
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow zeolite microspheres as a nest for enzymes: a new route to hybrid heterogeneous catalysts†
Valentin
Smeets
 a,
Walid
Baaziz
b,
Ovidiu
Ersen
b,
Eric M.
Gaigneaux
a,
Cédric
Boissière
c,
Clément
Sanchez
c and
Damien P.
Debecker
*a
a,
Walid
Baaziz
b,
Ovidiu
Ersen
b,
Eric M.
Gaigneaux
a,
Cédric
Boissière
c,
Clément
Sanchez
c and
Damien P.
Debecker
*a
aInstitute of Condensed Matter and Nanosciences (IMCN), UCLouvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504 CNRS – Université de Strasbourg, 23 rue du Loess, 67034 Strasbourg Cedex 2, France
cLaboratoire Chimie de la Matière Condensée de Paris (LCMCP), Sorbonne Université, CNRS, Collège de France, PSL Research University, 4 Place Jussieu, F-75005 Paris, France
First published on 9th December 2019
Abstract
In the field of heterogeneous catalysis, the successful integration of enzymes and inorganic catalysts could pave the way to multifunctional materials which are able to perform advanced cascade reactions. However, such combination is not straightforward, for example in the case of zeolite catalysts for which enzyme immobilization is restricted to the external surface. Herein, this challenge is overcome by developing a new kind of hybrid catalyst based on hollow zeolite microspheres obtained by the aerosol-assisted assembly of zeolite nanocrystals. The latter spheres possess open entry-ways for enzymes, which are then loaded and cross-linked to form cross-linked enzyme aggregates (CLEAs), securing their entrapment. This controlled design allows the combination of all the decisive features of the zeolite with a high enzyme loading. A chemo-enzymatic reaction is demonstrated, where the structured zeolite material is used both as a nest for the enzyme and as an efficient inorganic catalyst. Glucose oxidase (GOx) ensures the in situ production of H2O2 subsequently utilized by the TS-1 zeolite to catalyze the epoxidation of allylic alcohol toward glycidol. The strategy can also be used to entrap other enzymes or combination of enzymes, as demonstrated here with combi-CLEAs of horseradish peroxidase (HRP) and glucose oxidase. We anticipate that this strategy will open up new perspectives, leveraging on the spray-drying (aerosol) technique to shape microparticles from various nano-building blocks and on the entrapment of biological macromolecules to obtain new multifunctional hybrid microstructures.
Introduction
Following the green chemistry principles,1 sustainable synthesis routes are highly sought-after, not necessarily to maximize conversions and yields, but rather to minimize energy consumption and waste production.2 In this context, the integration of enzymatic and heterogeneous catalysis offers great potential for the development of new industrially relevant cascade processes.3,4 Examples of such combination are currently flourishing.5–9 For such chemo-enzymatic reactions, it appears attractive to develop efficient bifunctional hybrid catalysts that combine both the chemo and bio–catalytic activity on the same solid material.10–12 However, finding a suitable operational window in terms of reaction conditions is challenging because enzymes – unlike most inorganic catalysts – usually work in aqueous conditions under mild conditions of temperature and pH.13 Importantly, the preparation of an actual chemo-enzymatic heterogeneous catalyst itself is complicated because the two species are often somewhat incompatible. For example, combining a zeolite catalyst with an enzyme in the same multifunctional solid is problematic: zeolite micropores cannot accommodate the enzyme and a covalent grafting on the external surface of the zeolite crystals is ineffective. In the present work, we developed an original synthetic approach allowing to combine an enzyme with a zeolite to form a single chemo-enzymatic heterogeneous catalyst active in a cascade reaction. As a first proof of concept, we focus on a zeolite-catalyzed olefin epoxidation for which the oxidant is produced by an enzyme.Epoxides belong to an important class of chemical compounds as they are active intermediates for the production of high added value products in the pharmaceutical sector as well as polymers and adhesives.14 Many epoxidation reactions are run in the liquid phase in the presence of an oxidizing agent and of a solid catalyst, typically a titanosilicate (Ti–SiO2) – with Ti atoms in tetrahedral coordination in the silica framework.15 Among the available catalysts and oxidants, titanosilicalite-1 (TS-1) and hydrogen peroxide represent the most successful epoxidation processes at the industrial scale. On the first hand, TS-1 zeolite features tremendous performance for the epoxidation of lower olefins (e.g. propylene, allyl alcohol, allyl chloride, etc.),16 even in water and at moderately low temperature. On the second hand, hydrogen peroxide is considered as one of the greenest oxidants, since it produces water as the only by-product. However, industrially, H2O2 is currently produced via a non-green and centralized anthraquinone-based process.17 H2O2 is also intrinsically unstable: transportation and storage of peroxo compounds have been the cause of important damage in the chemical industry.
As an alternative, H2O2 could be produced in situ by an enzyme, such as glucose oxidase (GOx, E.C 1.1.3.4), using molecular oxygen and β-D-glucose as bio-sourced and renewable substrates.18 Immobilizing the enzyme onto a solid support (e.g. by adsorption, covalent binding, encapsulation)19–21 usually allows stabilizing the enzyme against heat and organic solvents as well as to recover it at the end of the reaction.22,23 Additional motivations for enzyme immobilization include the perspective of performing enzymatic reactions in continuous flow processes24,25 and implementing multi-enzymatic cascade reactions.26,27
Moving further, we aim at developing a hybrid chemo-enzymatic catalyst by combining the enzyme with a carrier that is itself catalytically active. Such hybrid catalyst – combining both the activity of a heterogeneous catalyst and of an enzyme in a single reusable solid – would be very attractive for various one-pot tandem catalytic systems3,28,29 where one reaction is carried out by an inorganic catalyst and the other one is catalyzed by an enzyme. However, only very few studies report such true hybrid heterogeneous catalysts showing a chemo-enzymatic activity.30–33 In 2010, Vennestrøm et al. proposed the chemo-enzymatic formation of epoxides by tandem catalysis using free GOx and TS-1 zeolite (Scheme 1).31 The direct immobilization of the enzyme on the surface of the microporous TS-1via covalent anchoring was also attempted but the epoxide production was limited to traces, due to the low enzyme loading, and to the poor enzyme stability against experimental conditions. In fact, TS-1 possesses a low external surface area and a low silanol content that limit the number of anchoring sites for the enzyme.
 | ||
| Scheme 1 Reaction pathway for the chemo-enzymatic combination of glucose oxidase (GOx) and inorganic TS-1 epoxidation catalyst (TS-1) with in situ production of H2O2. In the present work, the zeolite (blue) and the enzyme (green) are combined in a single bifunctional catalyst. | ||
A first way to increase the enzyme loading could be to immobilize it onto a Ti–SiO2 epoxidation catalyst possibly prepared by a bottom-up sol–gel approach to generate larger pores.34,35 Such mesostructured materials may possess a much larger accessible surface area for the enzyme coupled with a higher silanol content, allowing to increase the enzyme loading. Yet, in the presence of water (which is required for the enzyme) such amorphous mixed oxides rapidly deactivate.36,37 Therefore, TS-1 zeolite should be preferred as host for the enzyme, since the crystallinity and intrinsic hydrophobicity of this material protect the Ti active sites against deactivation when carrying out reaction in aqueous conditions.38 However, the microporosity of TS-1 limits the enzyme loading to the external surface, falling into a dilemma. A possible solution could be to use large hollow zeolite crystals39 as cargos to load large amounts of enzyme; yet these structures do not offer entry ways for enzymes to be loaded in the central cavity.
