
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget-activated transcription for the amplified sensing of protease biomarkers†
Fang
Liu
a,
Min
Yang
a,
Wenlu
Song
a,
Xingyu
Luo
a,
Rui
Tang
a,
Zhixi
Duan
b,
Wenyuan
Kang
a,
Shiyi
Xie
a,
Qingqing
Liu
a,
Chunyang
Lei
*a,
Yan
Huang
 a,
Zhou
Nie
*a and
Shouzhuo
Yao
a
a,
Zhou
Nie
*a and
Shouzhuo
Yao
a
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan Provincial Key Laboratory of Biomacromolecular Chemical Biology, Hunan University, Changsha 410082, P. R. China. E-mail: niezhou.hnu@gmail.com; cylei@hnu.edu.cn
bDepartment of Orthopedics, The Second Xiangya Hospital, Central South University, Changsha 410011, Hunan, P. R. China
First published on 10th February 2020
Abstract
Signal amplification is an effective way to achieve sensitive analysis of biomarkers, exhibiting great promise in biomedical research and clinical diagnosis. Inspired by the transcription process, here we present a versatile strategy that enables effective amplification of proteolysis into nucleic acid signal outputs in a homogeneous system. In this strategy, a protease-activatable T7 RNA polymerase is engineered as the signal amplifier and achieves 3 orders of magnitude amplification in signal gain. The versatility of this strategy has been demonstrated by the development of sensitive and selective assays for protease biomarkers, such as matrix metalloproteinase-2 (MMP-2) and thrombin, with sub-picomole sensitivity, which is 4.3 × 103-fold lower than that of the standard peptide-based method. Moreover, the proposed assay has been further applied in the detection of MMP-2 secreted by cancer cells, as well as in the assessment of MMP-2 levels in osteosarcoma tissue samples, providing a general approach for the monitoring of protease biomarkers in clinical diagnosis.
Introduction
Sensitive analysis of nucleic acid and protein biomarkers is of vital importance in the fields of biomedical research and clinical diagnosis.1–4 Signal amplification has been proved to be an effective method to achieve high sensitivity.5–8 During the flow of genetic information within a biological system, DNA encoded information is effectively amplified by replication and transcription.9,10 Inspired by this naturally occurring process in organisms, many signal amplification techniques have been developed for the sensitive analysis of biomarkers. For instance, by mimicking the semiconservative replication of DNA, Mullis developed the polymerase chain reaction (PCR) that enables exponentially amplified detection of target nucleic acids based on thermal cycling.11,12 Owing to advantages such as good sensitivity, high specificity and easy accessibility, the PCR and its variants have become the most widely used technologies for the amplified detection of nucleic acids.13 However, transcription,14 the first step of gene expression, in which a segment of DNA is transcribed into multiple RNAs by RNA polymerase (RNAP), has been less utilized in exploiting in vitro signal amplification approaches compared to replication. Taking advantage of the primer-independent isothermal signal amplification mechanism in transcription, several studies have employed transcription for amplified detection of nucleic acids and small molecules by modulating the accessibility of the promoter sequence to RNAP.15–19 Despite the significance of this progress, the engineering of RNAP for signal transduction and amplification has not yet attracted enough attention. By engineering RNAP with target-responsive capacity, more widespread applications of transcription in developing effective signal amplification strategies can be envisaged.Proteases have profound implications in modulating the initiation, progression and metastasis of most cancers.20–22 For example, matrix metalloproteinases (MMPs) are involved in extracellular matrix degradation and are overexpressed in many solid tumours, including melanoma and colorectal, breast, lung, ovarian, and prostate cancers.23–25 Therefore, abnormally expressed proteases have been considered promising candidate targets to leverage in both cancer therapeutics and diagnostics.24,26 Immunoassays enable sensitive detection of protease biomarkers in biological samples, but usually suffer from false positive results caused by the inability of antibodies to distinguish proteases from their inactive precursors.27,28 Despite the fact that peptide-based fluorometric assays can avoid this issue, the lack of signal amplification mechanisms for each proteolysis event leads to unsatisfactory sensitivity.29–34 These limitations highlight the need for a sensitive assay giving unambiguous results that reflect the protease activities accurately. In synthetic biology, proteases from tobacco etch virus (TEV) and hepatitis C virus (HCV) are widely used as switch modules to regulate the transcription of reporter genes for the construction of biological circuits in live cells.35–37 Considering the connection between proteases and transcription in these processes, we envisaged amplification of the existence of protease biomarkers into multiple RNA outputs based on in vitro transcription.
