for an efficient HER.
Based on the above observation, we put forward that hydroxide modification and element incorporation can help realize a synergistic modulation for accelerating both water dissociation and hydrogen ad-desorption steps in the HER. And a new class of noble-metal free catalysts, M-MoPOx-based nanomaterials in situ modified with hydroxide species on the surface (M(OH)x/M-MoPOx, M = Ni, Co, Fe, Mn), served as a platform to elaborate our proposed view. Combining experiments with DFT calculations, we demonstrated that on the one hand, hydroxide modification generates more active sites in catalysts for water dissociation, thus accelerating the Volmer step during the HER process. On the other hand, oxygen incorporation can regulate the electronic structure of M-MoP, resulting in modulated M–Had energetics. More interestingly, we also revealed that the interaction between M(OH)x and M-MoPOx further enhances their catalytic abilities, i.e., M(OH)x and M-MoPOx components in M(OH)x/M-MoPOx catalysts exhibit greater abilities in catalysing water dissociation and hydrogen ad-desorption steps than their corresponding single component catalysts, respectively. Benefiting from the hydroxide modification, element incorporation and component interaction, efficient HER activity in an alkaline environment was achieved, proving the validity of coupling water dissociation and hydrogen ad-desorption steps for efficient alkaline hydrogen evolution.
Results and discussion
The typical preparation procedure of the hydroxide species modified M-MoPOx based nanomaterials is schematically described in Scheme S1.† Taking Ni(OH)2/NiMoPOx/NF as the representative example, a flower-like structure composed of regular NiMoO4 nanocuboids on the surface of Ni foam was first acquired via a facile and scalable hydrothermal method.15 Field-emission scanning electron microscopy (FESEM) indicates that the NiMoO4 nanocuboids uniformly grown on Ni foam have an average width of about 300 nm and a very smooth surface (Fig. S1a†), and the 3D-network structure of Ni foam is of great benefit for loading the active species. Then the as-prepared NiMoO4/NF was transformed into NiMoPOx/NF by phosphatizing with NaH2PO2 at 400 °C for 2 h under an Ar gas flow. No obvious change in the morphology can be observed when NiMoO4/NF was converted into NiMoPOx/NF (Fig. S1b–d†), while the crystal structure changes from the high-crystallinity NiMoO4 phase (JCPDS no. 86-0361) to the Ni2P4O12 phase (JCPDS no. 76-1557) (Fig. 1b). As depicted in Fig. S1e,† the NiMoPOx nanocuboids are composed of numerous nanocrystallites with a small size of about 10 nm, uniformly embedded in the amorphous phase. High-resolution TEM (HRTEM) confirmed the crystalline nature of the nanocrystallites. The lattice fringes with an interplanar distance of 0.23 nm in Fig. S1f† correspond to the (−133) planes of Ni2P4O12. Energy dispersive X-ray spectra (Fig. S1g†) show that Ni, Mo, P and O are distributed over the NiMoPOx nanocuboids. The atomic contents of Ni, Mo, P and O are 12.22, 13.66, 14.33 and 59.80, respectively (Fig. S1h†), and the exact chemical formula of MMoPOx is NiMo1.12P1.17O4.89. Considering the P-XRD results and EDX-mapping results, it can be speculated that the NiMoPOx material contains crystalline Ni2P4O12 and an amorphous Mo-containing compound.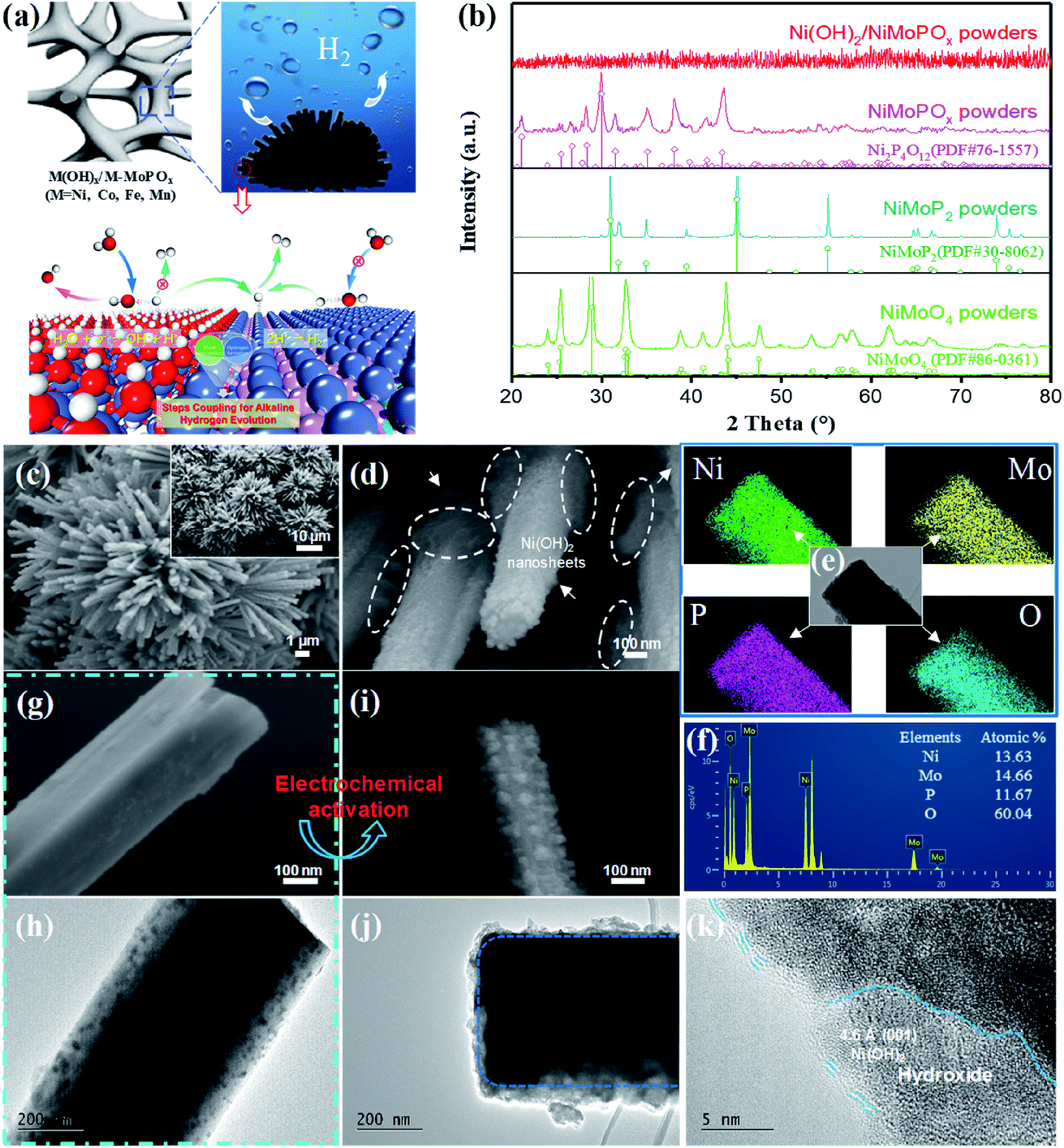 | ||