Here, we propose a new strategy (Scheme 2) for the preparation of a hybrid catalyst that allows to combine all the features of the zeolite (TS-1) with a high loading of enzyme (GOx). The method is based on the aerosol-assisted40–42 assembly of TS-1 nanocrystals to form hollow zeolite microspheres with a hierarchical porosity and high epoxidation activity in water-based media. Hierarchical zeolites43–50 have already been identified as possible candidates for enzyme immobilization51,52 but never exploited in chemo-enzymatic catalysis. The presented aerosol method allows preparing hierarchical hollow zeolite structures using an easy, continuous, and scalable aerosol process.53 The spheres can be loaded with the enzymes which are then cross-linked54,55 to secure their entrapment and avoid their subsequent leaching.56,57 Thus, the hollow microspheres are used both as epoxidation catalysts and as vessels for the enzyme. More importantly, we demonstrate that this material synthesis strategy allows to easily tune the enzyme loading, thereby providing a versatile method to envisage the one-pot production of valuable molecules. The strategy can also be used to entrap other enzymes and combinations of enzymes, which paves the way to more intricate chemo-enzymatic cascade processes.
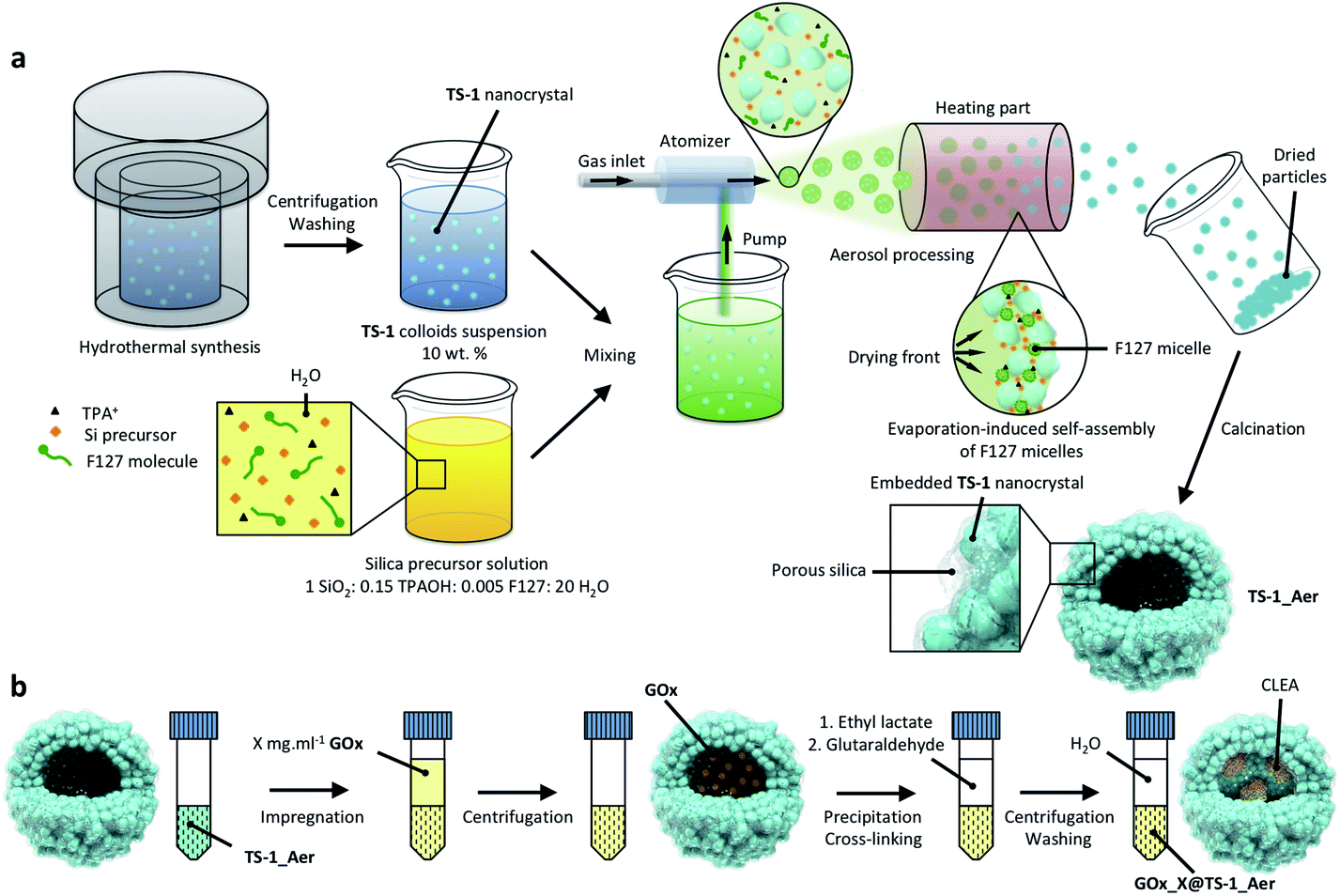 | ||
| Scheme 2 Schematic representation of the synthesis strategy used for (a) the preparation of hollow zeolite microspheres formed by the aggregation of TS-1 crystals using the aerosol method, (b) the immobilization of glucose oxidase on the aerosol-made material by the formation of entrapped cross-linked enzyme aggregates (CLEAs). | ||
Results and discussion
The inorganic catalyst – called “TS-1_Aer” – was prepared by an aerosol technique from a TS-1 colloids suspension58 (ca. 10 wt%) and a clear silica precursor solution containing a Si precursor as well as pluronic F127 and tetrapropylammonium cations (TPA+) templates. As depicted in Scheme 2a, the TS-1 suspension supplemented with Si precursor and with templates is sprayed in the form of an aerosol and dried rapidly upon contact with a flow of hot air (experimental details can be found in the ESI†). The two mixtures were mixed so that the final composition of the solid material is 50 vol% TS-1 and 50 vol% porous silica. As a reference material, TS-1 nanocrystals were isolated by centrifugation of the 10 wt% suspension, followed by drying in air and calcination. This reference catalyst is denoted “TS-1”. ICP-AES analysis showed that the experimental bulk Ti content (mol Ti/(mol Ti + mol Si) × 100%) of TS-1 was 1.5%, close to the nominal one (1.8%). Owing to the additional presence of the silica binder, the Ti content in TS-1_Aer was lower (1.1%).XPS analyses were conducted in order to investigate the surface composition and to probe the quality of the Ti dispersion. In TS-1, the surface Ti content was 1.3%, close to the bulk composition. From the Ti 2p3/2 peak decomposition59,60 (Fig. S7, ESI†), only 10% of the total Ti content is imputable to extra-framework Ti–O–Ti (458.5 eV) whereas a large majority of Ti species formed Ti–O–Si oxo-bridges (460.0 eV). The maximum absorption band at ca. 220 nm in the DRUV-vis spectrum of TS-1 indicates that these dispersed species are in a tetrahedral coordination sphere (Fig. 1a).61 The latter 4-coordination sites are known to be the active species in epoxidation.38 From the minor contribution above 250 nm and the data from XPS, we surmise that the small amount of extra-framework Ti species – with higher coordination numbers (5- and 6-) – were incorporated in the form of oligomeric Ti species or amorphous TiO2 nano-domains.62 However, no evidence of the presence of a TiO2 crystalline phase, such as anatase – having a characteristic absorption band at ca. 330 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63 – was found. This observation was further supported by the PXRD pattern of TS-1, which did not show the signature peak of anatase at ca. 25° (Fig. 1b).64
63 – was found. This observation was further supported by the PXRD pattern of TS-1, which did not show the signature peak of anatase at ca. 25° (Fig. 1b).64
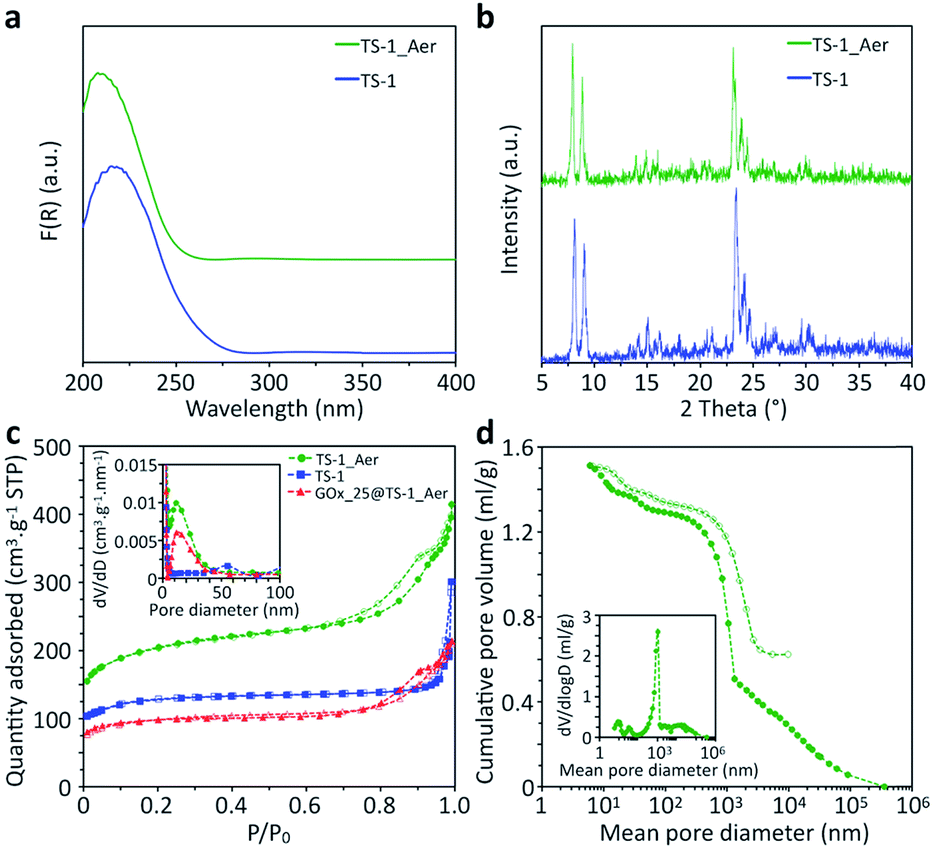 | ||
| Fig. 1 (a) DRUV-vis spectra (F(R) is the Kubelka–Munk Function) and (b) PXRD patterns of TS-1 and TS-1_Aer, (c) N2 adsorption–desorption isotherms of the TS-1, TS-1_Aer and GOx_25@TS-1_Aer catalysts, (d) Hg intrusion–extrusion isotherms of TS-1_Aer. Pore size distributions (PSDs) based on the adsorption/intrusion branch are shown in inset. Full symbols are used for the adsorption/intrusion isotherms and empty symbols are used for the desorption/extrusion isotherms. | ||
In TS-1_Aer, Ti atoms were still detected at the solid surface by XPS, although the signal from Ti electrons was partially masked by the surrounding silica phase (Fig. S7†). DRUV-vis spectrum of TS-1_Aer is analogous to that of TS-1 (Fig. 1a). Similar to TS-1, TS-1_Aer presented the typical MFI crystal structure58 measured by PXRD analysis (Fig. 1b). Those results confirmed that both Ti speciation and crystallinity of TS-1 were well preserved after the aerosol processing.
SEM-FEG images (Fig. 2a and b, see also Fig. S8†) show that single particles of TS-1_Aer – with size in the 0.5–8 μm range (Fig. S9†) – display a spherical shape. Embedded TS-1 crystals – with individual dimension of 100–150 nm – are visible at the surface along with the porous silica phase, showing pore size of ca. 10 nm (inset Fig. 2b) as well as interparticular voids between TS-1 crystals – with a mean size of 40–60 nm. Such results were confirmed by STEM Electron Tomography experiments realized on representative TS-1_Aer particles of ca. 3 μm and ca. 1.2 μm (Fig. 2c–e and S10,† respectively). Fig. 2d presents a typical (xy) slice extracted from the 3D volume, (Fig. 2d) which shows clearly the hollow character of a catalyst particle of ca. 3 μm (3D model animation is available in ESI†). The central void part was found to be about 1.2 μm in diameter (and about 400 nm for the particle of ca. 1.2 μm, Fig. S10c†). The porous silica shell is formed by individual crystals displaying a faceted morphology with a mean size of ca. 120 nm, consistent with the initial size of the TS-1 zeolite nanocrystals (Fig. S10d†).