To this end, a transcription-inspired signal amplification strategy that enables amplification of each proteolysis event into appreciable signals is required. On account of the inhibition of T7 RNA polymerase (T7 RNAP) by T7 lysozyme (T7 LYS),35,38–41 here we constructed a protease-activatable T7 RNAP (denoted as PR) by tethering the two proteins through a peptide linker containing the substrate sequence of a tumour-related protease. In the PR, T7 RNAP is in the inactive state, and its transcription activity is suppressed by the bound T7 LYS due to the formation of an intramolecular complex between them. Once the linker peptide is cleaved by the target protease, the intramolecular interaction between the two proteins is disrupted, thereby leading to the liberation of an active T7 RNAP and subsequent transcription (Scheme 1). The transcribed RNA is designed to form specific secondary structures (e.g., G-quadruplexes (G4))42 that can be lit up by specific organic fluorescent dyes.43,44 Taking advantage of the high efficiency of T7 RNAP-mediated transcription14 and the excellent specificity of G4 RNA–dye interaction,43 sensitive detection of the activity of target protease biomarkers is achieved based on the PR-based assay (PRA). Moreover, the PR can be readily fine-tuned by changing the linker peptide according to different proteases, which provides a versatile signal transduction and amplification strategy for the sensitive analysis of protease biomarkers, exhibiting promising potential in clinical diagnosis.
 | ||
| Scheme 1 Design and mechanism of the amplification of proteolysis into nucleic acid outputs. | ||
Results and discussion
To demonstrate the feasibility of the PR-based signal amplifier, tumour-related MMP-2 protease was chosen as the target model, and a PR responding to MMP-2 (PRMMP-2) was rationally designed. In the PRMMP-2, a catalytically inactivated T7 LYS was fused to the N-terminus of T7 RNAP by a flexible linker containing an MMP-2 cleavage site (GPL|GVRG).45 In order to investigate the effect of the linker on the interaction between T7 RNAP and T7 LYS, the structure of PRMMP-2 was subjected to molecular dynamics (MD) simulation to obtain the conformational stability of the protein. MD simulations were performed for up to 50 ns using GRO MACS 2016 (Fig. S1†). To describe the high amplitude concerted motion in a trajectory, principal components (PCs) extracted with a cosine content of 0.28, PC1 and PC2 for individual trajectories, were utilized to generate free energy landscape (FEL) maps.46 The coordinates from the FEL maps with a minimum energy cluster at 33.9 ns were used to retrieve the low energy representative structure (Fig. 1a). The retrieved structure of PRMMP-2 (blue in Fig. 1b) was highly similar to that of the T7 RNAP/T7 LYS complex without the linker (yellow in Fig. 1b),47 which theoretically revealed that the introduction of the linker has a negligible effect on the inhibition of T7 RNAP by T7 LYS.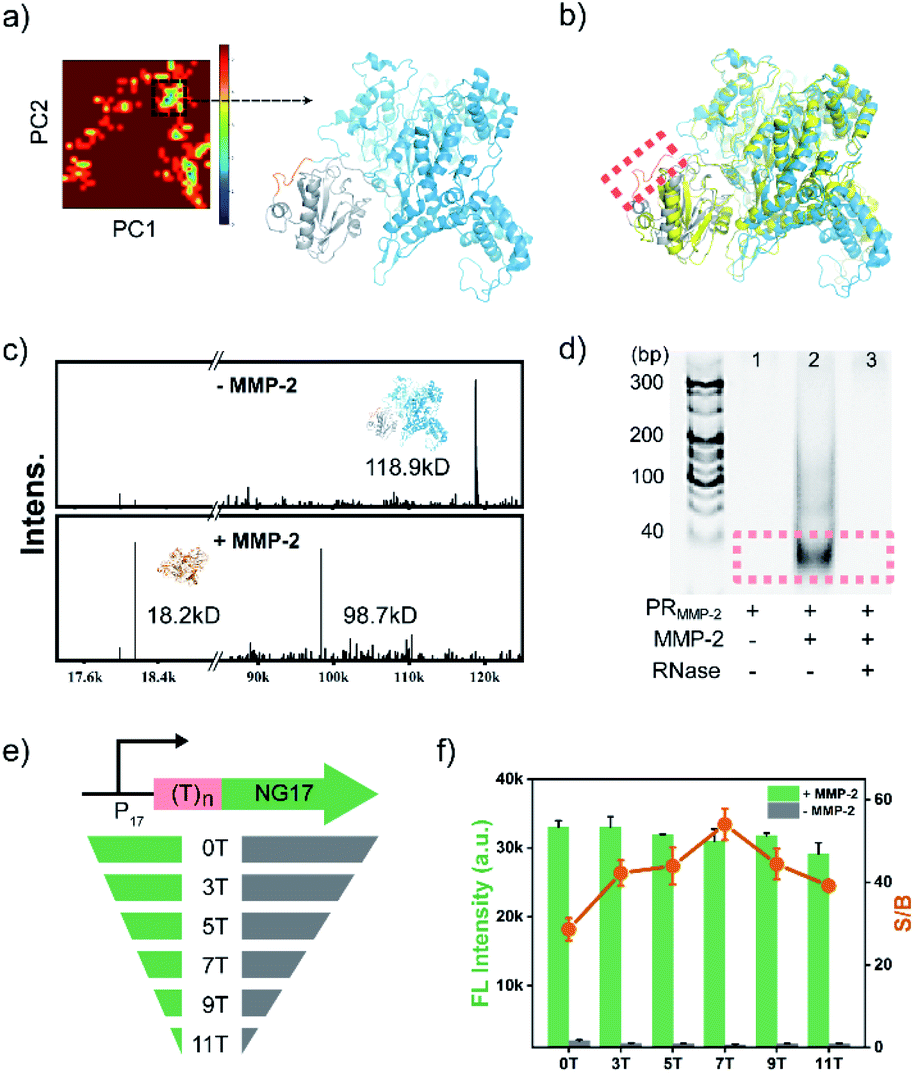 | ||
| Fig. 1 (a) Principal component analysis (PCA) depicting a low energy basin along with the representative structure. T7 RNAP and T7 LYS are coloured in gray and blue, respectively. (b) The structure of PRMMP-2 (blue) aligned with the FEL representatives of T7 RNAP/T7 LYS complex (code: 1ARO, shown in yellow). The linker is highlighted by the dashed box. (c) Characterization of the PRMMP-2 by MALDI-TOF MS. PRMMP-2, 7.5 μM and MMP-2, 500 nM. (d) Characterization of transcription products under different conditions by PAGE. All the samples were treated with DNase I (5 U) to digest the template DNA. (e) Schematic representation of a DNA template containing a spacer sequence with different lengths. (f) Effect of the length of the spacer sequence on the signal to background ratio. rNTP, 5.0 mM; DNA template, 1.0 μM; ThT, 2.0 μM; PRMMP-2, 0.75 μM; and MMP-2, 50 nM. | ||