| Fig. 1 (a) Schematic illustration of the microstructure of M(OH)x/M-MoPOx. (b) P-XRD patterns of NiMoO4, NiMoPOx and Ni(OH)2/NiMoPOx powders scraped from Ni foam. (c and d) SEM images, (e) elemental mapping and (f) EDX spectrum of Ni(OH)2/NiMoPOx. Morphology comparison of (g and h) NiMoPOx and (i and j) Ni(OH)2/NiMoPOx on the same scale. (k) HR-TEM image of Ni(OH)2/NiMoPOx. | ||
After electrochemical transformation in 1 M NaOH, tiny and flexible Ni(OH)2 nanosheets were generated through redox of Ni2P4O12, and were uniformly attached to the surface of pre-synthesized NiMoPOx nanocuboids to produce Ni(OH)2/NiMoPOx/NF hybrid nanocuboids (named Ni(OH)2/NiMoPOx). Fig. S2a† displays the CV curves at different scans for the presented NiMoPOx/NF. Obviously, the current density of NiMoPOx/NF continuously increases with CV scans until 8000 cycles, as well as the area of the redox zone (−0.15 to 0.2 V). These results reveal that the catalytic properties of NiMoPOx/NF change constantly with the CV scans. On the basis of the general electrochemical redox states of elements under potential bias,33 it is speculated that the Ni species on the surface of NiMoPOx/NF varied during electrochemical activation. The growing area of the redox zone implies that more Ni species were involved in the reaction during the redox switching scan. In addition, the existence of PO43− ions in the electrolytes used for electrochemical activation of M-MoPOx was detected by UV-vis-NIR spectroscopy (Fig. S3a†). XRD results show that a crystal structure transition occurred after 8000 cycles, as crystalline Ni2P4O12 disappeared and NiMoPOx/NF became an amorphous structure (Fig. 1b). It has been reported that Co-based phosphate would leach out and transform into hydroxide during the electrochemical redox process in alkaline solution,34 which shows us that an in situ transformation of crystalline Ni2P4O12 into amorphous Ni(OH)2 occurred on the NiMoPOx/NF surface via the following pathways:
| Ni2P4O12 + 12OH− = 2Ni(OH)2 + P4O124− + 4H2O | (1) |
Electron microscopy observations together with X-ray photon spectroscopy (XPS) of NiMoPOx/NF prove the evolution of local structures during electrochemical activation. SEM images (Fig. 1c and d) reveal that the morphology of NiMoPOx/NF is maintained in activated Ni(OH)2/NiMoPOx/NF, while a large number of nanosheets appear on the surface of NiMoPOx/NF. The diameter of Ni(OH)2/NiMoPOx/NF nanocuboids gets smaller and their surface becomes more rough than that of NiMoPOx/NF (Fig. 1g–j). Although the mapping images (Fig. 