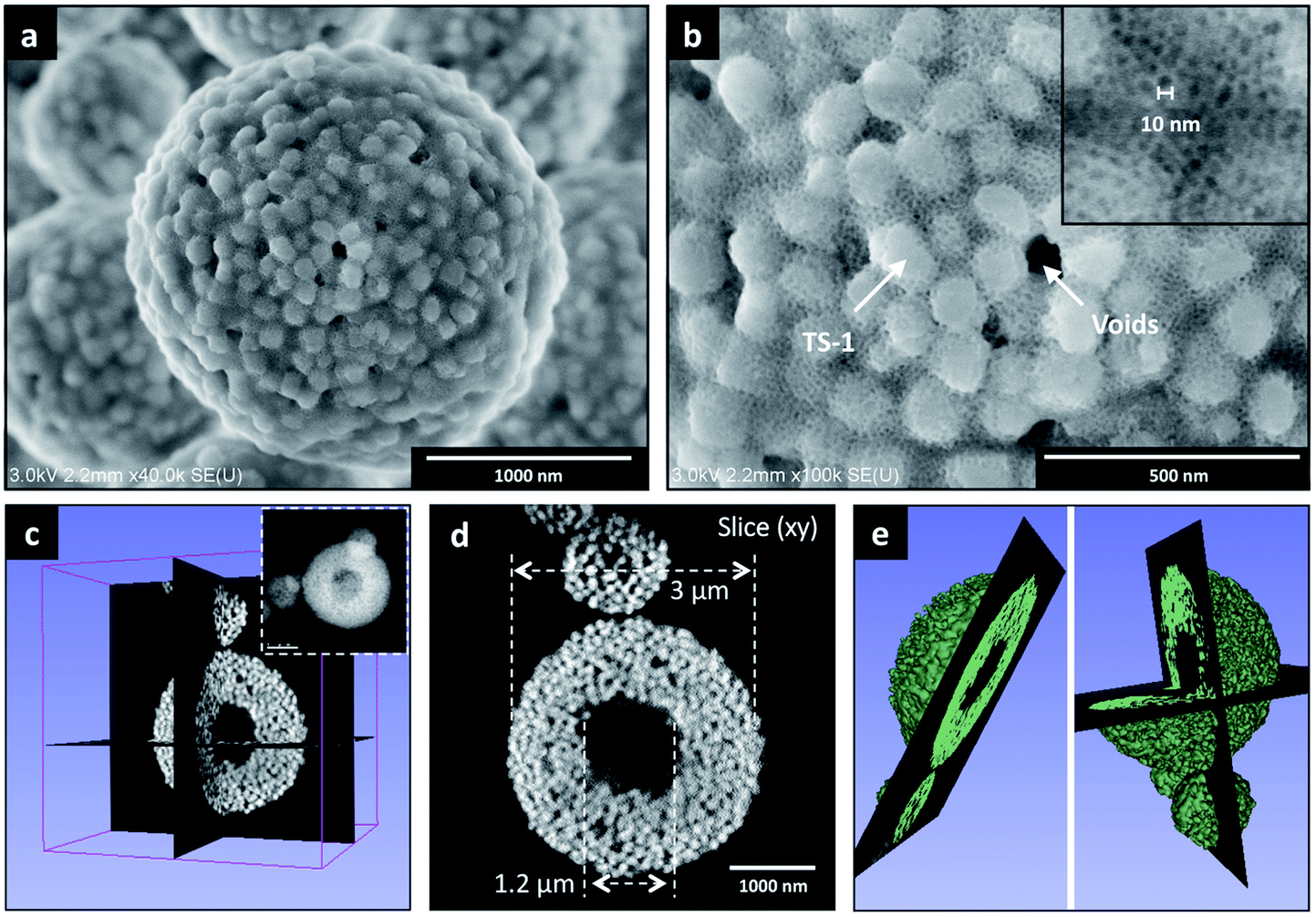 | ||
| Fig. 2 (a) SEM-FEG image of TS-1_Aer, (b) magnified image showing TS-1 crystals and ca. 10 nm mesopores from the porous silica phase (inset). Additional SEM-FEG images are presented in Fig. S8.† (c) 3D volume reconstructed by STEM Electron Tomography of one TS-1_Aer particle of ca. 3 μm in size (present inset), (d) typical (xy) slice extracted from the 3D volume illustrating the presence of a central cavity and (e) the corresponding 3D model (video animation available in ESI†). | ||
N2 adsorption–desorption isotherms of TS-1 and TS-1_Aer are shown in Fig. 1c, along with the adsorption pore size distributions (PSDs). As expected, TS-1 solely displays micropores (Vμ = 0.15 cm3 g−1) and interparticular voids which contribute to the total pore volume (VP = 0.43 cm3 g−1). For this material, the external specific surface area is 130 m2 g−1 according to the t-plot method (Fig. S11†). In comparison, a pore volume of 0.58 cm3 g−1 (with Vμ = 0.18 cm3 g−1) is obtained for TS-1_Aer and the external specific surface area reaches 320 m2 g−1. This is associated with the presence of mesopores in the silica binder, as also evidenced by the shape of the type IV isotherm (see Fig. 1c). Adsorption PSD in Fig. 1c confirms the presence of mesopores in the 10–20 nm range. This pore size is related to the templating effect of F127 micelles, swollen due to the additional incorporation of TPA+ cations.35,65 Thus the shell of the hollow spheres is porous enough to allow for the diffusion of individual GOx, whose size is 6.0 × 5.2 × 7.8 nm3.66
To have a better insight into the macroporosity of the aerosol-made catalyst, a Hg porosimetry experiment was carried out. The inset of Fig. 1d shows two contributions at 10 and 40 nm, which are in agreement with data from the N2 physisorption analysis (Fig. 1c) and SEM-FEG images (inset of Fig. 2b), respectively. The intrusion isotherm shows also an increase of the cumulative pore volume corresponding to pore diameters between 0.3 and 1.5 μm, which can be assigned to the hollow interior of the spheres, as also indicated by the STEM Electron Tomography experiments (Fig. 2d and S10c†). Interstices derived from the packing of the spheres are most likely found in the region between 1 and 10 μm, and represent approximately 26% of the intrusion volume according to the Mayer–Stowe model.67 Combining data from N2 physisorption and Hg porosimetry, the total pore volume – taking into account the three levels of micro-, meso- and macro-porosity – was ca. 1.3 cm3 g−1.
The catalytic performance of TS-1_Aer for the Ti-catalyzed conversion of allyl alcohol in water (45 °C) was compared to TS-1. A hot filtration test was carried out, confirming that all the activity was attributable only to the solid (Fig. S12†). The catalytic activity of TS-1_Aer (approximated by the kinetic constant k calculated by an initial rate analysis, Fig. 3a) is lower than that of TS-1. However, after normalization by the amount of TS-1 crystals to account for the presence of the silica binder (mass fraction of TS-1 in the materials is ca. 80 wt%), the two catalysts were shown to have roughly the same specific rate constant (ca. 2.5 × 10−5 s−1 (gTS-1 L−1)), showing that the intrinsic activity of TS-1 crystals in TS-1_Aer is fully preserved. This is correlated with the fact that the Ti speciation and catalyst crystallinity are completely maintained after aerosol processing. Besides, like TS-1, TS-1_Aer remained fully selective toward the epoxide product (no formation of glycerol after 3 h of reaction). A recyclability study showed that the activity was preserved over three consecutive tests (Fig. 3b).