Encouraged by the results of the computational assessment, the gene of PRMMP-2 was cloned and expressed in E. coli to obtain the PRMMP-2 fusion protein (Fig. S2†). The purified PRMMP-2 was incubated with MMP-2, and the enzymatic hydrolysates were analysed by SDS-PAGE and MALDI-TOF MS. Two new bands with faster migration rates appeared in the gel electrophoresis image (Fig. S3†). Moreover, new intense peaks at 18.2 kD and 98.7 kD that correspond to the molecular weights of T7 LYS and T7 RNAP proteins, respectively, were detected in the MS spectra (Fig. 1c). Therefore, these results confirmed the efficient cleavage of the PRMMP-2 protein by MMP-2. Then, we tested whether the MMP-2-mediated cleavage of PRMMP-2 can trigger the transcription activity of T7 RNAP. MMP-2-pretreated PRMMP-2 was mixed with the DNA template and rNTPs in transcription buffer, and the product was analysed by gel electrophoresis. The band of transcribed RNA was detected in the gel electrophoresis image, whereas there was no RNA band in the control group without the addition of MMP-2, suggesting the high-efficiency inhibition of T7 RNAP in PRMMP-2, as well as the effective activation of transcription activity by MMP-2 (Fig. 1d). Taken together, we have demonstrated that the protease activity of MMP-2 can be amplified into RNA outputs based on the designed PRMMP-2.
To convert the RNA outputs into detectable signals, the sequence of the transcribed RNA was designed as the counterpart of NG17 DNA,42 a G4 motif. Using Thioflavin T (ThT), a dye that enhances fluorescence more than 1000-fold upon binding to G4,43,44 as the indicator, the generation of transcribed RNA can be reflected by monitoring the green fluorescence of the system at 495 nm. On the basis of this design, the secondary structure of the transcribed RNA was characterized by circular dichroism (CD) spectroscopy. A positive band at 269 nm and a negative one at 243 nm were observed in the CD spectrum of transcribed RNA, which validated the formation of parallel G4 topologies48 (Fig. S4†). Having verified the formation of G4 RNA as designed, ThT was added to generate fluorescence signals. As shown in Fig. S5,† MMP-2 mediated cleavage induced a drastic fluorescence enhancement that was approximately 28-fold as high as that of the control group without MMP-2. These results suggested that a sensitive fluorometric assay for the detection of MMP-2 activity has been preliminarily established. However, the background fluorescence of the control was a bit higher than expected, which would have a negative effect on the detection sensitivity. Further study revealed that the background fluorescence was mainly due to the coexistence of PRMMP-2, template DNA and rNTPs (Fig. S6†). Previous studies have indicated that the T7 RNAP/T7 LYS complex can still bind to the T7 promoter sequence and generate abortive RNAs having a length of 8–10 nucleotides.44 Therefore, the PRMMP-2 could catalyse the generation of small fragments containing G-rich sequences, which are prone to forming intermolecular G4s and result in background fluorescence.49 To avoid the background fluorescence caused by the formation of intermolecular G4s, a spacer sequence of (dT)n was introduced between the promoter region and the NG17 region in the template DNA (Fig. 1e). As the length of the spacer increased from 0 to 7 nt, the background fluorescence decreased gradually, and the highest ratio of signal to background (S/B) was calculated to be 54 (Fig. 1f). Additionally, the effects of other parameters, such as the concentrations of PRMMP-2 and magnesium ions, were also investigated and optimized to improve the detection performance (Fig. S7 and S8†).