1e) show that the elements are still uniformly distributed over the nanocuboids, the decreased atomic content of the P element measured by EDX spectroscopy (Fig. 1f) further reveals the dissolution of P-species during electrochemical activation. The HR-TEM image (Fig. 1k) verifies the existence of newly formed amorphous Ni(OH)2 nanosheets with a short-order (001) plane on the Ni(OH)2/NiMoPOx/NF surface and the disappearance of crystalline Ni2P4O12 nanoparticles inside the catalyst. The variation of element valences and their contents on the catalyst surface was analysed by XPS (Fig. 2). A higher Ni content in XPS than in EDX in contrast to the lower P and Mo contents in XPS than in EDX (Table S1,†Fig. 1f) demonstrates the relatively outer position of Ni and inner position of P and Mo in the transformed nanostructure. Ni species have three states (Fig. 2a), i.e. Ni0 (851.8 eV), Ni2+ (855.9 eV), and Ni3+ (858.5 eV). Compared with NiMoPOx/NF, Ni(OH)2/NiMoPOx/NF has a higher content of Ni2+ species and a decreased Ni3+ content. Meanwhile, an additional peak of OH− groups is observed in the O 1s spectrum of Ni(OH)2/NiMoPOx/NF, indicating the new formation of Ni(OH)2 on the surface of the activated catalysts (Fig. 2d). The sharply decreased P and Ni3+ content together with the intensively increased Ni2+ content measured by XPS further confirms that the Ni2P4O12 first leached out and the Ni ions are then re-deposited to form the hydroxide during the electrochemical redox process. The overall element contents of Ni(OH)2/NiMoPOx in Fig. 1f reveal that the core component is amorphous NiMoPOx. The relative contents of Mo species with different valences are stably retained in the Ni(OH)2/NiMoPOx/NF hybrids, verifying that the amorphous NiMoPOx below the thin Ni(OH)2 layer retains its original state after electrochemical activation. Based on the above evidence, Ni(OH)2/NiMoPOx/NF is shown to be composed of amorphous NiMoPOx in the core and a thin Ni(OH)2 layer on its surface. Beyond Ni(OH)2/NiMoPOx/NF, a series of 3D-nanocuboid hybrids composed of other 3d metals (Co, Fe, and Mn) were obtained. SEM images show that CoMoPOx/NF and MnMoPOx/NF also have a nanocuboid-like structure while the structure of FeMoPOx/NF is nanoflake-like (Fig. S4†). The element valences and contents of M-MoPOx and activated M(OH)x/M-MoPOx were measured by XPS (Fig. S4–S7 and Table S2†). The variation trend of element content in M-MoPOx and corresponding M(OH)x/M-MoPOx is similar to that of NiMoPOx, demonstrating the universality of electrochemical activation in modulating the catalyst surface structure.
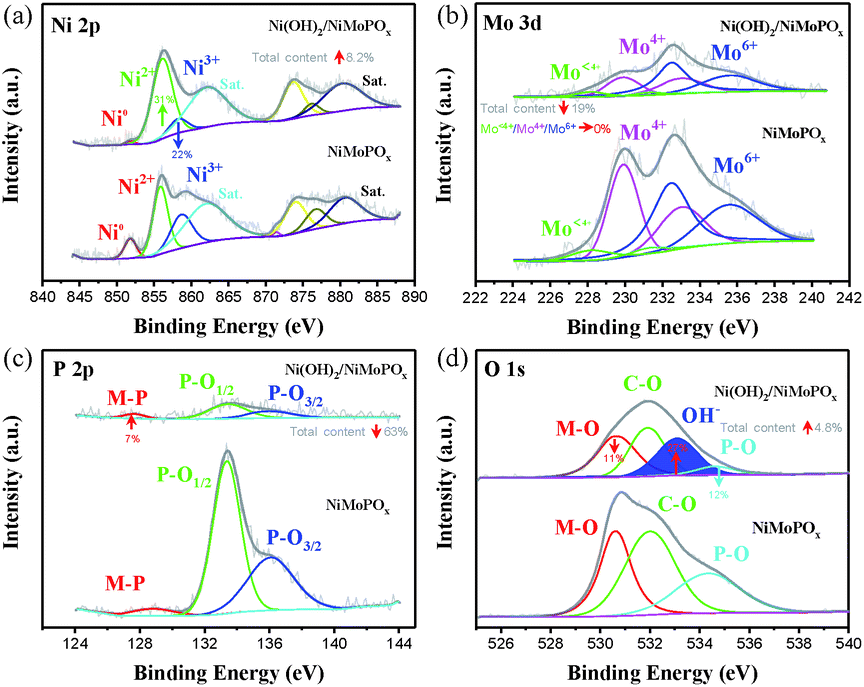 | ||
| Fig. 2 XPS spectra of NiMoPO4 and Ni(OH)2/NiMoPOx; (a) Ni 2p region, (b) Mo 3d region, (c) P 2p region and (d) O 1s region. | ||