 | ||
| Fig. 3 (a) Kinetic data for the Ti-catalyzed conversion of allyl alcohol into glycidol in H2O using aq. 30 wt% H2O2 as oxidizing agent. In inset is shown the corresponding initial rate analysis, (b) recyclability study of the TS-1 and TS-1_Aer catalysts. Data were collected in the initial stage of the reaction (45 min reaction time) and the catalytic activity (expressed in terms of glycidol production per g of catalyst) relative to the first test was calculated for each catalyst. Experimental conditions: T = 45 °C, [CATA] = 5 g L−1, [allyl alcohol] = 0.9 M, [H2O2] = 0.18 M. | ||
The enzyme was entrapped in TS-1_Aervia an impregnation-precipitation-cross-linking method, leading to the formation of cross-linked enzyme aggregates (CLEAs)68 inside of the hollow sphere particles (Scheme 2b, see also the detailed experimental procedure in ESI†). In this procedure, ethyl lactate is used as a precipitating agent, forcing the enzyme molecules to come closer to each other in the microparticles and allowing an efficient cross-linking upon the subsequent addition of glutaraldehyde. The obtained hybrid bifunctional catalyst was denoted “GOx_X@TS-1_Aer”, where X stands for the concentration of the enzymatic solution used during the impregnation step (in mg mL−1). The volume of solution that was used to impregnate the solid particles after elimination of the excess by centrifugation was in the 1.3–1.4 cm3 g−1 range, which is very close to the cumulative pore volume of ca. 1.3 cm3 g−1 estimated by N2-physisorption and Hg porosimetry. We therefore surmise that only a small fraction of the impregnation solution filled the interstitial voids between the packed spheres while most of the enzymes were inside the microsphere cavities (see below).
In the range of enzyme concentrations tested (i.e. 2.5–25 mg mL−1) – corresponding to nominal enzyme loadings of 5–50 mgGOx g−1 (one example of calculation is given in ESI†) – the immobilization yield was between 75 and 100%, as illustrated in Fig. S13.† This result shows that the final enzyme loading can be simply controlled by choosing the enzyme concentration in the solution. For example, the enzyme loading in GOx_25@TS-1_Aer was 37 mgGOx g−1 (vs. 50 mgGOx g−1 nominal loading). As a comparison, the enzyme loading in a material prepared without precipitation and cross-linking agents (GOx_25@TS-1_Aer_b, see ESI†) was 6 mgGOx g−1 only, showing that most of the enzyme was lost in the washing fractions in this case (Bradford assay). This result is a strong evidence of the major role of the precipitation and cross-linking steps on the heterogeneization of the enzyme and thus on the enzymatic activity of the hybrid material. Indeed, the curves in Fig. 4a show a ca. 8 times higher activity for GOx_25@TS-1_Aer compared to GOx_25@TS-1_Aer_b, consistent with the lower enzyme loading in the latter material. A filtration test on GOx_25@TS-1_Aer showed that more than 99% of the enzymatic activity could be assigned to the solid (Fig. S14†). Also, no leaching was detected even after prolonged storage of the catalyst, as verified by a Bradford test.
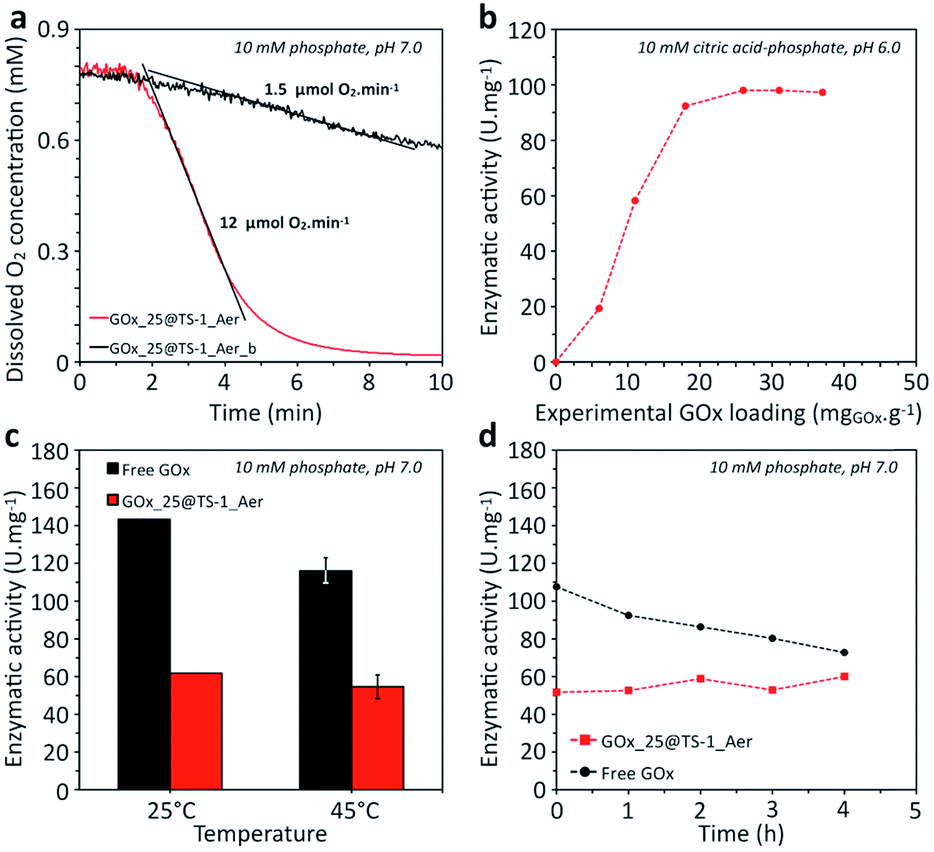 | ||
| Fig. 4 (a) Kinetic data for the GOx catalyzed consumption of oxygen using GOx_25@TS-1_Aer and GOx_25@TS-1_Aer_b (5 mg of solid catalyst in 50 mL), (b) comparison of the specific enzymatic activity in hybrid catalysts prepared with various enzyme loadings, (c) comparison of the specific enzymatic activity of free and immobilized GOx at 25 and 45 °C; the error bars represent standard deviations and are calculated on a triplicate, (d) stability against time at 45 °C of free and immobilized GOx. Experimental conditions: T = 45 °C, [D-glucose] = 200 mM. The specific enzymatic activity is expressed in U mg−1; the mass of GOx in the hybrid catalyst is deduced from the experimental enzyme loading. | ||
As opposed to the scenario where the enzyme would be grafted at the external surface of the zeolite, our method offers high flexibility in terms of enzyme loading because the loading can be tuned simply by adapting the enzyme concentration in the impregnation step. The hybrid catalysts prepared with various loadings of GOx reached different levels of specific enzymatic activity (Fig. 4b). The specific activity was constant at high loadings but dropped at low enzyme loading. Presumably, the cross-linking was too high in these solids, resulting in more constrained enzyme conformation with lower activity.