It is noteworthy that T7 RNAP-mediated RNA synthesis occurs with high efficiency,50,51 which means that the T7 RNAP molecule activated by each MMP-2-mediated cleavage enables the generation of multiple outputs of fluorescent RNA/dye complexes through transcription, thereby leading to an effective signal gain (Fig. 2a). In contrast, each MMP-2-mediated cleavage only could restore the emission of one fluorophore in the conventional peptide-based assay. Then, the fluorescence signals of the PRA and peptide-based assay (using the same substrate peptide as in PRMMP-2) in response to different concentrations of MMP-2 were investigated (Fig. S9 and S10†). Here, F/F0 is defined as the fluorescence response signal, where F and F0 are the fluorescence intensities of each system at the maximum emission wavelength in the presence and absence of MMP-2, respectively. The fluorescence response signals of 1.0 nM MMP-2 from the PRA and the peptide-based assay were determined to be 19.8 and 1.2 (Fig. 2b), respectively, demonstrating efficient signal gain in the PRA. For the PRA, a linear relationship between the fluorescence response signal and the logarithmic value of the MMP-2 concentration was obtained in the range from 0.3 pM to 7.0 nM (R2 = 0.990) with a limit of detection (LOD, 3σ) of 0.07 pM (Fig. 2c). In contrast, the linear range of the peptide-based assay was obtained in the range of 1.0–30 nM with an LOD of 0.3 nM. Therefore, the LOD of the PRA was about 4285-fold lower than that of the peptide-based assay, providing a much more sensitive method for the detection of MMP-2. Next, the specificity of the PRA toward MMP-2 was investigated by testing the potential interference from MMPs with similar substrate specificity in biological samples,52 including MMP-3, MMP-8 and MMP-9. The catalytic activities of the three MMPs were validated using commercialized assay kits (Fig. S11†). None of these competing MMPs were able to induce significant fluorescence signals even though higher concentrations of them were tested (Fig. S12†). Taken together, the results of these in vitro experiments suggested the good specificity of the PRA toward MMP-2.
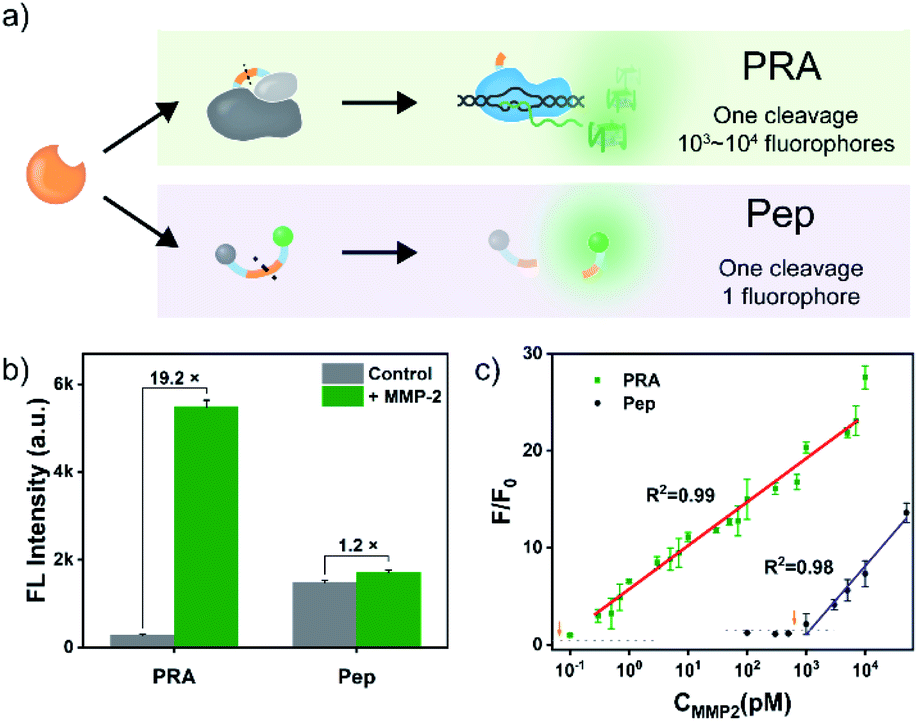 | ||
| Fig. 2 (a) Schematic presentation of the comparison of the PRA with the peptide-based assay (Pep) in response to MMP-2. (b) MMP-2 (1.0 nM) activated fluorescence responses in the PRA and the peptide-based assay (Pep), respectively. (c) Calibration curve of the PRA and Pep in response to MMP-2 at various concentrations. The dotted horizontal lines indicate the fluorescence signals for LOD estimation. | ||
A major advantage of the PRA, aside from the excellent sensitivity, is that its versatility is facile to implement by tuning the linker peptide in PR using the substrate of target proteases. Thrombin is a protease that plays a pivotal role in the process of thrombosis, platelet activation, and various cardiovascular diseases.53,54 To validate the versatility of PRA, a PRThr for thrombin was constructed by using a flexible peptide linker containing the thrombin cleavage site (LVPR|GS, Fig. S13†).54 The design was also verified as feasible by computational assessment (Fig. 3a), and a PRA for thrombin was proposed in the same way as for MMP-2. The PRA also showed high sensitivity (S/B = 45, Fig. 3b) and good specificity (Fig. S14 and S15†) toward thrombin, demonstrating the generality of the PRA in protease biomarker sensing.