The catalytic performance of the synthesized electrocatalysts in the alkaline HER was evaluated in N2-saturated 1.0 M NaOH. The polarization curves of Ni(OH)2/NiMoPOx/NF along with those of NiMoPOx/NF, NiMoP2/NF, NiMoO4/NF, Ni foam, and commercial Pt/C loaded on NF (20 wt% Pt/C/NF) without iR correction are shown in Fig. 3a. NiMoP2/NF exhibits a good HER activity, while Ni foam exhibits relatively poor HER activity, and NiMoO4/NF and Ni(OH)2/NF are even worse. As expected, the O incorporation greatly improves the HER performance of NiMoPOx/NF due to optimized hydrogen binding energy (Had).35 Furthermore, the generation of Ni(OH)2 on the NiMoPOx nanocuboids results in a sharply improved HER reactivity, which even outperformed that of the benchmark Pt/C. In the iR corrected polarization curves (Fig. 3b), Ni(OH)2/NiMoPOx/NF exhibits a rapid enhancement of the cathodic current along with negative potential and surpasses the current density of Pt/C when the overpotential exceeds 65 mV. Moreover, to drive a current density of 10 mA cm−2, Ni(OH)2/NiMoPOx/NF needs an extremely low overpotential of 51 mV, which is markedly lower than that of NiMoO4/NF (234 mV), NiMoPOx/NF (77 mV), and the most advanced HER electrocatalysts reported recently, such as Ni(OH)2/MoS2 (80 mV),36 NiCo2Px/CF (58 mV),37 Cu NDs/Ni3S2 NTs–CFs (128 mV)38 and CoMoP@C (81 mV)39 (more details in Table S6†). To obtain a larger cathodic current density of 100 mA cm−2, Ni(OH)2/NiMoPOx/NF needs an overpotential of 72 mV, approaching half the overpotential of 20% Pt/C/NF (154 mV, Table S3†). Such an excellent HER activity of Ni(OH)2/NiMoPOx/NF is impressive and shows its potential for industrial application.
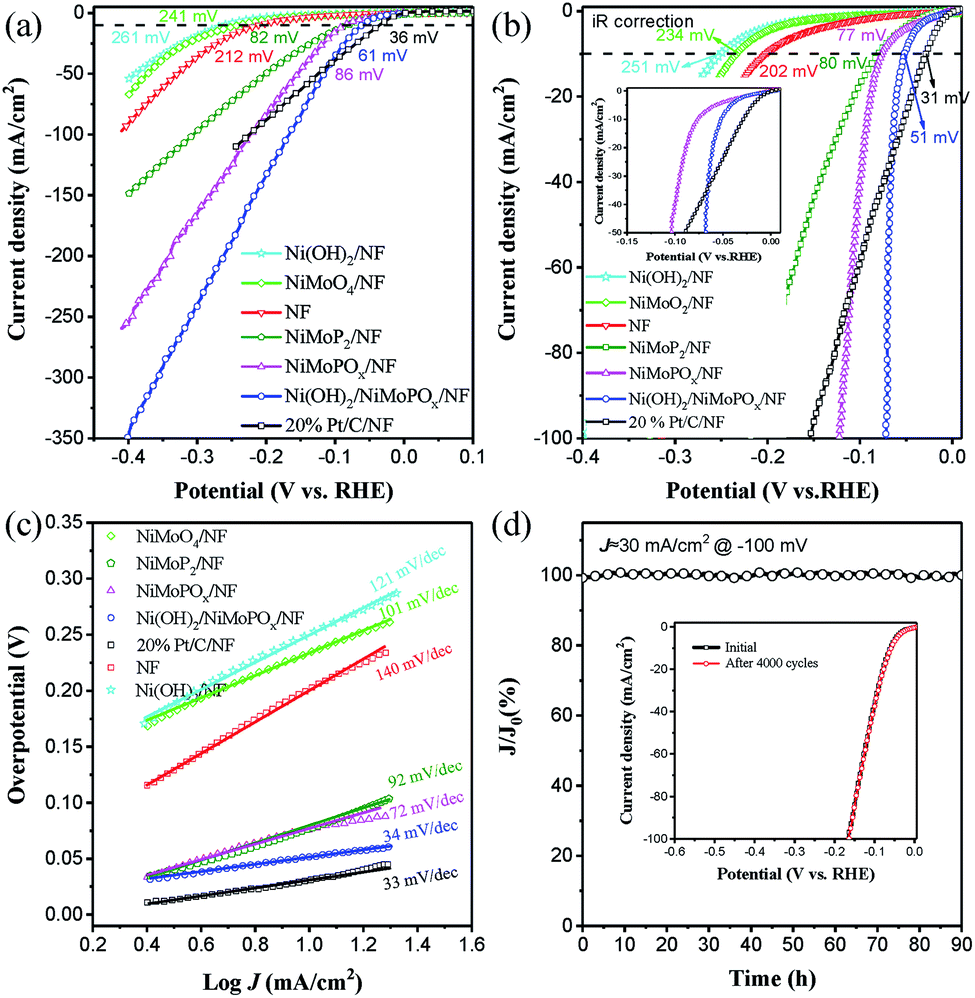 | ||
| Fig. 3 (a) LSV curves, (b) LSV curves with iR correction and (c) Tafel slopes of NiMoOx/NF, NiMoPOx/NF, Ni(OH)2/NiMoPOx/NF, 20% Pt/C/NF and NF with 5 mV s−1 in 1 M NaOH. (d) Chronopotentiometry curve of Ni(OH)2/NiMoPOx/NF at a constant potential of −100 mV for 90 hours. The inset in (d) is the LSV curves of Ni(OH)2/NiMoPOx/NF before and after 4000 CV scans at −0.3 V ∼ +0.2 V vs. RHE at 100 mV s−1. | ||
The Tafel plots of these electrocatalysts were measured to provide a deep insight into the HER reaction pathways on the electrocatalysts (Fig. 3c, Table S3†). The Tafel slope of Ni(OH)2/NiMoPOx/NF is 33 mV dec−1, much lower than that of Ni foam (140 mV dec−1), NiMoO4/NF (101 mV dec−1), NiMoPOx/NF (72 mV dec−1), and 20 wt% Pt/C/NF (34 mV dec−1). Such a low value of the Tafel slope suggests that the HER kinetics on Ni(OH)2/NiMoPOx/NF is determined by the Tafel step rather than by the Volmer step. Furthermore, the electrochemical impedance spectroscopy results (EIS, Fig. S8a†) of Ni(OH)2/NiMoPOx/NF further show a faster HER kinetics process than that on the NiMoPOx/NF. Compared with the NiMoPOx/NF, which undergoes the Volmer–Heyrovsky mechanism during the HER process, it is easy to infer that the formation of Ni(OH)2 on its surface significantly accelerates the HER reaction kinetics by optimizing the water dissociation step. In addition, the electrochemical specific surface area (ECSA, Fig. S8b and S9†) of Ni(OH)2/NiMoPOx/NF is 56.4 mF cm−2, which is similar to that of NiMoPOx/NF (48.6 mF cm−2), revealing that the enhancement of the HER activity is not attributed to the enlargement of the ECSA, but the improvement of the intrinsic activity.