The activity of the immobilized enzyme in GOx_25@TS-1_Aer was compared to the free enzyme (Fig. 4c). The results showed a residual activity of ca. 50% for the immobilized GOx. This activity drop should be attributed to the cross-linking procedure which is known to lower the accessibility to individual active sites (using our kinetic modeling – ESI, Fig. S18† – lower k and higher kmO values were obtained for the aggregated enzymes as compared to the free enzyme). However, entrapped CLEAs were also shown to exhibit higher thermal stability as compared to the free enzyme. Indeed, the time-dependent evolution of the enzymatic activity upon incubation at 45 °C showed a decrease of the activity for the free enzyme (estimated half-life of ca. 8 h), whereas the immobilized enzyme showed no sign of deactivation in the same time interval (Fig. 4d).69
SEM analysis showed that the immobilization step did not affect the catalyst spherical morphology. Besides, the catalytic activity of GOx_25@TS-1_Aer for the Ti-catalyzed conversion of allyl alcohol showed only a ca. 20% decrease compared to TS-1_Aer (Fig. 3a). This slight drop possibly results from the mass fraction of organics (ca. 10% w/w, see Fig. S4†), including small enzyme aggregates, in the hybrid material. This is evidenced by the loss of textural properties in GOx_25@TS-1_Aer (external specific surface area = 130 m2 g−1; VP = 0.30 cm3 g−1; Vμ = 0.09 cm3 g−1; see Fig. 1c) as compared to TS-1_Aer.
In conditions depicted in Fig. 5 – without addition of H2O2 – the inorganic catalyst TS-1_Aer is totally inactive for the epoxidation reaction. With TS-1_Aer and free GOx (Fig. 5a), the chemo-enzymatic cascade reaction took place: without addition of H2O2, the epoxide was formed with a yield of 18% (relative to glucose) and 63% selectivity (as estimated by the mass balance, and further confirmed by HPLC). It should be reminded that the selectivity is governed by the instability of the epoxide in the reaction conditions; spontaneous ring-opening is inevitable for long reaction times. Switching to the true hybrid heterogeneous catalyst, GOx_25@TS-1_Aer (Fig. 5b), glycidol production reached a value of 37 mM – corresponding to a yield of 23% and a glucose conversion of 34% (69% selectivity for the epoxide). The slight increase in performance as compared to the case where the enzyme is free is possibly due to confinement effect, implying that the H2O2 concentration close to the zeolite nano-crystals is higher when using the hybrid and can be consumed faster. The reported glycidol production (mM range) is much higher than the traces reported for a simple grafting of the enzyme on the external surface of TS-1.31 Enzyme leaching was shown to be limited to less than 1% during the one-pot reaction, as demonstrated by a Bradford assay carried out on filtrated reaction medium (after 24 h). The reproducibility of the data presented in Fig. 5b was confirmed by synthesizing a new hybrid catalyst following the whole same protocol and performing the same reaction (Fig. S15†). In the repeated experiment, the glucose conversion reached 28%, whereas the epoxide yield was of 20% (73% selectivity). It can be observed, however, that the H2O2 production seems to peak at an early stage of the experiment. This occurs both for the free enzyme and for the hybrid. In fact, GOx is known to be irreversibly deactivated by its product H2O2,70 meaning that the epoxidation reaction is not fueled with additional H2O2 after a few hours of operation.
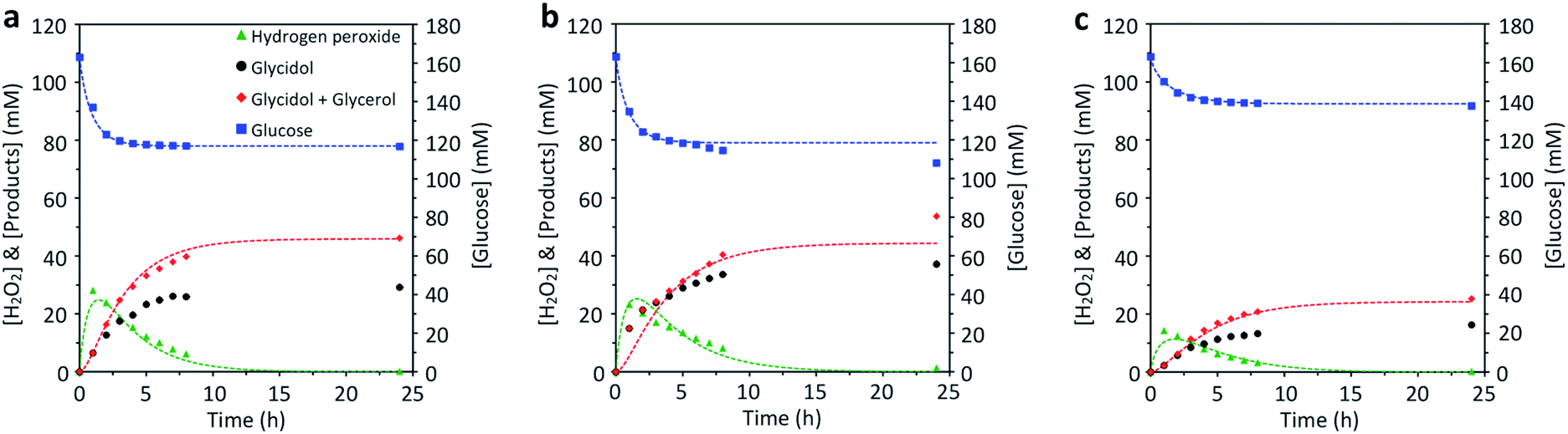 | ||
| Fig. 5 Chemo-enzymatic epoxidation of allyl alcohol with (a) free GOx and TS-1_Aer, (b) GOx_25@TS-1_Aer, and (c) GOx_2.5@TS-1_Aer. The amount of free enzyme in experiment (a) was adjusted so as to have the same enzymatic activity as in the hybrid GOx_25@TS-1_Aer catalyst (experiment (b)). Dashed lines represent a fitted mathematical model (see Table S1†). Experimental conditions: T = 45 °C, [D-glucose] = 0.16 M, [CATA] = 5 g L−1, [allyl alcohol] = 0.9 M, 24 h reaction time. | ||
The epoxide production obtained with the hybrid catalyst could be tuned by simply changing the enzyme loading in the hybrid catalyst. This is shown by testing the GOx_2.5@TS-1_Aer catalyst (Fig. 5c), the latter having a ca. 6 times lower enzyme loading (Fig. S13†) and ca. 5 times lower specific enzymatic activity (Fig. 4b) compared to GOx_25@TS1_Aer. In this case, only 16 mM of glycidol was formed after 24 h (10% yield, 64% selectivity); the glucose conversion was of 16%. The lack of proportionality between the intrinsic GOx activity in GOx_2.5@TS-1_Aer and GOx_25@TS-1_Aer and the corresponding final glucose conversion in the chemo-enzymatic reaction indicates that the conversion in the presence of the GOx_25@TS-1_Aer catalyst is mostly limited by the oxygen supply, suggesting that even higher epoxide production could be reached by improving the oxygen mass transfer rate in the medium.