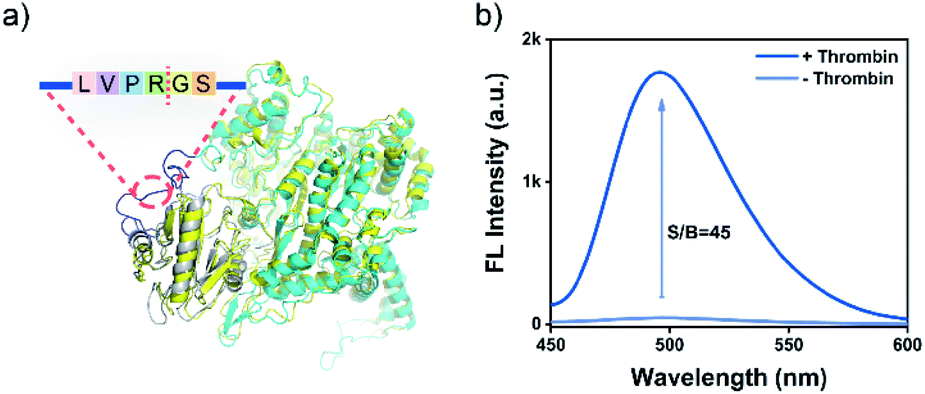 | ||
| Fig. 3 (a) The structure of PRThr (cyan) aligned with the PCA representatives of the T7 RNAP/T7 LYS complex (code: 1ARO, yellow). (b) Fluorescence emission spectra for the PRThr-based PRA in the presence or absence of thrombin (50 nM). PRThr, 0.75 μM; rNTP, 5.0 mM; DNA template, 1.0 μM; and ThT, 2.0 μM. | ||
Next, the practical performance of the PRA in the detection of proteases in biological samples was assessed. To this end, the PRA was employed to determine the activities of MMP-2 secreted by different cells. Human hepatocyte cells (LO2) and human hepatoma carcinoma cells (HepG2) were cultured in 6-well plates at different cell amounts (10 to 104 cells), and activities of secreted MMP-2 in the culture medium were analysed by the PRA (Fig. 4a). As shown in Fig. 4b, there was a strong correlation between the fluorescence response and the amount of HepG2 cells, and the signal of MMP-2 secreted by 10 cancer cells could be detected. In contrast, negligible fluorescence responses were observed in the group of LO2 cells. These results indicated high levels of MMP-2 secreted by HepG2 cells, which agreed well with the levels of MMP-2 secreted from the two cell lines quantified by western blotting (Fig. 4c) and the peptide-based assay (Fig. S16†).
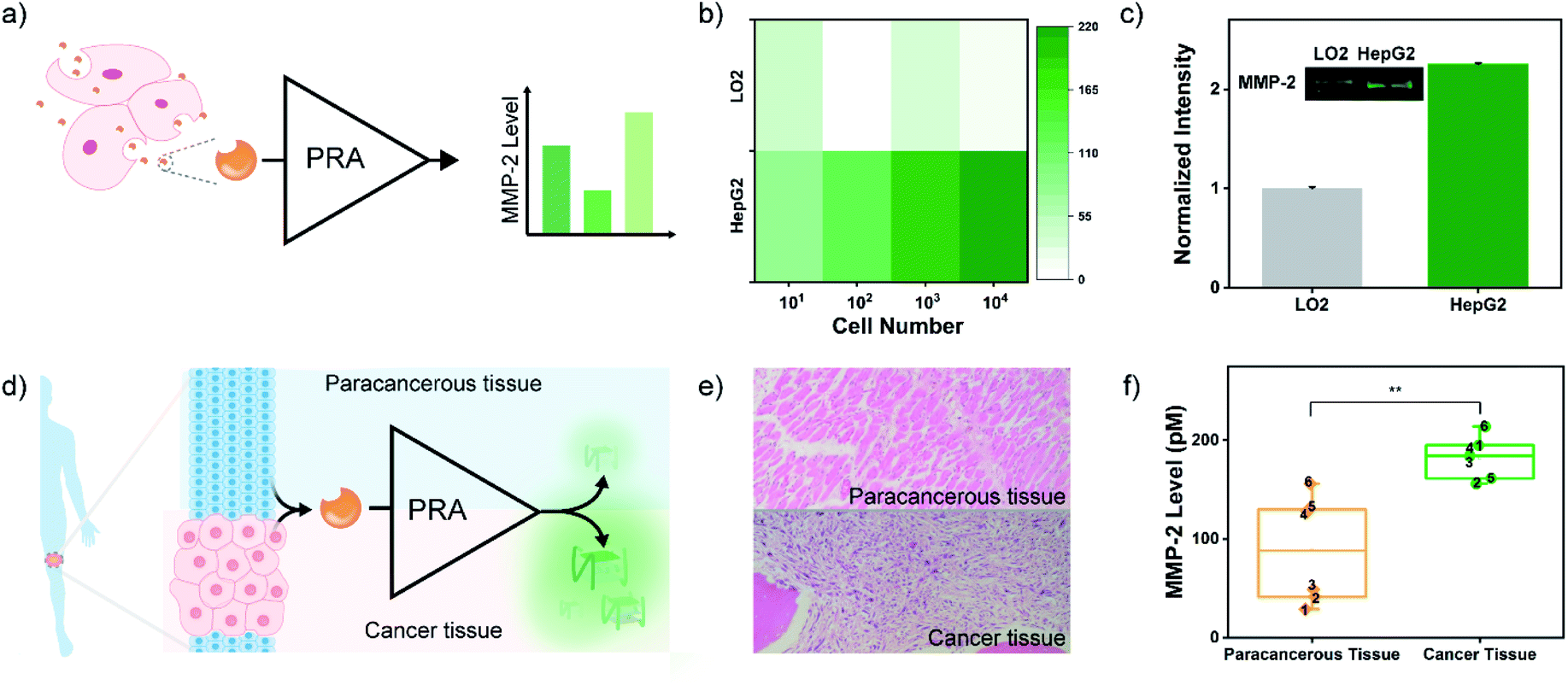 | ||
| Fig. 4 (a) Schematic of the measurement process of MMP-2 activity in a cell culture supernatant. (b) Heat map of the fluorescence response of cell culture supernatants at different numbers of LO2 and HepG2, respectively. (c) Western blot assay of secreted MMP-2 in the culture medium of LO2 and HepG2 cells. (d) Scheme of the assessment of MMP-2 activity in clinical tissue samples by the PRA. (e) Pathological sections of paracancerous tissue and cancer tissue from one patient with osteosarcoma. Original magnification, ×200. (f) MMP-2 levels in the OS and paracancerous tissue samples from 6 OS patients determined by the PRA. The concentration of total protein is 10 mg mL−1. **p < 0.01. | ||