Stability is another important evaluation index for the performance of catalysts in practical applications, especially in strongly alkaline media. The long-term stability of the best catalyst Ni(OH)2/NiMoPOx/NF was studied by continuous electrolysis at an overpotential of 100 mV for over 90 h. A steady current curve without notable degradation was observed during the 90 h aging test in an alkaline environment (Fig. 3d). Besides, the LSV curves (inset in Fig. 3d) of Ni(OH)2/NiMoPOx/NF show almost no change after 4000 CV cycles, revealing that Ni(OH)2/NiMoPOx/NF possesses good corrosion resistance in a wider potential region. The Ni(OH)2/NiMoPOx/NF also exhibits a comparable HER activity and stability to the industrial PtRuNiP/Ni electrode when integrated into a practical water electrolyzer (Fig. S10†). It is not difficult to understand the excellent stability of Ni(OH)2/NiMoPOx/NF: (i) the unique three-dimensional flower-like structures comprising regular nanocuboids can provide patulous architectures and open spaces that are beneficial to promote the release of generated gas bubbles. (ii) The in situ growth technology enhances the mechanical adhesion between catalysts and substrates. (iii) Moreover, the hydroxides formed on the surface of the catalyst and appropriate phosphate species can serve as protective agents to enhance the stability of Ni(OH)2/NiMoPOx/NF in alkaline solution.40,41
In order to confirm the synergistic effect of the hybrid structure for coupling the water dissociation and hydrogen ad-desorption processes during the alkaline HER, we studied the structural properties and HER activities of NiMoPOx, Ni(OH)2/NiMoPOx and Ni(OH)2/NiMoPOx-acid in 1 M HClO4 and 1 M NaOH (Fig. 4a, b and S11†). As for the Raman spectrum of the NiMoPOx sample, the absorption bands at ∼200, 330, 710, 880 and 960 cm−1 can be assigned to the phosphomolybdate anion.42,43 After electrochemical activation, two new broad bands at 365 and 470 cm−1 appear, corresponding to the Eg(T) mode of the Ni-OH lattice vibration and the A1g(T) mode due to υNi-OH, respectively.44,45 The band at 550 cm−1 is consistent with the structural defects.46,47 These results indicate the generation of amorphous Ni(OH)2 on the surface of NiMoPOx during electrochemical activation. The band of P–O–P for Ni(OH)2/NiMoPOx is red-shifted in comparison with that for the NiMoPOx, due to the interaction between Ni(OH)2 and NiMoPOx. The Ni(OH)2/NiMoPOx was then treated by acid etching in 0.5 M H2SO4 to remove the generated Ni(OH)2 on its surface and its Raman spectrum changes back to that of the NiMoPOx sample, revealing that only the surfacial component of NiMoPOx changes but the inside does not change during electrochemical activation.