In order to demonstrate the applicability of the present strategy to other enzymes, CLEAs of horseradish peroxidase (HRP) were entrapped in TS-1_Aer while keeping a nominal enzyme loading of 50 mg g−1 (HRP_25@TS-1_Aer). A colorimetric assay with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and H2O2 as substrates demonstrated that the HRP was successfully immobilized in the solid and remained active (Fig. 6, see also Fig. S6a–d†). Moving further, combi-CLEAs made of HRP and GOx (75:25 w/w, 50 mg g−1 nominal enzyme loading) could be effectively formed inside the microspheres (HRP/GOx_25@TS-1_Aer). Without external addition of H2O2, the reaction occurred, illustrating the possibility to carry out multi-enzymatic cascade reactions (here GOx produces H2O2 and HRP uses it to oxidize ABTS, see Fig. 6a and S6e–h†). These results confirm that the strategy presented here could be exploited to design multifunctional hybrid solids hosting multi-enzyme systems that could be applied in more complex cascade reactions or for example also as sensors and detection devices.
 | ||
| Fig. 6 Colorimetric assay of HRP using ABTS as substrate. (a) Kinetic data for the formation of ABTS+˙ catalyzed by HRP in HRP_25@TS-1_Aer and HRP/GOx_25@TS-1_Aer. Filtration tests proved that the enzymatic activity was located in the solid (see one example for HRP/GOx_25@TS-1_Aer in the figure, see also Fig. S6†). (b) Evolution of the color of the solution as a function of the production of ABTS+˙ catalyzed by HRP in HRP_25@TS-1_Aer. Experimental conditions: 10 mM citric acid-phosphate pH 6.0, tests carried out at room temperature (ca. 23 °C). The detailed experimental procedures are presented in the ESI.† | ||
Conclusions
In conclusion, we report an innovative and effective approach for combining zeolite and enzyme catalysts in the same bifunctional solid and for exploiting them in a one-pot cascade chemo-enzymatic reaction. Leveraging on the aerosol process, hollow zeolite microspheres can be shaped by the aggregation of crystalline nano-building blocks of TS-1 zeolites cemented by a thin porous silica layer. These one-pot generated hollow microspheres are used as cargos for glucose oxidase. The GOx enzyme is loaded in the microsphere and then trapped in the form of cross-linked enzyme aggregates, thereby providing a good control on the enzyme loading, promoting enzyme stability, and preventing enzyme leaching. When this hybrid catalyst is put at work, a cascade chemo-enzymatic reaction is demonstrated: GOx produces H2O2in situ and the latter is then used by TS-1 to catalyze the epoxidation of the olefin. The conversion and selectivity obtained with these hybrid catalysts are much higher than the traces of products obtained using a simple grafting on zeolite crystals. This study constitutes one of the rare examples of chemo-enzymatic reaction carried out with a single bifunctional hybrid catalyst, and more precisely the first example of an effective hybrid catalyst based on an enzyme and a zeolite. The catalyst preparation strategy based on the formation of hollow particles is allegedly applicable to other catalytic nano-building units (nanocrystals of zeolites, clays, MOFs, COFs, nanoparticles, etc.) to allow their combination with enzymes, multi-enzymes or other biomacromolecules, thus opening new opportunities for the preparation of multifunctional hybrid materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the ‘Communauté française de Belgique’ for the financial support through the ARC programme (15/20-069). F. Devred and E. Niyigena are acknowledged for the technical and logistical support. V. Smeets is thankful to F. R. S.–F. N. R. S. for his FRIA PhD grant. The authors thank the unité de chimie des nanomatériaux (UCNM) from UNamur for the access to UV-Vis equipment and Hg porosimeter. The authors thank the Francqui Foundation for the chair, which made it possible for Prof. Sanchez to visit Belgium. FEGSEM instrumentation was facilitated by the Institut des Matériaux de Paris Centre (IMPC FR2482) and was funded by UPMC, CNRS and by the C'Nano projects of the Région Ile-de-France. Prof. Patrick Gerin is gratefully acknowledged for providing access to the optical oxygen sensor.References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2009, 39, 301–312 RSC.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, 2007 Search PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12 CrossRef.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318–325 CrossRef.
- T. Görbe, K. P. J. Gustafson, O. Verho, G. Kervefors, H. Zheng, X. Zou, E. V. Johnston and J.-E. Bäckvall, ACS Catal., 2017, 7, 1601–1605 CrossRef.
- P. O'Brien, D. Lopez-Tejedor, R. Benavente and J. M. Palomo, ChemCatChem, 2018, 10, 4992–4999 CrossRef.
- H. Huang, C. A. Denard, R. Alamillo, A. J. Crisci, Y. Miao, J. A. Dumesic, S. L. Scott and H. Zhao, ACS Catal., 2014, 4, 2165–2168 CrossRef CAS.
- W. Zhang, E. Fernández-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55 CrossRef CAS PubMed.
- M. Frey, L. Seyidova, D. Richard and P. Fongarland, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.04.056.
- A. Gimbernat, M. Guehl, M. Capron, N. Lopes Ferreira, R. Froidevaux, J.-S. Girardon, P. Dhulster, D. Delcroix and F. Dumeignil, ChemCatChem, 2017, 9, 2080–2084 CrossRef CAS.
- F. Dumeignil, M. Guehl, A. Gimbernat, M. Capron, N. L. Ferreira, R. Froidevaux, J.-S. Girardon, R. Wojcieszak, P. Dhulster and D. Delcroix, Catal. Sci. Technol., 2018, 8, 5708–5734 RSC.
- P. N. R. Vennestrøm, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 249–258 CrossRef.
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC.
- M. Bohnet, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- S. T. Oyama, Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, Elsevier, 2011 Search PubMed.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71–83 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed.
- L. Cao and R. D. Schmid, Carrier-bound Immobilized Enzymes: Principles, Application and Design, Wiley-VCH, Weinheim, 1st edn, 2005 Search PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- C. Chan, L. Sepunaru, S. V. Sokolov, E. Kätelhön, N. P. Young and R. G. Compton, Chem. Sci., 2017, 8, 2303–2308 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- J. Huo, J. Aguilera-Sigalat, S. El-Hankari and D. Bradshaw, Chem. Sci., 2015, 6, 1938–1943 RSC.
- L. van den Biggelaar, P. Soumillion and D. P. Debecker, Catalysts, 2017, 7, 54 CrossRef.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- K. S. Rabe, J. Müller, M. Skoupi and C. M. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 13574–13589 CrossRef CAS PubMed.
- X. Lian, Y.-P. Chen, T.-F. Liu and H.-C. Zhou, Chem. Sci., 2016, 7, 6969–6973 RSC.
- M. Filice and J. M. Palomo, ACS Catal., 2014, 4, 1588–1598 CrossRef CAS.
- Y. Lin, L. Wu, Y. Huang, J. Ren and X. Qu, Chem. Sci., 2015, 6, 1272–1276 RSC.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C.-W. Tai and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006–14010 CrossRef PubMed.