Osteosarcoma (OS) is the most common primary malignant bone cancerous tumour in children and adolescents.55 OS has high malignant and metastatic potentials, and MMP-2 is one of the most vital enzymes deeply involved in metastasis of the cancer cells.56 Thus, determination of MMP-2 levels in OS tissues will help understand the process of cellular motility and invasion and migration of cancer cells (metastasis), thereby improving the prognosis and long-term survival. Herein, the MMP-2 levels in the OS tissues and paracancerous tissues of 6 OS patients were analysed by the PRA with the approval of the local institutional review boards (Fig. 4d). Crude proteins were extracted from OS and paracancerous tissues that were pathologically diagnosed (Fig. 4e). And the expressions of MMP-2 in these samples were confirmed by western blotting (Fig. S17†). Then, MMP-2 activities of the samples were evaluated by the PRA. As shown in Fig. 4f and S18,† elevated fluorescence signals of MMP-2 activity were detected in all cancer tissues compared to paracancerous tissues. However, some paracancerous tissues also exhibited a high level of MMP-2 activity, which might be associated with the high metastatic potential of OS,55 implying the potential of the PRA in the early diagnosis of metastasis.
Conclusions
In summary, by engineering protease-activatable T7 RNAP (PR) as the signal amplifier to transduce and amplify proteolysis into multiple RNA outputs, we developed a PR-based assay (PRA) for rapid and ultrasensitive detection of protease activity. Taking advantage of the effective transcription catalysed by T7 RNAP, the versatility of the PRA has been demonstrated not only in the monitoring of MMP-2 secreted by cancer cells, but also in the assessment of MMP-2 levels in OS tissues, exhibiting promising applications in clinical diagnosis. Given the predictable hybridization and programmable design nature of nucleic acids, the output RNA transduced by PR can be further coupled with aptamers, dynamic DNA/RNA nanotechnology, and even a CRISPR/Cas system.57–60 Therefore, aside from the fields of bioassay and biosensing, the potential applications of the proposed signal amplifier in intelligent response-type molecule recognition, nucleic acid circuits and gene editing, are also worth exploring.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21725503, 21974038, 21575038 and 21675044), the Young Top-notch Talent for Ten Thousand Talent Program, and the Fundamental Research Funds for the Central Universities.Notes and references
- L. Wu and X. Qu, Chem. Soc. Rev., 2015, 44, 2963–2997 RSC.
- R. Qian, Y. Cao, L. Zhao, Z. Gu and Y. Long, Angew. Chem., Int. Ed., 2017, 56, 4802–4805 CrossRef CAS PubMed.