 | ||
| Fig. 4 (a) Raman spectra and (b) comparison between activities for the HER, expressed as the overpotential required for a 10 mA cm−2 current density, in 1 M HClO4 and 1 M NaOH of the NiMoPOx, Ni(OH)2/NiMoPOx and Ni(OH)2/NiMoPOx-acid; (c) LSV curves of M(Ni, Co, Fe, Mn)-MoPOx/NF and their activated M(OH)2/M-MoPOx/NF electrodes without iR correction; (d) comparison with M(Ni, Co, Fe, Mn)-MoPOx/NF electrocatalysts before and after electrochemical activation in alkaline solutions. | ||
It should be pointed out that the HER activity at high pH values is determined using M–Had bond strength and the energy required for water dissociation simultaneously. While in acid electrolyte, the HER activity can only be determined using M–Had bond strength, as the H protons are abundant.48 Therefore, the alkaline activities of HER catalysts are always worse than their acid activities due to the additional energy required for water dissociation. As shown in Fig. 4b, the observed activities of the three catalysts in alkaline solutions are obviously lower than those in acid solutions. The alkaline HER activity of Ni(OH)2/NiMoPOx is significantly superior to that of NiMoPOx in alkaline electrolyte, and is similar to that of NiMoPOx in acid electrolyte. When the generated Ni(OH)2 on the surface of Ni(OH)2/NiMoPOx is removed by acid etching, the HER activity of Ni(OH)2/NiMoPOx-acid in both acid and alkaline electrolytes reduces back to that of the original NiMoPOx. These results confirm that the obstruction of water dissociation for NiMoPOx in alkaline electrolyte can be effectively resolved by the generated Ni(OH)2.
The HER activities of other 3d M(OH)x/M-MoPOx (M = Ni, Co, Fe, Mn) hybrids were systemically investigated to confirm the universality of coupling water dissociation and hydrogen ad-desorption for excellent alkaline HER activity. The HER polarization curves in 1 M NaOH are shown in Fig. 4c. A clear alkaline HER activity trend is observed in the order of Fe < Mn < Co < Ni for both M-MoPOx catalysts and the derived M(OH)x/M-MoPOx hybrids. Besides, all these derived M(OH)x/M-MoPOx hybrids exhibit an obvious improvement in activity to different degrees compared with the original M-MoPOx catalysts (Fig. 4d). This result reveals that the in situ formed hydroxide is commonly useful for improving the HER activity of these M-MoPOx-based catalysts. The Tafel slopes, EIS spectra, ECSA and ECSA-normalized HER polarization curves of these catalysts were further measured (Fig. S12–S14, Table S4†). The Tafel slopes of Ni(OH)2/NiMoPOx/NF and Co(OH)2/CoMoPOx/NF sharply reduce from around 70 mV dec−1 to below 40 mV dec−1 as well as that of Mn(OH)x/MnMoPOx/NF, revealing that the sluggish Volmer step (i.e. water dissociation) was thoroughly accelerated by the newly formed M(OH)x on the catalyst surface. The relatively larger Tafel slope of Fe(OH)x/FeMoPOx/NF is ascribed to the very strong adsorption of the Fe based hydroxide. The much smaller reaction resistances (Rct) of these M(OH)x/M-MoPOx catalysts further confirm the faster HER kinetics process on the hybrid catalysts. Therefore, it is speculated that the hybridized M(OH)x side is favourable for producing H* and OH−via accelerating the water dissociation step (H2O → H* + OH− + e−).49,50 On the other hand, all these M-MoPOx catalysts are weak in water dissociation, and the distinct difference in their HER activities should be ascribed to the ability of catalyzing the H2 ad-desorption step. Despite the strong water dissociation ability of Ni(OH)2, the poor HER activity of the Ni(OH)2/NF compared with that of NiMoPOx and Ni(OH)2/NiMoPOx further confirms the crucial role of the hydrogen ad-desorption step in the overall HER process. Based on the experimental results above, we can easily assume that superior alkaline HER catalysts should be capable of catalysing both water dissociation and hydrogen ad-desorption steps efficiently.