- P. N. R. Vennestrøm, E. Taarning, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 943–945 CrossRef.
- X. Zhang, L. Jing, F. Chang, S. Chen, H. Yang and Q. Yang, Chem. Commun., 2017, 53, 7780–7783 RSC.
- X. Li, X. Cao, J. Xiong and J. Ge, Small, 2019 DOI:10.1002/smll.201902751.
- D. P. Debecker, Chem. Rec., 2018, 18, 662–675 CrossRef CAS PubMed.
- V. Smeets, C. Boissière, C. Sanchez, E. M. Gaigneaux, E. Peeters, B. F. Sels, M. Dusselier and D. P. Debecker, Chem. Mater., 2019, 31, 1610–1619 CrossRef CAS.
- G. Deo, A. M. Turek, I. E. Wachs, D. R. C. Huybrechts and P. A. Jacobs, Zeolites, 1993, 13, 365–373 CrossRef CAS.
- V. Smeets, L. Ben Mustapha, J. Schnee, E. M. Gaigneaux and D. P. Debecker, Mol. Catal., 2018, 452, 123–128 CrossRef CAS.
- M. G. Clerici, Kinet. Catal., 2015, 56, 450–455 CrossRef CAS.
- C. Pagis, A. R. Morgado Prates, D. Farrusseng, N. Bats and A. Tuel, Chem. Mater., 2016, 28, 5205–5223 CrossRef CAS.
- D. P. Debecker, S. Le Bras, C. Boissière, A. Chaumonnot and C. Sanchez, Chem. Soc. Rev., 2018, 47, 4112–4155 RSC.
- C. Boissiere, D. Grosso, A. Chaumonnot, L. Nicole and C. Sanchez, Adv. Mater., 2011, 23, 599–623 CrossRef CAS PubMed.
- D. P. Debecker, M. Stoyanova, F. Colbeau-Justin, U. Rodemerck, C. Boissière, E. M. Gaigneaux and C. Sanchez, Angew. Chem., Int. Ed., 2012, 51, 2129–2131 CrossRef CAS PubMed.
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- J. Zhou, Z. Hua, W. Wu, Z. Liu, Y. Zhu, Y. Chen and J. Shi, Dalton Trans., 2003, 2011(40), 12667–12669 Search PubMed.
- A. Dong, Y. Wang, Y. Tang, N. Ren, Y. Zhang and Z. Gao, Chem. Mater., 2002, 14, 3217–3219 CrossRef CAS.
- Z. Guo, G. Xiong, L. Liu, P. Li, L. Hao, Y. Cao and F. Tian, J. Porous Mater., 2016, 23, 407–413 CrossRef CAS.
- G. Majano, A. Restuccia, M. Santiago and J. Pérez-Ramírez, CrystEngComm, 2012, 14, 5985–5991 RSC.
- L.-H. Chen, X.-Y. Li, G. Tian, Y. Li, J. C. Rooke, G.-S. Zhu, S.-L. Qiu, X.-Y. Yang and B.-L. Su, Angew. Chem., Int. Ed., 2011, 50, 11156–11161 CrossRef CAS.
- N. Masoumifard, R. Guillet-Nicolas and F. Kleitz, Adv. Mater., 2018, 30, 1704439 CrossRef PubMed.
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, Microporous Mesoporous Mater., 2020, 293, 109801 CrossRef CAS.
- V. R. R. Marthala, M. Friedrich, Z. Zhou, M. Distaso, S. Reuss, S. A. Al-Thabaiti, W. Peukert, W. Schwieger and M. Hartmann, Adv. Funct. Mater., 2015, 25, 1832–1836 CrossRef CAS.
- A. Corma, V. Fornes and F. Rey, Adv. Mater., 2002, 14, 71–74 CrossRef CAS.
- C. Niu, M. Liu, X. Gao, X. Ye, Y. Wen, Y. Liu and X. Wang, ACS Omega, 2019, 4, 4397–4404 CrossRef CAS PubMed.
- R. A. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- H. Yamaguchi, Y. Kiyota and M. Miyazaki, Catalysts, 2018, 8, 174 CrossRef.
- D. Jung, M. Paradiso and M. Hartmann, J. Mater. Sci., 2009, 44, 6747 CrossRef CAS.
- J. Lee, J. Kim, J. Kim, H. Jia, M. I. Kim, J. H. Kwak, S. Jin, A. Dohnalkova, H. G. Park, H. N. Chang, P. Wang, J. W. Grate and T. Hyeon, Small, 2005, 1, 744–753 CrossRef CAS PubMed.
- A. Thangaraj, M. J. Eapen, S. Sivasanker and P. Ratnasamy, Zeolites, 1992, 12, 943–950 CrossRef CAS.
- B. M. Reddy, B. Chowdhury and P. G. Smirniotis, Appl. Catal., A, 2001, 211, 19–30 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- G. Berlier, V. Crocellà, M. Signorile, E. Borfecchia, F. Bonino and S. Bordiga, in Structure and Reactivity of Metals in Zeolite Materials, ed. J. Pérez Pariente and M. Sánchez-Sánchez, Springer International Publishing, Cham, 2018, pp. 91–154 Search PubMed.
- L. Marchese, E. Gianotti, V. Dellarocca, T. Maschmeyer, F. Rey, S. Coluccia and J. M. Thomas, Phys. Chem. Chem. Phys., 1999, 1, 585–592 RSC.
- K. Madhusudan Reddy, S. V. Manorama and A. Ramachandra Reddy, Mater. Chem. Phys., 2003, 78, 239–245 CrossRef.
- K. Thamaphat, P. Limsuwan and B. Ngotawornchai, Kasetsart J.: Nat. Sci., 2008, 42, 357–361 Search PubMed.
- S. Pega, C. Boissière, D. Grosso, T. Azaïs, A. Chaumonnot and C. Sanchez, Angew. Chem., Int. Ed., 2009, 48, 2784–2787 CrossRef CAS PubMed.
- A. Y. Khan, S. B. Noronha and R. Bandyopadhyaya, Biochem. Eng. J., 2014, 91, 78–85 CrossRef CAS.
- P. A. Webb, C. Orr and M. I. Corporation, Analytical methods in fine particle technology, Micromeritics Instrument Corporation, 1997 Search PubMed.
- R. Schoevaart, M. W. Wolbers, M. Golubovic, M. Ottens, A. P. G. Kieboom, F. van Rantwijk, L. a. M. van der Wielen and R. A. Sheldon, Biotechnol. Bioeng., 2004, 87, 754–762 CrossRef CAS PubMed.
- R. R. Nair, P. Demarche and S. N. Agathos, New Biotechnol., 2013, 30, 814–823 CrossRef CAS PubMed.
- K. Kleppe, Biochemistry, 1966, 5, 139–143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional figures, detailed description of the mathematical model. See DOI: 10.1039/c9sc04615a |
| This journal is © The Royal Society of Chemistry 2020 |