- S. J. Kim, S. J. Choi, J. S. Jang, H. J. Cho and I. D. Kim, Acc. Chem. Res., 2017, 50, 1587–1596 CrossRef CAS PubMed.
- L. Gao, L. Li, X. Wang, P. Wu, Y. Cao, B. Liang, X. Li, Y. Lin, Y. Lu and X. Guo, Chem. Sci., 2015, 6, 2469–2473 RSC.
- C. Li, W. Liu, J. Hu and C. Zhang, Chem. Sci., 2019, 10, 8675–8684 RSC.
- M. Liu, Q. Zhang, D. Chang, J. Gu, J. D. Brennan and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6142–6146 CrossRef CAS PubMed.
- R. Duan, X. Lou and F. Xia, Chem. Soc. Rev., 2016, 45, 1738–1749 RSC.
- M. S. Reid, X. C. Le and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 11856–11866 CrossRef CAS PubMed.
- M. J. Davey and M. O'Donnell, Curr. Opin. Chem. Biol., 2000, 4, 581–586 CrossRef CAS.
- B. Alberts, Nature, 2003, 421, 427–431 CrossRef PubMed.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CrossRef CAS PubMed.
- M. Rahman, M. Uddin, R. Sultana, A. Moue and M. Setu, Anwer Khan Mod. Med. Coll. J., 2013, 4, 30–36 CrossRef.
- F. Liu, H. Liu, Y. Liao, J. Wei, X. Zhou and D. Xing, Biosens. Bioelectron., 2015, 74, 778–785 CrossRef CAS PubMed.
- G. L. Hager, J. G. McNally and T. Misteli, Mol. Cell, 2009, 35, 741–753 CrossRef CAS PubMed.
- C. Y. Yu, B. C. Yin, S. Wang, Z. Xu and B. C. Ye, Anal. Chem., 2014, 86, 7214–7218 CrossRef CAS PubMed.
- X. Li, F. Zheng and R. Ren, Chem. Commun., 2015, 51, 11976–11979 RSC.
- K. Zhang, K. Wang, X. Zhu and M. Xie, Chem. Commun., 2017, 53, 5846–5849 RSC.
- J. Cao, Y. Yao, K. Fan, G. Tan, W. Xiang, X. Xia, S. Li, W. Wang and L. Zhang, Sci. Adv., 2018, 4, eaau4602 CrossRef CAS PubMed.
- Z. Wang, Y. Luo, X. Xie, X. Hu, H. Song, Y. Zhao, J. Shi, L. Wang, G. Glinsky, N. Chen, R. Lal and C. Fan, Angew. Chem., Int. Ed., 2018, 57, 972–976 CrossRef CAS PubMed.
- M. Lee, R. Fridman and S. Mobashery, Chem. Soc. Rev., 2004, 33, 401 RSC.
- S. D. Mason and J. A. Joyce, Trends Cell Biol., 2011, 21, 228–237 CrossRef CAS PubMed.
- M. Vizovisek, R. Vidmar, M. Drag, M. Fonovic, G. S. Salvesen and B. Turk, Trends Biochem. Sci., 2018, 43, 829–844 CrossRef CAS PubMed.
- C. Chang and Z. Werb, Trends Cell Biol., 2001, 11, S37–S43 CrossRef CAS PubMed.
- R. Roy, J. Yang and M. A. Moses, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- A. I. Anzellotti and N. P. Farrell, Chem. Soc. Rev., 2008, 37, 1629–1651 RSC.
- J. Mu, F. Liu, M. S. Rajab, M. Shi, S. Li, C. Goh, L. Lu, Q. H. Xu, B. Liu, L. G. Ng and B. Xing, Angew. Chem., Int. Ed., 2014, 53, 14357–14362 CrossRef CAS PubMed.
- D. Martinez, M. Munera, J. F. Cantillo, J. Wortmann, J. Zakzuk, W. Keller, L. Caraballo and L. Puerta, Int. J. Mol. Sci., 2019, 20, 3025 CrossRef CAS PubMed.
- D. Feng, F. Tian, W. Qin and X. Qian, Chem. Sci., 2016, 7, 2246–2250 RSC.
- J. W. Choi, H. Lee, G. Lee, Y. R. Kim, M. J. Ahn, H. J. Park, K. Eom and T. Kwon, Theranostics, 2017, 7, 2878–2887 CrossRef CAS PubMed.
- E. J. Kwon, J. S. Dudani and S. N. Bhatia, Nat. Biomed. Eng., 2017, 1, 0054 CrossRef CAS PubMed.
- T. Ma, Y. Hou, J. Zeng, C. Liu, P. Zhang, L. Jing, D. Shangguan and M. Gao, J. Am. Chem. Soc., 2018, 140, 211–218 CrossRef CAS PubMed.
- H. Bui, C. W. Brown 3rd, S. B. White, S. A. Diaz, M. H. Stewart, K. Susumu, E. Oh, M. G. Ancona, E. R. Goldman and I. L. Medintz, Small, 2019, 15, e1805384 CrossRef PubMed.
- L. Yin, H. Sun, H. Zhang, L. He, L. Qiu, J. Lin, H. Xia, Y. Zhang, S. Ji, H. Shi and M. Gao, J. Am. Chem. Soc., 2019, 141, 3265–3273 CrossRef CAS PubMed.
- B. C. Dickinson, M. S. Packer, A. H. Badran and D. R. Liu, Nat. Commun., 2014, 5, 5352 CrossRef CAS PubMed.
- J. Pu, I. Chronis, D. Ahn and B. C. Dickinson, J. Am. Chem. Soc., 2015, 137, 15996–15999 CrossRef CAS PubMed.
- B. C. Dickinson, Nat. Methods, 2018, 15, 489–490 CrossRef CAS PubMed.
- B. A. Moffatt and F. W. Studier, Cell, 1987, 49, 221–227 CrossRef CAS PubMed.
- X. Cheng, X. Zhang, J. W. Pflugrath and F. W. Studier, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4034–4038 CrossRef CAS PubMed.
- D. Jeruzalmi and T. A. Steitz, J. Mol. Biol., 1997, 274, 748–756 CrossRef CAS PubMed.
- X. Zhang and F. W. Studier, J. Mol. Biol., 1997, 269, 10–27 CrossRef CAS PubMed.
- V. Kuryavyi, L. A. Cahoon, H. S. Seifert and D. J. Patel, Structure, 2012, 20, 2090–2102 CrossRef CAS PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- Z. Liu, X. Luo, Z. Li, Y. Huang, Z. Nie, H. H. Wang and S. Yao, Anal. Chem., 2017, 89, 1892–1899 CrossRef CAS PubMed.
- Z. Wang, X. Li, D. Feng, L. Li, W. Shi and H. Ma, Anal. Chem., 2014, 86, 7719–7725 CrossRef CAS PubMed.
- S. Borkotoky and A. Murali, BMC Struct. Biol., 2017, 17, 7 CrossRef PubMed.
- D. Jeruzalmi and T. A. Steitz, EMBO J., 1998, 17, 4101–4113 CrossRef CAS PubMed.
- A. C. Bhasikuttan and J. Mohanty, Chem. Commun., 2015, 51, 7581–7597 RSC.
- J. Zhu, L. Zhang, S. Dong and E. Wang, Chem. Sci., 2015, 6, 4822–4827 RSC.
- S. Ulrich and E. T. Kool, Biochemistry, 2011, 50, 10343–10349 CrossRef CAS PubMed.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D. K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- U. Eckhard, P. F. Huesgen, O. Schilling, C. L. Bellac, G. S. Butler, J. H. Cox, A. Dufour, V. Goebeler, R. Kappelhoff, U. A. D. Keller, T. Klein, P. F. Lange, G. Marino, C. J. Morrison, A. Prudova, D. Rodriguez, A. E. Starr, Y. Wang and C. M. Overall, Matrix Biol., 2016, 49, 37–60 CrossRef CAS PubMed.
- L. Lin, S. Liu, Z. Nie, Y. Chen, C. Lei, Z. Wang, C. Yin, H. Hu, Y. Huang and S. Yao, Anal. Chem., 2015, 87, 4552–4559 CrossRef CAS PubMed.
- H. T. Ta, N. Arndt, Y. Wu, H. J. Lim, S. Landeen, R. Zhang, D. Kamato, P. J. Little, A. K. Whittaker and Z. P. Xu, Nanoscale, 2018, 10, 15103–15115 RSC.
- M. Kansara, M. W. Teng, M. J. Smyth and D. M. Thomas, Nat. Rev. Cancer, 2014, 14, 722–735 CrossRef CAS PubMed.
- M. H. Chien, W. J. Lee, Y. C. Yang, P. Tan, K. F. Pan, Y. C. Liu, H. C. Tsai, C. H. Hsu, Y. C. Wen, M. Hsiao and K. T. Hua, Cancer Lett., 2018, 433, 86–98 CrossRef CAS PubMed.
- M. Kushwaha, W. Rostain, S. Prakash, J. N. Duncan and A. Jaramillo, ACS Synth. Biol., 2016, 5, 795–809 CrossRef CAS PubMed.
- M. Li, M. Zheng, S. Wu, C. Tian, D. Liu, Y. Weizmann, W. Jiang, G. Wang and C. Mao, Nat. Commun., 2018, 9, 2196 CrossRef PubMed.
- J. Valero, N. Pal, S. Dhakal, N. G. Walter and M. Famulok, Nat. Nanotechnol., 2018, 13, 496–503 CrossRef CAS PubMed.
- J. D. Munzar, A. Ng and D. Juncker, Chem. Soc. Rev., 2019, 48, 1390–1419 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04692e |
| This journal is © The Royal Society of Chemistry 2020 |