To gain further insights into the origin of the hybrid heterostructure for the excellent HER activity, DFT calculations on the H2O dissociation step (ΔG(H2O), Volmer step) and the H ad-desorption step (ΔG(H), Tafel step) were performed (Fig. 5a). The energy change in the Volmer reaction (ΔGR, Fig. 5b) for Ni(OH)2/NiMoPOx (0.60 eV) is lower than that of Ni*MoPOx (1.31 eV) and Ni*(OH)2 (1.38 eV), revealing that the Ni*(OH)2/NiMoPOx hybrid is more favourable for H2O dissociation thermodynamically. Moreover, the water dissociation kinetic energy barrier (ΔGTS) of Ni*(OH)2/NiMoPOx dramatically decreases from the 2.89 eV of Ni*MoPOx to 1.01 eV, suggesting that the sluggish Volmer step on NiMoPOx was greatly accelerated after the in situ generation of the Ni(OH)2 component. As for the concomitant Tafel step (Fig. 5c), the Ni(OH)2 has a very negative ΔG(H) (−0.37 eV), indicating a very high H adsorption strength, while all these NiMoP based catalysts present modulated ΔG(H) closer to the thermoneutral. The ΔG(H) of Ni*MoPOx is further decreased by 0.04 eV when O atoms are doped into the Ni*MoP, suggesting that the incorporation of O can modulate the H adsorption on the surface of the NiMoP catalyst. Interestingly, the final Ni(OH)2/Ni*MoPOx obtains an optimal ΔG(H) of 0.14 eV, which is even close to the absolute value of ΔG(H) for Pt* (−0.09 eV).51,52 Note that the water dissociation kinetic energy barrier (ΔGTS) and hydrogen adsorption free energy (ΔG(H)) of Ni(OH)2/NiMoPOx are superior to the ΔGTS of pure Ni(OH)2 and the ΔG(H) of pure NiMoPOx. This result reveals that the interaction between the two components in Ni(OH)2/NiMoPOx could further enhance their catalytic ability, i.e., the Ni(OH)2 and NiMoPOx components in Ni(OH)2/NiMoPOx catalysts possess greater ability to catalyze the water dissociation and hydrogen ad-desorption steps, respectively. Thus, the two types of active sites required for alkaline hydrogen evolution coexist adjacently in the Ni(OH)2/NiMoPOx catalysts, providing a new and smoother mechanism for hydrogen evolution via the synergy of Ni(OH)2 and NiMoPOx components. That is, H* is formed by H2O* dissociation on the metal hydroxide first and then transferred to the adjacent NiMoPOx sites to form H2. These results combined with experimental observations reveal that the excellent HER activity of Ni(OH)2/NiMoPOx is attributed to the synergistic effect in the hybrid components.
 | ||
| Fig. 5 (a) Chemisorption models of H and OH intermediates on the surfaces of NiMoPOx and the Ni(OH)2/NiMoPOx hybrid; calculated adsorption energy diagram of (b) the water dissociation step and (c) hydrogen ad-desorption for Ni(OH)2, NiMoPOx and the Ni(OH)2/NiMoPOx hybrid. The symbol * in the sample name represents the active site for DFT calculations. Color codes: Mo, cyan; P, pink; Ni, blue; O, red; H, white. | ||
Conclusions
In summary, synergistic modulation of both Volmer and Tafel steps for alkaline hydrogen evolution was achieved in NiMoP nanocuboids by hydroxide modification and element incorporation. By in situ modification with the hydroxide, the HER catalysts are endowed with the strong ability to dissociate water. Besides, the M–Had energetics of the NiMoP catalyst was optimized by oxygen incorporation, accelerating the hydrogen ad-desorption process. The interaction between the two components in the Ni(OH)2/NiMoPOx further enhances their individual catalytic ability. Therefore, the optimized oxygen incorporated NiMoP catalyst with a thin hydroxide layer modification possesses high reactivities for both water dissociation and hydrogen ad-desorption steps, thus exhibiting a superior HER performance in an alkaline environment. As demonstrated, the optimal Ni(OH)2/NiMoPOx possesses a remarkable HER activity with a low overpotential of 51 mV at 10 mA cm−2 as well as an excellent long-term stability for over 90 h, making it a prominent alternative for Pt-based catalysts. This work provides a direction to design efficient electrocatalysts by strengthening the reactivity of each elementary step.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially sponsored by the National Natural Science Foundation of China (Grant nos. 21822803, and 91834301). Special thanks to Lianqiao Tan and Jiao Yang for their contributions during the revision process.Notes and references
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS
- L. Peng and Z. Wei, Prog. Chem., 2018, 14–28 Search PubMed
- L. Peng, S. A. S. Syed and Z. Wei, Chin. J. Catal., 2018, 39, 1575–1593 CrossRef CAS
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC
- H. Wang and L. Gao, Curr. Opin. Electrochem., 2018, 7, 7–14 CrossRef CAS
- L. Peng, X. Zheng, L. Li, L. Zhang, N. Yang, K. Xiong, H. Chen, J. Li and Z. Wei, Appl. Catal., B, 2019, 245, 122–129 CrossRef CAS
- X. Zhang, F. Zhou, W. Pan, Y. Liang and R. Wang, Adv. Funct. Mater., 2018, 1804600, DOI:10.1002/adfm.201804600
- L. Yang, L. Zeng, H. Liu, Y. Deng, Z. Zhou, J. Yu, H. Liu and W. Zhou, Appl. Catal., B, 2019, 249, 98–105 CrossRef CAS
- Y. Huang, J. Hu, H. Xu, W. Bian, J. Ge, D. Zang, D. Cheng, Y. Lv, C. Zhang, J. Gu and Y. Wei, Adv. Energy Mater., 2018, 8, 1800789 CrossRef
- Y. Huang, J. Ge, J. Hu, J. Zhang, J. Hao and Y. Wei, Adv. Energy Mater., 2018, 8, 1701601 CrossRef
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef
- L. Peng, Y. Nie, L. Zhang, R. Xiang, J. Wang, H. Chen, K. Chen and Z. Wei, ChemCatChem, 2017, 9, 1588–1593 CrossRef CAS
- K. Xiong, L. Li, L. Zhang, W. Ding, L. S. Peng, Y. Wang, S. G. Chen, S. Y. Tan and Z. D. Wei, J. Mater. Chem. A, 2015, 3, 1863–1867 RSC
- Z. Hao, S. Yang, J. Niu, Z. Fang, L. Liu, Q. Dong, S. Song and Y. Zhao, Chem. Sci., 2018, 9, 5640–5645 RSC
- R. Xiang, Y. Duan, L. Peng, Y. Wang, C. Tong, L. Zhang and Z. Wei, Appl. Catal., B, 2019, 246, 41–49 CrossRef CAS
- Z. Liu, L. Zhao, Y. Liu, Z. Gao, S. Yuan, X. Li, N. Li and S. Miao, Appl. Catal., B, 2019, 246, 296–302 CrossRef CAS
- X. Xiao, L. Tao, M. Li, X. Lv, D. Huang, X. Jiang, H. Pan, M. Wang and Y. Shen, Chem. Sci., 2018, 9, 1970–1975 RSC
- L. Su, X. Cui, T. He, L. Zeng, H. Tian, Y. Song, K. Qi and B. Y. Xia, Chem. Sci., 2019, 10, 2019–2024 RSC
- Y. Huang, X. Song, J. Deng, C. Zha, W. Huang, Y. Wu and Y. Li, Appl. Catal., B, 2019, 245, 656–661 CrossRef CAS
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 36, 5308–5309 CrossRef PubMed
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed
- A. M. Appel, D. L. Dubois and M. R. Dubois, J. Am. Chem. Soc., 2005, 127, 12717–12726 CrossRef CAS
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J. Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC
- A. B. Laursen, S. Kegnæs, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 RSC
- F. Safizadeh, E. Ghali and G. Houlachi, Int. J. Hydrogen Energy, 2015, 40, 256–274 CrossRef CAS
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed
- Z. H. Deng, J. Wang, Y. Nie and Z. D. Wei, J. Power Sources, 2017, 352, 26–33 CrossRef CAS
- M. Gong, D.-Y. Wang, C.-C. Chen, B.-J. Hwang and H. Dai, Nano Res., 2015, 9, 28–46 CrossRef
- G. Q. Shi and H.-Y. Wang, Acta Phys.-Chim. Sin., 2018, 34, 22–35 Search PubMed
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC
- J. Ryu, N. Jung, J. H. Jang, H.-J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef CAS
- L. Zhang, X. Wang, X. Zheng, L. Peng, J. Shen, R. Xiang, Z. Deng, L. Li, H. Chen and Z. Wei, ACS Appl. Energy Mater., 2018, 1, 5482–5489 CAS
- B. Zhang, J. Liu, J. Wang, Y. Ruan, X. Ji, K. Xu, C. Chen, H. Wan, L. Miao and J. Jiang, Nano Energy, 2017, 37, 74–80 CrossRef CAS
- R. Zhang, X. Wang, S. Yu, T. Wen, X. Zhu, F. Yang, X. Sun, X. Wang and W. Hu, Adv. Mater., 2016, 29, 1605502 CrossRef PubMed
- J. X. Feng, J. Q. Wu, Y. Tong and G. R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS PubMed
- Y. Y. Ma, C. X. Wu, X. J. Feng, H. Q. Tan, L. K. Yan, Y. Liu, Z. Kang, E. Wang and Y. G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC
- Y. Yan, B. Y. Xia, X. M. Ge, Z. L. Liu, A. Fisher and X. Wang, Chem.–Eur. J., 2015, 21, 18062–18067 CrossRef CAS PubMed
- L. Peng, J. Wang, Y. Nie, K. Xiong, Y. Wang, L. Zhang, K. Chen, W. Ding, L. Li and Z. Wei, ACS Catal., 2017, 7, 8184–8191 CrossRef CAS
- C. Stinner, R. Prins and T. Weber, J. Catal., 2000, 191, 438–444 CrossRef CAS
- D. Boudlich, L. Bih, M. E. H. Archidi, M. Haddad, A. Yacoubi, A. Nadiri and B. Elouadi, J. Am. Ceram. Soc., 2002, 85, 623–630 CrossRef CAS
- P. Hermet, L. Gourrier, J.-L. Bantignies, D. Ravot, T. Michel, S. Deabate, P. Boulet and F. Henn, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235211 CrossRef
- J.-L. Bantignies, S. Deabate, A. Righi, S. Rols, P. Hermet, J.-L. Sauvajol and F. Henn, J. Phys. Chem. C, 2008, 112, 2193–2201 CrossRef CAS
- M. C. Biesinger, B. P. Payne, L. W. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS
- J. W. Lee, T. Ahn, D. Soundararajan, J. M. Ko and J.-D. Kim, Chem. Commun., 2011, 47, 6305–6307 RSC
- R. Subbaraman and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., 2012, 124, 12663–12666 CrossRef
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2016, 16, 70 CrossRef PubMed
- T. Liu, X. Ma, D. Liu, S. Hao, G. Du, Y. Ma, A. M. Asiri, X. Sun and L. Chen, ACS Catal., 2016, 7, 98–102 CrossRef
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04603h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |